
Abstract
The transcriptome from lupulin glands and associated bracts from cone tissue of hop (Humulus lupulus) c.v. ‘Cascade’, during three stages of development: early, mid, and late or near-harvest, was sequenced. Significant increases were found in expression patterns of many genes involved in the biosynthesis of bitter acids, xanthohumol, and volatile secondary metabolites or “hop oils” during the middle stage of cone development. The biosynthesis of thiol precursors responsible for popular “tropical fruit” flavors in beer is not well known, but homologs of genes hypothesized to be involved in this process tend to be up-regulated during the late stage in hop cones. More research needs to be performed to describe the pathway of thiol precursor biosynthesis in hops. Hierarchical clustering revealed overlap of samples taken from each developmental stage, likely due to non-uniform ripening of cones on the plant. It is proposed that the mid-stage of cone development is critical for the development of important flavor-producing secondary metabolites in hops, and this is supported by previous research describing concentrations of secondary metabolites. It is hypothesized that abiotic stress during the mid-stage of cone development may be quite detrimental to the bitter acid concentrations ultimately found in hops.
Introduction
Hop cones provide flavoring properties to beer, an industry valued at $116 billion in the United States in 2019. Hop flavors are derived from secondary metabolites produced in modified glandular trichomes and stored in the lupulin glands of the cones. They are usually categorized as resins or bitter acids, phenolics, thiol precursors, and volatile terpenoids or “essential oils.” Considerable work has been put into understanding the biosynthesis pathways of these compounds and while gaps still exist (e.g., thiol precursors), we now have a solid understanding of the genes involved in biosynthesis of hop secondary metabolites and their regulatory components ().
Figure 1. A schematic diagram of the known genes within the pathways for biosynthesis of important flavor compounds in hop. Question marks denote pathways that are yet unproven in the literature to our knowledge.
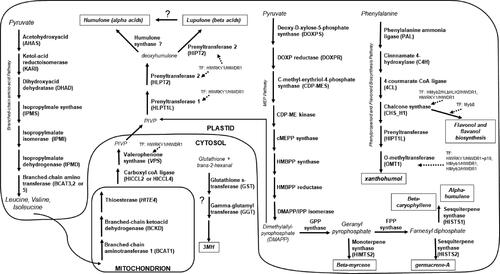
One major flavor component provided by hop cones is bitterness. This flavor is derived from the isomerization of alpha acids during the wort boil. Alpha and beta acids are classified as soft resins for their solubility in hexane,[Citation1] and consist of two groups of analogues (alpha acids: humulone, cohumulone, and adhumulone; beta acids: lupulone, colupulone, and adlupulone) with acyl side chains that differ according to their branched chain amino acid (BCAA) derivative (i.e., leucine, isoleucine, and valine). The BCAAs are derived from pyruvate via the chloroplastic BCAA biosynthesis pathway.[Citation2] The pathway producing isoleucine involves two enzymes (acetolactate synthase (ALS) and 2,3-dihydroxyisovalerate dehydratase (DHAD)), while the pathway to produce valine involves an intermediary step with an additional enzyme (ketol-acid reductoisomerase (KARI). The pathway to produce leucine involves the same pathway as valine but includes four additional enzymes: isopropylmalate synthase (IPMS), isopropylmalate dehydratase (IPMD), and isopropylmalate dehydrogenase (IPMDH). The final step in the BCAA biosynthesis pathway is catalyzed by the enzyme branched-chain amino transferase 2, 3, or 5 (BCAT2, 3, or 5). For bitter acid biosynthesis, the BCAAs are broken down in the mitochondria by a branched-chain amino transferase 1 (BCAT1)[Citation3] and a branched-chain keto-acid dehydrogenase (BCKDH), and finally to a branched-chained fatty acid by a thioesterase (HlTE4).[Citation4] This branched-chain fatty acid is transported to the cytosol by an unknown mechanism, where it is re-esterified by a carboxyl-CoA ligase (HlCCL2 or HlCCL4)[Citation4] and then converted by the enzyme valerophenone synthase (VPS) into phlorisovalerophenone (PIVP).[Citation5–7] The methyl-D-erythritol 4-phosphate (MEP) pathway in the chloroplast provides an alternative pathway for PIVP synthesis, however the mevalonate (MVA) pathway does not provide substantial PIVP.[Citation8–10] The PIVP is moved to the chloroplast via an unknown transporter mechanism, and prenylated by two membrane-associated prenyltransferases (HlPT-1/HlPT1L and HlPT-2)[Citation11–13] to the bitter acid precursor. In the final step of alpha acid synthesis, a deoxyhumulone hydroxylase, or humulone synthase, is thought to convert the alpha acid precursor to one of the alpha acid analogues. To our knowledge, this step remains unconfirmed, though there is evidence that an enzyme is involved,[Citation14] some authors have found putative proteins identified as humulone synthase using mass spectrometry,[Citation15] and two putative humulone synthase sequences from H. lupulus have been published in GenBank (KJ398144.1, KJ398145.1). It is not clear, however, if the GenBank sequences identified as putative humulone synthases are, in fact, involved in alpha acid biosynthesis. In the final step of beta acid synthesis, the precursor is prenylated a third time (HlPT-2).[Citation13] Some authors depict the biosynthesis of alpha acids as arising from beta acids,[Citation16] and de Keukeleire et al.[Citation17] find a decline in beta acid content near harvest that coincides with a rise in alpha acid content. This conversion of beta acids to alpha acids, however, also remains unconfirmed to our knowledge.
Other important compounds from the hop cones are prenylated flavonoids. Xanthohumol is the most common prenylflavonoid in cone lupulin glands, ranging from 0.2 to 1.1%[Citation17] depending on the cultivar. For biosynthesis of xanthohumol, the amino acid phenylalanine is converted to p-coumaroyl-CoA by the actions of phenylalanine ammonia-lyase or PAL1 and cinnamate 4-hydroxylase or C4H. The product p-coumaroyl-CoA is ligated by the enzyme 4-coumarate-CoA ligase (4CL), and then chalcone synthase (CHS_H1)[Citation7] catalyzes the condensation of p-coumaroyl-CoA and malonyl-CoA to naringenin chalcone. The open-ring configuration of this compound is stabilized by a chalcone isomerase-like protein (CHIL2),[Citation18] while the same prenyltransferase involved in bitter acid production, HlPT1L,[Citation18] converts chalconaringenin to desmethylxanthohumol (DMX). DMX is methylated by an o-methyltransferase (OMT1)[Citation10] to xanthohumol. Matousek et al. have established that the gene CHS_H1 is positively regulated by a MBW transcription factor complex made of proteins Myb2, bHLH2, and WDR1.[Citation19] The promoter region for OMT1 is activated by transcription factors HlWRKY1,[Citation20] HlMyb1,[Citation21] and s-HlMyb3,[Citation22] especially in combination with HlWDR1. Kocábek et al. found a homologous Myb8 transcription factor has a role in diverting metabolite flux away from prenylated flavonoids and toward biosynthesis of other flavonoids (such as quercetin and leucocyanidin).[Citation16]
Volatile terpenoids, isoprenoids, or “hop oils” include hundreds of characterized molecules, which in total represent 0.1 − 2% v/w of dried hop cones.[Citation23] Some of these compounds impart “floral,” “citrus,” or “piney” flavors on their own or as a result of the activity of yeast enzymes during fermentation.[Citation24] Most of the volatile terpenoids are monoterpene or sesquiterpene compounds that may be oxygenated, non-oxygenated hydrocarbons, or they may be sulphinated.[Citation25] These volatiles include the monoterpene myrcene, the sesquiterpenes alpha-humulene, beta-caryophyllene, monoterpene alcohols such as linalool, limonene and geraniol,[Citation24] and approximately 200 other compounds.[Citation1,Citation26] These compounds are produced from precursors derived from the MEP pathway and are then converted to geranyl diphosphate (GPP) by GPP synthase. Geranyl diphosphate is in turn converted to the monoterpene beta-myrcene by a monoterpene synthase (MTS2),[Citation9] or GPP is converted to farnesyl diphosphate (FPP) by squalene/phytoene synthase or farnesyl-PP synthase,[Citation27] and thence to humulene and caryophyllene by sesquiterpene synthase 1 (HlSTS1).[Citation9] Oxygenated volatile secondary metabolites include the monoterpene alcohols such as linalool, which are formed from GPP by S-linalool synthase.[Citation28]
Volatile thiols are considered responsible for popular “tropical fruit,” “black-currant,” “grapefruit,” or “passion fruit” flavors in beer. These compounds are synthesized during the fermentation process by yeast beta-lyase from non-volatile glutathionylated or cysteinylated 4-methyl-4-mercaptopental-2-one (4MMP or 4MSP), and glutathionylated or cysteinylated 3-mercaptohexan-1-ol (3MH or 3SH),[Citation29–31] and have a very low perception threshold in water (approximately 0.07 ng/L).[Citation32] The majority of the work on these compounds has been conducted in grapes and wine, but the precursors of thiol compounds have been identified in hop cones.[Citation33] Nearly all of the precursors identified have been 3MH conjugates, though some cultivars contain small amounts of glutathionylated-4MMP, and fewer contain cysteinylated-4MMP.[Citation33] ‘Cascade’ is known to contain the precursors cysteinylated-3MH, gluathionylated-3MH, as well as a small amount of cysteinylated-4MMP.[Citation33,Citation34] The biosynthesis of these precursor compounds in plants is not well understood.[Citation35] In grapes, it is hypothesized that biosynthesis occurs in the cytosol from the conjugation of glutathione and 2-hexenal via glutathione S-transferase (GST),[Citation36,Citation37] followed by conversion to S-3-(hexan-1-ol)-cysteine by membrane-associated gamma-glutamyl transferase carboxypeptidases (GGT).[Citation37]
Advantage was taken of the advanced state of understanding of the biosynthesis of agronomically important secondary metabolites in hops to track gene expression in cone lupulin glands and bracts. Cones were collected from cv. USDA Cascade at three time periods during the growing season (early, mid, late), and the transcriptome was sequenced from a collection of tissue including lupulin glands, bracts, and bracteoles. Critical periods of gene expression in the biosynthesis pathways of agronomically important secondary metabolites are described.
Experimental methods
Collection
Cones were collected from a single hill of cv. ‘Cascade’ growing at the USDA-ARS Hop Research fields in Corvallis, Oregon, U.S.A. Early cones were collected on August 3, 2018. Mid stage cones were collected two weeks later in mid-August, and late-stage cones were collected at harvest in late August. Cones were removed from the plant, wrapped in aluminum foil, and flash frozen in the field using liquid nitrogen before being returned and stored at −80 °C until processing.
RNA isolation and sequencing
To isolate RNA, a single bracteole with associated lupulin glands was ground in liquid N. Extraction buffer (2.42% w/v tris base, 1.27% w/v lithium chloride, 0.37% w/v EDTA, 1.5% w/v N-lauroylsarcosine, 1.0% w/v sodium dodecyl sulfate, 1.0% w/v deoxycholic acid) was added (600 µL) with beta-mercaptoethanol (31 µL per sample) and immediately vortexed. The samples were frozen at −80° C for 30 min, then thawed at 35-40 °C to ensure cell breakage. Potassium acetate (210 µL, 8.5 M and pH 6.0 − 6.5) was added and mixed by inverting. The tubes were incubated on ice for 15 min, then centrifuged at 1,950 x g for 5 min, then again at 12,200 x g for 5 min. The supernatant was transferred to new tubes and 500 µL TRIzol reagent was added. The tubes were vortexed and incubated at room temperature for 5 min. Chloroform (300 µL) was added, and the sample was mixed by inversion and centrifuged at 12,000 x g for 10 min at 4 °C. The tubes were incubated on ice for 3 min to maximize chloroform separation, and the aqueous layer was transferred into a new 2 mL tube. Room temperature isopropyl alcohol (0.7 volumes) was added, and the tube was mixed by inversion and incubated at −20 °C for 2+ hours. The tubes were centrifuged at 12,200 x g for 10 min, and the pellet was then washed twice with 80% ethanol, air dried fully, and re-suspended with 50 µL RNA-free water. For separation of DNA and RNA, 2 µL of 8 M LiCl (for a 2 M final concentration) was added, and the tubes were stored at 4 °C overnight. Tubes were then centrifuged at 12,200 x g for 10 min, the supernatant was discarded, and the pellet was washed with 80% ethanol. This step was repeated twice. The cleaned and dry pellet was resuspended in 20 µL formamide. Samples were tested for integrity and concentration on an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, U.S.A.).
For samples with RIN (RNA Integrity Numbers) values > 8, the TruSeq SR Cluster Kit v3-cBot-HS (Illumina, San Diego, CA, U.S.A.) was used to prepare 27 paired-end libraries. There are nine replicates for each development stage. Sequencing of 150 bp paired-end libraries took place on one lane on an Illumina HiSeq 3000 at the Oregon State University Center for Genome Research and Biocomputing.
RNA-seq alignment, transcript assembly, and preparation for differential expression
RNA-seq reads with cutadapt version 1.15[Citation38] were trimmed, requiring a minimum read length of 20 bp. Reads were aligned to the Cascade reference genome[Citation39] with STAR 2.7.1a.[Citation40] A genome assembly index with STAR “genomeGenerate” was built and then the reads were aligned to the reference assembly of Cascade with STAR “alignReads”.
The RNA-seq alignments were assembled into transcripts with StringTie v1.3.3b,[Citation41] using a merged set of de novo transcripts from cone, leaf, meristem, and stem, to guide the transcript assembly. To create the de novo merged reference transcript assembly of the four tissues, the first step was to create a de novo transcript assembly from all of the cone samples, and then a merge of this de novo cone transcript assembly with the de novo assembly of leaf, meristem, and stem tissues described in Padgitt-Cobb et al.[Citation39] The cuffmerge[Citation42] was used to merge all of the transcript assemblies. The goal was to maximize the detection of cone-specific transcripts that might not be present in other tissues. The “prepDE.py” script from StringTie was used to prepare the transcripts for differential expression with DESeq2.[Citation43] Commands from the pipeline can be found at the GitHub project page: https://github.com/padgittl/CascadeTimeCourse.
Data analysis
Functional annotations were established using Mercator4v.2.[Citation44] Many genes within the biosynthesis pathways for secondary metabolites have been experimentally confirmed and sequences from hop are available in the literature and on GenBank (https://www.ncbi.nlm.nih.gov/). In these cases, the transcript database was manually curated. Mercator4v2 annotations were confirmed for important secondary metabolite biosynthesis genes using a reverse BLAST against Genbank to confirm the gene identity. Differential gene expression analysis was conducted with DESeq2,[Citation43] and differentially expressed genes were defined as having an FDR adjusted p-value < 0.05, and a log-fold change value >2 or < −2. Normalized read counts were calculated using DESeq2, and heatmaps were created using ggplot2 in R v. 3.6.1 using scaled average values of the normalized read counts per cone developmental stage. Hierarchical clustering of samples was performed with FPKM values for each sample.
Results
A total of 362,769,990 reads from 27 libraries, with GC content of 46.05% on average, were sequenced (). An average of 13.44 reads per million were mapped, and the average uniquely mapped read percentage for all samples was 65.6%.
Table 1. Sequencing results of 27 libraries, showing the total number of raw input reads, the average length of mapped reads, as well as the percent of reads that were uniquely mapped, mapped multiple times, the percent of the total reads that were mapped to the reference genome, and the percent GC content of the library.
Some of the most abundant transcripts in the database appeared to encode ribosomal RNAs, a component of alpha-tubulin, the nuclear encoded small subunit of ribulose bisphosphate carboxylase (Rubisco), an adenosylmethione decarboxylase involved in cellular growth, a chloroplastic polyphenol oxidase, a number of components of the light-harvesting complex II (LHC-II), a fructose 1,6-bisphosphate aldolase, and an ATP-dependent Rubisco activase. Also among the most abundant transcripts was an o-methyltransferase (OMT) with strong similarity to OMT5, which has been suggested to be involved in lignin biosynthesis in hops.[Citation10] A transcript that was highly expressed during the early stage only, with no apparent expression in the mid or late stage, had strong similarity (e-value = 0) to a novel transcribed region from Arabidopsis thaliana (AT3G06365.1), which was down-regulated under osmotic stress.[Citation45]
Likely due to non-uniform ripening among cones on the bine, there was considerable overlap among development stages. There were 376 differentially expressed genes (DEGs) between the early and mid-stage of cone development (104 were up-regulated, 272 down-regulated), and 1,896 DEGs between the mid to late-stage (968 were up-regulated, 928 were down-regulated).
Expression of genes involved in bitter acid biosynthesis
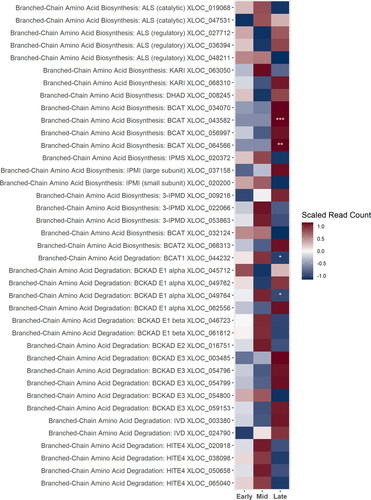
Genes involved in the branched-chain amino acid (BCAA) biosynthesis pathway were not strongly up-regulated during the early stage of cone development. More genes were up-regulated during the mid-stage, or alternatively during the late stage (). The gene putatively identified as BCAT1 involved in BCAA degradation in the mitochondria preceding bitter acid biosynthesis was up-regulated during the mid-stage, as were several genes putatively involved in the BCKAD complex. All genes putatively identified as HlTE4 responsible for converting the degraded BCAA to a branched-chain fatty acid for transport to the cytosol were also up-regulated during the mid-stage ().
Figure 2. A heatmap of gene expression patterns scaled across development stages for putative homologs of genes within the branched-chain amino acid (BCAA) biosynthesis and degradation pathways. The order of the genes from top to bottom represents the order of the pathway from the biosynthesis of the BCAAs to their degradation in the mitochondria as the precursors to the bitter acids. Gene acronyms are defined in . The XLOC numbers refer to the gene sequence available on hopbase.org. There are multiple XLOC numbers for many genes because there are multiple copies of the gene, errors in the assembly, or uncertainty in the annotation. Asterisks indicate expression levels are significantly different from the previous developmental stage with a BH-adjusted p-value < 0.05. * = logfold-change > 2 or < -2; ** = logfold-change > 4 or < -4; *** = logfold-change > 6 or < -6.
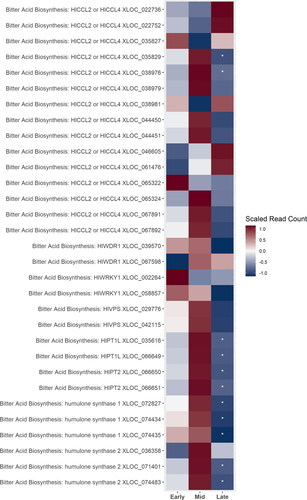
Most of the genes putatively involved in bitter acid synthesis were up-regulated during mid-stage cone development (). These included most of the genes putatively identified as HlCCL2 or 4, VPS, the two prenyltransferases, and the putative humulone synthases. Most of these genes were then strongly down-regulated during the late stage, though some transcripts putatively identified as CCL2 or 4 are up-regulated during the late stage. The genes identified as transcription factors for VPS and the prenyltransferases were up-regulated during the early- and mid-stage ().
Figure 3. A heatmap of gene expression patterns scaled across development stages for putative homologs of genes within the bitter acid biosynthesis pathway. The order of the genes from top to bottom represents the order of the pathway from the degradation of the BCAAs in the cytosol to the formations of the bitter acids in the plastid. Gene acronyms are defined in . The XLOC numbers refer to the gene sequence available on hopbase.org. There are multiple XLOC numbers for some genes because there are multiple copies of the gene, errors in the assembly, or uncertainty in the annotation. Asterisks indicate expression levels are significantly different from the previous developmental stage with a BH-adusted p-value < 0.05. * = logfold-change > 2 or < -2; ** = logfold-change > 4 or < -4; *** = logfold-change > 6 or < -6.
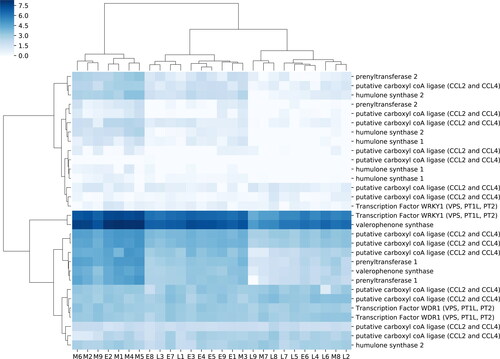
Hierarchical clustering of samples within the bitter acid synthesis pathway shows a strong pattern linking a VPS transcript and a WRKY1 transcription factor across samples, though the other transcription factors in the complex cluster with putative CCL2 or CCL4 transcripts (), which are involved in re-esterifying the degraded BCAA in the cytosol before conversion to PIVP by VPS. Clustering by samples (i.e., bracts and lupulin glands from a single cone) revealed significant overlap of the developmental stage. For example, the smallest cluster contained six samples from the mid-stage of cone development, and one sample from the early stage. Another cluster contained seven samples from the early stage, and two samples from the late stage of development. The final cluster contained seven samples from the late stage, two from the mid stage, and one from the early stage ().
Figure 4. A heatmap of gene expression patterns based on FPKM with hierarchical clustering of expression based on sample and transcript.
Expression of genes involved in prenylated flavonoid biosynthesis
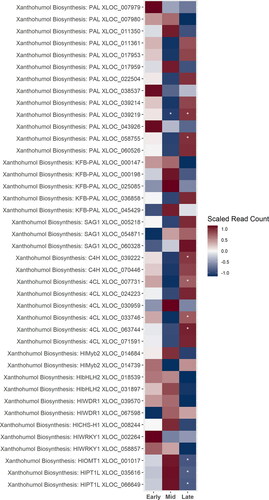
The final stages of xanthohumol biosynthesis include the hop-specific genes CHS_H1, HlOMT, and HlPT1L; these transcripts were up-regulated during the mid-stage of cone development, and the transcription factor complex for CHS_H1 and OMT1 was up-regulated during both the early- and mid-stages (). The pattern was less clear for the first steps in the pathway of xanthohumol biosynthesis, perhaps because of the greater number of putative homologs of the genes involved in p-coumaroylCoA biosynthesis. There are three putative PAL genes that were strongly up-regulated during the early-stage, another three that were up-regulated during the mid-stage, and seven that were up-regulated during the late-stage. Most of the genes that putatively code for Kelch-repeat F-box (KFB) proteins that act to post-translationally modify PAL by targeting it for degradation[Citation46] were up-regulated during the mid-stage.
Figure 5. A heatmap of gene expression patterns scaled across development stages for putative homologs of genes within the xanthohumol biosynthesis pathway. The order of the genes from top to bottom represents the order of the pathway from the initial conversion of phenylalanine to the biosynthesis of xanthohumol. Gene acronyms are defined in . The XLOC numbers refer to the gene sequence available on hopbase.org. There are multiple XLOC numbers for many genes because there are multiple copies of the gene, errors in the assembly, or uncertainty in the annotation. Asterisks indicate expression levels are significantly different from the previous developmental stage with a BH-adusted p-value < 0.05. * = logfold-change > 2 or < -2; ** = logfold-change > 4 or < -4; *** = logfold-change > 6 or < -6.
Expression of genes involved in volatile compounds
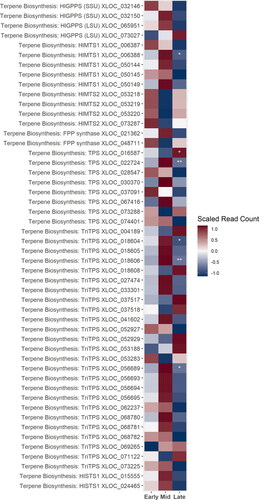
The volatile terpenoids or “hop oils” are synthesized from compounds derived from the plastidic MEP pathway. The MEP pathway was largely up-regulated during the mid-stage, though some copies of DXS and DXR were up-regulated during the early-stages. Likewise, many of the genes putatively identified as terpene synthases were up-regulated during mid-stage, including copies of HlGPPS, MTS1, FPP synthases, and various other non-specific terpene synthases genes. Interestingly, however, the gene responsible for synthesis of beta-myrene, MTS2, was up-regulated during the early stage ().
Figure 6. A heatmap of gene expression patterns that were significantly different in at least one development stage scaled across treatments for putative homologs of genes within the volatile secondary metabolites, or “hop oil” biosynthesis pathway. Not all putative homologs of genes within these pathways are shown for simplicity. XLOC numbers refer to the gene sequence available on hopbase.org. Asterisks indicate expression levels are significantly different from the previous developmental stage with a BH-adusted p-value < 0.05. * = logfold-change > 2 or < -2; ** = logfold-change > 4 or < -4; *** = logfold-change > 6 or < -6.
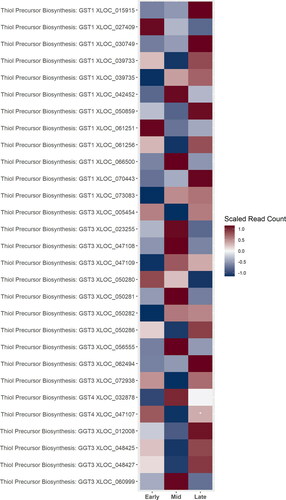
The genes involved in the biosynthesis of the thiol precursors are not well understood. Hop sequence homologs of genes putatively identified as involved in 3MH biosynthesis in grapes were identified, and there was no strong pattern in expression patterns based on the cone development stage. Many of the genes putatively identified as GGT3 were up-regulated during the late stage of cone development ().
Figure 7. A heatmap of gene expression patterns scaled across development stages for putative homologs of genes within the thiol precursor biosynthesis pathway. Gene acronyms are defined in . XLOC numbers refer to the gene sequence available on hopbase.org. Asterisks indicate expression levels are significantly different from the previous developmental stage with a BH-adusted p-value < 0.05. * = logfold-change > 2 or < -2; ** = logfold-change > 4 or < -4; *** = logfold-change > 6 or < -6.
Discussion
The expressed genes were sequenced along a time series based on cone development to track the expression of genes involved in the biosynthesis of important components of cone chemistry. Many genes involved in the biosynthesis of bitter acids and prenylated flavonoids were up-regulated during the mid-stage of cone development. Likewise, many genes involved in the biosynthesis of volatile secondary metabolites or “hop oils” were up-regulated during the mid-stage. This mid-stage of cone development thus appears to be the most critical period for production of important flavor compounds in hop cones. There was, however, some overlap in the clustering patterns of samples from each stage, demonstrating staggered ripening of individual cones on a single plant.
De Keukeleire et al. sampled cones for secondary metabolite content and identified 3 to 4 stages depending on the cultivar.[Citation17] Stage 1 cones were taken when the florescence first appeared on August 8, and stage 2 cones were taken approximately 10 days later. In some cultivars, they identified two different sized cones, which they distinguished as two distinct stages. The final stage cones were collected at harvest. They found alpha acids, beta acids, and prenylated flavonoids (desmethylxanthohumol and xanthohumol) in small concentrations at the onset of florescence, and beta acid concentrations tended to be higher than alpha acid concentrations. All compounds increased significantly during the middle stage, and in cultivars where they identified two different size classes at this period, secondary metabolite concentration increased significantly with the size of the cone. Increases in gene expression patterns during the mid-stage found in this study are corroborated by the chemistry results from de Keukeleire et al. that demonstrated increases in bitter acid content and prenylated flavonoids during the mid-stage of cone development.
Valerophenone synthase (VPS) is responsible for the conversion of the degraded BCAA to PIVP[Citation5–7] in the cytosol. Expression of VPS has been shown to correlate with total bitter acid content,[Citation47] and the gene is therefore considered a critical step within the biosynthesis pathway for bitter acids. Expression of VPS increases during the mid-stage of cone development, and it declines during the late stage, though the difference is not significant among samples. It is believed that this lack of statistical significance is due to staggered ripening of cones from different parts of the bine. A strong increase in expression patterns of the VPS transcription factor HlWRKY1[Citation20] was found during the early stage of cone development, preceding increases in VPS expression, and strong clustering of these two genes in our clustering analysis.
De Keukeleire et al.[Citation17] found an increase in alpha acid concentration and prenylated flavonoid concentrations at harvest, however they also found a small but significant decrease in beta acid concentrations. Close to harvest, we found a significant decrease in the expression levels of the gene putatively involved in the final step in alpha acid synthesis (the putative humulone synthase), which is not consistent with the observed increases in alpha acid concentrations.[Citation17] The protein concentrations or activity were not measured, and therefore our data are not conclusive, however it suggests that the genes identified as putative humulone synthases were not actually involved in (or are not the only mechanism involved) in the final stages of alpha acid biosynthesis. The gene(s) involved in the final step of alpha acid synthesis have not yet been verified experimentally to our knowledge; the sequences used to identify the humulone synthase were from the unpublished data available on NCBI GenBank, and it is not clear if they are, in fact, involved in alpha acid biosynthesis.
Nagel et al. found CHI-like (CHIL) transcripts were the most abundant EST in libraries from glandular trichomes.[Citation9] These proteins stabilize the open-ring formation of desmethylxanthohumol (DMX) and naringenin chalcone to enhance CHS activity, and CHIL2 was shown to increase DMX production by 2.3-fold.[Citation18] Our study sequenced lupulin glands, and also bract and bracteole tissue, and consequently identified a number of transcripts coding for LHC-II components, the nuclear-encoded small subunit of Rubisco, and Rubisco activase among the most abundant transcripts. In our dataset, the most abundant transcript had a strong similarity to a ribosomal RNA from Arabidopsis thaliana (AT3G41768.1). Chalcone isomerase-like transcripts were not among the most expressed transcripts in our dataset. Eight transcripts were identified that were significant matches for CHIL1 and CHIL2 (GenBank MG324004.1 and MG324005.1), and expression levels were similar for both transcripts. The highest expression of these occurred during the mid-stage development period, along with expression for other genes involved in xanthohumol biosynthesis.
The thiol-precursor molecules were lyased during fermentation by yeast enzymes to produce popular flavors in beer. The biosynthesis of these precursor molecules in hops is not yet understood but hop homologs of the genes putatively involved in thiol precursor biosynthesis in grapes were up-regulated during the late stage of cone development. Many of these genes are involved in abiotic and biotic stress response,[Citation36,Citation48] and could be up-regulated in response to high temperature and desiccation stress as the cone matures and dries. This could suggest that thiol-precursor concentrations could be highly dependent on harvest time, and premature harvest could result in low precursor concentrations, however further work should be conducted to identify the hop genes within this biosynthesis pathway.
There are many putative genes within the BCAA biosynthesis and degradation, flavonoid biosynthesis, and volatile secondary metabolite biosynthesis pathways that are up-regulated during the late stage. For some of these transcripts, particularly for genes that have a large number of putative transcripts annotated as that gene name, it could represent an incorrect annotation or assembly error. For example, there are 15 transcripts that are annotated as members of the CCL family (either CCL2 or CCL4), which are involved in re-esterifying the degraded branched chain amino acids in the cytosol. Five of these transcripts are up-regulated in the late stage of cone development, while eight are up-regulated in the mid stage, and two are up-regulated during the early stage. All of these transcripts were confirmed as CCL2 or CCL4 by individual reverse best BLAST searches, but it seems unlikely that ‘Cascade’ has 15 copies of these two genes. Our annotation and/or assembly was likely too conservative, in this case, and included other members of the CCL gene family, or multiple transcripts that should be collapsed into a single transcript. Likewise, a number of transcripts annotated as phenylalanine ammonia lyase (PAL), involved in flavonoid biosynthesis, are up-regulated at various stages of cone development. These transcripts were annotated based on homology to Arabidopsis thaliana sequences, and due to evolutionary divergence, likely captures other members of this gene family that have different functions in hop cones. However, without experimental evidence to confirm the in vitro function of these transcripts, we chose to present all transcripts with putative homology to genes in these pathways. In these cases, the conclusion was that the mid stage of cone development was critical for bitter acid and prenylated flavonoid biosynthesis based largely on the expression patterns in transcripts identified by homology to published hop genes (ex. HlVPS, HlOMT1, HlPT1L, and HlPT2), and we are therefore more confident in their annotation.
Conclusion
It was found that gene expression patterns for most of the genes involved in important secondary metabolites that produce flavor compounds in hops peaked during the mid-stage of cone development. In Oregon, U.S.A., this corresponded to about mid-August. Cone chemistry analysis from previous studies suggested that concentrations of these secondary metabolites also rise during this stage. This mid-stage of cone development, then, is a critical period for the production of flavor compounds in hops, though there was evidence that development is not uniform throughout the plant. This suggests that secondary metabolite concentrations are most sensitive to conditions such as low-water stress and high temperatures that cause declines in expression[Citation49] at any period of development, but particularly during this mid stage period. High temperature stress is difficult to avoid in the field, however low-water stress should be minimized in irrigated systems during this period.
Acknowledgments
RLE was supported by the Brewers Association and Washington State Department of Agriculture’s Specialty Crop Block Grant Program (K2298). LKPC is supported by an AFRI Predoctoral Fellowship (grant no. 2020-67034-31722) from the USDA National Institute of Food and Agriculture. We thank Jared Powell for help in the field. We thank Daniel Moore, Vicky Hollenbeck, and Nanci Adair of USDA-ARS for help optimizing RNA extractions and collecting samples. We thank Andrew Black of Oregon State University for bioinformatics help.
Disclosure statement
The authors have no known conflicts of interest.
Data availability statement
The data will be published at hopbase.org upon publication.
Additional information
Funding
Literature cited
- Neve, R. A. Hops; Chapman and Hall: London, 1991.
- Binder, S.; Knill, T.; Schuster, J. Branched-Chain Amino Acid Metabolism in Higher Plants. Physiol. Plant. 2007, 129, 68–78. DOI: 10.1111/j.1399-3054.2006.00800.x.
- Clark, S. M.; Vaitheeswaran, V.; Ambrose, S. J.; Purves, R. W.; Page, J. E. Transcriptome Analysis of Bitter Acid Biosynthesis and Precursor Pathways in Hop (Humulus lupulus). BMC Plant Biol. 2013, 13, 12. DOI: 10.1186/1471-2229-13-12.
- Xu, H.; Zhang, F.; Liu, B.; Huhman, D. V.; Sumner, L. W.; Dixon, R. A.; Wang, G. Characterization of the Formation of Branched Short-Chain Fatty Acid:CoAs for Bitter Acid Biosynthesis in Hop Glandular Trichomes. Mol. Plant. 2013, 6, 1301–1317. DOI: 10.1093/mp/sst004.
- Novák, P.; Krofta, K.; Matoušek, J. Chalcone Synthase Homologues from Humulus lupulus: Some Enzymatic Properties and Expression. Biologia Plant. 2006, 50, 48–54. DOI: 10.1007/s10535-005-0073-y.
- Okada, Y.; Ito, K. Cloning and Analysis of Valerophenone Synthase Gene Expressed Specifically in Lupulin Gland of Hop (Humulus lupulus L.). Biosci. Biotechnol. Biochem. 2001, 65, 150–155. DOI: 10.1271/bbb.65.150.
- Okada, Y.; Sano, Y.; Kaneko, T.; Abe, I.; Noguchi, H.; Ito, K. Enzymatic Reactions by Five Chalcone Synthase Homologs from Hop (Humulus lupulus L.). Biosci. Biotechnol. Biochem. 2004, 68, 1142–1145. DOI: 10.1271/bbb.68.1142.
- Goese, M.; Kammhuber, K.; Bacher, A.; Zenk, M. H.; Eisenreich, W. Biosynthesis of Bitter Acids in hops. A (13)C-NMR and (2)H-NMR study on the building blocks of humulone. Eur. J. Biochem. 1999, 263, 447–454. DOI: 10.1046/j.1432-1327.1999.00518.x.
- Wang, G.; Tian, L.; Aziz, N.; Broun, P.; Dai, X.; He, J.; King, A.; Zhao, P. X.; Dixon, R. A. Terpene Biosynthesis in Glandular Trichomes of Hop. Plant Physiol. 2008, 148, 1254–1266. DOI: 10.1104/pp.108.125187.
- Nagel, J.; Culley, L. K.; Lu, Y.; Liu, E.; Matthews, P. D.; Stevens, J. F.; Page, J. E. EST Analysis of Hop Glandular Trichomes Identifies an O-Methyltransferase that Catalyzes the Biosynthesis of Xanthohumol. Plant Cell. 2008, 20, 186–200. DOI: 10.1105/tpc.107.055178.
- Tsurumaru, Y.; Sasaki, K.; Miyawaki, T.; Momma, T.; Umemoto, N.; Yazaki, K. An Aromatic Prenyltransferase-like Gene HlPT-1 Preferentially Expressed in Lupulin Glands of Hop. Plant Biotechnol. 2010, 27, 199–204. DOI: 10.5511/plantbiotechnology.27.199.
- Tsurumaru, Y.; Sasaki, K.; Miyawaki, T.; Uto, Y.; Momma, T.; Umemoto, N.; Momose, M.; Yazaki, K. HlPT-1, a Membrane-Bound Prenyltransferase Responsible for the Biosynthesis of Bitter Acids in Hops. Biochem. Biophys. Res. Commun. 2012, 417, 393–398. DOI: 10.1016/j.bbrc.2011.11.125.
- Li, H.; Ban, Z.; Qin, H.; Ma, L.; King, A. J.; Wang, G. A Heteromeric Membrane-Bound Prenyltransferase Complex from Hop Catalyzes Three Sequential Aromatic Prenylations in the Bitter Acid Pathway. Plant Physiol. 2015, 167, 650–659. DOI: 10.1104/pp.114.253682.
- Fung, S.; Zuurbier, K. W. M.; Paniego, N. B.; Scheffer, J. J. C.; Verpoorte, R. Conversation of Deoxyhumulone into the Hop Alpha-Acid Humulone. Phytochemistry 1997, 44, 1047–1053. DOI: 10.1016/S0031-9422(96)00671-1.
- Champagne, A.; Boutry, M. A Comprehensive Proteome Map of Glandular Trichomes of Hop (Humulus lupulus L.) Female Cones: Identification of Biosynthetic Pathways of the Major Terpenoid-Related Compounds and Possible Transport Proteins. Proteomics 2017, 17, 1–10. DOI: 10.1002/pmic.201600411.
- Kocábek, T.; Mishra, A. K.; Matoušek, J.; Patzak, J.; Lomnická, A.; Khare, M.; Krofta, K. The R2R3 Transcription Factor HlMYB8 and Its Role in Flavonoid Biosynthesis in Hop (Humulus lupulus L.). Plant Sci. 2018, 269, 32–46. DOI: 10.1016/j.plantsci.2018.01.004.
- De Keukeleire, J.; Ooms, G.; Heyerick, A.; Roldan-Ruiz, I.; Van Bockstaele, E.; De Keukeleire, D. Formation and Accumulation of Alpha-Acids, Beta-Acids, Desmethylxanthohumol, and Xanthohumol during Flowering of Hops (Humulus lupulus L.). J. Agric. Food Chem. 2003, 51, 4436–4441. DOI: 10.1021/jf034263z.
- Ban, Z.; Qin, H.; Mitchell, A. J.; Liu, B.; Zhang, F.; Weng, J. K.; Dixon, R. A.; Wang, G. Noncatalytic Chalcone Isomerase-Fold Proteins in Humulus lupulus Are Auxiliary Components in Prenylated Flavonoid Biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E5223–E5232. DOI: 10.1073/pnas.1802223115.
- Matoušek, J.; Kocábek, T.; Patzak, J.; Füssy, Z.; Procházková, J.; Heyerick, A. Combinatorial Analysis of Lupulin Gland Transcription Factors from R2R3Myb, bHLH and WDR Families Indicates a Complex Regulation of Chs_H1 Genes Essential for Prenylflavonoid Biosynthesis in Hop (Humulus lupulus L.). BMC Plant Biol. 2012, 12, 27. DOI: 10.1186/1471-2229-12-27.
- Matoušek, J.; Kocábek, T.; Patzak, J.; Bříza, J.; Siglová, K.; Mishra, A. K.; Duraisamy, G. S.; Týcová, A.; Ono, E.; Krofta, K. The “Putative" Role of Transcription Factors from HlWRKY Family in the Regulation of the Final Steps of Prenylflavonid and Bitter Acids Biosynthesis in Hop (Humulus lupulus L.). Plant Mol. Biol. 2016, 92, 263–277. DOI: 10.1007/s11103-016-0510-7.
- Matoušek, J.; Vrba, L.; Novák, P.; Patzak, J.; De Keukeleire, J.; Škopek, J.; Heyerick, A.; Roldán-Ruiz, I.; De Keukeleire, D. Cloning and Molecular Analysis of the Regulatory Factor HlMyb1 in Hop (Humulus lupulus L.) and the Potential of Hop to Produce Bioactive Prenylated Flavonoids. J. Agric. Food Chem. 2005, 53, 4793–4798. DOI: 10.1021/jf050175y.
- Matoušek, J.; Kocábek, T.; Patzak, J.; Škopek, J.; Maloukh, L.; Heyerick, A.; Fussy, Z.; Roldán-Ruiz, I.; Keukeleire, D. D. HlMyb3, a Putative Regulatory Factor in Hop (Humulus lupulus L.), Shows Diverse Biological Effects in Heterologous Transgenotes. J. Agric. Food Chem. 2007, 55, 7767–7776. DOI: 10.1021/jf071153+.
- Aberl, A.; Coelhan, M. Determination of Volatile Compounds in Different Hop Varieties by Headspace-Trap GC/MS-in Comparison with Conventional Hop Essential Oil Analysis. J. Agric. Food Chem. 2012, 60, 2785–2792. DOI: 10.1021/jf205002p.
- Takoi, K. Flavor Hops Varieties and Various Flavor Compounds Contributing to Their “Varietal Aromas”: A Review. Tech. Q. Master Brew. Assoc. Am. 2019, 56, 113–123.
- Steenackers, B.; De Cooman, L.; De Vos, D. Chemical Transformations of Characteristic Hop Secondary Metabolites in Relation to Beer Properties and the Brewing Process: A Review. Food Chem. 2015, 172, 742–756. DOI: 10.1016/j.foodchem.2014.09.139.
- Bocquet, L.; Sahpaz, S.; Hilbert, J. L.; Rambaud, C.; Rivière, C. Humulus lupulus L., a Very Popular Beer Ingredient and Medicinal Plant: Overview of Its Phytochemistry, Its Bioactivity, and Its Biotechnology. Phytochem. Rev. 2018, 17, 1047–1090. DOI: 10.1007/s11101-018-9584-y.
- Okada, Y.; Sugimoto, M.; Ito, K. Molecular Cloning and Expression of Farnesyl Pyrophosphate Synthase Gene Responsible for Essential Oil Biosynthesis in Hop (Humulus lupulus). J. Plant Physiol. 2001, 158, 1183–1188. DOI: 10.1078/S0176-1617(04)70145-5.
- Pichersky, E.; Lewinsohn, E.; Croteau, R. Purification and Characterization of S-Linalool Synthase, an Enzyme Involved in the Production of Floral Scent in Clarkia breweri. Arch. Biochem. Biophys. 1995, 316, 803–807. DOI: 10.1006/abbi.1995.1107.
- Tominaga, T.; Des Gachons, C. P.; Dubourdieu, D. A New Type of Flavor Precursors in Vitis vinifera L. cv. Sauvignon Blanc: S-Cysteine Conjugates. J. Agric. Food Chem. 1998, 46, 5215–5219. DOI: 10.1021/jf980481u.
- Gros, J.; Tran, T. T. H.; Collin, S. Enzymatic Release of Odourant Polyfunctional Thiols from Cysteine Conjugates in Hop. J. Inst. Brew. 2013, 119, 221–227. DOI: 10.1002/jib.80.
- Kishimoto, T.; Morimoto, M.; Kobayashi, M.; Yako, N.; Wanikawa, A. Behaviors of 3-Mercaptohexan-1-ol and 3-Mercaptohexyl Acetate during Brewing Processes. J. Am. Soc. Brew. Chem. 2008, 66, 192–196. DOI: 10.1094/ASBCJ-2008-0702-01.
- Darriet, P.; Tominaga, T.; Lavigne, V.; Boidron, J.-N.; Dubourdieu, D. Identification of a Powerful Aromatic Component of Vitis vinifera L. var. Sauvignon Wines: 4-Mercapto-4-Methylpentan-2-One. Flavour Fragr. J. 1995, 10, 385–392. DOI: 10.1002/ffj.2730100610.
- Roland, A.; Viel, C.; Reillon, F.; Delpech, S.; Boivin, P.; Schneider, R.; Dagan, L. First Identification and Quantification of Glutathionylated and Cysteinylated Precursors of 3-Mercaptohexan-1-ol and 4-Methyl-4-Mercaptopentan-2-One in Hops (Humulus lupulus). Flavour Fragr. J. 2016, 31, 455–463. DOI: 10.1002/ffj.3337.
- Kishimoto, T.; Kobayashi, M.; Yako, N.; Iida, A.; Wanikawa, A. Comparison of 4-Mercapto-4-Methylpentan-2-One Contents in Hop Cultivars from Different Growing Regions. J. Agric. Food Chem. 2008, 56, 1051–1057. DOI: 10.1021/jf072173e.
- Lin, J.; Massonnet, M.; Cantu, D. The Genetic Basis of Grape and Wine Aroma. Hortic. Res. 2019, 6–81. DOI: 10.1038/s41438-019-0163-1
- Kobayashi, H.; Takase, H.; Suzuki, Y.; Tanzawa, F.; Takata, R.; Fujita, K.; Kohno, M.; Mochizuki, M.; Suzuki, S.; Konno, T. Environmental Stress Enhances Biosynthesis of Flavor Precursors, S-3-(Hexan-1-ol)-Glutathione and S-3-(Hexan-1-ol)-L-Cysteine, in Grapevine through Glutathione S-Transferase Activation. J. Exp. Bot. 2011, 62, 1325–1336. DOI: 10.1093/jxb/erq376.
- Thibon, C.; Cluzet, S.; Mérillon, J. M.; Darriet, P.; Dubourdieu, D. 3-Sulfanylhexanol Precursor Biogenesis in Grapevine Cells: The Stimulating Effect of Botrytis cinerea. J. Agric. Food Chem. 2011, 59, 1344–1351. DOI: 10.1021/jf103915y.
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. DOI: 10.14806/ej.17.1.200.
- Padgitt-Cobb, L. K.; Kingan, S. B.; Wells, J.; Elser, J.; Kronmiller, B.; Moore, D.; Concepcion, G.; Peluso, P.; Rank, D.; Jaiswal, P.; et al. A Draft Phased Assembly of the Diploid Cascade Hop (Humulus lupulus) Genome. Plant Genome. 2021, 14, e20072. DOI: 10.1002/tpg2.20072.
- Dobin, A.; Davis, C. A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T. R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. DOI: 10.1093/bioinformatics/bts635.
- Pertea, M.; Pertea, G. M.; Antonescu, C. M.; Chang, T. C.; Mendell, J. T.; Salzberg, S. L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. DOI: 10.1038/nbt.3122.
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D. R.; Pimentel, H.; Salzberg, S. L.; Rinn, J. L.; Pachter, L. Differential Gene and Transcript Expression Analysis of RNA-Seq Experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. DOI: 10.1038/nprot.2012.016.
- Love, M. I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550–521. DOI: 10.1186/s13059-014-0550-8.
- Schwacke, R.; Ponce-Soto, G. Y.; Krause, K.; Bolger, A. M.; Arsova, B.; Hallab, A.; Gruden, K.; Stitt, M.; Bolger, M. E.; Usadel, B. MapMan4: A Refined Protein Classification and Annotation Framework Applicable to Multi-Omics Data Analysis. Mol. Plant. 2019, 12, 879–892. DOI: 10.1016/j.molp.2019.01.003.
- Luhua, S.; Hegie, A.; Suzuki, N.; Shulaev, E.; Luo, X.; Cenariu, D.; Ma, V.; Kao, S.; Lim, J.; Gunay, M. B.; et al. Linking Genes of Unknown Function with Abiotic Stress Responses by High-Throughput Phenotype Screening. Physiol. Plant. 2013, 148, 322–333., DOI: 10.1111/ppl.12013.
- Zhang, X.; Gou, M.; Liu, C. J. Arabidopsis Kelch Repeat F-Box Proteins Regulate Phenylpropanoid Biosynthesis via Controlling the Turnover of Phenylalanine Ammonia-Lyase. Plant Cell. 2013, 25, 4994–5010. DOI: 10.1105/tpc.113.119644.
- Castro, C. B.; Whittock, L. D.; Whittock, S. P.; Leggett, G.; Koutoulis, A. DNA Sequence and Expression Variation of Hop (Humulus lupulus) Valerophenone Synthase (VPS), a Key Gene in Bitter Acid Biosynthesis. Ann. Bot. 2008, 102, 265–273. DOI: 10.1093/aob/mcn089.
- Gullner, G.; Komives, T.; Király, L.; Schröder, P. Glutathione S-Transferase Enzymes in Plant-Pathogen Interactions. Front. Plant Sci. 2018, 9, 1836. DOI: 10.3389/fpls.2018.01836.
- Eriksen, R. L.; Padgitt-Cobb, L. K.; Townsend, M. S.; Henning, J. A. Gene Expression for Secondary Metabolite Biosynthesis in Hop (Humulus lupulus L.) Leaf Lupulin Glands Exposed to Heat and Low-Water Stress. Sci. Rep. 2021, 11, 1–18.