
ABSTRACT
EURO-GANEX is an innovative hydrometallurgical process for the group separation of transuranic actinides from future spent nuclear fuels. A flowsheet test of a process upset (or ‘maloperation’) has been carried out involving the reduction in the concentration of the scrub acid feed to the extract-scrub contactors. The experimental test was completed in two stages. First, the extract-scrub contactors were run under normal flowsheet conditions before initiating the process upset by reducing the scrub acidity from 0.5 to 0.05 mol/L HNO3. At low acidity, a change in the online UV-Vis spectra of the solvent phase indicated the formation of hydrolyzed plutonium species or plutonium colloid formation. Surprisingly, however, this did not lead to the recycle and accumulation of plutonium in the extract-scrub contactors as expected indicating that the EURO-GANEX process is quite robust towards this process upset. Further spectroscopic investigations of the organic-phase species were also performed to characterize the conditions under which the hydrolyzed plutonium species form in the EURO-GANEX solvent as well as separate TODGA and DMDOHEMA phases. The spectroscopic studies supported the view that Pu(IV) hydrolyses at low acidity but the hydrolysis is limited by the organic ligands and is consequently reversible on raising the acidity again. The hydrolysis was also observed in separate TODGA and DMDOHEMA phases.
Introduction
Closing the nuclear fuel cycle with minor actinide (MA) partitioning and transmutation (P&T) as well as plutonium multi-recycling is an objective of sustainable advanced fuel cycles.[Citation1–9] While uranium and plutonium recycling can improve the utilization of natural resources, P&T of the MA enables a reduction of the transuranic burden and hence the radiotoxicity and long-term heat loading of radioactive wastes in the geological repository.[Citation6,Citation8,Citation10–15]
However, multi-recycling of transuranic (TRU) elements requires fast reactors capable of burning these fuels as well as advanced reprocessing and TRU fuel fabrication technologies for recycling. The development of advanced separation processes for the recovery and recycling of minor actinides is thus regarded as a key objective for future closed fuel cycles.[Citation2,Citation3,Citation5,Citation16–21] Two approaches are envisaged: the homogeneous and heterogeneous recycling routes. Heterogeneous recycling involves separate (U,Pu) and (MA) fuels distributed in different locations around the fast reactor core. Options for heterogeneous recycling generally involve separation processes for the minor actinides americium and curium that follow on from an initial PUREX-type process that recovers uranium, plutonium, and neptunium.[Citation22] Homogeneous recycling, on the other hand, recycles the actinides in a single (U,TRU) fuel homogeneously distributed in the fast reactor core. This requires a new process since the PUREX process, as used presently in the commercial-scale reprocessing of spent nuclear fuels (SNF),[Citation23,Citation24] cannot recover all the TRU as a single group. While this can be achieved with molten salt-based pyrochemical processes, particularly if the fast reactor uses metal fuels,[Citation25] new aqueous processes are still under heavy development. In Europe, these processes have been termed GANEX (Grouped ActiNide EXtraction) and are generally directed towards the reprocessing of high plutonium content mixed oxide (MOX) fuels; a recent review describes the different strategies in detail.[Citation26] GANEX is a hydrometallurgical process based on solvent extraction of actinides between aqueous nitric acid and an organic phase comprising a selective extracting agent, a co-extractant or phase modifier (if needed) and an inert kerosene diluent.[Citation17,Citation27–31]
EURO-GANEX flowsheet
There are variations of the GANEX process, but they commonly envisage a two-cycle process involving an initial separation of (bulk) uranium to reduce the amount of material to be processed and simplify the chemistry in the second cycle.[Citation17,Citation26] The aim of the GANEX second cycle is to recover the transuranic actinides (Np-Cm) together in a single fraction either by co-extraction with the lanthanides followed by selective stripping or by selective extraction.[Citation26] The former strategy is most well developed with two process flowsheets that have been developed and ‘hot’ tested with spent fuels, viz. “CEA-GANEX” and “EURO-GANEX” processes.[Citation30,Citation31] CEA-GANEX was developed by the Commissariat à l’Énergie Atomique et aux Énergies Alternatives (CEA, France), whereas EURO-GANEX has been developed under European Framework Programme project ACSEPT (ACtinide recycling by SEParation and Transmutation), SACSESS (Safety of ACtinide SEparation ProceSSes) and latterly by GENIORS (GENeration IV Integrated Oxide fuel Recycling Strategies).[Citation3,Citation4] An option to meet the alternative strategy based on selective extraction, “CHALMEX”, is being developed by Chalmers University (Sweden) but only through batch tests to date.[Citation32,Citation33] A variation on EURO-GANEX that produces separate (Np,Pu) and (Am,Cm) products for heterogeneous recycling has also been reported.[Citation34]
The hot test of the EURO-GANEX cycle, with spent nuclear fuel, has been reported and shown to deliver very good actinide recoveries and good decontamination from most fission products, including lanthanides. The development process and underlying chemistry are described in earlier references,[Citation35–37] but briefly EURO-GANEX uses a combination of two amidic extractants to co-extract actinides and lanthanides in oxidation states III, IV, and VI. The extractants, N,N,N’,N’-tetra-n-octyl-diglycolamide (TODGA) and N,N’-dimethyl-N,N’-dioctyl-2-(2-hexylethoxy) malonamide (DMDOHEMA), were selected based on their combined ability to cope with at least 10 g/L Pu loading at 6 mol/L HNO3 without formation of a third phase, which is a recognized process safety and operability hazard.[Citation38] A combination of hydrophilic complexing agents, i.e. a sulphonated 2,6-bis(1,2,4-triazin-3-yl)-pyridine (SO3-Ph-BTP or ‘sulphonated-BTP’[Citation39]) and acetohydroxamic acid (AHA),[Citation40] are then employed to selectively strip the trivalent and tetravalent actinides from the organic phase, leaving the lanthanides in the solvent. Hexavalent neptunium (NpO22+) is also stripped but by reduction to the less organic soluble pentavalent oxidation state (NpO2+) with AHA.[Citation36] Other key features of the EURO-GANEX flowsheet as demonstrated in the hot test[Citation31] are:
Compatibility with short residence time centrifugal contactors that are commonly regarded as next-generation solvent extraction equipment.[Citation41]
Ability to manage at least 10 g/L Pu in the initial highly active (HA) feed; consistent with mixed oxide fuel reprocessing.
Use of a hold back agent trans-1,2-diaminocyclohexane-N,N,N’,N’-tetraacetic acid (CDTA) in the initial extract-scrub to suppress extraction of zirconium and palladium.[Citation42]
Safety and process upsets
The development of advanced actinide separation processes is generally focused upon achieving the desired performance; essentially, high recoveries of actinides to minimize losses to the waste streams and good enough decontamination factors (DF) from fission product species to obtain suitable products for finishing and economic fuel manufacture.[Citation19]
However, the safety of the proposed processes is also an important consideration in the design of advanced actinide separation processes.[Citation43] Consequently, it is essential to underpin the normal operating envelope of the flowsheet and assess the impact of any potential process changes upon the process performance.[Citation43,Citation44] Ideally, flowsheet conditions are optimized such that the process can cope with minor deviations (e.g. in flow rates, concentrations, temperature) without suffering a detrimental impact upon the overall performance, but it is also important to understand the effects more significant process upsets (or ‘maloperations’) can have. Undesirable effects include:
Degradation of the process performance. In some cases, the reduction in DF may result in out-of-specification products, while in other cases, the mis-routing (‘break through’) of radioactive species to raffinate streams may give rise to hazards in downstream plants.
Process operability issues, e.g. entrainment of one phase in the other or third-phase formation.
Cycling and accumulation of metal ions within a contactor (e.g. mixer-settler battery, pulsed column or centrifugal contactor cascade) leading to a radiological, criticality hazard or further misrouting.[Citation44]
The impact and severity of any given process upset will depend upon the precise conditions as well as the extent and duration of the process deviation. Clearly, understanding the causes of potential process upsets and how they evolve over time is vital to underpinning the safety assessment of any proposed flowsheet.[Citation44]
Due to the sensitivity of process upsets to the precise process conditions, it is not possible to adopt a purely experimental approach to assess all potential scenarios for a flowsheet. Instead, the development of dynamic process models is a more practical approach, which enables the sensitivity of the flowsheet over a wide variety of conditions to be investigated.[Citation44,Citation45] However, the provision of accurate models that can predict the performance of a flowsheet outside its normal operating envelope requires experimental data to develop the model first and then to validate it.
Process safety assessment
One of the objectives of the SACSESS project was to perform a process safety review of the EURO-GANEX flowsheet under normal and potential off-normal conditions.[Citation46] For this purpose, a safety assessment methodology was developed based on the experience of the project collaborators across Europe.[Citation47] This methodology was then utilized to carry out a review of the EURO-GANEX flowsheet[Citation31] based on a workshop style approach. The output from this review[Citation47] identified potential hazards associated with the process and recommended areas for further research with a view to further understanding these issues.
Areas identified for further research included safety of process reagents; effects of radiolytic and hydrolytic degradation of the solvent, including solvent clean-up; potential for gas generation; effects of temperature on the process; crude formation and process upsets due to changes in or complete losses of reagent concentrations or flows.
More specifically, several process upsets were identified based on deviations in flow rates/concentrations of reagent feeds in the extract-scrub section of the EURO-GANEX flowsheet. In some cases, it was anticipated that these will lead to a misrouting of species and degradation of the recoveries and/or DFs in the product streams, while other maloperations have the potential to lead to oversaturation of the solvent and third-phase formation. One of the key process upsets identified involved a decrease in the nitric acid concentration of the scrub acid feed in the EURO-GANEX flowsheet, similar to that reported for the “i-SANEX” flowsheet.[Citation45] Under these conditions there is a potential for plutonium and other metal ions to be backwashed from the solvent, with subsequent re-extraction by the higher acidity in the extract contactors, leading to the recycling and accumulation of plutonium (and/or other actinides) within the process across the extract-scrub section. Figure S1 (Electronic Supplementary Information, ESI) illustrates schematically how such a process upset would proceed over time (exemplar data from simulation of a hypothetical tributyl phosphate-based solvent extraction process, provided for illustrative purposes only). With the loss of scrub acid, the solute (metal ion) is seen to start accumulating in the scrub section where the organic phase containing the solute extracted from a high acidity solution meets a very low acidity aqueous phase, causing the solute to be backwashed into the aqueous phase, only for it to be re-extracted in the high acidity region at the feed stage. Over time, this peak grows and moves along the contactor eventually breaking through into the aqueous raffinate. Consequently, such a scenario was selected for experimental testing. This scenario was selected on the basis that:
It was a suitably severe test of the flowsheet with potentially significant impacts on a future safety case
It was a realistic scenario (change to one reagent flow only)
It focused on the extract-scrub section (scenarios involving the stripping section were deferred since optimization of the stripping section to replace the sulphonated-BTP with a CHONFootnote1 ligand is in progress)[Citation48,Citation49]
It could be tested with a surrogate feed (i.e. a feed with representative concentrations of components prepared in the laboratory but without the high gamma radiation field associated with hot tests on spent fuels)
Therefore, a flowsheet trial was proposed to investigate the potential cycling and accumulation of plutonium and americium in the extract-scrub contactors resulting from this possible process upset. This report summarizes the results from the experimental trial designed to assess the impact of a low scrub acid feed upon the extract-scrub contactors in the EURO-GANEX flowsheet. The results obtained in the flowsheet trial were then supplemented by some additional spectroscopic studies with a view to characterising the species involved.
Experimental methods
General approach
In brief, the approach to testing this process upset was to run the primary extract-scrub section of the EURO-GANEX flowsheet (based on the flowsheet reported in ref.[Citation31]) to steady-state operation under normal conditions. Then, the process would be upset by the replacement of the “scrub acid” with a lower concentration feed and the flowsheet run until either a new steady state was reached or the evolution of the process upset was clearly observed, e.g. by break-through of plutonium and americium into the aqueous raffinate. The flowsheet was to be tested in laboratory scale centrifugal contactors (1 cm rotor diameter) in a radiochemical glove box. Various previous papers have described the facility, equipment and procedures for testing flowsheets in the centrifugal contactor cascades in our laboratories in detail.[Citation29,Citation34,Citation50–53] At the end of the trial, profile samples of both the aqueous and organic phases were taken. Online UV-Vis absorption spectroscopy was used to monitor key species in real time during the flowsheet trial. Details specific to this flowsheet are given in the following subsections.
Note that all radioactive materials such as plutonium and americium were handled in a nuclear licenced facility in designated fume hoods or glove boxes working under approved procedures.
Flowsheet
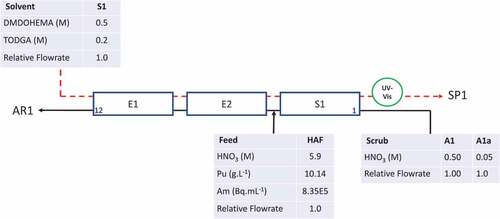
The configuration of the flowsheet was based on the hot test.[Citation31] The compositions of the feeds and flow rates are summarized in for the normal (scrub flow A1) and off-normal (scrub flow A1a) operations. Only the extract-scrub section of the flowsheet was set up for this trial and this comprises three banks of contactors, each with four stages, giving a total of eight stages extract (E1 and E2) and four stages of scrub (S1). The active feed (HAF) is introduced between stages 4 and 5. Plutonium and americium are extracted from the active feed by a counter-current flow of solvent introduced at stage 12 (S1). Under normal flowsheet conditions the loaded solvent was then contacted with a low acid scrub feed of 0.5 mol/L HNO3 introduced at stage 1 (A1). To initiate the process upset A1 was reduced to 0.05 mol/L HNO3 (A1a).
Figure 1. Configuration for flowsheet test of EURO-GANEX cycle with normal (A1) and reduced (A1a) scrub acid feeds.
Analysis
Online UV-Vis spectroscopy was used to monitor the plutonium loading of the solvent product (SP1) from the extract-scrub contactors. A flow cell with a 5 mm path length was positioned in the SP1 product line and connected to a Zeiss diode array spectrometer via 600 µm core optical fibres. By measuring the absorbance of Pu(IV) at selected wavelengths (526 and 791 nm) it was possible to follow the run-up to steady state under normal flowsheet conditions and observe the effect upon plutonium concentration throughout the test. (Note that the organic-phase spectra, including how the selected peaks change with conditions, are discussed in detail in the “Discussion” section).
Samples of SP1 were analyzed by γ-spectroscopy to determine the plutonium and americium concentrations under normal and off-normal conditions. Samples of the aqueous raffinate (AR1) were submitted for analysis by α-spectrometry to determine the residual plutonium concentration in AR1.
The aqueous and solvent profile samples were also analyzed by γ-spectroscopy to determine the plutonium and americium concentrations across the extract-scrub contactors. The acidity profiles of the aqueous and solvent samples were also determined by potentiometric titration against calibrated NaOH in 1 mol/L potassium fluoride (KF) medium. The KF medium was chosen as F− ions will complex metal ions, thereby avoiding bias to the results due to metal ion hydrolysis; the results were corrected for bias from the medium by appropriate calibrations.
Uncertainties in α and γ spectroscopies have been estimated as ca. ±10%, although they may be greater close to limits of detection.[Citation34] This uncertainty includes all contributing errors to the analytical procedures, including analytical errors such as small changes in geometries, sampling (particularly from radiochemical glove boxes), dilutions, etc.
Feed preparation
Scrub acid feed (A1)
A scrub acid feed of 0.5 mol/L HNO3 was prepared to attain steady-state conditions under normal conditions. Two additional scrub acid feeds at 0.1 mol/L and 0.05 mol/L HNO3 were also prepared for running the flowsheet under maloperation conditions.
Solvent feed (S1)
The solvent feed was prepared by dilution of TODGA and DMDOHEMA with Exxsol D80 diluent to obtain a final solvent composition of 0.2 mol/L TODGA (Technocomm, UK) and 0.5 mol/L DMDOHEMA (Technocomm, UK). The solvent was used as prepared without any further treatment or washing.
HA feed (HAF)
An active feed containing 10 g/L Pu in 5.9 mol/L HNO3 was prepared by dilution of a concentrated lab stock solution. The solution also contained 241Am from 241Pu decay (8.31E5 Bq/mL). As the main objective was to observe the effect of a low acid maloperation upon the behaviour of plutonium and americium, no further elements were added to the HAF. (This obviously represents a simplification of the system).
Rig operation
During the trial, the extract-scrub contactors were operated at 4500 rpm. Initially, the contactors were flooded with non-active aqueous feeds, i.e. A1 and a non-active substitute feed (5.9 mol/L HNO3) for the HAF. Once the aqueous phase was observed to exit from the product outlets, the solvent feed (S1) was started. Start-up with non-active feeds established the required solvent/aqueous ratio and acid profiles in the contactors and ensured stable operations before introduction of the HAF. Once the aqueous and solvent phases were observed to exit from their respective product outlets, the substitute feed was changed over to the HAF.
At approximately 1 h intervals, the rotor speeds and product flow rates were measured and samples of AR1 and SP1 products taken. The flowsheet run-up to steady-state operation was observed by monitoring the plutonium loading of the solvent with online UV-Vis spectroscopy. Upon reaching steady state, the flowsheet was operated for a further 2 h under normal conditions before the nitric acid concentration of the scrub acid was reduced to initiate the process upset. Initially, the scrub acid feed was reduced to 0.1 mol/L HNO3 to confirm this did not affect the hydrodynamic stability. After operating under these conditions for 30 min the scrub acid feed was then reduced to 0.05 mol/L HNO3 (A1a) and operated under the upset conditions for a further 3 h.
Results
General flowsheet performance
In general, the flowsheet performed well under normal conditions, achieving near-complete extraction of plutonium and americium in the extract-scrub contactors, as anticipated based on the previous tests.[Citation29,Citation31,Citation34]
Measurement of the product flow rates confirmed that feed flow rates were stable throughout the trial. Rotor speeds also remained stable within the operating range of 4500 rpm ±5 rpm during the trial. Good phase separation was observed throughout the trial, and all product samples were observed to be free from entrainment. No hydrodynamic issues or additional phases were observed under either normal or off-normal conditions.
Solvent Product (SP1)
The initial run-up to and operation under both steady-state and upset conditions were followed by online UV-Vis spectroscopy and analysis of the solvent product (SP1) by γ-spectroscopy. Changes in absorbance for selected absorption maxima at 791 and 526 nm during the course of the trial are illustrated in . The online analysis indicates that the plutonium concentration in the solvent reached steady state after approximately 1 h of operation with the HAF and remained constant while operating under normal flowsheet conditions. The online UV-Vis results were confirmed by γ-analysis of the solvent product ( and ). These also show that steady state was achieved approximately 1 h after switching from the inactive feed to the HAF. The solvent samples obtained under normal flowsheet conditions show a higher than expected concentration of americium, with recoveries of 120% when compared to the initial active feed. This was attributed to residual americium contamination present in the rig from an earlier trial where high (g/L) levels of americium were processed.[Citation52] Despite the higher than expected recovery of americium, the concentration in the solvent remained constant under normal operating conditions, discounting the possibility of this being caused by the recycle and subsequent breakthrough of americium (). The results also showed that a plutonium concentration between 10.4 and 11.4 g/L was obtained under normal flowsheet conditions. This gives a mass balance of 102–112% based on the plutonium concentration in the active feed.
Figure 2. Online UV-Vis analysis of solvent product (SP1); note 5 min moving average plotted to smooth fluctuations in measured absorbances) and data normalized to absorbance values at end of the test. Online data compared to off-line γ-spectroscopy data from (normalized to HAF for Pu and Am). Vertical lines mark the start of the maloperation and the time for changing the HAF pump.
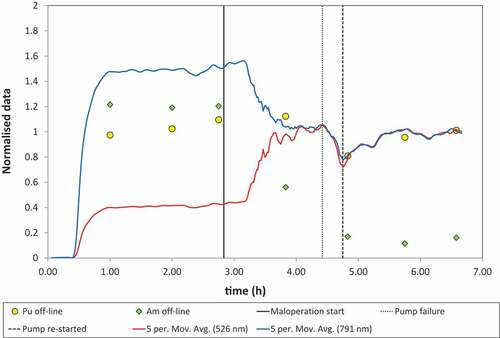
Table 1. Plutonium and americium concentration in SP1 (measured by γ-spectroscopy).
After operating at steady state for 2 h, the process upset was initiated by reducing the HNO3 concentration of the scrub acid in stages, first to 0.1 mol/L and then to 0.05 mol/L HNO3. This phased approach was taken to ensure that this did not cause hydrodynamic problems with operation of the rig (i.e. poor-phase separation and entrainment of aqueous in the solvent product). Unexpected changes were then observed in the measured absorbances at 791 and 526 nm, with an increase at 526 nm and decrease at 791 nm (the causes of this are due to spectral shifts that are explained in the “Plutonium speciation in the EURO-GANEX (TODGA + DMDOHEMA) solvent” section). The fluctuation in the measurements also increased. Note that the dip at 4.8 h was caused by a temporary loss of the active feed due to the HAF pump failing. However, on rectifying the failure, the absorbances recovered to the new steady-state values. The changes in absorbance were also accompanied by a visible change in the colour of the solvent product from orange-brown to green.
In contrast, there was a rapid decrease in the americium concentration of the solvent product under the low acid off-normal conditions. The γ-spectroscopy measurements on SP1 showed that under these conditions, only 15% of americium present in the feed was routed with the solvent with approximately 85% of the americium entering the contactors being recycled in the extract-scrub contactor bank.
Aqueous Raffinate (AR1)
Due to the low residual concentration of plutonium in AR1 the two final samples obtained under both normal and upset conditions were analyzed by α-spectrometry (). The recovery of plutonium in the extract contactors, based upon the activity of239/240Pu in the HAF and AR1 samples, exceeds 99.99% throughout the trial. The activity for 239/240Pu in AR1 remains at a low level under both normal and process upset conditions and shows that (the minimal) losses of plutonium to the aqueous raffinate are not affected by the low acid feed. Americium concentration in AR1 was also very low but due to interferences was not quantifiable by α-spectrometry. The aqueous profile sample showed that the americium content of the aqueous phase is significantly lower than 100 Bq/mL by the final stages of the extract contactor (i.e. >99.98% recovery). Therefore, americium has also not broken through into the aqueous raffinate.
Table 2. Alpha spectrometry results for aqueous raffinate (AR1) and calculated percentage plutonium recoveries.
Profile results
Upon completion of the trial, samples of the aqueous and solvent phases were taken from each stage of the extract-scrub contactors to determine the final concentration profiles for acid, plutonium, and americium across the contactors.
To assess the impact of the process upset conditions, the profile data are compared with results from a previous trial[Citation34] where the extract-scrub contactors were operated under near-identical flowsheet conditions (feed acidity, scrub acidity, feed plutonium concentration and S/A ratio in extract-scrub contactors were all the same. Flow rates were 25% lower in the previous test). The only notable difference being that the profile for Am under normal flowsheet conditions was obtained with a feed containing a significantly higher level of americium (5 MBq/mL) than this trial, although both represent low concentrations overall (and it contained other fission products elements as well as 0.1 mol/L CDTA).
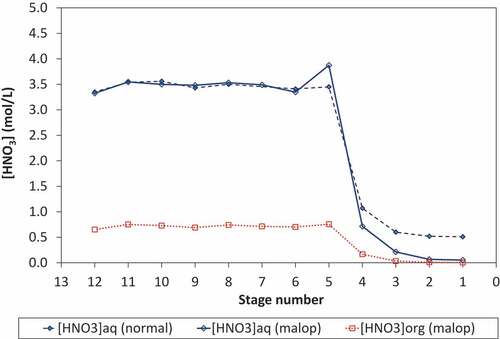
The acid profiles of the extract–scrub contactors obtained under normal and process upset conditions were obtained (). As expected, a reduction in the scrub acidity leads to a corresponding reduction in the acidity of the scrub contactors (stages 1–4). However, it is evident that this has little impact upon the aqueous acid profile of the extract contactors which show a consistent nitric acid concentration of 3.5 mol/L HNO3 in stages 5–12.
Figure 3. Acid profiles for normal and process upset conditions.
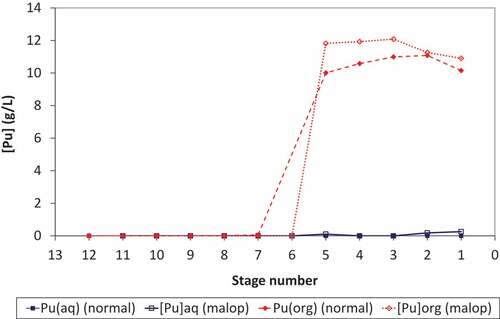
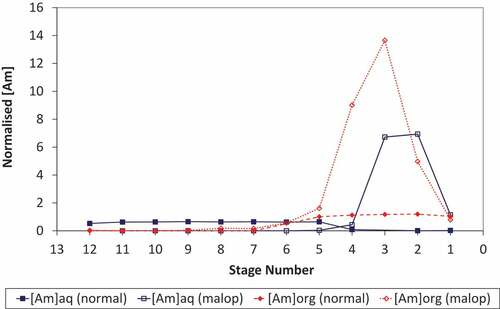
The concentration profiles for plutonium obtained under normal and upset conditions show that upon reduction of the scrub acidity to 0.05 mol/L the plutonium loading of the solvent in the extract-scrub contactors remains almost constant at approximately 10 g/L (). Therefore, under these conditions, the reduction in scrub acidity has had little impact upon the extraction and routing of plutonium in the EURO-GANEX flowsheet. Comparing the organic-phase samples obtained from each stage upon completion of the trial showed a clear colour change from green (at stage 1) to orange-brown at stage 5 as the acidity increases across stages 1–5 of the extract-scrub contactors and then colourless by stage 12 of the extract section (E1) where the plutonium content is very low.
Figure 4. Plutonium profiles under normal and process upset conditions.
The concentration profiles obtained for americium under normal and process upset conditions differ (). Under normal flowsheet conditions, americium is rapidly extracted at the feed stage and gives a constant concentration in the solvent phase across stages 1–5. It should be noted that the profile for americium under normal conditions was obtained with an active feed containing a higher concentration of americium (5 MBq/mL) than the maloperation trial and so should be used to compare the shape of the concentration profiles but not the magnitude (hence data normalized to the respective HAF concentrations are presented in ). Reducing the nitric acid concentration of the scrub acid results in significant recycle of americium in the flowsheet. It is evident that reducing the acidity of the scrub acid feed leads to backwashing of americium into the aqueous phase in stages 1–3. Subsequent re-extraction of americium at higher acidity in the extract contactor results in significant recycle between the extract and scrub banks. Consequently, a peak in the americium concentration profile is observed in the aqueous and solvent phase at stages 2 and 3, respectively. Over the duration of this maloperation, the recycle produces a peak americium concentration in the solvent that is over an order of magnitude higher than the initial active feed.
Figure 5. Americium profiles under normal and process upset conditions; profiles normalized to the respective HAF and overlaid to illustrate the accumulation under process upset conditions (n.b. initial Am concentrations in the HAF were not the same).
Discussion
Brief review of plutonium colloid formation and extraction
The so-called plutonium polymer or colloid, with its striking emerald-green colour, has been known since the earliest days of plutonium chemistry[Citation54–56] and its formation and properties (in the aqueous phase) have been studied extensively (e.g. see[Citation57–61]). Regions of stability have been defined[Citation62,Citation63] and it has been shown that colloidal plutonium can be identified by the characteristic UV-Vis absorption spectrum (see ESI Figure S2). Lloyd and Haire[Citation64] found the primary particles were <20 Å in size and were most stabilized at a nitrate-to-plutonium ratio of 0.8 (and ≤0.1 mol/L), this value being consistent with 1:1 association with surface facing plutonium atoms in the face centred cubic structure. Colloidal plutonium forms when Pu(IV) hydrolyzes at low acidity and the hydrolysis products polymerize. Aging of plutonium colloids causes dehydration and it becomes very difficult to reverse the process. The change during aging was considered to be due to conversion of amorphous to crystalline particles rather than particle growth. The usual model of Pu(IV) polymerization is hydrolysis of the solvated cation to give hydroxyl (OH) bridged long-chain polymeric species (olation) that can dehydrate on aging or heating to oxo-bridged forms (oxolation), which are far harder to depolymerize, and eventually evolve to PuO2.[Citation65]
Walther et al.[Citation66] reexamined the UV-vis absorption spectra of hydrolyzed Pu(IV). They noted that the strong absorptions in the low wavelength (high energy) end of the spectra (<500 nm) are indicative of plutonium colloid and attributed to Rayleigh scattering of suspended particles. They deconvoluted Pu(IV) colloid or polymer spectra in perchloric acid and showed that distinctive bands at 620 and 740 nm are due to these species, not monomeric Pu(OH)3+.
Recent studies have shown that the plutonium colloids can form monodisperse, nanometer-sized, surface-stabilized dioxide particles such as nanoclusters of [Pu38O56Cl54(H2O)8]14- with no evidence for Pu-OH or dioxo (PuO2x+) groups.[Citation67] Dissolution of the red crystals in 2 mol/L LiCl gave a green solution with a UV/vis absorption spectrum consistent with that of plutonium (IV) colloid. The data indicated that the anion was preserved in solution and it corresponded to the functional definition of plutonium colloid taken from Lloyd and Haire[Citation64] (ESI Figure S2). The direct oxolation was thus suggested as the mechanism of hydrolysis rather than via olation.[Citation67] Further investigations found evidence that the surface chlorides were labile when reacted with 6 mol/L HCl and, although there was a colour change from green to red, the [Pu38O56]40+ core remained the same while the number of chlorides decreased to 42.[Citation68] The UV/vis spectrum changed also. The green plutonium colloid species was extracted by trichloroacetic acid (TCA) in octanol (DPu ≈10 on extraction from 0.04 mol/L HNO3 into 1 mol/L TCA) and it was shown that the Pu-O core remained intact in the organic phase. Further clusters were reported by Sigmon and Hixon based around the [Pu38O56]40+ core as well as [Pu22O28(OH)4]28+ and [Pu16O19(OH)4]22+.[Citation69] As well as the proportion of hydroxyl groups decreasing, the Pu-OH bond lengths decreased as the cluster size increased from Pu16 to Pu22 to Pu38 units. Therefore, this was proposed as evidence of the olation to oxolation mechanism. Another study, based on extended X-ray absorption fine structure (EXAFS) found smaller plutonium clusters of 5–6 Pu atoms in freshly prepared colloids and 3–4 atoms in 5-year-aged colloids.[Citation70] Recently, Tamain[Citation71] and Chupin[Citation72] have reported the characterization of water-soluble plutonium (IV) hexamers in which a [Pu6(OH)4O4]12+ cluster is stabilized by 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) or by acetate ligands. OH and O atoms form μ3 bridging ligands and, in the former complex, four DOTA molecules coordinate the four equatorial Pu(IV) atoms where each ligand complexes one Pu(IV) with two O atoms from two of its carboxylates. One carboxylate is bridging and one is not coordinated.
There are few reports of plutonium colloids in an organic phase. Plutonium polymer was found to be non-extractable into dibutyl carbitol, trifluorothiophenoyl acetone, and TBP but very well extracted by 5% dibutyl phosphate.[Citation55] It is also extracted with organophosphorus molecules dihexyl-N,N-diethylcarbamolymethylphosphonate (DHDECMP), octylphenyl-N,N-diisobutylcarbamoylmethylphosphine oxide (OΦD(IB)CMPO) and trioctyl phosphine oxide (TOPO) from 0.04 and 7 mol/L nitric acid solutions (although extraction with TOPO was possibly due to the 1% dioctylphosphoric acid present).[Citation73] Stripping was difficult although sodium carbonate was effective. Both the green colour of the organic phase and the UV-Vis absorption spectra (not included in ref.[Citation73]) confirmed that the polymeric form was retained in the organic phase after extraction. Similar findings were reported for the extraction of aged plutonium polymer into HDHoEP (bis(hexoxyethyl) hydrogen phosphate) and H2MEHP (2-ethylhexyl dihydrogen phosphate).[Citation74] The polymer was retained in the organic phases and were shown by SANS (small-angle neutron scattering) to be rods with the organic molecules wrapped around them, solvating them in the benzene diluent.
Plutonium speciation in the EURO-GANEX (TODGA + DMDOHEMA) solvent
It was seen that despite almost no effect on plutonium extraction, the reduction of the acidity resulted in a significant change in the online UV-Vis absorbances obtained for the solvent product () and that there is an associated colour change. Clearly, this implied some speciation changes in the organic phase. Therefore, additional UV-Vis spectroscopic studies were undertaken to investigate the underlying causes.
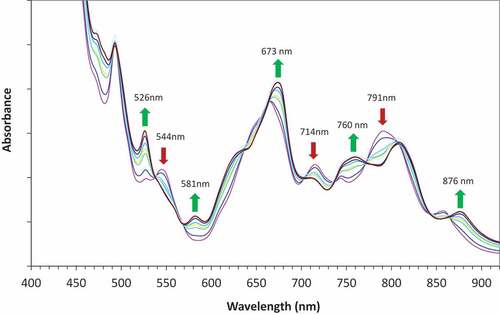
First, a loaded solvent phase was prepared by contacting fresh organic phase (0.2 mol/L TODGA/0.5 mol/L DMDOHEMA in OK) with a 10 g/L Pu solution in 3 mol/L HNO3 at an S/A ratio of one. The loaded solvent phase was then washed four times with 0.5 mol/L HNO3 to reduce the acid concentration of the solvent to levels expected in the solvent product from the extract-scrub contactors of the GANEX flowsheet under normal operating conditions. An aliquot of the loaded solvent was then pipetted into separate vials and contacted with a range of dilute nitric acid solutions, from 0.01 to 0.2 mol/L HNO3, to assess the impact of contact with low acidity solutions. All scrubbing tests were performed at ambient glovebox temperature (22°C) with a contact time of 2 min and S/A ratio of 1. At each scrub acid concentration <0.5 mol/L, the solvent phase was observed to change colour from orange-brown to green, while the aqueous phase remained colourless. The results indicated that there was little significant backwashing of plutonium from the solvent phase even with a scrub acidity as low as 0.01 mol/L HNO3. There were no obvious issues (e.g. entrainment, third phase, slow disengagement) in any of the experiments. As discussed, the bright green colour of plutonium solutions in low acid conditions is typically associated with the formation of plutonium colloid.[Citation55] It was thus concluded that the formation of a green coloured solvent phase under low acid concentration is indicative of a change in the plutonium speciation and potentially formation of ‘colloidal’ plutonium. Therefore, UV-Vis spectra of the plutonium loaded solvent phase were obtained to try and identify the species present in the solvent in each of the conditions studied. Under normal conditions ([HNO3] >0.5 mol/L) the solvent phase exhibits a characteristic Pu(IV) spectrum ().[Citation75] At low acid concentration, there is a significant shift in the position and intensity of the absorption maxima. Consequently, there are changes in the extinction coefficients at the wavelengths that were used to monitor the plutonium loading of the solvent in that lead to changes in measured absorbances despite there being no significant change in plutonium concentration. With the realizsation that there were speciation changes occurring in the organic phase during the trial, some full UV-vis spectra from the flowsheet trial were recorded during the trial; these are given in and confirm that the speciation changes occurring in the trial are the same as those observed in the batch experiments. Pu(IV) is known to form colloids in solutions at low acidity. However, since no solids were observed and the spectrum is not the same to that previously reported for colloidal plutonium ( and ESI Figure S2), it was hypothesized that the change in the plutonium spectra with nitric acid concentration could be due to the formation of a hydrolyzed plutonium species that remains soluble in the organic phase and that does not proceed to form polymeric plutonium colloid(s).
Figure 6. Comparison of UV-Vis spectra for plutonium in the GANEX solvent, contacted with high and low acidity solutions, and aqueous plutonium colloid (reproduced from ref.[Citation64]).
![Figure 6. Comparison of UV-Vis spectra for plutonium in the GANEX solvent, contacted with high and low acidity solutions, and aqueous plutonium colloid (reproduced from ref.[Citation64]).](/cms/asset/4e36ed47-aef2-428f-86e1-cb8303fd48c5/lsei_a_2136488_f0006_oc.jpg)
Figure 7. Change in the online UV-Vis spectra of Pu in SP1 during the flowsheet trial (spectra taken between 3.17 h (pink) and 3.83 h (brown) from start of trial).
Further batch solvent extraction experiments were performed using the solvent product derived from the process upset flowsheet test. These experiments were performed to assess the acidity at which (a) formation of the hydrolyzed plutonium species (colloid) was observed and (b) to assess whether this process is reversible and so will not affect the later stripping section.
A portion of the loaded solvent obtained under normal operations was diluted with fresh solvent (preequilibrated with 0.5 mol/L HNO3) to obtain a plutonium concentration of 5 g/L. Aliquots of the loaded solvent were taken and contacted with an equal volume of HNO3 (from 0.01 to 0.5 mol/L) to determine the concentration at which formation of the hydrolyzed plutonium species occurs. The phases were contacted for 5 min before separating. The solvent phase was then analyzed by UV-Vis spectroscopy.
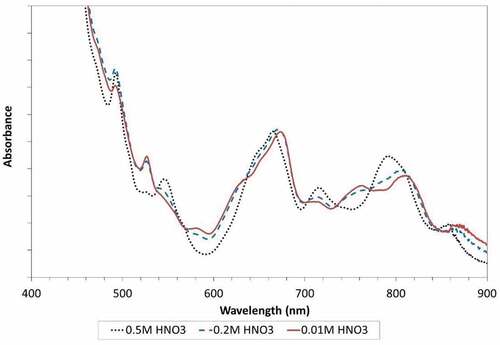
Effects of decreasing scrub acid concentration on the UV-Vis spectra of the loaded solvent are shown in . There is an obvious shift in the wavelength and intensity of the absorption maxima as the acidity of the scrub solution is decreased, which indicates a change in the plutonium speciation of the solvent phase. The characteristic Pu(IV) absorption maxima were observed to shift or decrease in intensity. It was clear that this change in speciation occurred between the scrub acid concentration of 0.5 and 0.2 mol/L HNO3. The changes in the spectra evolved gradually as the acidity of the solvent phase decreased following contact with progressively lower concentration scrub acid feeds down to 0.01 mol/L HNO3.
Figure 8. Change in Pu(IV) UV-vis spectra with decreasing scrub acid concentration (spectra for 0.1 and 0.05 mol/L HNO3 are not shown).
The reversibility of the reaction was also investigated using the solvent product obtained under maloperation conditions (containing the hydrolyzed species). Aliquots of the solvent were taken and contacted with an equal volume of HNO3 to determine the effect upon the UV-Vis spectrum of the species present in the solvent phase. The aqueous and solvent phases were contacted for 5 min at ambient glovebox temperature (22°C) and, after allowing the phases to separate, the spectra of the solvent phases were recorded.
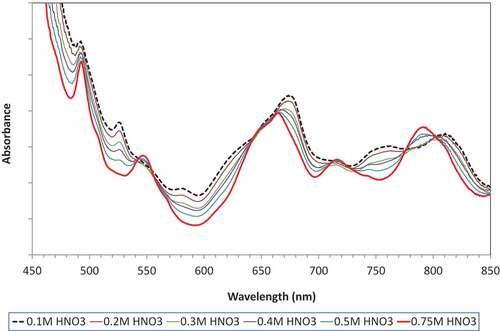
The UV-Vis spectra () clearly showed that the change in plutonium speciation was reversible. As the acidity of the solvent was increased through contact with aqueous phases with progressively higher nitric acid concentration there was a change in position and intensity of the absorption maxima until the characteristic Pu(IV) spectrum was obtained. The results showed that the change in speciation was not instantaneous once a threshold acid concentration is reached but was a gradual transition between the characteristic Pu(IV) spectra and the hydrolyzed Pu(IV) species. The presence of isosbestic points in the spectrum at 568, 769, and 860 nm indicates that the stoichiometry of the conversion between the species present in the solvent phase remained constant for the duration of these batch tests.[Citation76]
Figure 9. Change in UV-vis spectra of hydrolyzed Pu(IV) species following contact with aqueous solutions of increasing acid concentration.
Based on the literature it appears that while this may be the first example of extraction of the Pu(IV) polymer by amidic molecules, there are a few precedents for the solvent extraction of plutonium polymer[Citation55,Citation73,Citation74] and growing evidence that nano-clusters of specific and limited sizes can be formed,[Citation67–69,Citation71,Citation72] under specific conditions at least, similar to that hypothesized to be occurring here in the organic phase.
Plutonium speciation in (separate) TODGA and DMDOHEMA organic phases
Some additional experiments were performed to determine whether the spectral changes with low acidity could be attributed to the TODGA or DMDOHEMA ligands in the mixed EURO-GANEX solvent. Since, as Pu(IV) causes precipitation with TODGA at low concentrations (<5 g/L Pu),[Citation35] spectra of 0.5 mol/L TODGA were recorded at 3 g/L Pu(IV). Spectra were recorded following extractions from high (5 mol/L) and medium (3 mol/L) initial aqueous HNO3 alongside those following twice scrubbing the latter organic phases with 0.05 mol/L HNO3.
The spectra of Pu(IV) in 0.5 mol/L TODGA after extractions from 3 and 5 mol/L HNO3 were identical and in agreement with that reported previously by Liu et al.[Citation75] Significant changes were evident after scrubbing twice with 0.05 mol/L HNO3 ().
Figure 10. Pu(IV) spectra in (a) 0.5 mol/L TODGA phases and (b) 0.5 mol/L DMDOHEMA phases.
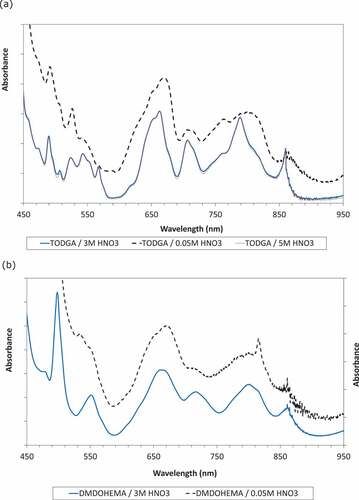
The spectra of Pu(IV) in 0.5 mol/L DMDOHEMA after extractions from 3 and 5 mol/L HNO3 were also identical. Changes are also observed in these spectra after scrubbing twice with 0.05 mol/L HNO3, including the appearance of a small sharp peak at 817 nm ().
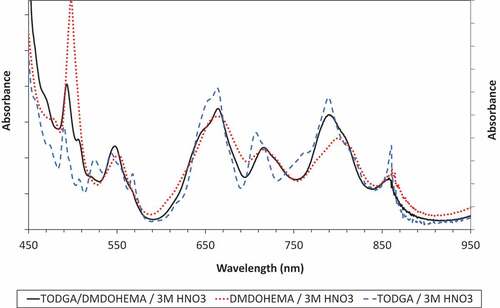
Comparing spectra at the higher acidity () indicates that the Pu(IV) spectrum in the EURO-GANEX solvent is a mix of the individual TODGA and DMDOHEMA spectra, as would be expected if both ligands are acting as extractants for Pu(IV). Only one organic-phase complex is found for all three solvent compositions at medium to high acidities.
Figure 11. Comparing Pu(IV) spectra from 0.5 mol/L TODGA, 0.5 mol/L DMDOHEMA, 0.2 mol/L TODGA + 0.5 mol/L DMDOHEMA after extraction from 3 mol/L HNO3.
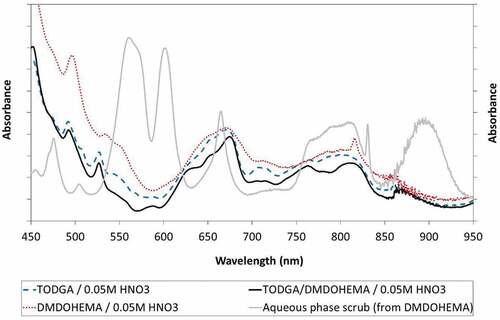
Comparing spectra taken after scrubbing with 0.05 mol/L HNO3 () indicates that the Pu(IV) spectrum in the EURO-GANEX solvent is more similar to that in TODGA; although the spectrum in DMDOHEMA is not too dissimilar, implying the same effect in all three solvent compositions is occurring. The interesting observation, however, was that in the solutions with DMDOHEMA some Pu(IV) was scrubbed to the aqueous phase and apparently disproportionated to Pu(III) (double peaks 550–650 nm) and Pu(VI) (peak at 830 nm).[Citation77] It is possible some Pu(VI) remained in the organic phase (peak 817 nm[Citation78]) but there was little evidence of Pu(III) in the organic phase. Therefore, it appears that while DMDOHEMA is important to the solvent to prevent precipitation at high Pu loadings, TODGA is important in fully retaining Pu(IV) in the organic phase at the low acidities where this process upset was tested. So at low acidities, it seems both TODGA and DMDOHEMA ligands are still acting as extractants for Pu(IV), but there is a speciation change as Pu(IV) hydrolyzes which is to an extent limited by the organic medium, i.e. solid colloids are not visibly formed nor do the spectra fully resemble those in ESI Figure S2.
Figure 12. Comparing Pu(IV) spectra from 0.5 mol/L TODGA, 0.5 mol/L DMDOHEMA, 0.2 mol/L TODGA + 0.5 mol/L DMDOHEMA after scrubbing with 0.05 mol/L HNO3. Also with the 0.05 mol/L HNO3 aqueous phase after scrubbing 0.5 mol/L DMDOHEMA.
Moreover, it is noted that the spectra show similar changes to those observed by Tamain[Citation71] and Chupin[Citation72] who reported Pu(IV) hexamers stabilized by organic ligands (ESI Figure S3), but as with the Pu(IV) colloid () and [Pu38O56Cl54(H2O)8]14- (ESI Figure S2) the spectra do not fully agree so the structure of the species observed in TODGA and DMDOHEMA solutions cannot be identified based on UV-Vis spectra alone.
Finally, partial least squares fitting was applied, scaling for the reduced plutonium concentration in TODGA (3 g/L cf. 5 g/L in DMDOHEMA), the aim being to combine the individual TODGA and DMDOHEMA spectra to compare with the EURO-GANEX spectrum, see ESI Figure S4. It is concluded that the contribution to the EURO-GANEX spectrum lies largely in the favour of complexation by TODGA rather than DMDOHEMA, implying that DMDOHEMA, which is present in greater concentration (0.5 mol/L cf. 0.2 mol/L TODGA) acts more as a phase modifier in this system. However, the residuals (inset ESI Figure S4), show some fluctuation that is probably indicative of a contribution from ternary complexes that have not been included in the fitting.
Conclusions
An experimental flowsheet trial has been successfully completed to assess the impact of a reduced scrub acid feed upon plutonium and americium in the EURO-GANEX flowsheet. This was identified as a potentially serious process upset during a safety review.
Surprisingly, a significant reduction in the scrub acid feed concentration, from 0.5 to 0.05 mol/L HNO3, did not result in the backwashing and recycle of plutonium in the extract-scrub contactors. A constant plutonium loading of the solvent was maintained under both normal and maloperation conditions. Moreover, a significant change in the UV-Vis spectrum of Pu(IV) was observed under the low acid conditions of the process upset. However, the species produced at low acidity are still retained in the solvent phase and did not lead to the misrouting or precipitation of plutonium. This was confirmed by the low losses of plutonium to the aqueous raffinate, <0.003%, obtained for both normal and off-normal flowsheet conditions. These results demonstrated the EURO-GANEX flowsheet is robust to the recycle or misrouting of plutonium for process upsets involving significant deviations of the scrub acidity.
Although routing of plutonium was not affected under the conditions of this process upset, the reduction in the scrub acidity led to the significant accumulation of americium. Analysis of the solvent product shows that under these conditions approximately 85% of the americium present in the active feed was being recycled between the extract and scrub sections of the flowsheet. By the end of the trial, the peak americium concentration in the solvent phase at stage 2 was over an order of magnitude higher than the original active feed.
Similar studies of other potential process upsets are required to fully define the operating envelope of the EURO-GANEX process but process models are really needed to undertake sensitivity studies before committing to further experimental trials. With regard to the specific scenario described here, extending the process upset to a full simulant (including lanthanides and fission products) to assess whether increased solvent saturation with other species affects the hydrodynamics (third phase) or extraction and routing of plutonium, minor actinides, and lanthanides would be useful.
Spectroscopic studies to characterize the species produced at low acidity suggested the formation of a hydrolyzed plutonium species or colloidal plutonium (formation of plutonium colloid in nitric acid solution occurs at <0.2 mol/L HNO3 for 10 g/L Pu solutions[Citation62]). It was shown that the species formed during the trial was the same as formed in batch tests and that it was reversible on raising the acidity again. This reversibility suggested the onward polymerization of the hydrolyzed plutonium species to colloidal plutonium is inhibited by the solvent shell around a plutonium-hydroxide core, some precedents for this concept exist already.[Citation67,Citation71,Citation72] The roles of both the TODGA and DMDOHEMA extractants were explored and it was found that the same hydrolysis occurs with solutions of the separate extractants although TODGA displays a greater role fully retaining Pu(IV) in the EURO-GANEX organic phase at low acidity. Further characterization of the hydrolyzed Pu(IV) species, using complementary methods such as EXAFS, in these amidic extractants is of fundamental interest.
LSEI_2136488_Supplementary_Material
Download PDF (604 KB)Acknowledgments
Financial support for this research was provided by the European Commission via the project SACSESS (FP7-Fission-2012 grant agreement no. 323282). Additional financial support was provided by the UK Nuclear Decommissioning Authority, Sellafield Ltd and NNL Core Science “ARIS” theme. Further support (for spectroscopic studies) was provided by the £46m Advanced Fuel Cycle Programme (AFCP) as part of the UK Department for Business, Energy and Industrial Strategy’s (BEIS) £505m Energy Innovation Programme. The authors thank C. Carrigan, J. Holt, C. Campbell, B. McLuckie (NNL) for technical assistance during the test and D. Woodhead and K. Wallace (NNL) for provision of Figure S1 (ESI).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplemental material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/07366299.2022.2136488.
Additional information
Funding
Notes
1 CHON indicates that the ligand contains only carbon, hydrogen, oxygen, and nitrogen atoms and so should be fully decomposable to gases.
References
- Management of Spent Nuclear Fuel and Its Waste, the European Academies’ Science Advisory Council (EASAC), Brussels, 2014, pp. 1–40.
- Baron, P., National Programmes in Chemical Partitioning: A Status Report, 5245, Nuclear Energy Agency, Organisation for Economic Cooperation and Development, 2010, pp. 1–120.
- Bourg, S.; Geist, A.; Adnet, J.-M.; Rhodes, C.; Hanson, B. C. Partitioning and Transmutation Strategy R&D for Nuclear Spent Fuel: The SACSESS and GENIORS Projects. EPJ Nucl. Sci. Technol. 2020, 6, 35. DOI: 10.1051/epjn/2019009.
- Bourg, S.; Hill, C.; Caravaca, C.; Rhodes, C.; Ekberg, C.; Taylor, R.; Geist, A.; Modolo, G.; Cassayre, L.; Malmbeck, R., et al. ACSEPT–Partitioning Technologies and Actinide Science: Towards Pilot Facilities in Europe. Nucl. Eng. Des. 2011, 241, 3427–3435. DOI: 10.1016/j.nucengdes.2011.03.011.
- Collins, E. D.; DelCul, G. D.; Spencer, B. B.; Jubin, R. T.; Maher, C.; Kim, I.-T.; Lee, H.; Fedorov, Y. S.; Saprykin, V. F.; Beznosyuk, V. I., et al. State-Of-The-Art Report on the Progress of Nuclear Fuel Cycle Chemistry, NEA No, 7267; OECD-NEA: Paris, France, 2018; pp. 1–300.
- Eynde, G. V. D.; Pedoux, S.; Trtilek, R.; Fritz, L.; Evans, C.; Mathonnière, G.; Werf, J. V. D.; Lucibello, P.; Suzuki, K.; Sano, T., et al. Strategies and Considerations for the Back End of the Fuel Cycle, NEA 7469; Paris, France: OECD-NEA, 2021; pp. 1–72.
- Poinssot, C.; Boullis, B.; Bourg, S. Role of Recycling in Advanced Nuclear Fuel Cycles. In Reprocessing and Recycling of Spent Nuclear Fuels; Taylor, R. J., Ed.; Woodhouse Publishing Ltd: Oxford, 2015; pp. 27-48.
- Taylor, R.; Bodel, W.; Stamford, L.; Butler, G. A Review of Environmental and Economic Implications of Closing the Nuclear Fuel Cycle. Part 1: Wastes and Environmental Impacts. Energies. 2022, 15, 1433. DOI: 10.3390/en15041433.
- Taylor, R.; Bodel, W.; Butler, G. A Review of Environmental and Economic Implications of Closing the Nuclear Fuel Cycle-Part Two: Economic Impacts. Energies. 2022, 15, 2472. DOI: 10.3390/en15072472.
- González-Romero, E. M. Impact of Partitioning and Transmutation on the High Level Waste Management. Nucl. Eng. Des. 2011, 241, 3436–3444. DOI: 10.1016/j.nucengdes.2011.03.030.
- Grenèche, D.; Quiniou, B.; Boucher, L.; Delpech, M.; Gonzalez, E.; Alvarez, F.; Cunado, M. A.; Serrano, A.; Cormenzana, J. L.; Kuckshinrichs, W., et al. RED-IMPACT: Impact of Partitioning, Transmutation and Waste Reduction Technologies on the Final Nuclear Waste Disposal. Synthesis Report Volume 15; Forschungszentrum Julich GmbH: Julich, 2008; pp. 1–187.
- Nishihara, K.; Nakayama, S.; Morita, Y.; Oigawa, H.; Iwasaki, T. Impact of Partitioning and Transmutation on LWR High-Level Waste Disposal. J. Nucl. Sci. Technol. 2008, 45, 84–97. DOI: 10.1080/18811248.2008.9711418.
- Nishihara, K.; Oigawa, H.; Nakayama, S.; Ono, K.; Shiotani, H. Impact of Partitioning and Transmutation on High-Level Waste Disposal for the Fast Breeder Reactor Fuel Cycle. J. Nucl. Sci. Technol. 2010, 47, 1101–1117. DOI: 10.1080/18811248.2010.9720977.
- Salvatores, M.; Palmiotti, G. Radioactive Waste Partitioning and Transmutation Within Advanced Fuel Cycles: Achievements and Challenges. Prog. Part. Nucl. Phys. 2011, 66, 144–166. DOI: 10.1016/j.ppnp.2010.10.001.
- Serp, J.; Poinssot, C.; Bourg, S. Assessment of the Anticipated Environmental Footprint of Future Nuclear Energy Systems. Evidence of the Beneficial Effect of Extensive Recycling. Energies. 2017, 10(9), 1445. DOI: 10.3390/en10091445.
- Baron, P.; Cornet, S. M.; Collins, E. D.; DeAngelis, G.; Del Cul, G.; Fedorov, Y.; Glatz, J. P.; Ignatiev, V.; Inoue, T.; Khaperskaya, A., et al. A Review of Separation Processes Proposed for Advanced Fuel Cycles Based on Technology Readiness Level Assessments. Prog. Nucl. Energy. 2019, 117, 103091. DOI: 10.1016/j.pnucene.2019.103091.
- Modolo, G.; Geist, A.; Miguirditchian, M. Minor Actinide Separations in the Reprocessing of Spent Nuclear Fuels: Recent Advances in Europe. In Reprocessing and Recycling of Spent Nuclear Fuels; Taylor, R. J., Ed.; Woodhouse Publishing Ltd: Oxford, 2015; pp. 245-288.
- Moyer, B. A.; Lumetta, G. J.; Mincher, B. J. 11 - Minor Actinide Separation in the Reprocessing of Spent Nuclear Fuels: Recent Advances in the United States. In Reprocessing and Recycling of Spent Nuclear Fuel; Taylor, R., Ed.; Woodhead Publishing: Oxford, 2015; pp. 289–312.
- Nash, K. L.; Lumetta, G. J. Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment; Woodhouse Publishing Ltd: Oxford, 2011; pp. 1–492.
- Natarajan, R. 9 - Reprocessing of Spent Fast Reactor Nuclear Fuels. In Reprocessing and Recycling of Spent Nuclear Fuel; Taylor, R., Ed.; Woodhead Publishing: Oxford, 2015; pp. 213–243.
- Poinssot, C.; Rostaing, C.; Greandjean, S.; Boullis, B. Recycling the Actinides, the Cornerstone of Any Sustainable Nuclear Fuel Cycles. Procedia Chem. 2012, 7, 349–357. DOI: 10.1016/j.proche.2012.10.055.
- Geist, A.; Adnet, J.-M.; Bourg, S.; Ekberg, C.; Galán, H.; Guilbaud, P.; Miguirditchian, M.; Modolo, G.; Rhodes, C.; Taylor, R. An Overview of Solvent Extraction Processes Developed in Europe for Advanced Nuclear Fuel Recycling, Part 1 — Heterogeneous Recycling. Sep. Sci. Technol. 2020, 56, 1866–1881. DOI: 10.1080/01496395.2020.1795680.
- Denniss, I. S.; Jeapes, A. P. Reprocessing Irradiated Fuel. In The Nuclear Fuel Cycle; Wilson, P. D., Ed.; Oxford Science Publications: Oxford, 1996; pp. 116-137.
- Herbst, R. S.; Baron, P.; Nilsson, M. Standard and Advanced Separation: PUREX Processes for Nuclear Fuel Reprocessing. In Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment; Nash, K. L. Lumetta, G. J., Eds.; Woodhouse Publishing Ltd: Oxford, 2011; pp. 141–175.
- Lewin, R. G.; Harrison, M. T. 15 - International Developments in Electrorefining Technologies for Pyrochemical Processing of Spent Nuclear Fuels. In Reprocessing and Recycling of Spent Nuclear Fuel; Taylor, R., Ed.; Woodhead Publishing: Oxford, 2015; pp. 373–413.
- Lyseid Authen, T.; Adnet, J.-M.; Bourg, S.; Carrott, M.; Ekberg, C.; Galán, H.; Geist, A.; Guilbaud, P.; Miguirditchian, M.; Modolo, G., et al. An Overview of Solvent Extraction Processes Developed in Europe for Advanced Nuclear Fuel Recycling, Part 2 — Homogeneous Recycling. Sep. Sci. Technol. 2022, 57, 1724–1744. DOI: 10.1080/01496395.2021.2001531.
- Aneheim, E.; Ekberg, C.; Fermvik, A.; Foreman, M. R. S. J.; Grűner, B.; Hájková, Z.; Kvičalová, M. A TBP/BTBP-Based GANEX Separation Process—part 2: Ageing, Hydrolytic, and Radiolytic Stability. Solvent Extr. Ion Exch. 2011, 29, 157–175. DOI: 10.1080/07366299.2011.539462.
- Aneheim, E.; Ekberg, C.; Fermvik, A.; Foreman, M. R. S. J.; Retegan, T.; Skarnemark, G. A TBP/BTBP-Based GANEX Separation Process. Part 1: Feasibility. Solvent Extr. Ion Exch. 2010, 28, 437–458. DOI: 10.1080/07366299.2010.480930.
- Carrott, M.; Bell, K.; Brown, J.; Geist, A.; Gregson, C.; Hères, X.; Maher, C.; Malmbeck, R.; Mason, C.; Modolo, G., et al. Development of a New Flowsheet for Co-Separating the Transuranic Actinides: The “EURO-GANEX” Process. Solvent Extr. Ion Exch. 2014, 32, 447–467. DOI: 10.1080/07366299.2014.896580.
- Miguirditchian, M.; Roussel, H.; Chareyre, L.; Baron, P. HA Demonstration in the Atalante Facility of the GANEX 2nd Cycle for the Grouped TRU Extraction. In Global 2009; American Nuclear Society: Paris, France, 2009; p. 9378.
- Malmbeck, R.; Magnusson, D.; Bourg, S.; Carrott, M.; Geist, A.; Hérès, X.; Miguirditchian, M.; Modolo, G.; Müllich, U.; Sorel, C., et al. Homogenous Recycling of Transuranium Elements from Irradiated Fast Reactor Fuel by the EURO-GANEX Solvent Extraction Process. Radiochim. Acta. 2019, 107, 917–929. DOI: 10.1515/ract-2018-3089.
- Lyseid Authen, T.; Wilden, A.; Halleröd, J.; Schneider, D.; Kreft, F.; Modolo, G.; Ekberg, C. Batch Tests for Optimisation of Solvent Composition and Process Flexibility of the CHALMEX FS-13 Process. Solvent Extr. Ion Exch. 2021, 39, 1–17. DOI: 10.1080/07366299.2020.1797988.
- Lyseid Authen, T.; Wilden, A.; Schneider, D.; Kreft, F.; Modolo, G.; StJ Foreman, M. R.; Ekberg, C. Batch Flowsheet Test for a GANEX-Type Process: The CHALMEX FS-13 Process. Solvent Extr. Ion Exch. 2021, 40, 189–202. DOI: 10.1080/07366299.2021.1890372.
- Carrott, M.; Maher, C.; Mason, C.; Sarsfield, M.; Taylor, R. TRU-SANEX”: A Variation on the EURO-GANEX and I-SANEX Processes for Heterogeneous Recycling of Actinides Np-Cm. Sep. Sci. Technol. 2016, 51, 2198–2213. DOI: 10.1080/01496395.2016.1202979.
- Brown, J.; McLachlan, F.; Sarsfield, M. J.; Taylor, R. J.; Modolo, G.; Wilden, A. Plutonium Loading of Prospective Grouped Actinide Extraction (GANEX) Solvent Systems Based on Diglycolamide Extractants. Solvent Extr. Ion Exch. 2012, 30, 127–141. DOI: 10.1080/07366299.2011.609378.
- Carrot, M. J.; Gregson, C. R.; Taylor, R. J. Neptunium Extraction and Stability in the GANEX Solvent: 0.2 M TODGA/0.5 M DMDOHEMA/Kerosene. Solvent Extr. Ion Exch. 2013, 31, 463–482. DOI: 10.1080/07366299.2012.735559.
- Carrott, M.; Geist, A.; Hères, X.; Lange, S.; Malmbeck, R.; Miguirditchian, M.; Modolo, G.; Wilden, A.; Taylor, R. Distribution of Plutonium, Americium and Interfering Fission Products Between Nitric Acid and a Mixed Organic Phase of TODGA and DMDOHEMA in Kerosene, and Implications for the Design of the “EURO-GANEX” Process. Hydrometallurgy. 2015, 152, 139–148. DOI: 10.1016/j.hydromet.2014.12.019.
- Plaue, J.; Gelis, A.; Czerwinski, K. Plutonium Third Phase Formation in the 30% TBP/Nitric Acid/Hydrogenated Polypropylene Tetramer System. Solvent Extr. Ion Exch. 2006, 24, 271–282. DOI: 10.1080/07366290600646814.
- Geist, A.; Müllich, U.; Magnusson, D.; Kaden, P.; Modolo, G.; Wilden, A.; Zevaco, T. Actinide(iii)/Lanthanide(iii) Separation via Selective Aqueous Complexation of Actinides(iii) Using a Hydrophilic 2,6-Bis(1,2,4-Triazin-3-Yl)-Pyridine in Nitric Acid. Solvent Extr. Ion Exch. 2012, 30, 433–444. DOI: 10.1080/07366299.2012.671111.
- Carrott, M. J.; Fox, O. D.; Maher, C. J.; Mason, C.; Taylor, R. J.; Sinkov, S. I.; Choppin, G. R. Solvent Extraction Behavior of Plutonium (IV) Ions in the Presence of Simple Hydroxamic Acids. Solvent Extr. Ion Exch. 2007, 25, 723–745. DOI: 10.1080/07366290701634560.
- Hanson, B. 6 - Process Engineering and Design for Spent Nuclear Fuel Reprocessing and Recycling Plants. In Reprocessing and Recycling of Spent Nuclear Fuel; Taylor, R., Ed.; Woodhead Publishing: Oxford, 2015; pp. 125–151.
- Sypula, M.; Wilden, A.; Schreinemachers, C.; Malmbeck, R.; Geist, A.; Taylor, R.; Modolo, G. Use of Polyaminocarboxylic Acids as Hydrophilic Masking Agents for Fission Products in Actinide Partitioning Processes. Solvent Extr. Ion Exch. 2013, 3, 748–764. DOI: 10.1080/07366299.2012.700591.
- Williams, L. G. 4 - Safety and Security Issues in the Reprocessing and Recycling of Spent Nuclear Fuels for Advanced Fuel Cycles. In Reprocessing and Recycling of Spent Nuclear Fuel; Taylor, R., Ed.; Woodhead Publishing: Oxford, 2015; pp. 63–90.
- Sutton, A., Initial Recovery of the Magnox Reprocessing Plant Following a Loss of Reductant Feed in the Solvent Extraction Process, in: International Conference on Nuclear Criticality Safety 2015 Curran Associates, Inc., Red Hook, New York, Charlotte, North Carolina, USA 2015, pp. 113–122.
- McLachlan, F.; Taylor, R.; Whittaker, D.; Woodhead, D.; Geist, A. Modelling of Innovative SANEX Process Maloperations. Procedia Chem. 2016, 21, 109–116. DOI: 10.1016/j.proche.2016.10.016.
- Geist, A.; Taylor, R.; Ekberg, C.; Guilbaud, P.; Modolo, G.; Bourg, S. The SACSESS Hydrometallurgy Domain — an Overview. Procedia Chem. 2016, 21, 218–222. DOI: 10.1016/j.proche.2016.10.031.
- Bourg, S.; Guilbaud, P.; Mendes, E.; Ekberg, C.; Gibilaro, M.; Soucek, P.; Modolo, G.; Geist, A.; Boo, E.; Duplantier, B., et al., SACSESS: Final Report, SACSESS – R01.3 – rev 0, CEA, France, 2016, pp. 1–35.
- Macerata, E.; Mossini, E.; Scaravaggi, S.; Mariani, M.; Mele, A.; Panzeri, W.; Boubals, N.; Berthon, L.; Charbonnel, M.-C.; Sansone, F., et al. Hydrophilic Clicked 2,6-Bis-Triazolyl-Pyridines Endowed with High Actinide Selectivity and Radiochemical Stability: Toward a Closed Nuclear Fuel Cycle. J. Am. Chem. Soc. 2016, 138, 7232–7235. DOI: 10.1021/jacs.6b03106.
- Wilden, A.; Schneider, D.; Paparigas, Z.; Henkes, M.; Kreft, F.; Geist, A.; Mossini, E.; Macerata, E.; Mariani, M.; Gullo, M. C., et al. Selective Actinide(iii) Separation Using 2,6-Bis[1-(Propan-1-Ol)-1,2,3-Triazol-4-Yl]pyridine (PyTri-Diol) in the Innovative-SANEX Process: Laboratory Scale Counter Current Centrifugal Contactor Demonstration. Radiochim. Acta. 2022, 110, 515–525. DOI: 10.1515/ract-2022-0014.
- Taylor, R.; Bourg, S.; Glatz, J.-P.; Modolo, G. Development of Actinide Separation Processes for Future Nuclear Fuel Cycles in Europe. Nucl. Future. 2015, 11, 38–43.
- Bell, K. The PuMa Lab at NNL. Nucl. Future. 2012, 8, 35–39.
- Brown, J.; Campbell, C.; Carrigan, C.; Carrott, M.; Greenough, K.; Maher, C.; McLuckie, B.; Mason, C.; Gregson, C.; Griffiths, T., et al. Americium and Plutonium Purification by Extraction (The AMPPEX Process): Development of a New Method to Separate 241am from Aged Plutonium Dioxide for Use in Space Power Systems. Prog. Nucl. Energy. 2018, 106, 396–416. DOI: 10.1016/j.pnucene.2018.02.008.
- Baker, A.; Fells, A.; Carrott, M. J.; Maher, C. J.; Hanson, B. C. Process Intensification of Element Extraction Using Centrifugal Contactors in the Nuclear Fuel Cycle. Chem. Soc. Rev. 2022, 51, 3964–3999. DOI: 10.1039/D2CS00192F.
- Connick, R. E.; Kasha, M.; McVey, W. H.; Sheline, G. E. Spectrophotometric Studies of Plutonium in Aqueous Solution. In The Transuranium Elements; Seaborg, G. T., Katz, J. J. Manning, W. M., Eds.; McGraw-Hill Book Company: New York, 1949; pp. 559–601.
- Ockenden, D. W.; Welch, G. A. 653. The Preparation and Properties of Some Plutonium Compounds. Part V. Colloidal Quadrivalent Plutonium. J. Chem. Soc. (Resumed) 1956, 3358–3363. DOI:10.1039/jr9560003358.
- Hindman, J. C. Ionic and Molecular Species of Plutonium in Solution. In The Actinide Elements; Seaborg, G. T. Katz, J. J., Eds.; McGraw-Hill Book Company: USA, Ann Arbor, 1954; pp. 301–370.
- Cleveland, J. M., The Chemistry of Plutonium, In, American Nuclear Society, La Grange Park, 1979, pp. 396–409.
- Neck, V.; Altmaier, M.; Fanghänel, T. Solubility of Plutonium Hydroxides/Hydrous Oxides Under Reducing Conditions and in the Presence of Oxygen. C. R. Chim. 2007, 10, 959–977. DOI: 10.1016/j.crci.2007.02.011.
- Clark, D. L.; Hecker, S. S.; Jarvinen, G. D.; Neu, M. P. Plutonium. In Actinide and Transactinide Elements; Morss, L. R., Edelstein, N. M., Fuger, J. Katz, J. J., Eds.; Springer: AA Dordrecht, 2006; pp. 813–1264.
- Cleveland, J. M. Solution Chemistry of Plutonium. In Plutonium Handbook; Wick, O. J., Ed.; American Nuclear Society: La Grange Park, 1980; pp. 403–520.
- Altmaier, M.; Gaona, X.; Fellhauer, D.; Clark, D. L.; Runde, W. H.; Hobart, D. E. Aqueous Solution and Coordination Chemistry of Plutonium. In Plutonium Handbook; Clark, D. L., Geeson, D. A. Hanrahan, R. J., Eds.; American Nuclear Society: La Grange Park, 2019; pp. 1543–1726.
- Brunstad, A. Polymerization and Precipitation of Plutonium(iv) in Nitric Acid. Ind. Eng. Chem. 1959, 51, 38–40. DOI: 10.1021/ie50589a031.
- Paviet-Hartmann, P.; Senentz, G. Prevention of Pu(iv) Polymerization in a Purex-Based Process. In Global 2007; American Nuclear Society: Boise, Idaho, 2007; pp. 1865–1869.
- Lloyd, M. H.; Haire, R. G. The Chemistry of Plutonium in Sol-Gel Processes. Radiochim. Acta. 1978, 25, 139–148. DOI: 10.1524/ract.1978.25.34.139.
- Scoazec, H.; Pasquiou, J. Y.; Germain, M., Some Plutonium IV Polymers Properties in PUREX Process, in: I.Chem.E. Symposium Series No.119, Rugby, UK: Institution of Chemical Engineers, 1990, pp. 221–233.
- Walther, C.; Cho, H. R.; Marquardt, C. M.; Neck, V.; Seibert, A.; Yun, J. I.; Fanghänel, T. Hydrolysis of Plutonium(iv) in Acidic Solutions: No Effect of Hydrolysis on Absorption-Spectra of Mononuclear Hydroxide Complexes. Radiochim. Acta. 2007, 95, 7–16. DOI: 10.1524/ract.2007.95.1.7.
- Soderholm, L.; Almond, P. M.; Skanthakumar, S.; Wilson, R. E.; Burns, P. C. The Structure of the Plutonium Oxide Nanocluster [Pu38o56cl54(h2o)8]14−. Angew. Chem. Int. Ed. 2008, 47, 298–302. DOI: 10.1002/anie.200704420.
- Wilson, R. E.; Skanthakumar, S.; Soderholm, L. Separation of Plutonium Oxide Nanoparticles and Colloids. Angew. Chem. Int. Ed. 2011, 50, 11234–11237. DOI: 10.1002/anie.201105624.
- Sigmon, G. E.; Hixon, A. E. Extension of the Plutonium Oxide Nanocluster Family to Include {pu16} and {pu22}. Chem. Eur. J. 2019, 25, 2463–2466. DOI: 10.1002/chem.201805605.
- Ekberg, C.; Larsson, K.; Skarnemark, G.; Ödegaard-Jensen, A.; Persson, I. The Structure of Plutonium(IV) Oxide as Hydrolysed Clusters in Aqueous Suspensions. Dalton Trans. 2013, 42, 2035–2040. DOI: 10.1039/C2DT32185H.
- Tamain, C.; Dumas, T.; Guillaumont, D.; Hennig, C.; Guilbaud, P. First Evidence of a Water-Soluble Plutonium(iv) Hexanuclear Cluster. Eur. J. Inorg. Chem. 2016, 2016, 3536–3540. DOI: 10.1002/ejic.201600656.
- Chupin, G.; Tamain, C.; Dumas, T.; Solari, P. L.; Moisy, P.; Guillaumont, D. Characterization of a Hexanuclear Plutonium(iv) Nanostructure in an Acetate Solution via Visible–Near Infrared Absorption Spectroscopy, Extended X-Ray Absorption Fine Structure Spectroscopy, and Density Functional Theory. Inorg. Chem. 2022, 61, 4806–4817. DOI: 10.1021/acs.inorgchem.1c02876.
- Muscatello, A. C.; Navratil, J. D.; Killion, M. E. Solvent Extraction of Plutonium (IV) Polymer by Dihexyl-N, N-Diethyl-Carbamoylmethylphosphonate (DHDECMP. Sep. Sci. Technol. 1983, 18, 1731–1746. DOI: 10.1080/01496398308056124.
- Thiyagarajan, P.; Diamond, H.; Soderholm, L.; Horwitz, E. P.; Toth, L. M.; Felker, L. K. Plutonium(iv) Polymers in Aqueous and Organic Media. Inorg. Chem. 1990, 29, 1902–1907. DOI: 10.1021/ic00335a028.
- Liu, Q.; Zhou, J.; Zhu, L.; Zhang, Y.; Li, D.; Yang, S.; Tian, G. Extraction of Actinides Including Neptunyl (V) by Asymmetrical N,N’-Dimethyl-N,N’-Dioctyl-Diglycolamide in Comparison with Symmetrical N,N,N’,N’-Tetraoctyl-Diglycolamide—sterically Structural Insight into Actinide Separations†. Solvent Extr. Ion Exch. 2020, 38, 485–495. DOI: 10.1080/07366299.2020.1765493.
- McNaught, A. D.; Wilkinson, A. IUPAC. Compendium of Chemical Terminology, 2nd Ed. (The “Gold Book”); Blackwell Scientific Publications: Oxford, 1997 (Online version (2019-) created by S. J. Chalk). DOI: 10.1351/goldbook
- Wilson, R.; Hu, Y.; Nitsche, H., Detection and Quantification of Pu(III, IV, V, and VI) Using a 1.0-meter Liquid Core Waveguide, in, Lawrence Berkeley National Laboratory, 2005. https://escholarship.org/uc/item/15k4s3p3.
- Lumetta, G. J.; Heller, F. D.; Hall, G. B.; Asmussen, S. E.; Sinkov, S. I. Optical Spectroscopic Investigation of Hexavalent Actinide Ions in N-Dodecane Solutions of Tri-Butyl Phosphate. Solvent Extr. Ion Exch. 2021, 39, 56–73. DOI: 10.1080/07366299.2020.1805051.