Abstract
The sirtuin family of histone deacetylases (HDACs) was named after their homology to the Saccharomyces cerevisiae gene silent information regulator 2 (Sir2). In the yeast, Sir2 has been shown to mediate the effects of calorie restriction on the extension of life span and high levels of Sir2 activity promote longevity. Like their yeast homologs, the mammalian sirtuins (SIRT1‐7) are class III HDACs and require NAD+ as a cofactor to deacetylate substrates ranging from histones to transcriptional regulators. Through this activity, sirtuins are shown to regulate important biological processes ranging from apoptosis, adipocyte and muscle differentiation, and energy expenditure to gluconeogenesis. We review here the current knowledge regarding the role of sirtuins in metabolism, longevity, and discuss the possible therapeutic applications that could result from the understanding of their function in different organs and pathologies.
| Abbreviations | ||
| SIR2 | = | silent information regulator 2 |
| HAT | = | histone acetyltransferase |
| HDAC | = | histone deacetylase |
| CR | = | calorie restriction |
| PKB/AKT | = | protein kinase B |
| GDP | = | guanosine diphosphate |
| GTP | = | guanosine triphosphate |
| NAD+ | = | nicotinamide adenine dinucleotide |
| NADH | = | reduced form of NAD+ |
| Pol II | = | RNA polymerase II |
| TFIIB | = | transcription factor IIB |
| TFIIE | = | transcription factor IIE |
| Daf‐16 | = | ‘dauer’ larvae transcription factor‐16 |
| FOXO | = | forkhead box subgroup ‘O’ transcription factor |
| HML | = | homothallic mating‐type loci left |
| HMR | = | homothallic mating‐type loci right |
| NFκB | = | nuclear factor kappa B transcription factor |
| PGC‐1α | = | peroxisome proliferator‐activated receptor gamma (PPARγ) coactivator‐1α |
Introduction
Histone acetylation is the main type of covalent histone modification and is achieved by a class of enzymes termed histone acetyltransferases (HATs) Citation1. HATs acetylate histones on lysines, whereas histone deacetylation involves another family of enzymes, the histone deacetylases (HDACs) (Citation2, a review). These HDACs are classified in three groups on the basis of their homology with the yeast Saccharomyces cerevisiae HDACs: RPD3 (group I), HDA1 (group II), and Sir2 (group III). Class I and II enzymes are inhibited by trichostatin (TSA), whereas class III HDACs are not inhibited by TSA and are NAD+‐dependent Citation3,4, suggesting a regulation of Sir2 molecules by the metabolic state of the cells (reviewed in Citation5). Since the Sir2 family of proteins are also able to exert their enzymatic activity not only on histones but also on numerous other proteins, such as the transcriptional regulators, they are involved in many cellular processes, ranging in yeast from gene silencing, DNA repair, progression of the cell cycle, to the control of ageing.
The Sir2 protein first deacetylates histones and then localizes with the yeast protein Sir4, to form a tight silencing complex binding to the telomeres Citation6. The localization of Sir2 complex to the histone tails, initially identified to induce transcriptional repression of the silent mating type loci HML and HMR (homothallic mating‐type loci left and right, respectively) Citation7,8, produces chromatin silencing, and a recent study reported that the Sir2 complex reduces the promoter occupancy by the transcription factors IIB and IIE (TFIIB, TFIIE) and the RNA polymerase II (Pol II) preventing hence the proper assembly of the preinitiation complex of the transcriptional machinery Citation9. Most interesting was the fact that overexpression of the gene encoding the Sir2 protein leads to a decrease in histone acetylation Citation10 and an increase in life span in yeast Citation11, in the nematode Caenorhabditis elegansCitation12 and in metazoans Citation13. Likewise, Sir2‐activating compounds (STACs), such as resveratrol, present in the skin of red grapes, promote longevity in yeast Citation14 and other organisms ranging from the worm and drosophila Citation15 to the mouse Citation16. On the other hand, both mutations of the Sir2 gene and pharmacological inhibition of Sir2 protein by nicotinamide induces an acceleration of ageing in yeast Citation17. Although the functions of Sir2 have been relatively well established in yeast and C. elegans, their function in mammals remains rather elusive. This review will give an overview of the role of the sirtuin family of proteins in different species and their potential contribution to disease.
SIRT1/Sir2, caloric restriction, metabolism, neurodegeneration, and longevity
In the last few years, an increasing number of studies from S. cerevisiae, C. elegans, Drosophila melanogaster, and mouse models have linked caloric restriction (CR) and metabolism with longevity. The matter has been extensively reviewed by Bordone and Guarente Citation18; in this section we give an overview of how SIRT1, the mammalian Sir2 homolog, which is the best characterized sirtuin family member, and CR could affect metabolism and longevity in humans. To better understand the mechanisms by which CR extends life span, it is instructive to take a close look at studies carried out in various species.
Studies based on non‐mammalian models
Studies in Saccharomyces cerevisiae
In yeast, reduction in glucose levels in the media, mimicking CR, results in a substantial extension in life span, which is Sir2p‐ and NAD+‐dependent, since mutants for Sir2p and nicotinate phosphoribosyl transferase (NPT1), an enzyme required for NAD+ formation, failed to reproduce this effect Citation19. In the same study, mutants for components of the glucose‐signaling pathway, such as the GTP‐GDP (guanosine triphosphate‐guanosine diphosphate) exchange factor CDC25, the glucose sensing receptors gpr1 and gpa2, and hexokinase, the first enzyme in the glycolytic pathway, mimicked the CR‐mediated increase in longevity. This life span extension due to CR was attributed to an increase in respiration Citation20. Indeed a mutation in the gene encoding the cytochrome c1 CYT1 is no longer able to promote CR‐mediated longevity, suggesting that blocking the mitochondrial electron transport chain and respiration prevents life span extension. Moreover, overexpression of Hap4 in yeast, a gene known to cause a switch from fermentation to respiration, yielded a 35% extension in life span under normal glucose conditions. It is well established that respiration increases the NAD+/NADH ratio through oxidation of NADH by the NADH dehydrogenase Citation21.
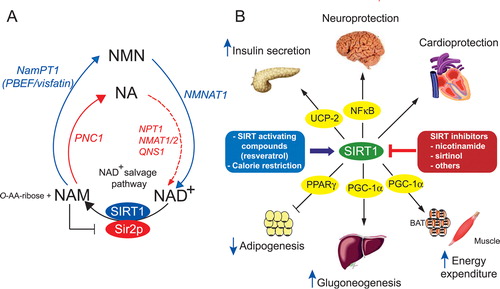
The Sir2p‐mediated conversion of NAD+ leads to the formation of nicotinamide (NAM), a powerful Sir2/SIRT1 inhibitor Citation17 and O‐acetyl‐ADP‐ribose (O‐AA‐ribose) Citation22 (). Recent evidence demonstrated an active role of O‐AA‐ribose in the regulation of gene silencing by the Sir2/3/4 assembly complex Citation23. In yeast, an alternative route to synthesize NAD+, other than its de novo production from the amino acid tryptophan, and which starts from nicotinic acid (NA), is present under the form of the NAD+ salvage pathway. In the NAD+ salvage pathway, NAM obtained from NAD+ cleavage is deaminated to NA by the nicotinamidase PNC1. NA formed through the actions of PNC1 then undergoes a series of reactions giving successively rise to nicotinate mononucleotide (NAMN), nicotinate adenine dinucleotide (NAAD), and NAD+, that are catalyzed by the enzymes nicotinate phosphoribosyl transferase (NPT1), nicotinate mononucleotide adenyltransferases (NMAT1/2), and NAD+ synthetase (QNS1), respectively Citation24. The newly recycled NAD+ then serves again as a cofactor for Sir2p‐mediated deacetylation ().
Multiple protagonists of the NAD+ salvage pathway participate in life span extension in yeast. Furthermore, an intense debate about whether Sir2p‐dependent CR life span extension could be due to a depletion in the noncompetitive Sir2p inhibitor NAM, rather than a modification in the NAD+/NADH ratio, is still ongoing. Interestingly, CR increased the expression of PNC1 and hence PNC1 activity, which is required and sufficient to extend life span in yeast through Sir2p activation. Conversely, mutations in the yeast PNC1 gene accelerate cellular ageing Citation25. These observations hence suggested that NAM depletion, through the activation of PNC1, is sufficient to activate Sir2 and increase longevity in yeast. Moreover, mutation of PNC1 and overexpression of Nnt1, a NAM methyltransferase which reduced the excess NAM levels, restored the CR‐induced life span extension in PNC1 mutants, suggesting that other factors (such as NADH lowering) might play a role in CR‐mediated life span extension Citation26. Other components of the NAD+ salvage pathway, like NPT1, the rate‐limiting enzyme in the NAD+ biosynthesis, have also been shown to regulate life span in yeast. Increased dosage of NPT1, but not of NMAT, enhanced the total cellular NAD+ levels and enhanced the transcriptional activity of the catalytic domain of Sir2p thereby extending yeast life span Citation27, whereas NPT1 mutations yielded a defect in silencing at silent mating‐type loci, telomeres, and rDNA Citation28. Overexpression of two related mitochondrial NADH dehydrogenases, Nde1 and Nde2, decreased NADH levels without changing NAD+ levels, but still increased yeast life span both under normal glucose culture conditions (2%) and CR conditions (0.5%), suggesting that reducing the quantity of the competitive Sir2p inhibitor NADH might also contribute to the CR‐induced increase in yeast longevity Citation26. Interestingly, Hst2, a Sir2 homolog that contributes to rDNA stability, is another mediator of CR‐induced life span extension in yeast, indicating that other Sir2 homologs could regulate longevity under CR conditions Citation29. Taken together, these data suggest that Sir2 requires NAD+ to deacetylate proteins, and manipulations of the salvage pathway that converge on enhanced NAD+ availability induce Sir2‐dependent life span extension in the budding yeast Citation24. Under CR other pathways, such as a decrease in the competitive Sir2 inhibitor NADH (by increasing Nde activity) or the noncompetitive Sir2 inhibitor NAM (by increasing PNC1 activity), could contribute to control Sir2 activity. Furthermore, a Sir2‐independent CR‐mediated life span extension mechanism exists that is mediated via Hst2.
Three important factors are thought to participate in Sir2‐mediated longevity. First, one of the consequences of increased Sir2 activity is the significant reduction in the number of extrachromosomal circular DNA repeats (ERCs) Citation11, whose accumulation is deleterious to yeast and accelerates ageing Citation30, probably through sequestration of transcription factors essential for their replication. The second mechanism that contributes to the yeast life span acts via the fermentable growth medium‐induced (FGM) pathway. Under fermentation conditions, glucose and other nutrients activate a yet unknown pathway that activates Sch9, a kinase related to the mammalian protein kinase B (PKB)/AKT, a negative regulator of the stress resistance genes. This results in the subsequent accumulation of reactive oxygen species (ROS), thereby accelerating ageing Citation31. Interestingly, mutation of Sch9 yields a substantial increase in yeast life span probably through increasing stress resistance Citation32,33 (). It is at present not known whether this pathway is similar to Daf‐16/FOXO signaling, which participates in the control of ageing in other organisms, and whether Sch9 mutations affect ERCs formation. Finally, yeast evolving in a high osmolarity live much longer than those in a balanced osmolarity medium. A high osmolarity shock activates a subset of osmotic responsive genes that change the metabolism to favor glycerol biosynthesis thereby generating more NAD+, the Sir2 cofactor necessary to mediate longevity Citation34.
Figure 1. Sir2/SIRT1 and signaling pathways in different species. In yeast Sir2p activity is increased through increased expression of the NAD+ salvage pathway enzyme, nicotinamidase PNC1, but also by increasing the NAD+/NADH ratio (or lowering NADH levels) in response to calorie restriction (CR). Mutation of the glucose sensing receptors Gpr1 and Gpa2 mimics the CR effects on Sir2p, which inhibit the formation of the deleterious extrachromosomal circular DNA repeats (ERCs) and contribute to life span extension in yeast. The glucose and nutrients fermentation pathway activates the AKT‐related kinase Sch9 which induces oxidative stress and participates in yeast ageing. Inactivation of this pathway promotes longevity in yeast. The longevity pathway is conserved amongst worms, flies and mammals. Activation of the insulin growth factor receptor IGFR (Daf‐2/dIGFR) activates the insulin receptor substrate IRS (p65/CHICO), which stimulates phosphoinositol‐3 kinase PI3K (AGE‐1/dPI3K), which in turn phosphorylates the PKB/AKT kinase. AKT phosphorylates and inactivates FOXO (Daf‐16/dFOXO). Inactivation of the insulin receptor pathway promotes longevity in worms, drosophila, and mammals through increased activity of FOXO. Sir2/SIRT1 activation induces life span extension through interaction with Daf‐16/FOXO factors. Gpr‐1 = G‐protein coupled receptor‐1; Gpa‐2 = G‐alpha protein‐2; FGM = Fermentable growth medium; PNC1 = pyrazinamidase/Nicotinamidase 1; Sir2p = yeast silent information regulator 2; NADH = reduced form of nicotinamide adenine dinucleotide; Sch9 = Saccharomyces cerevisiae protein kinase 9; ERCs = extrachromosomal circular DNA repeats; Daf‐16 = ‘dauer’ larvae transcription factor‐16; AGE‐1 = worm homolog of the phosphoinositol‐3 kinase; AKT = protein kinase B; Rpd3 = reduced potassium dependence 3 (class II histone deacetylase); IGF‐1 = insulin growth factor‐1; FOXO = forkhead box subgroup ‘O’ transcription factor; IRS = insulin receptor substrate; CHICO = drosophila homolog of IRS; SIRT1 = sirtuin 1.
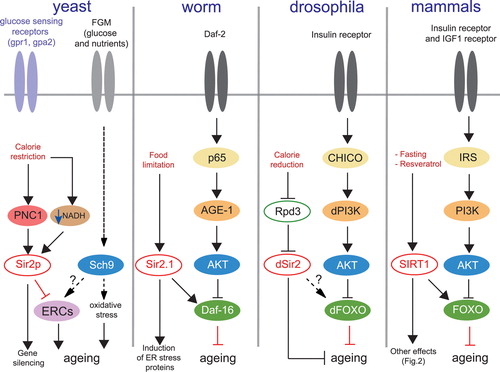
Figure 2. A: yeast and mammalian NAD+ salvage pathways. Yeast (red) Sir2p utilizes the cofactor NAD+ to deacetylate proteins and in this reaction produces nicotinamide (NAM) and O‐acetyl‐ADP‐ribose (O‐AA‐ribose). NAM is deaminated by the nicotinamidase PNC1 to form nicotinic acid (NA). NA will give rise successively to nicotinate mononucleotide (NAMN), nicotinate adenine dinucleotide (NAAD) and nicotinamide adenine dinucleotide (NAD+) by the enzymes nicotinate phosphoribosyl‐transferase (NPT1), nicotinate mononucleotide adenyltransferase (NMAT), and nicotinamide adenine dinucleotide (NAD) synthetase (QNS), respectively (red arrows). In mammals (blue) NAM is recycled to NAD+ in two steps through the formation of nicotinamide mononucleotide (NMN) by means of the NAM phosphoribosyltransferase NamPT (PBEF/visfatin) and nicotinamide mononucleotide adenyltransferase (NMNAT) (blue arrows). B: SIRT1 protective functions in metabolism and diseases. SIRT1 can be regulated positively by CR and SIRT‐activating compounds and negatively by SIRT inhibitors. SIRT1 activation induces survival of cardiomyocytes, protects neurons from cell death, and favors insulin secretion by repressing the uncoupling protein 2 (UCP2). SIRT1 decreases white adipocyte tissue formation through repression of PPARγ and promotes gluconeogenesis in response to fasting through PGC‐1α, and stimulates mitochondrial biogenesis in the brown adipose tissue (BAT) and the muscle through activation of PGC‐1α. NA = nicotinic acid; NAD+ = nicotinamide adenine dinucleotide; NAM = nicotinamide; NMN = nicotinamide mononucleotide; PNC1 = pyrazinamidase/Nicotinamidase 1; NamPT1 = nicotinamide phosphoribosyl transferase 1; PBEF = pre‐B cell enhancing factor; NPT1 = nicotinate phosphoribosyl transferase 1; NMAT1/2 = nicotinate mononucleotide adenyltransferase 1/2; QNS = NAD synthetase; NMNAT1 = nicotinamide mononucleotide adenyltransferase; O‐AA‐ribose = O‐acetyl‐ADP‐ribose; UCP‐2 = uncoupling protein 2; NFκB = nuclear factor kappa B; PPARγ = peroxisome proliferator‐activated receptor gamma; PGC‐1α = peroxisome proliferator‐activated receptor gamma coactivator‐1 alpha; BAT = brown adipose tissue; SIRT1 = sirtuin 1.
Studies in Caenorhabditis elegans and Drosophila melanogaster
In C. elegans, the yeast Sir2p ortholog, Sir2.1, extends life span through the forkhead transcription factor Daf‐16 (homolog of mammalian FOXO) signaling pathway. Loss of function studies of components of the Daf‐2 (homolog of the mammalian insulin receptor) signaling pathway, such as in Daf‐2 or the homolog of the mammalian phosphatidyl‐inositol‐3 kinase (PI3K), AGE‐1, extend the worm's life span () Citation35,36. This pathway controls the entry of worms in ‘dauer’, a larval developmental state of growth arrest that is induced upon food limitation, and which is a feature of young larvae before reproductive maturation (reviewed in Citation37). Activation of Daf‐2/AGE‐1 signaling results in the phosphorylation of AKT kinase that sequesters Daf‐16 in the cytoplasm resulting in its inactivation Citation38,39. The long‐lived mutants require the entry of Daf‐16 into the nucleus to activate target genes necessary for dauer formation. Duplication of chromosomal regions containing Sir2.1 in the nematode extends life span by up to 50%. Interestingly, the Sir2.1‐duplicated strains carrying a mutated Daf‐16 displayed the same decreased life span as observed in Daf‐16 mutants, proving that Sir2.1 acts upstream of Daf‐16. In addition, Sir2.1‐duplicated strains do not further extend life span of Daf‐2 mutants indicating that extra copies of Sir2.1 promote longevity through the Daf‐2/Daf‐16 signaling pathway Citation12. A direct interaction between Sir2.1 and Daf‐16, facilitated by the nematode 14‐3‐3 protein, furthermore suggests that these three proteins physically interact Citation40,41.
Also dSir2, the drosophila Sir2 ortholog, controls life span extension under CR conditions, and mutants that remove or decrease dSir2 levels are no longer able to promote the CR‐mediated longevity. The dSir2‐mediated life extension seems to work in the same pathway as the Rpd3 histone deacetylase, since Rpd3 is thought to negatively regulate dSir2, and CR induces a decrease in Rpd3 expression Citation13. Moreover, long‐lived Rpd3 mutant flies display high dSir2 expression levels Citation42. Interestingly, flies deficient in the homolog of the mammalian insulin receptor substrate (IRS) protein, CHICO, show a significant increase in life span Citation43. Recent studies showed that activation of dFOXO in the fly brain and/or fat body extends life span and inhibits the endogenous insulin‐dependent signaling in the fat body Citation44,45. Although both CR and downregulation of the insulin pathway participate in life span extension, it is still not proven that the two pathways work in concert and converge on dFOXO to promote longevity in drosophila.
Studies in mammals
The mammalian NAD+ salvage pathway is different from the yeast pathway. NAM is directly transformed to nicotinamide mononucleotide (NMN) by the enzyme nicotinamide phosphoribosyltransferase (NamPT) Citation46, which then yields NAD+ through the action of nicotinamide mononucleotide adenyl transferase (NMNAT) (). NamPT was found to be the equivalent of the pre‐B‐cell‐enhancing factor (PBEF), a protein that stimulated B‐cell colony formation Citation47, and visfatin, an adipokine expressed in visceral fat with glucose‐lowering properties Citation48. Although some evidence exists that NamPT activates SIRT1 transcriptional activity Citation27, NamPT overexpression in mice would be necessary to determine whether NamPT is the functional homolog of yeast PNC1 in life span extension.
The role of the insulin‐signaling pathway in the determination of mammalian life span has been studied in insulin‐like growth factor receptor (IGF‐R)‐deficient mice. Although IGF‐R−/− mice die shortly after birth, IGF‐R+/− animals are viable, display reduced IGF‐R levels, and live longer than IGF‐R+/+ littermates. IGF‐R+/− animals exhibit low AKT kinase activity suggesting an increase in FOXO activity, reproducing features of the Daf‐16‐mediated longevity pathway in C. elegans. In addition, IGF‐R+/− mice are more resistant to oxidative stress. This phenotype was, however, gender‐dependent since only IGF‐R+/− female mice displayed a significant increase in life span, whereas IGF‐R+/− males showed a modest, non‐significant increase in longevity Citation49. Mice in which the insulin receptor was specifically inactivated in the adipose tissue also live longer and are protected against age‐related obesity and its subsequent metabolic abnormalities Citation50.
In mammals, SIRT1 has been linked with metabolic control. The importance of SIRT1 in the nutrient control of glucose homeostasis was demonstrated to involve the modulation of the acetylation status and hence the stimulation of the activity of the metabolic coregulator peroxisome proliferator‐activated receptor gamma (PPARγ) coactivator‐1α (PGC‐1α). In the liver, SIRT1 in a complex including the hepatocyte nuclear factor‐4 (HNF‐4) deacetylates and activates PGC‐1α, promoting gluconeogenesis following fasting Citation51. SIRT1 also functions together with PGC‐1α, beyond the liver. Indeed, activated PGC‐1α promotes mitochondrial function in the skeletal muscle and the brown adipose tissue, leading to enhanced energy expenditure, increased exercise performance, and protection from diet‐induced insulin resistance and hepatosteatosis Citation16,Citation52. Although these studies suggest that PGC‐1α is a privileged partner for SIRT1, other signaling pathways are also solicited (). For instance, in white adipose tissue, where the activation of SIRT1 by resveratrol decreased fat accumulation and its inhibition resulted in triglyceride accumulation, it was suggested that SIRT1 activation repressed the nuclear receptor PPARγ Citation53, an effect that, in addition to SIRT1, also involved the nuclear receptor corepressor (NcoR) and silencing mediator SMRT Citation54. A recent study also emphasized the role of the endothelial nitric oxide synthase (eNOS) signaling in CR‐mediated increases in mitochondrial biogenesis and life span in mice. CR animals showed an increase in mitochondrial biogenesis that correlated with an increase in the expression of SIRT1, PGC‐1α and eNOS in various tissues. Interestingly, these effects were completely inhibited in eNOS−/− animals in which life span was significantly reduced Citation55.
SIRT1 has also been demonstrated to augment insulin secretion in response to glucose in pancreatic β‐cells of β‐cell‐specific SIRT1‐overexpressing (BESTO) transgenic mice Citation56. This response was accompanied by a decrease in the expression of the uncoupling protein‐2 (UCP‐2) that could increase ATP production in the β‐cells of BESTO transgenic mice. UCPs uncouple oxygen consumption from ATP generation by allowing leakage of protons (H+), thereby participating in the reduction of ROS generation. The SIRT1‐mediated decrease in UCP‐2 expression impedes H+ ‘leakage’ and allows a more efficient coupling of electron transport with the ATP production. Likewise, UCP‐2−/− mice also exhibit a similar phenotype with improved glucose tolerance and enhanced insulin secretion Citation57. SIRT1 achieves this effect on UCP‐2 expression by directly binding to and repressing UCP‐2 gene transcription in pancreatic β‐cells Citation58. SIRT1 can also form a complex with FOXO1 and the promyelocytic leukemia protein PML to activate two insulin transcription factors, NeuroD and MafA, which may protect the pancreatic β‐cell pathway from oxidative damage Citation59. An approach to increase SIRT1 dosage in pancreatic β‐cells could hence be of potential interest to maintain a healthy β‐cell function in diabetic patients.
SIRT1 and SIRT3 deacetylate acetyl coenzyme A synthetase (AceCS). AceCS catalyzes the formation of acetyl‐CoA from acetate, coenzyme A, and ATP. Whereas SIRT1 has been shown to deacetylate the cytoplasmic AceCS1, whose activity controls acetyl‐CoA levels in the cytoplasm for fatty acid synthesis, SIRT3 deacetylates the mitochondrial AceCS2 which regulates acetyl‐CoA‐requiring pathways in the mitochondria such as the tricarboxylic acid cycle Citation60. Since acetate metabolism is impaired in diabetes and ageing, it is legitimate to speculate on the potential role of SIRT1 and SIRT3 in the pathophysiology of these diseases through the regulation of AceCS1 and AceCS2. Further validation of these observations in in vivo models is still required.
A central role for SIRT1 in disease of the central nervous system has also been highlighted in animal models. In the Wallerian degeneration slow (Wlds) mouse model, SIRT1 activation protects axons against neuronal injury. This Wlds mouse bears in fact a dominant mutation producing an overexpressed chimeric Wlds protein composed of the ubiquitin assembly protein Ufd2a and the NAD+ salvage pathway enzyme NMNAT1. Decreasing SIRT1 activity reduces the axonal protection originally observed, whereas SIRT1 activation by resveratrol decreases the axonal degeneration after neuronal injury. This suggests that the neuroprotection in the Wlds mouse model is achieved by increasing the neuronal NAD+ reserve and/or SIRT1 activity Citation61. Furthermore it has been reported that the SIRT1 agonist resveratrol protects C. elegans neurons expressing a fragment of the Huntington disease‐associated protein huntingtin and mammalian neurons from mutant polyglutamine cytotoxicity in a HdhQ111 knock‐in mouse model of Huntington disease Citation62. In addition, SIRT1 activation significantly decreases neuronal cell death induced by amyloid‐beta (Aβ) peptides through inhibition of NFκB signaling Citation63. Specific brain hSIRT1 overexpression in transgenic animals induces a significant increase in the α‐secretase activity, an enzyme that cleaves the amyloid precursor peptide (APP) within the Aβ peptide, favoring thereby the nonamyloidogenic pathway of the APP processing Citation64. In addition, a recent study demonstrated the protective effect of CR against Alzheimer's disease‐type brain amyloidosis in monkeys. Monkeys maintained on CR diet had significantly reduced contents of Aβ peptides in the temporal cortex that correlated with enhanced SIRT1 concentrations as compared to monkeys under normal diet Citation65. From these studies, it became clear that pharmacological strategies aiming at activating SIRT1 would impede Aβ peptide deposition in the brain and prevent the development of Alzheimer's disease.
From the animal studies discussed above, it was suggested that SIRT1 could contribute to the pathogenesis of some complex diseases. SIRT1 could hence be considered as a serious candidate target for therapeutic interventions in metabolic and neurodegenerative disorders. In line with this hypothesis, genetic variants (SNPs) in the human SIRT1 gene have been shown to be tightly associated with energy expenditure Citation52. We predict that future human studies will link SIRT1 even more tightly with metabolic and neurodegenerative diseases, stimulating the development of therapeutics for their treatment.
Emerging functions for the other sirtuins
SIRT2
The tubulin‐deacetylase protein SIRT2 Citation66 has been demonstrated to be downregulated in human gliomas, which are amongst the most frequent malignant brain tumors Citation67. The SIRT2 gene maps at chromosome 19q13.2, a region frequently deleted in human gliomas. Furthermore, ectopic expression of SIRT2 in a glioma cell line decreased colony formation suggesting a potential tumor suppressor role of SIRT2. This could be explained by the fact that SIRT2 plays an important role in the control of mitotic exit in the cell cycle where increased SIRT2 activity severely delays cell cycle progression through mitosis Citation68, but also by the fact that SIRT2 acts as a mitotic checkpoint protein that prevents chromosomal instability and the formation of hyperploid cells in the early metaphase Citation69. Further studies such as SIRT2 brain‐specific inactivation in genetically engineered mice should bring an insight into the mechanism and the role of SIRT2 in the pathophysiology of this aggressive type of brain cancer. Very recently, SIRT2 was described as an oligodendroglial cytoplasmic protein, localized to the outer and juxtanodal loops in the myelin sheath, that decreases cell differentiation through α‐tubulin deacetylation suggesting a potential implication in myelinogenesis Citation70.
SIRT3
The mitochondrial SIRT3 deacetylase Citation71,72 has been linked with adaptative thermogenesis. SIRT3 expression is induced in mice in both white and brown adipose tissue (BAT) by CR and in BAT upon cold exposure. SIRT3 furthermore activates known mitochondrial genes such as PGC‐1α and UCP‐1 suggesting an important role of SIRT3 in thermogenesis Citation73. In addition, the SIRT3 gene has been linked to longevity. In fact, the genotype defined by the G477T polymorphism in exon 3, was associated with survivorship Citation74. Furthermore, a VNTR (a 72‐bp repeat core) polymorphism in intron 5 of the SIRT3 gene, that acts as an allele‐specific enhancer activity on a reporter gene, was associated with longevity in male subjects Citation75. This VNTR might represent the functional variant that accounts for the association between the silent marker G477T and longevity in old male subjects.
SIRT4
Although the mitochondrial expressed protein SIRT4 has a conserved sirtuin domain, it seems not to possess in vitro deacetylase activity Citation76. However, SIRT4 ADP‐ribosylates and inhibits the mitochondrial glutamate dehydrogenase (GDH). GDH controls amino acid‐stimulated insulin secretion by regulating glutamine and glutamate oxidative metabolism. Inhibition of GDH activity by SIRT4 decreases insulin secretion in mouse pancreatic β‐cells in response to amino acids Citation76. Interestingly, the two different sirtuins SIRT1 and SIRT4 seem to work in opposite directions to control insulin secretion. Furthermore SIRT4 expression is downregulated in response to CR in β‐cells, which is opposite to the regulation of SIRT1 during CR. More studies are still needed to see whether SIRT4 (and SIRT1) can be integrated in the pathophysiology of type 1 and 2 diabetes that both are characterized by alterations in insulin secretion.
SIRT6
Whereas little is known about the activity of SIRT5, SIRT6 is suggested to control genomic DNA stability and DNA repair. SIRT6−/− mice die prematurely and display severe defects including important lymphopenia, loss of subcutaneous fat, decreased bone mineral density, metabolic defects with a profound imbalance in glucose homeostasis and decreased IGF‐1 levels. The phenotype of the SIRT6−/− mice mimics multiple pathologies found in elderly humans Citation77. Although SIRT6 was originally described as an exclusive ADP‐ribosyltransferase Citation78, it was recently demonstrated that SIRT6 deacetylates histones and the DNA repair enzyme, DNA polymerase β (polβ) in vitroCitation77. An effect on DNA repair was hence proposed to explain the phenotype in SIRT6−/− mice, and it was suggested that SIRT6 could play an essential role in maintaining organ integrity as animals age.
SIRT7
SIRT7 is the only sirtuin to be localized in the nucleolus and is a component of the RNA polymerase I (Pol I) transcriptional machinery. SIRT7 interacts with RNA Pol I and histones, and positively regulates the transcription of rDNA during transcriptional elongation, which accounts for about 60% of total transcription in metabolically active cells in mammals Citation79. SIRT7 overexpression increases Pol I‐mediated transcription in a NAD+‐dependent manner, whereas SIRT7 inhibition induces a decrease in the transcription of Pol I and its association with rDNA Citation80. Depletion of SIRT7 stopped cell proliferation and triggered apoptosis. It was suggested that SIRT7 may regulate rDNA transcription by sensing cellular NAD+ levels and that diet‐induced changes in NAD+/NADH ratio might modulate SIRT7 to link the cellular energy status with rRNA synthesis and ribosome production. It is worth noting that SIRT1 negatively regulates RNA Pol I through deacetylation of TAFI68 Citation81, which goes opposite of SIRT7 action.
Table I. Expression pattern, cellular distribution, and function of sirtuin deacetylases. Sirtuins are differentially expressed in different organs based on their transcripts.
Concluding remarks
In the last decade, sirtuin biology has come a long way from their original description as yeast NAD+‐dependent class III HDACs, that control yeast life span. In mammals, seven orthologs of Sir2 have been identified, SIRT1 to 7, and the exact biological function of most of these sirtuins still remains only partially characterized. Of particular interest is the fact that SIRT1 not only deacetylates histones to mediate gene silencing, but is able to interact with and deacetylate some well known transcriptional regulators thereby modulating specifically various biological processes. Hence modulating the expression of SIRT1 or its activity, by using sirtuin‐activating compounds such as resveratrol, will have pleiotropic effects. SIRT1 activation reduces fat accumulation and adipocyte differentiation through repression of the activity of the adipogenic nuclear receptor PPARγ. SIRT1 also promotes mitochondrial function and energy expenditure and consequently protects mice from diet‐induced obesity, through deacetylation and subsequent activation of PGC‐1α in the skeletal muscle and in the brown adipose tissue. The SIRT1/PGC‐1α interaction is also important in the liver, where SIRT1 activation upon fasting induces gluconeogenesis and prevents against hepatosteatosis. In addition, SIRT1 significantly enhances insulin secretion in the pancreatic β‐cells. In combination, these studies illustrate that SIRT1 is a major modulator of metabolism. SIRT1 activation also seems to be endowed with neuroprotective activities as suggested from the study of models of Huntington or Alzheimer's disease. Furthermore, other sirtuins might play important roles in some diseases, as illustrated by SIRT2, which is downregulated in human gliomas. Obviously, more studies, in animal models and humans, are still needed to define the exact role of sirtuins in the pathophysiology of human diseases. It can, however, be predicted that therapeutic interventions aiming at activating or blocking sirtuins, depending on the context, will one day become helpful in the treatment of human diseases.
Acknowledgements
We thank greatly members of the Auwerx laboratory for critical reading of the manuscript and helpful discussions. This work was supported by grants of the Centre National de la Recherche Scientifique (CNRS); Institut National pour la Science et la Recherche Médicale (INSERM); National Institutes of Health (NIH); the European Union (EU) and the Hôpitaux Universitaires de Strasbourg.
References
- Chen H., Lin R. J., Xie W., Wilpitz D., Evans R. M. Regulation of hormone‐induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell 1999; 98: 675–86
- Courey A. J., Jia S. Transcriptional repression: the long and the short of it. Genes Dev 2001; 15: 2786–96
- Imai S., Johnson F. B., Marciniak R. A., McVey M., Park P. U., Guarente L. Sir2: an NAD‐dependent histone deacetylase that connects chromatin silencing, metabolism, and aging. Cold Spring Harb Symp Quant Biol 2000; 65: 297–302
- Imai S., Armstrong C. M., Kaeberlein M., Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD‐dependent histone deacetylase. Nature 2000; 403: 795–800
- Lin S. J., Guarente L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr Opin Cell Biol 2003; 15: 241–6
- Hoppe G. J., Tanny J. C., Rudner A. D., Gerber S. A., Danaie S., Gygi S. P., et al. Steps in assembly of silent chromatin in yeast: Sir3‐independent binding of a Sir2/Sir4 complex to silencers and role for Sir2‐dependent deacetylation. Mol Cell Biol 2002; 22: 4167–80
- Ivy J. M., Klar A. J., Hicks J. B. Cloning and characterization of four SIR genes of Saccharomyces cerevisiae. Mol Cell Biol 1986; 6: 688–702
- Rine J., Herskowitz I. Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics 1987; 116: 9–22
- Chen L., Widom J. Mechanism of transcriptional silencing in yeast. Cell 2005; 120: 37–48
- Braunstein M., Rose A. B., Holmes S. G., Allis C. D., Broach J. R. Transcriptional silencing in yeast is associated with reduced nucleosome acetylation. Genes Dev 1993; 7: 592–604
- Kaeberlein M., McVey M., Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 1999; 13: 2570–80
- Tissenbaum H. A., Guarente L. Increased dosage of a sir‐2 gene extends lifespan in Caenorhabditis elegans. Nature 2001; 410: 227–30
- Rogina B., Helfand S. L. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A 2004; 101: 15998–6003
- Howitz K. T., Bitterman K. J., Cohen H. Y., Lamming D. W., Lavu S., Wood J. G., et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003; 425: 191–6
- Wood J. G., Rogina B., Lavu S., Howitz K., Helfand S. L., Tatar M., et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004; 430: 686–9
- Baur J. A., Pearson K. J., Price N. L., Jamieson H. A., Lerin C., Kalra A., et al. Resveratrol improves health and survival of mice on a high‐calorie diet. Nature 2006; 444: 337–42
- Bitterman K. J., Anderson R. M., Cohen H. Y., Latorre‐Esteves M., Sinclair D. A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem 2002; 277: 45099–107
- Bordone L., Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol 2005; 6: 298–305
- Lin S. J., Defossez P. A., Guarente L. Requirement of NAD and SIR2 for life‐span extension by calorie restriction in Saccharomyces cerevisiae. Science 2000; 289: 2126–8
- Lin S. J., Kaeberlein M., Andalis A. A., Sturtz L. A., Defossez P. A., Culotta V. C., et al. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 2002; 418: 344–8
- Bakker B. M., Overkamp K. M., van Maris A. J., Kotter P., Luttik M. A., van Dijken J. P., et al. Stoichiometry and compartmentation of NADH metabolism in Saccharomyces cerevisiae. FEMS Microbiol Rev 2001; 25: 15–37
- Tanner K. G., Landry J., Sternglanz R., Denu J. M. Silent information regulator 2 family of NAD‐dependent histone/protein deacetylases generates a unique product, 1‐O‐acetyl‐ADP‐ribose. Proc Natl Acad Sci U S A 2000; 97: 14178–82
- Liou G. G., Tanny J. C., Kruger R. G., Walz T., Moazed D. Assembly of the SIR complex and its regulation by O‐acetyl‐ADP‐ribose, a product of NAD‐dependent histone deacetylation. Cell 2005; 121: 515–27
- Anderson R. M., Bitterman K. J., Wood J. G., Medvedik O., Cohen H., Lin S. S., et al. Manipulation of a nuclear NAD+ salvage pathway delays aging without altering steady‐state NAD+ levels. J Biol Chem 2002; 277: 18881–90
- Anderson R. M., Bitterman K. J., Wood J. G., Medvedik O., Sinclair D. A. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 2003; 423: 181–5
- Lin S. J., Ford E., Haigis M., Liszt G., Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev 2004; 18: 12–6
- Revollo J. R., Grimm A. A., Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem 2004; 279: 50754–63
- Sandmeier J. J., Celic I., Boeke J. D., Smith J. S. Telomeric and rDNA silencing in Saccharomyces cerevisiae are dependent on a nuclear NAD(+) salvage pathway. Genetics 2002; 160: 877–89
- Lamming D. W., Latorre‐Esteves M., Medvedik O., Wong S. N., Tsang F. A., Wang C., et al. HST2 mediates SIR2‐independent life‐span extension by calorie restriction. Science 2005; 309: 1861–4
- Sinclair D. A., Guarente L. Extrachromosomal rDNA circles—a cause of aging in yeast. Cell 1997; 91: 1033–42
- Longo V. D. The Ras and Sch9 pathways regulate stress resistance and longevity. Exp Gerontol 2003; 38: 807–11
- Fabrizio P., Pozza F., Pletcher S. D., Gendron C. M., Longo V. D. Regulation of longevity and stress resistance by Sch9 in yeast. Science 2001; 292: 288–90
- Fabrizio P., Liou L. L., Moy V. N., Diaspro A., SelverstoneValentine J., Gralla E. B., et al. SOD2 functions downstream of Sch9 to extend longevity in yeast. Genetics 2003; 163: 35–46
- Kaeberlein M., Andalis A. A., Fink G. R., Guarente L. High osmolarity extends life span in Saccharomyces cerevisiae by a mechanism related to calorie restriction. Mol Cell Biol 2002; 22: 8056–66
- Kimura K. D., Tissenbaum H. A., Liu Y., Ruvkun G. daf‐2, an insulin receptor‐like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 1997; 277: 942–6
- Morris J. Z., Tissenbaum H. A., Ruvkun G. A phosphatidylinositol‐3‐OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 1996; 382: 536–9
- Burnell A. M., Houthoofd K., O'Hanlon K., Vanfleteren J. R. Alternate metabolism during the dauer stage of the nematode Caenorhabditis elegans. Exp Gerontol 2005; 40: 850–6
- Ogg S., Paradis S., Gottlieb S., Patterson G. I., Lee L., Tissenbaum H. A., et al. The Fork head transcription factor DAF‐16 transduces insulin‐like metabolic and longevity signals in C. elegans. Nature 1997; 389: 994–9
- Lin K., Dorman J. B., Rodan A., Kenyon C. daf‐16: An HNF‐3/forkhead family member that can function to double the life‐span of Caenorhabditis elegans. Science 1997; 278: 1319–22
- Berdichevsky A., Viswanathan M., Horvitz H. R., Guarente L. C. elegans SIR‐2.1 interacts with 14‐3‐3 proteins to activate DAF‐16 and extend life span. Cell 2006; 125: 1165–77
- Wang Y., Oh S. W., Deplancke B., Luo J., Walhout A. J., Tissenbaum H. A. C. elegans 14‐3‐3 proteins regulate life span and interact with SIR‐2.1 and DAF‐16/FOXO. Mech Ageing Dev 2006; 127: 741–7
- Rogina B., Helfand S. L., Frankel S. Longevity regulation by Drosophila Rpd3 deacetylase and caloric restriction. Science 2002; 298: 1745
- Clancy D. J., Gems D., Harshman L. G., Oldham S., Stocker H., Hafen E., et al. Extension of life‐span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 2001; 292: 104–6
- Hwangbo D. S., Gershman B., Tu M. P., Palmer M., Tatar M. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature 2004; 429: 562–6
- Giannakou M. E., Goss M., Junger M. A., Hafen E., Leevers S. J., Partridge L. Long‐lived Drosophila with overexpressed dFOXO in adult fat body. Science 2004; 305: 361
- Rongvaux A., Shea R. J., Mulks M. H., Gigot D., Urbain J., Leo O., et al. Pre‐B‐cell colony‐enhancing factor, whose expression is up‐regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur J Immunol 2002; 32: 3225–34
- Samal B., Sun Y., Stearns G., Xie C., Suggs S., McNiece I. Cloning and characterization of the cDNA encoding a novel human pre‐B‐cell colony‐enhancing factor. Mol Cell Biol 1994; 14: 1431–7
- Fukuhara A., Matsuda M., Nishizawa M., Segawa K., Tanaka M., Kishimoto K., et al. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science 2005; 307: 426–30
- Holzenberger M., Dupont J., Ducos B., Leneuve P., Geloen A., Even P. C., et al. IGF‐1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003; 421: 182–7
- Bluher M., Kahn B. B., Kahn C. R. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 2003; 299: 572–4
- Rodgers J. T., Lerin C., Haas W., Gygi S. P., Spiegelman B. M., Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC‐1alpha and SIRT1. Nature 2005; 434: 113–8
- Lagouge M., Argmann C., Gerhart‐Hines Z., Meziane H., Lerin C., Daussin F., et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC‐1alpha. Cell 2006; 127: 1109–22
- Picard F., Auwerx J. PPAR(gamma) and glucose homeostasis. Annu Rev Nutr 2002; 22: 167–97
- Picard F., Kurtev M., Chung N., Topark‐Ngarm A., Senawong T., Machado De Oliveira R., et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR‐gamma. Nature 2004; 429: 771–6
- Nisoli E., Tonello C., Cardile A., Cozzi V., Bracale R., Tedesco L., et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 2005; 310: 314–7
- Moynihan K. A., Grimm A. A., Plueger M. M., Bernal‐Mizrachi E., Ford E., Cras‐Meneur C., et al. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose‐stimulated insulin secretion in mice. Cell Metab 2005; 2: 105–17
- Zhang C. Y., Baffy G., Perret P., Krauss S., Peroni O., Grujic D., et al. Uncoupling protein‐2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 2001; 105: 745–55
- Bordone L., Motta M. C., Picard F., Robinson A., Jhala U. S., Apfeld J., et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol 2006; 4: e31
- Kitamura Y. I., Kitamura T., Kruse J. P., Raum J. C., Stein R., Gu W., et al. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab 2005; 2: 153–63
- Hallows W. C., Lee S., Denu J. M. Sirtuins deacetylate and activate mammalian acetyl‐CoA synthetases. Proc Natl Acad Sci U S A 2006; 103: 10230–5
- Araki T., Sasaki Y., Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 2004; 305: 1010–3
- Parker J. A., Arango M., Abderrahmane S., Lambert E., Tourette C., Catoire H., et al. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat Genet 2005; 37: 349–50
- Chen J., Zhou Y., Mueller‐Steiner S., Chen L. F., Kwon H., Yi S., et al. SIRT1 protects against microglia‐dependent amyloid‐beta toxicity through inhibiting NF‐kappaB signaling. J Biol Chem 2005; 280: 40364–74
- Qin W., Yang T., Ho L., Zhao Z., Wang J., Chen L., et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem 2006; 281: 21745–54
- Qin W., Chachich M., Lane M., Roth G., Bryant M., de Cabo R., et al. Calorie restriction attenuates Alzheimer's disease type brain amyloidosis in Squirrel monkeys (Saimiri sciureus). J Alzheimers Dis 2006; 10: 417–22
- North B. J., Marshall B. L., Borra M. T., Denu J. M., Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+‐dependent tubulin deacetylase. Mol Cell 2003; 11: 437–44
- Hiratsuka M., Inoue T., Toda T., Kimura N., Shirayoshi Y., Kamitani H., et al. Proteomics‐based identification of differentially expressed genes in human gliomas: down‐regulation of SIRT2 gene. Biochem Biophys Res Commun 2003; 309: 558–66
- Dryden S. C., Nahhas F. A., Nowak J. E., Goustin A. S., Tainsky M. A. Role for human SIRT2 NAD‐dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol Cell Biol 2003; 23: 3173–85
- Inoue T., Hiratsuka M., Osaki M., Yamada H., Kishimoto I., Yamaguchi S., et al. SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene 2007; 26: 945–57
- Li W., Zhang B., Tang J., Cao Q., Wu Y., Wu C., et al. Sirtuin 2, a mammalian homolog of yeast silent information regulator‐2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating alpha‐tubulin. J Neurosci 2007; 27: 2606–16
- Onyango P., Celic I., McCaffery J. M., Boeke J. D., Feinberg A. P. SIRT3, a human SIR2 homologue, is an NAD‐dependent deacetylase localized to mitochondria. Proc Natl Acad Sci U S A 2002; 99: 13653–8
- Schwer B., North B. J., Frye R. A., Ott M., Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide‐dependent deacetylase. J Cell Biol 2002; 158: 647–57
- Shi T., Wang F., Stieren E., Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem 2005; 280: 13560–7
- Rose G., Dato S., Altomare K., Bellizzi D., Garasto S., Greco V., et al. Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp Gerontol 2003; 38: 1065–70
- Bellizzi D., Dato S., Cavalcante P., Covello G., Di Cianni F., Passarino G., et al. Characterization of a bidirectional promoter shared between two human genes related to aging: SIRT3 and PSMD13. Genomics 2007; 89: 143–50
- Haigis M. C., Mostoslavsky R., Haigis K. M., Fahie K., Christodoulou D. C., Murphy A. J., et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006; 126: 941–54
- Mostoslavsky R., Chua K. F., Lombard D. B., Pang W. W., Fischer M. R., Gellon L., et al. Genomic instability and aging‐like phenotype in the absence of mammalian SIRT6. Cell 2006; 124: 315–29
- Liszt G., Ford E., Kurtev M., Guarente L. Mouse Sir2 homolog SIRT6 is a nuclear ADP‐ribosyltransferase. J Biol Chem 2005; 280: 21313–20
- Grummt I., Pikaard C. S. Epigenetic silencing of RNA polymerase I transcription. Nat Rev Mol Cell Biol 2003; 4: 641–9
- Ford E., Voit R., Liszt G., Magin C., Grummt I., Guarente L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev 2006; 20: 1075–80
- Muth V., Nadaud S., Grummt I., Voit R. Acetylation of TAF(I)68, a subunit of TIF‐IB/SL1, activates RNA polymerase I transcription. EMBO J 2001; 20: 1353–62