Abstract
Mood disorders, including major depression and bipolar disorder, remain a major unmet medical need as current antidepressant and mood stabilizing therapies require chronic treatment for efficacy and are not effective in all patients. Multiple deficits, including cell atrophy and loss, have been observed in limbic and cortical brain regions of patients with mood disorders and in stressed animals. It is thought that antidepressant and mood stabilizing medications restore these deficits by reestablishing proper patterns of gene expression and function. In support of this hypothesis, numerous changes in gene expression and activity have been observed in limbic and cortical brain regions of mood disorder patients, and thymoleptic therapies have been shown to reciprocally regulate many of these changes. These findings have implicated four main signaling pathways in the pathophysiology and/or treatment of mood disorders, namely the cyclic‐AMP, phosphoinositol, mitogen‐activated protein kinase, and glycogen synthase kinase signaling cascades. Below we review this literature, and discuss potential targets for novel antidepressant and mood stabilizing drug design that are highlighted by these findings.
| Abbreviations | ||
| 5‐HT | = | serotonin |
| ADT | = | antidepressant |
| AKT | = | v‐akt murine thymoma viral oncogene homolog |
| AMPA | = | α‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐ propionic acid |
| ATP | = | adenosine triphosphate |
| BAD | = | BCL2‐antagonist of cell death |
| BDNF | = | brain‐derived neurotrophic factor |
| BPD | = | bipolar disorder |
| CAMK | = | Ca2+‐calmodulin‐regulated kinase |
| cAMP | = | cyclic adenosine monophosphate |
| CREB | = | cAMP regulatory element binding protein |
| DAG | = | diacylglyceral |
| DUSP | = | dual specificity phosphatase |
| ECS | = | electroconvulsive seizure |
| ERK | = | extracellular signal‐regulatedkinase |
| FGF2 | = | fibroblast growth factor 2 |
| GSK | = | glycogen synthase kinase |
| HDAC | = | histone deacetylase |
| IGF‐I | = | insulin‐like growth factor I |
| IMPase | = | inositol monophosphatase |
| IP3 | = | inositol tris‐phosphate |
| IPPase | = | inositol polyphosphate 1‐phosphatase |
| MAOIs | = | monoamine oxidase inhibitors |
| MAPK | = | mitogen‐activated protein kinase |
| MARKS | = | myristoylated alanine‐rich C kinase substrate |
| MDD | = | major depressive disorder |
| NE | = | norepinephrine |
| NGF | = | nerve growth factor |
| NT‐3 | = | neurotrophin‐3 |
| PDE | = | phosphodiesterase |
| PIP2 | = | phosphatidylinositol bisphosphate |
| PKA | = | protein kinase A |
| PKC | = | protein kinase C |
| PLC | = | phospholipase C |
| PP1 | = | protein phosphatase 1 |
| PP2A | = | protein phosphatase 2A |
| SMIT | = | sodium/myo‐inositol cotransporter 1 |
| SNRI | = | selective norepinephrine reuptake inhibitor |
| SSRI | = | selective serotonin reuptake inhibitor |
| STEP | = | striatal enriched protein tyrosine phosphatase |
| TCAs | = | tricyclic antidepressants |
| TCF/LEF | = | T cell‐specific transcription factor, lymphoid enhancer factor |
| VEGF | = | vascular endothelial growth factor |
| VPA | = | valproate |
Introduction
Mood disorders, including major depression and bipolar disorder, are among the most prevalent and disabling disorders affecting today's society. It is estimated that 12%–17% of individuals suffer from mood disorders at some point in their lifetime, costing the United States alone 100 billion dollars each year in lost productivity and treatment expenses Citation1–6. Major depressive disorder (MDD) is characterized by chronic depressed mood, the inability to experience pleasure, withdrawal of interest, feelings of worthlessness, and suicidal tendencies Citation7. Bipolar disorder (BPD) is characterized by two distinct stages: depressive episodes with symptoms similar to MDD, and manic episodes characterized by a hyperaroused state, increased motor activity, racing thoughts, impaired judgment, and decreased sleep requirements Citation7. While numerous antidepressants (ADTs) and mood stabilizing drugs have been developed, therapeutic response to any of these treatments requires weeks or months of treatment and is realized in only a subset (∼60%–70%) of patients Citation8–12. Thus, mood disorders remain a major unmet medical need and the focus of intense study for the development of faster acting, broadly efficacious treatments Citation9.
Currently available chemical ADTs include monoamine oxidase inhibitors (MAOIs), tricyclic antidepressants (TCAs), and selective serotonin or norepinephrine reuptake inhibitors (SSRIs, SNRIs, respectively), and function to elevate levels of the monoamines serotonin (5‐HT) and/or norepinephrine (NE) in the synaptic cleft Citation9. A variety of medications are used to treat bipolar disorder, including lithium, select anticonvulsants such as valproate (VPA) and carbamazepine, and antipsychotics. A nonchemical approach, electroconvulsive therapy (ECT) has proven to be both a highly effective antidepressant and antimanic therapy Citation13. However, delayed therapeutic onset is associated with each of these therapies, leading to the hypothesis that the efficacy of ADTs and mood stabilizers requires restoration of neural networks altered in the brains of mood disorder patients which is achieved by reestablishing proper patterns of gene expression and function Citation14,15. In support of this hypothesis, numerous structural and biochemical changes have been observed in the brains of patients with MDD or BPD, many of which are reversed by ADTs or mood stabilizing agents.
Multiple morphological and cellular deficits have been observed in the brains of patients with MDD or BPD (for review, see Citation16–18). Postmortem and imaging studies of patients with MDD have detected reductions in neuronal arbors, glial size and number, cerebral blood flow, and glucose metabolism in limbic and cortical brain regions, although not all studies have observed these deficits Citation16–18. Similarly, reduced overall volume, neuronal size and glial numbers have been observed in limbic and cortical brain regions of BPD patients relative to healthy controls Citation16–18. Stress, a known precipitator of mood disorders in humans, causes atrophy, cell loss and reductions in hippocampal neurogenesis in limbic brain structures in animal models, and these types of cellular deficits could contribute to the atrophy observed in MDD and BPD Citation19.
Conversely, chronic administration of ADTs and mood stabilizers produces an overall enhancement of cell number, growth, and resiliency in limbic and cortical brain regions Citation17. ADTs or mood are reported to reduce or even reverse hippocampal atrophy in depressed or posttraumatic stress disorder patients Citation20,21. Chronic lithium treatment increases gray matter volume in BPD patients, and VPA or lithium administration prevents the reductions in prefrontal cortex gray matter observed in BPD patients Citation22,23. In rodent models, chemical and nonchemical ADTs block the stress‐induced atrophy of hippocampal neurons and increase the proliferation and survival of newborn neurons in hippocampus Citation24–28. Lithium also increases neurogenesis in rodent hippocampus Citation29.
Research to uncover the biochemical alterations that result in the anatomical and behavior phenotypes of MDD and BPD, as well as the biochemical mechanisms whereby thymoleptics compensate for or restore the defects observed in the brains of MDD and BPD patients, is facilitating the development of new and potentially more efficacious ADTs and mood stabilizing agents. In this review, we will focus our discussion on four intracellular signaling pathways that have been intensely studied for their contributions to mood disorders and/or the actions of ADTs and mood stabilizers. These are the cyclic adenosine monophosphate (cAMP) and phosphoinositol second messenger systems, the mitogen‐activated protein kinase (MAPK) pathway, and the glycogen synthase kinase‐3 (GSK‐3) signaling pathway. The findings discussed below begin to unravel the complex molecular underpinnings of mood disorders and have resulted in multiple preclinical and clinical studies of novel pharmacological strategies for the treatment of MDD and BPD.
Key messages
Alterations in intracellular signaling pathways have been observed in the brains of patients with mood disorders and/or in response to antidepressant and mood stabilizing agents.
These pathways include the cyclic‐AMP, phosphoinositol, ERK mitogen‐activated kinase, and glycogen synthase kinase signaling cascades, and present multiple potential drug targets for the treatment of mood disorders.
The cAMP second messenger system
The cAMP second messenger system is directly coupled to monoamine signaling through G‐protein‐coupled receptors and is thought to play a central role in mood regulation () Citation30,31. There are several 5‐HT and adrenoreceptor (AR) subtypes that regulate the cAMP system: 5‐HT4, 5‐HT5A, 5‐HT6, 5‐HT7, and β‐ARs stimulate, while 5‐HT1A, 5‐HT1B, 5‐HT1D and 5‐HT1E, 5‐HT1F and α2‐AR inhibit cAMP production Citation32–34. These receptors are coupled to adenylyl cyclase (AC), the enzyme that catalyses the formation of cAMP from ATP via stimulatory Gs and inhibitory Gi/o proteins Citation35. cAMP regulates central nervous system (CNS) function primarily by activating protein kinase A (PKA). PKA phosphorylates numerous substrates to regulate many aspects of neurobiology such as gene transcription, neurotransmitter receptor efficacy, and neurotransmitter release Citation36,37. One major substrate of PKA in the CNS is Ser‐133 of the cAMP regulatory element binding protein (CREB). Ser‐133 phosphorylation activates CREB to promote the transcription of CRE‐containing genes that regulate cell proliferation, development, plasticity, and survival Citation38 (Tanis et al., unpublished observations). Phosphodiesterases (PDEs) downregulate the cAMP system by degrading cAMP to AMP, and the cAMP system is tightly controlled by adjusting the equilibrium between cAMP production and degradation through the regulation of ACs and PDEs Citation35,Citation39,40.
Figure 1. The cAMP second messenger system. Chemical antidepressants upregulate 5‐HT and NE signaling to activate the cAMP signaling cascade. cAMP production results in the activation of CREB‐mediated gene transcription. PKA activates CREB both directly by phosphorylation and indirectly by triggering Ca2+ influx and the subsequent activation of CAMK and ERK. CREB triggers the expression of genes that promote cell proliferation, growth and resiliency, effects that could contribute to the ADT‐like effects observed in behavioral models. Molecules/proteins/genes altered in mood disorder patients or shown to regulate behavior in animal models of depression/ADT response are colored according to their associated effects (green = ADT, blue = prodepressive, yellow = antimanic, red = promanic, see online version for colour).
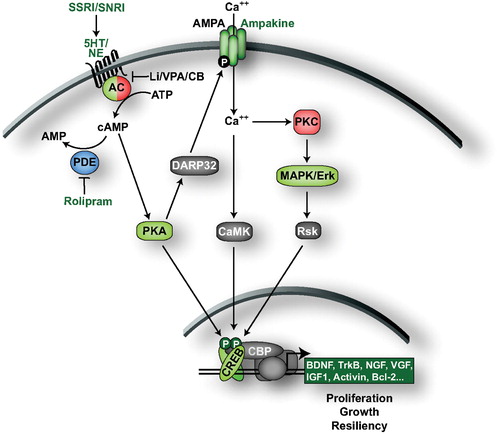
Postmortem studies have demonstrated that the cAMP system is downregulated in the brains of depressed patients. Decreased AC activity and CREB expression have been observed in cortical regions of the brains of MDD patients Citation41–43. Moreover, reduced cAMP binding to PKA, PKA activity, and CREB phosphorylation were observed in fibroblasts isolated from MDD patients relative to normal subjects Citation44,45. Conversely, antidepressants activate multiple components of the cAMP signaling cascade. Chronic administration of tricyclic ADTs or electroconvulsive seizures (ECS) was shown to enhance G‐protein‐mediated activation of AC in rat cortex and hypothalamus membranes Citation46,47. Chronic administration of ECS, the MAOI tranylcypromine, and the SNRI desipramine were shown to alter the subcellular localization of PKA and increase its localized activity in rat frontal cortex Citation48,49. Long‐term administration of a diverse set of ADTs (SSRIs, SNRIs, MAOIs, ECS) increased the expression of PDE4 A and B in rat frontal cortex and PDE4B expression in striatum potentially to compensate for the changes in AC activity produced by ADTs Citation50,51. Blockade of PDE4 with rolipram or papaverine increases the expression of CRE‐containing genes that elicit ADT efficacy, such as CREB and brain‐derived neurotrophic factor (BDNF), and rolipram has antidepressant efficacy in both preclinical and clinical trials Citation31,Citation52,53. Taken together, these studies strongly indicate that the cAMP system is downregulated in depressed subjects, and that pharmacological upregulation of cAMP signaling produces ADT effects.
Mood stabilizers are also known to regulate the cAMP system. A large body of literature demonstrates that lithium elevates basal AC activity but attenuates receptor‐stimulated AC activation in both preclinical and clinical studies (for review see Citation54). VPA is also reported to decrease the cAMP response to stimuli in cultured cells Citation55. Carbamazepine inhibits AC activity to reduce cAMP levels and inhibits CREB phosphorylation following pharmacological activation of ACs by forskolin Citation56. Combined, these studies suggest that while an overall decrease in the cAMP system is observed in MDD, the cAMP system may be overresponsive to cellular stimuli in BPD patients Citation54.
As mentioned above, CREB is a major mediator of the cellular responses to cAMP production, and induces the expression of CRE‐containing genes in response to elevated cAMP levels. A variety of studies indicate that CREB is an important regulator of behavioral phenotypes in animal models of depression and ADT response. Multiple CREB target genes have been shown to produce positive effects on these behaviors, including BDNF, nerve growth factor (NGF), VGF (nonacronymic), IGF‐I, activin, and B cell lymphoma‐2 proteins (Bcl‐2) Citation57–62. CREB levels were found to be decreased in postmortem cortex of depressed patients, but increased in patients receiving ADTs at time of death Citation41. Similarly in rodents, a diverse set of chemical and nonchemical ADTs increase CREB expression and phosphorylation in hippocampus Citation52,Citation63,64. Overexpression of CREB in rat hippocampus produces ADT effects in the forced swim and learned helplessness paradigms. These are referred to as behavioral despair models of depression and are responsive to ADTs. In the forced swim test, a rat or mouse is placed in a container of water and the immobility time or latency to immobility is recorded as a measure of helplessness. ADT decreases the immobility time. In the learned helplessness test a rodent is exposed to an inescapable stress, such as footshock, which results in despair that is measured in this case by an inability to learn to escape in subsequent testing. This escape deficit is reversed by ADT. There are limitations to these models, but the finding that CREB expression in the hippocampus decreases immobility and escape deficits clearly demonstrates ADT‐like effects Citation65.
While monoamine signaling is directly coupled to CREB activation through PKA, ADTs may also activate CREB via phosphorylation by Ca2+‐calmodulin‐regulated kinases (CAMKs) and/or the MAP kinase ERK (). Both CAMKIV and ERK are activated in rat frontal cortex by ADTs Citation66, possibly via the dopamine and cAMP-regulated phosphoprotein of 32 kDa (DARPP32). DARPP32 is phosphorylated and activated by PKA, leading to blockade of protein phosphatase‐1 (PP1). Indeed, activation of the 5‐HT neurotransmitter system by SSRI ADTs activates DARPP32, and results in increased phosphorylation of AMPA subunit GluA1, an event that enhances AMPA conductance Citation67–69. The ADT effects of AMPA potentiators may also be due to regulation of CREB‐mediated gene transcription via activation of CAMKs Citation67.
In contrast to the above studies, overexpression of CREB in the nucleus accumbens produces a depressive‐like response in the forced swim test and learned helplessness paradigm, and overexpression of a dominant negative form of CREB in the nucleus accumbens produces an ADT effect in these models Citation70,71. These region‐specific effects of CREB may be explained by the neural networks controlled by these brain regions and/or by CREB activating a different set of transcripts in these brain regions. Indeed, we have observed significant differences in CREB occupancy at gene promoters in hippocampus and striatum (Tanis et al., unpublished observations).
The ERK MAPK signaling pathway
Another central regulator of behavior and CREB activity is the mitogen‐activated protein kinase (MAPK) signaling pathway () Citation72. The membrane‐associated small guanosine triphosphatase, RAS (RAS GTPase) activates the RAF serine/threonine kinase to phosphorylate and activate MAPK kinase (MEK) which in turn phosphorylates and activates ERK. Activation of ERK results in subsequent phosphorylation of numerous proteins in multiple cellular compartments. For example, upon translocation to the nucleus ERK phosphorylates and activates multiple transcription factors. ERK also phosphorylates and activates ribosomal S6 kinase (RSK) family kinases to phosphorylate and activate the transcription factor CREB.
Figure 2. The ERK MAPK signaling pathway. Neurotrophic and growth factor receptors engage the RAF‐MEK‐ERK MAPK signaling cascade to regulate transcription factors including CREB. Activation of the ERK cascade by numerous neurotrophic/growth factors produces ADT effects in animal models. Increased Ca2+ levels triggered, for example by the glutamatergic system, also activate ERK signaling through PKC. The ERK‐CREB signaling pathway is downregulated by several phosphatases, including PP1, PP2A, DUSPs and STEP. CREB triggers the expression of genes that promote cell proliferation, growth, and resiliency, effects that could contribute to the ADT‐like effects observed in behavioral models. Molecules/proteins/genes altered in mood disorder patients or shown to regulate behavior in animal models of depression/ADT response are colored according to their associated effects (green = ADT, blue = prodepressive, yellow = antimanic, red = promanic, see online version for colour).
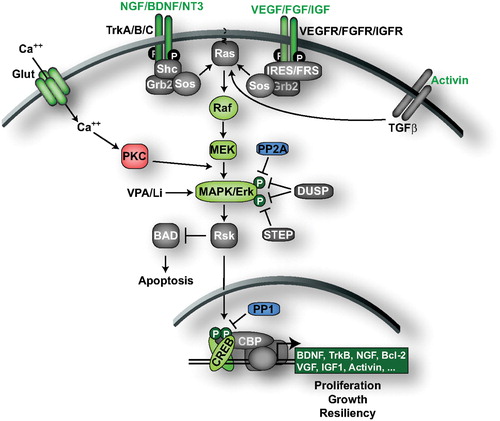
Numerous studies have implicated the ERK cascade in the etiology and treatment of mood disorders. Postmortem studies indicate that the brains of individuals who committed suicide have reduced B‐RAF and ERK abundance and activity Citation73–75. Conversely, rodent studies demonstrate that the ERK signaling cascade is activated by ADTs including fluoxetine and ECS Citation76–78. Similarly, the mood stabilizers lithium and VPA increase the activity of ERK, RSK, and CREB in cortex and hippocampus, and valproate was shown to promote ERK pathway‐dependent cortical neuronal growth and hippocampal neurogenesis in rodent models Citation79–81. Lithium and VPA also increase phosphorylation of the RSK substrate BAD Citation79.
Pharmacological manipulations of ERK signaling also affect behavior in models of depression and antidepressant response. Systemic blockade of the MAPK pathway in mice with the MEK inhibitor PD184161 produced depressive‐like behavior in the forced swim test, tail suspension test, and learned helplessness paradigms, and blocked the effects of the SNRI desipramine and the SSRI fluoxetine in the forced swim test Citation82. PD184161 also acutely increased locomotor activity, a manic‐like effect Citation82. Similarly, locomotor activity is increased by another MEK inhibitor SL327, and lithium blocks this effect, suggesting that regulation of ERK signaling may be important for the antimanic effects of lithium Citation79.
The MAPK signaling pathway has been heavily studied for mediating the antidepressant effects of neurotrophic factors and growth factors (for review see Citation162). Concordant with the reduced levels of B‐RAF and ERK, postmortem studies of depressed patients who committed suicide have revealed reduced levels of brain‐derived neurotrophic factor (BDNF), its receptor TrkB, neurotrophin‐3 (NT‐3), fibroblast growth factor 2 (FGF2) and FGF receptors Citation74,75,Citation83–86. Similarly in animal models, stress decreases expression of BDNF, NT‐3, nerve growth factor (NGF), the neurotrophin receptors p75, TrkA, TrkB, and TrkC, as well as vascular endothelial growth factor (VEGF) and its receptor Flk‐1 Citation16,Citation87,88. Conversely, an extensive list of chemical and nonchemical ADTs have been shown to increase the expression of neurotrophic/growth factors in rodent brain, including BDNF, VEGF, and insulin‐like growth factor I (IGF‐I) Citation89–94. Further, the decreases in BDNF, FGF2 and FGF receptors observed by postmortem gene profiling of depressed brains are normalized by ADTs Citation75,Citation83,Citation86,Citation95–98. Lithium and VPA were also shown to increase BDNF expression in rodents Citation79,Citation99. Supporting the hypothesis that neurotrophic/growth factor signaling plays a significant role in the treatment of depression, BDNF infusion into rodent midbrain, hippocampus, or ventricles produces ADT‐like effects (for review see Citation100). Like CREB, however, BDNF produces depressive phenotypes in the rodent ventral tegmental area demonstrating region‐specific effects similar to CREB Citation101,102. Intracranial infusions of NT‐3, IGF‐I, VEGF and activin A or B also produce antidepressant effects in rodents Citation92,Citation103–105.
Neurotrophic/growth factors are directly coupled to MAP kinase signaling (). Activation of neurotrophic/growth factor receptors induces receptor autophosphorylation forming docking sites for adapter proteins that recruit the guanine‐nucleotide exchange factor SOS, resulting in GTP loading of RAS and activation of the ERK signaling cascade. Indeed, the ERK signaling pathway is required for the antidepressant effects of BDNF. Activating phosphorylation of ERK is induced following infusion of BDNF into rat hippocampus, and the specific MEK inhibitor U0126 blocks BDNF‐induced ERK phosphorylation and ADT effects Citation103. In CREB‐deficient mice, the expression of BDNF is not increased by desipramine, providing convincing evidence that CREB underlies the upregulation of BDNF by ADTs Citation106.
A critical determinant of BDNF signaling is the dephosphorylation and inactivation of MEK, ERK and CREB. This occurs via a number of different protein phosphatases, including the dual specificity phosphatases (DUSPs). These enzymes are particularly important for inactivation of MAPK signaling because they inactivate/dephosphorylate both the activating threonine and tyrosine phosphorylation sites within these enzymes (). Of the 10 DUSPs identified, DUSP 1, 2, 4, 6, 7, and 9 are known or expected to have high affinity for ERK dephosphorylation Citation83,Citation107,108. Interestingly the expression levels of DUSP 1, 2, and 6 were found increased in rat frontal cortex and hippocampus after acute or chronic ECS suggesting that DUSPs may offset the enhancement of BDNF‐ERK‐CREB signaling following ADTs Citation77. The serine/threonine phosphatase PP2A can dephosphorylate the activating threonine phosphorylation site of ERK Citation109, and the Ser/Thr phosphatase PP1 dephosphorylates CREB Citation110. These phosphatases may also counteract the upregulation of BDNF‐ERK‐CREB signaling by ADTs, as preliminary studies in our laboratory demonstrated that the PP2A and PP1 inhibitor okadaic acid enhances the antidepressant effects of low‐dose BDNF infusions into rat hippocampus in the learned helplessness paradigm (Shirayama Y, Duman RS, unpublished observations). The compilation of the above findings strongly indicates that the RAS‐ERK‐CREB signaling pathway positively regulates behavior in models of depression and antidepressant response.
The phosphoinositol cycle
In addition to activating the cAMP and MAPK pathways, a wide variety of receptor tyrosine kinases (such as Trks) and G‐protein‐coupled receptors (such as 5‐HT2) activate phospholipase C (PLC) to hydrolyze phosphatidylinositol 4,5‐bisphosphate (PIP2) into inositol tris‐phosphate (IP3) and diacylglycerol (DAG) () Citation111,112. IP3 promotes the release of Ca2+ from intracellular stores, resulting in the activation of CAMK, and the subsequent activating phosphorylation of CREB. Ca2+ release, as well as DAG, activate multiple protein kinase C (PKC) isoforms which then translocate to their active sites where they associate with isoform‐specific proteins such as receptors for activated C‐kinase (RACKs). PKC isoforms phosphorylate a diverse set of substrates to regulate neurotransmission, neuronal excitability, development, and gene expression Citation113,114.
Figure 3. Phosphoinositol signaling and depletion by mood stabilizers. Following activation by growth factor and G‐protein‐coupled receptors, PLC catalyzes the production of IP3 and DAG to trigger release of Ca2+ from intracellular stores, and the activation of PKC to regulate neurotransmission, synaptic plasticity, and development. Mood stabilizers inhibit multiple aspects of this pathway, including influx of myoinositol through SMIT, recycling of IP3 into PIP2, and the activity of PKC. Molecules/proteins/genes altered in mood disorder patients or shown to regulate behavior in animal models of depression/ADT response are colored according to their associated effects (green = ADT, blue = prodepressive, yellow = antimanic, red = promanic, see online version for colour).
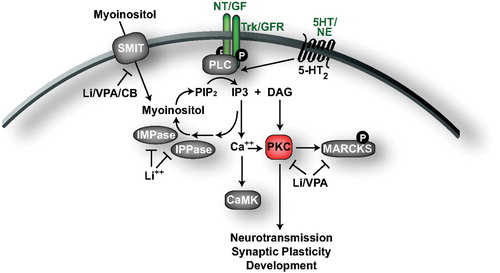
The phosphoinositol cycle was the first intracellular pathway implicated in the action of mood stabilizers (). In 1971 lithium was shown to decrease myoinositol levels in rat cerebral cortex, and this was later correlated with an increase in myoinositol‐1‐phosphate Citation115,116. Lithium administration has also been shown to reduce myoinositol levels in the right frontal lobe of patients with BPD Citation117. IP3 is metabolized by three sequential hydrolysis steps to yield inositol, which is then reincorporated into phosphatidylinositol to regenerate PIP2. Lithium directly inhibits inositol polyphosphate 1‐phosphatase (IPPase) and inositol monophosphatase (IMPase), two enzymes required for the recycling of IP3 to inositol. This results in the observed elevation in levels of myoinositol‐1‐phosphate (the substrate of IMPase), and the reduced levels of myoinositol. Like lithium, VPA administration decreases myoinositol and increases myoinositol‐1‐phosphate in rat brain Citation118. Lithium, VPA, and carbamazepine reduce the activity and expression of the high‐affinity myoinositol transporter SMIT in astrocyte cultures Citation119. Lithium, VPA, and carbamazepine also increase the spread of growth cones and inhibit their collapse, an effect overcome by exogenous myoinositol or pharmacological elevation of IP3, further indicating that each of these drugs downregulates the phosphoinositol cycle Citation120. In addition, chronic lithium was shown to increase the expression of the IMPase IMPA1 and myoinositol‐1‐phosphate synthase in mouse hippocampus Citation121. While these studies indicate that mood stabilizers alter the phosphoinositol cycle, it remains unclear whether inositol depletion contributes to the therapeutic mechanism of mood stabilizers. For an in‐depth discussion on this subject, the reader is referred to an excellent review by A. J. Harwood Citation122.
As mentioned above, PKC is a central functional effector of the phosphoinositol cycle, and several studies have implicated PKC in the actions of mood stabilizers. Lithium reduces the translocation of PKC to the cell membrane in rat cortex, and an increased association between PKC and RACK‐1 has been observed in the brains of patients with BPD Citation123,124. Chronic lithium also decreases the expression of select PKC isoforms, namely PKCα and PKCε, and the hippocampal expression of myristoylated alanine‐rich C kinase substrate (MARCKS), a major substrate of PKC Citation125–127. Similarly, VPA was shown to reduce PKC activity, the expression of PKCα and PKCε, and MARCKS expression in cultured neurons Citation128.
The GSK‐3 signaling pathway
The GSK‐3 signaling pathway has been strongly implicated in the actions of both ADTs and mood stabilizers (see Gould and Manji, for an excellent in‐depth review Citation129). GSK‐3 activity is linked to the activation of transcription factors that promote cell cycle arrest and apoptosis () Citation130–132. Conversely, GSK‐3 activity is directly or indirectly linked to the downregulation of β‐catenin, Jun, CREB, heat shock factor‐1 (HSF‐1), and nuclear factors of activated T cell (NFAT), transcription factors that regulate cell proliferation, growth, resiliency, and immune responses Citation132,133. One of the best‐characterized substrates of GSK‐3 is β‐catenin, which modulates cell adhesion in the cytoplasm and also translocates to the nucleus to activate transcription of T cell‐specific transcription factor, lymphoid enhancer factor (TCF/LEF) target genes. Phosphorylation of β‐catenin by GSK targets it for ubiquitin‐dependent degradation, downregulating TCF/LEF gene transcription.
Figure 4. GSK‐3 signaling and mood stabilization. GSK‐3 regulates multiple transcription factors to trigger cell cycle arrest and apoptosis, while inhibiting the transcription of genes that promote cell proliferation, growth and resiliency. Wnt glycoproteins inhibit GSK‐3 activity through interactions with Disheveled while receptor tyrosine kinases and select G‐protein‐coupled receptors activate PI3K, which activates AKT to phosphorylate and inhibit GSK‐3. AKT also inhibits apoptosis through phosphorylation of Bad, a proapoptotic protein. Molecules/proteins/genes altered in mood disorder patients or shown to regulate behavior in animal models of depression/ADT response are colored according to their associated effects (green = ADT, blue = prodepressive, yellow = antimanic, red = promanic, see online version for colour).
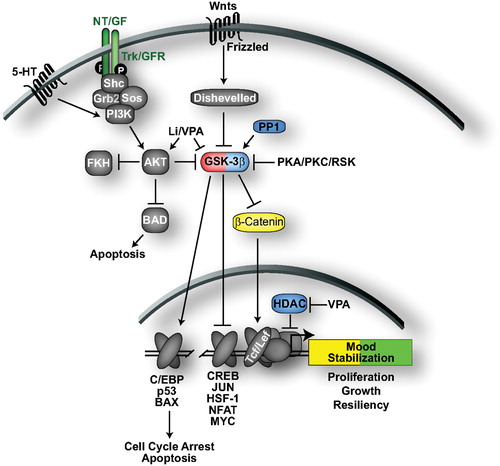
Numerous cellular stimuli converge to downregulate GSK‐3, which is generally active under basal conditions (). Wnt glycoproteins bind to Frizzled receptors, which leads to regulation of Disheveled and inhibition of GSK‐3 Citation132. Multiple kinases, including AKT, PKA, PKC, and RSK, phosphorylate GSK‐3 to inhibit its kinase activity in response to cellular stimuli Citation132. For example, activation of neurotrophic factor, growth factor, and select G‐protein‐coupled receptors (such as 5‐HT1A) recruits and activates PI3K Citation134,135. PI3K activates AKT that then phosphorylates and inactivates GSK‐3. GSK‐3 is reactivated following dephosphorylation by PP1 Citation136.
GSK‐3 activity is downregulated by a wide variety of ADTs and mood stabilizers, including lithium, VPA, ECS, MAOIs, SSRIs, tricyclics, and antipsychotics. Lithium is a direct inhibitor of GSK‐3 at therapeutic concentrations Citation137,138. Lithium also activates AKT and PKC, which phosphorylate and inactivate GSK‐3 Citation139–141. Indeed, numerous studies have found that therapeutic doses of lithium decrease GSK‐3 activity in rodent brain (reviewed in Citation129). In concert with the reduction of GSK‐3 activity, lithium administration has been shown to increase β‐catenin levels in rat cortex Citation142. VPA has also been shown to induce AKT activity, inhibitory phosphorylation of GSK‐3, and increased levels of β‐catenin in cell culture and in rodent brain Citation139,Citation143–145. Increased serotonin activity, mediated by a diverse set of ADTs, including SSRIs, MAOIs, and tricyclic ADTs, has also been shown to increase GSK‐3 inhibitory phosphorylation Citation145,146. ECS increases AKT activity, GSK‐3 phoshorylation, β‐catenin levels and the expression of Wnt‐2 Citation147–149. The antiphsychotics clozapine, haloperidol, and risperidone increase GSK‐3 expression and phosphorylation and β‐catenin levels in rodent brain Citation150,151.
Direct inhibition of GSK‐3 activity elicits ADT and antimanic properties in animal models. Systemic administration of selective GSK‐3 inhibitors, or heterozygous deletion of GSK‐3 produced ADT effects in the forced swim test Citation152–154. Amphetamine induces hyperactivity in rodents and is one of the most popular and reproducible models for antimanic efficacy. Interestingly, amphetamine reduces AKT activity and GSK‐3 phosphorylation Citation155. Further, pharmacological inhibition of GSK‐3 attenuates amphetamine‐induced hyperactivity, and amphetamine‐induced hyperactivity is reduced in gsk‐3β+/− mice Citation153,Citation155.
Summary and implications for drug design
In summary, the need for more efficacious, faster acting ADTs and mood stabilizers has driven intense research to uncover the molecular underpinnings of mood disorders and their current treatments. We have highlighted multiple intracellular signaling pathways discovered by this research effort to be disrupted in the brains of mood disorder patients or modified by thymoleptic drugs. These findings present several novel targets for ADT and mood stabilizing drug design, and compounds directed against several of these targets are already in development.
For example, PDEs represent an attractive target for pharmacological enhancement of cAMP signaling that is downregulated in depressed patients. Indeed, blockade of PDE4 with rolipram has antidepressant efficacy in preclinical and clinical trials Citation156. However, the potential for the use of first‐generation PDE inhibitors is limited due to common side effects, including nausea and emesis Citation156. A second‐generation of more tolerable PDE4 inhibitors that cross the blood‐brain barrier is being developed for neurological disorders, including Alzheimer's disease, mild cognitive impairment, and depression, but the ADT efficacy of these compounds has not yet been reported Citation39,40.
Another attractive area for drug design is the MAPK‐ERK pathway, as a variety of stimuli that activate this pathway produce positive effects in behavioral models of depression/ADT response. Potential drug targets include inhibition of the phosphatases PP1, PP2A, STEP, and DUSPs that downregulate MAP kinases and CREB‐mediated gene transcription. Indeed, our preliminary studies indicate that the PP1/PP2A inhibitor okadaic acid enhances the antidepressant effects of low‐dose BDNF infusions into rat hippocampus (Shirayama Y, Duman RS, unpublished observations). Studies are currently underway in our laboratory using selective phosphatase knockout mice to determine which phosphatase isoforms are the best targets for ADT drug development.
It may also be possible to target transcriptional regulators to produce mood enhancing patterns of gene transcription. For example, a recent study demonstrated that downregulation of histone deacetylase 5 (HDAC5) contributes to the ADT effects of imipramine in rodents Citation157. Interestingly, VPA is a direct inhibitor of HDAC activity at therapeutic concentrations () Citation158. Combined, these studies suggest that HDACs are potential targets for ADT drug design.
As described above, lithium and VPA downregulate PKC signaling. The PKC inhibitor tamoxifen has shown antimanic efficacy in initial clinical studies, although larger, placebo‐controlled studies are still underway Citation159. Future preclinical and clinical studies with more specific PKC inhibitors are also needed to fully determine the efficacy and tolerability of PKC inhibition for the treatment of mood disorders.
GSK‐3 is an attractive target for thymoleptic drug development as a broad set of ADTs and mood stabilizers have been shown to inhibit GSK‐3 signaling, and direct inhibition of GSK‐3 activity elicits ADT and antimanic properties in animal models Citation129. Selective small‐molecule inhibitors of GSK‐3 are rapidly being developed for the treatment of a variety of human disorders including diabetes, Alzheimer's disease, stroke, and inflammatory conditions, and these compounds may provide ADT and/or mood stabilizing efficacy Citation160,161.
Finally, extracellular stimuli and receptors that regulate cAMP, MAPK, phosphoinositol or GSK‐3 signaling pathways, as well as downstream targets of these pathways, present numerous additional possibilities for thymoleptic drug development. Thus, elucidation of the molecular signaling pathways that underlie mood disorders has enabled numerous novel therapeutic approaches for the treatment of these illnesses. However, when targeting these intracellular signaling pathways for therapeutic applications it is important to keep in mind that they each function in multiple biological processes throughout the body, and systemic regulation of any of these pathways could lead to adverse side effects. It may be possible to avoid these difficulties by developing strategies to regulate these pathways in specific brain regions or neuronal circuits. For example, it will likely be beneficial to target brain region‐specific phosphatase, PKC, or PDE isoforms for the treatment of mood disorders. Although more studies are needed to determine the efficacy and tolerability of targeting intracellular signaling pathways for the treatment of mood disorders, our ever‐increasing molecular understanding of these disorders is presenting an encouraging future for the treatment and prevention of these widespread illnesses that affect the lives of more than 10% of the population.
Acknowledgements
This work is supported by USPHS grants MH45481 and 2 PO1 MH25642, a Veterans Administration National Center Grant for PTSD, and by the Connecticut Mental Health Center.
References
- Kessler R. C., Berglund P., Demler O., Jin R., Koretz D., Merikangas K. R., et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS‐R). JAMA 2003; 289: 3095–105
- Wittchen H. U., Knauper B., Kessler R. C. Lifetime risk of depression. Br J Psychiatry Suppl 1994; 16–22
- Pincus H. A., Pettit A. R. The societal costs of chronic major depression. J Clin Psychiatry 2001; 62(Suppl 6)5–9
- Stewart W. F., Ricci J. A., Chee E., Hahn S. R., Morganstein D. Cost of lost productive work time among US workers with depression. JAMA 2003; 289: 3135–44
- Greenberg P. E., Kessler R. C., Birnbaum H. G., Leong S. A., Lowe S. W., Berglund P. A., et al. The economic burden of depression in the United States: how did it change between 1990 and 2000?. J Clin Psychiatry 2003; 64: 1465–75
- Murray C. J., Lopez A. D. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 1997; 349: 1498–504
- Goodwin F., Jamison K. R. Manic Depressive Illness. Oxford University Press, New York 1990
- Nelson J. C. A review of the efficacy of serotonergic and noradrenergic reuptake inhibitors for treatment of major depression. Biol Psychiatry 1999; 46: 1301–8
- Wong M. L., Licinio J. From monoamines to genomic targets: a paradigm shift for drug discovery in depression. Nat Rev Drug Discov 2004; 3: 136–51
- Montgomery S. A. Why do we need new and better antidepressants?. Int Clin Psychopharmacol 2006; 21(Suppl 1)S1–10
- Geddes J. R., Burgess S., Hawton K., Jamison K., Goodwin G. M. Long‐term lithium therapy for bipolar disorder: systematic review and meta‐analysis of randomized controlled trials. Am J Psychiatry 2004; 161: 217–22
- Zarate C. A., Jr., Quiroz J. A. Combination treatment in bipolar disorder: a review of controlled trials. Bipolar Disord 2003; 5: 217–25
- Mukherjee S., Sackeim H. A., Schnur D. B. Electroconvulsive therapy of acute manic episodes: a review of 50 years' experience. Am J Psychiatry 1994; 151: 169–76
- D'Sa C., Duman R. S. Antidepressants and neuroplasticity. Bipolar Disord 2002; 4: 183–94
- Yamada M., Higuchi T. Antidepressant‐elicited changes in gene expression Remodeling of neuronal circuits as a new hypothesis for drug efficacy. Prog Neuropsychopharmacol Biol Psychiatry 2005; 29: 999–1009
- Duman R. S., Monteggia L. M. A neurotrophic model for stress‐related mood disorders. Biol Psychiatry 2006; 59: 1116–27
- Manji H., Duman R. S. Impairments of neuroplasticity and cellular resilience in severe mood disorders: implications for the development of novel therapeutics. Psychopharmacol Bull 2001; 35: 5–49
- Manji H. K., Drevets W. C., Charney D. S. The cellular neurobiology of depression. Nat Med 2001; 7: 541–7
- Duman R. Depression: A case of neuronal life and death?. Biol Psychiatry 2004; 56: 140–45
- Sheline Y., Gado M. H., Kraemer H. C. Untreated depression and hippocampal volume loss. Am J Psychiatry 2003; 160: 1516–8
- Vermetten E., Vythilingam M., Southwick S. M., Charney D. S., Bremner J. D. Long‐term treatment with paroxetine increases verbal declarative memory and hippocampal volume in posttraumatic stress disorder. Biol Psychiatry 2003; 54: 693–702
- Moore G., Bebchuk J. M., Wilds I. B., Chen G., Manji H. K., Menji H. K. Lithium‐induced increase in human brain grey matter. Lancet 2000; 356: 1241–2
- Drevets W. Neuroimaging studies of mood disorders. Biol Psychiatry 2000; 48: 813–29
- Malberg J., Eisch A. J., Nestler E. J., Duman R. S. Chronic antidepressant treatment increases neurogenesis in adult hippocampus. J Neurosci 2000; 20: 9104–10
- Czeh B., Michaelis T., Watanabe T., Frahm J., de Biurrun G., van Kampen M., et al. Stress‐induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc Natl Acad Sci U S A 2001; 98: 12796–801
- Madsen T., Treschow A., Bengzon J., Bolwig T. G., Lindvall O., Tingström A. Increased neurogenesis in a model of electroconvulsive therapy. Biol Psychiatry 2000; 47: 1043–49
- Manev H., Uz T., Smalheiser N. R., Manev R. Antidepressants alter cell proliferation in the adult brain in vivo and in neural cultures in vitro. Eur J Pharmacol 2001; 411: 67–70
- Santarelli L., Saxe M., Gross C., Surget A., Battaglia F., Dulawa S., et al. Requirement of hippocampal neurogenisis for the behavioral effects of antidepressants. Science 2003; 301: 805–09
- Chen G., Rajkowska G., Du F., Seraji‐Bozorgzad N., Manji H. K. Enhancement of hippocampal neurogenesis by lithium. J Neurochem 2000; 75: 1729–34
- Perez J., Tardito D., Mori S., Racagni G., Smeraldi E., Zanardi R. Abnormalities of cAMP signaling in affective disorders: implication for pathophysiology and treatment. Bipolar Disord 2000; 2: 27–36
- Duman R. Novel therapeutic approaches beyond the serotonin receptor. Biol Psychiatry 1998; 44: 324–35
- Adayev T., Ranasinghe B., Banerjee P. Transmembrane signaling in the brain by serotonin, a key regulator of physiology and emotion. Biosci Rep 2005; 25: 363–85
- Zheng M., Zhu W., Han Q., Xiao R. P. Emerging concepts and therapeutic implications of beta‐adrenergic receptor subtype signaling. Pharmacol Ther 2005; 1083): 257–68
- Aantaa R., Marjamaki A., Scheinin M. Molecular pharmacology of alpha 2‐adrenoceptor subtypes. Ann Med 1995; 27((4))439–49
- Kamenetsky M., Middelhaufe S., Bank E. M., Levin L. R., Buck J., Steegborn C. Molecular details of cAMP generation in mammalian cells: a tale of two systems. J Mol Biol 2006; 362: 623–39
- Brandon E. P., Idzerda R. L., McKnight G. S. PKA isoforms, neural pathways, and behaviour: making the connection. Curr Opin Neurobiol 1997; 7: 397–403
- Nguyen P. V., Woo N. H. Regulation of hippocampal synaptic plasticity by cyclic AMP‐dependent protein kinases. Prog Neurobiol 2003; 71: 401–37
- Lonze B. E., Ginty D. D. Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002; 35: 605–23
- Dyke H. J., Montana J. G. Update on the therapeutic potential of PDE4 inhibitors. Expert Opin Investig Drugs 2002; 11: 1–13
- Houslay M. D., Schafer P., Zhang K. Y. Keynote review: phosphodiesterase‐4 as a therapeutic target. Drug Discov Today 2005; 10: 1503–19
- Dowlatshahi D., MacQueen G. M., Wang J. F., Young L. T. Increased temporal cortex CREB concentrations and antidepressant treatment in major depression. Lancet 1998; 352: 1754–5
- Dowlatshahi D., MacQueen G. M., Wang J. F., Reiach J. S., Young L. T. G protein‐coupled cyclic AMP signaling in postmortem brain of subjects with mood disorders: effects of diagnosis, suicide, and treatment at the time of death. J Neurochem 1999; 73: 1121–6
- Cowburn R. F., Marcusson J. O., Eriksson A., Wiehager B., O'Neill C. Adenylyl cyclase activity and G‐protein subunit levels in postmortem frontal cortex of suicide victims. Brain Res 1994; 633: 297–304
- Manier D., Eiring A., Shelton R. C., Sulser F. β‐Adrenoceptor‐linked protein Kinase A (PKA) activity in human fibroblasts from normal subjects and from patients with major depression. Neuropsychopharm 1996; 15: 556–61
- Manier D. H., Shelton R. C., Ellis T. C., Peterson C. S., Eiring A., Sulser F. Human fibroblasts as a relevant model to study signal transduction in affective disorders. J Affect Disord 2000; 61: 51–8
- Menkes D. B., Rasenick M. M., Wheeler M. A., Bitensky M. W. Guanosine triphosphate activation of brain adenylate cyclase: enhancement by long‐term antidepressant treatment. Science 1983; 129: 65–7
- Ozawa H., Rasenick M. M. Chronic electroconvulsive treatment augments coupling of the GTP‐binding protein Gs to the catalytic moiety of adenylyl cyclase in a manner similar to that seen with chronic antidepressant drugs. J Neurochem 1991; 56: 330–8
- Perez J., Tinelli D., Brunello N., Racagni G. cAMP‐dependent phosphorylation of soluble and crude microtubule fractions of rat cerebral cortex after prolonged desmethylimipramine treatment. Eur J Pharmacol 1989; 172: 305–16
- Nestler E., Terwilliger R. Z., Duman R. S. Chronic antidepressant administration alters the subcellular distribution of cAMP‐dependent protein kinase in rat frontal cortex. J Neurochem 1989; 53: 1644–7
- Ye Y., Conti M., Houslay M. D., Faroqui S. M., Chen M., O'Donnell J. M. Noradrenergic activity differentially regulates the expression of rolipram‐sensitive, high‐affinity cyclic AMP phosphodiesterase (PDE4) in rat brain. J Neurochem 1997; 69: 2397–404
- Takahashi M., Terwilliger R., Lane S., Mezes P. S., Conti M., Duman R. S. Chronic antidepressant administration increases the expression of cAMP phosphodiesterase 4A and 4B isoforms. J Neurosci 1999; 19: 610–18
- Nibuya M., Nestler E. J., Duman R. S. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci 1996; 16: 2365–72
- Fujimaki K., Morinobu S., Duman R. S. Administration of a cAMP phosphodiesterase 4 inhibitor enhances antidepressant‐induction of BDNF mRNA in rat hippocampus. Neuropsychopharmacology 2000; 22: 42–51
- Gould T. D., Quiroz J. A., Singh J., Zarate C. A., Manji H. K. Emerging experimental therapeutics for bipolar disorder: insights from the molecular and cellular actions of current mood stabilizers. Mol Psychiatry 2004; 9: 734–55
- Chen G., Manji H. K., Wright C. B., Hawver D. B., Potter W. Z. Effects of valproic acid on beta‐adrenergic receptors, G‐proteins, and adenylyl cyclase in rat C6 glioma cells. Neuropsychopharmacology 1996; 15: 271–80
- Chen G., Pan B., Hawver D. B., Wright C. B., Potter W. Z., Manji H. K. Attenuation of cyclic AMP production by carbamazepine. J Neurochem 1996; 67: 2079–86
- Deogracias R., Espliguero G., Iglesias T., Rodriguez‐Pena A. Expression of the neurotrophin receptor trkB is regulated by the cAMP/CREB pathway in neurons. Mol Cell Neurosci 2004; 26: 470–80
- Fukuchi M., Tabuchi A., Tsuda M. Transcriptional regulation of neuronal genes and its effect on neural functions: cumulative mRNA expression of PACAP and BDNF genes controlled by calcium and cAMP signals in neurons. J Pharmacol Sci 2005; 98: 212–18
- McCauslin C. S., Heath V., Colangelo A. M., Malik R., Lee S., Mallei A., et al. CAAT/Enhancer‐binding Protein {delta} and cAMP‐response Element‐binding Protein Mediate Inducible Expression of the Nerve Growth Factor Gene in the Central Nervous System. J Biol Chem 2006; 281: 17681–8
- Thomas M. J., Umayahara Y., Shu H., Centrella M., Rotwein P., McCarthy T. L. Identification of the cAMP response element that controls transcriptional activation of the insulin‐like growth factor‐I gene by prostaglandin E2 in osteoblasts. J Biol Chem 1996; 271: 21835–41
- Tanimoto K., Yoshida E., Mita S., Nibu Y., Murakami K., Fukamizu A. Human activin betaA gene. Identification of novel 5' exon, functional promoter, and enhancers. J Biol Chem 1996; 271: 32760–9
- Pugazhenthi S., Miller E., Sable C., Young P., Heidenreich K. A., Boxer L. M., et al. Insulin‐like growth factor‐I induces bcl‐2 promoter through the transcription factor cAMP‐response element‐binding protein. J Biol Chem 1999; 274: 27529–35
- Jeon S. H., Seong Y. S., Juhnn Y. S., Kang U. G., Ha K. S., Kim Y. S., et al. Electroconvulsive shock increases the phosphorylation of cyclic AMP response element binding protein at Ser‐133 in rat hippocampus but not in cerebellum. Neuropharmacology 1997; 36: 411–4
- Thome J., Sakai N., Shin K. H., Steffen C., Zhang Y‐J., Impey S., et al. cAMP response element‐mediated gene transcription is upregulated by chronic antidepressant treatment. J Neurosci 2000; 20: 4030–6
- Chen A‐H., Shirayama Y., Shin K‐H., Neve R. L., Duman R. S. Expression of the cAMP response element binding protein (CREB) in hippocampus produces antidepressant effect. Biol Psychiatry 2001; 49: 753–62
- Tiraboschi E., Tardito D., Kasahara J., Moraschi S., Pruneri P., Gennarelli M., et al. Selective phosphorylation of nuclear CREB by fluoxetine is linked to activation of CaM kinase IV and MAP kinase cascades. Neuropsychopharmacology 2004; 29: 1831–40
- Alt A., Nisenbaum E. S., Bleakman D., Witkin J. M. A role for AMPA receptors in mood disorders. Biochem Pharmacol 2006; 71: 1273–88
- Svenningsson P., Tzavara E. T., Liu F., Fienberg A. A., Nomikos G. G., Greengard P. DARPP‐32 mediates serotonergic neurotransmission in the forebrain. Proc Natl Acad Sci U S A 2002; 99: 3188–93
- Svenningsson P., Tzavara E. T., Witkin J. M., Fienberg A. A., Nomikos G. G., Greengard P. Involvement of striatal and extrastriatal DARPP‐32 in biochemical and behavioral effects of fluoxetine (Prozac). Proc Natl Acad Sci U S A 2002; 99: 3182–7
- Newton S. S., Thome J., Wallace T. L., Shirayama Y., Schlesinger L., Sakai N., et al. Inhibition of cAMP response element‐binding protein or dynorphin in the nucleus accumbens produces an antidepressant‐like effect. J Neurosci 2002; 22: 10883–90
- Pliakas A., Carlson R. R., Neve R. L., Konradi C., Nestler E. J., Carlezon W. A. Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated CREB expression in the nucleus accumbens. J Neurosci 2001; 21: 7397–403
- Duman R. Role of neurotrophic factors in the etiology and treatment of mood disorders. Neuromolecular Med 2004; 5: 11–25
- Dwivedi Y., Rizavi H. S., Conley R. R., Pandey G. N. ERK MAP kinase signaling in post‐mortem brain of suicide subjects: differential regulation of upstream Raf kinases Raf‐1 and B‐Raf. Mol Psychiatry 2006; 11: 86–98
- Dwivedi Y., Rizavi H. S., Roberts R. C., Conley R. C., Tamminga C. A., Pandey G. N. Reduced activation and expression of ERK1/2 MAP kinase in the post‐mortem brain of depressed suicide subjects. J Neurochem 2001; 77: 916–28
- Karege F., Vaudan G., Schwald M., Perroud N., La Harpe R. Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Brain Res Mol Brain Res 2005; 136: 29–37
- Mercier G., Lennon A. M., Renouf B., Dessouroux A., Ramauge M., Courtin F., et al. MAP kinase activation by fluoxetine and its relation to gene expression in cultured rat astrocytes. J Mol Neurosci 2004; 24: 207–16
- Kodama M., Russell D. S., Duman R. S. Electroconvulsive seizures increase the expression of MAP kinase phosphatases in limbic regions of rat brain. Neuropsychopharm 2005; 30: 360–71
- Bhat R. V., Engber T. M., Finn J. P., Koury E. J., Contreras P. C., Miller M. S., et al. Region‐specific targets of p42/p44MAPK signaling in rat brain. J Neurochem 1998; 70: 558–71
- Einat H., Yuan P., Gould T. D., Li j., Du J., Zhang L., et al. The role of the extracellular signal‐regulated kinase signaling pathway in mood modulation. J Neurosci 2003; 23: 7311–16
- Hao Y., Creson T., Zhang L., Li P., Yuan P., Gould T. D., et al. Mood stabilizer valproate promotes ERK pathway‐dependent cortical neuronal growth and neurogenesis. J Neurosci 2004; 24: 6590–9
- Yuan P., Juong L. D., Jiang Y. M., Gutkind J. S., Manji H. K., Chen G. The mood‐stabilizer valproic acid activates mitogen‐activated protein kinases and promotes neurite growth. J Biol Chem 2001; 19: 19
- Duman C. H., Schlesinger L., Kodama M., Russell D. S., Duman R. S. A role for MAP kinase signaling in behavioral models of depression and antidepressant treatment. Biol Psychiatry 2007; 61: 661–70
- Dwivedi Y., Rizavi H. S., Conley R. R., Roberts R. C., Tamminga C. A., Pandey G. N. Altered gene expression of brain‐derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry 2003; 60: 804–15
- Karege F., Perret G., Bondolfi G., Schwald M., Bertschy G., Aubry J. M. Decreased serum brain‐derived neurotrophic factor levels in major depressed patients. Psychiatry Res 2002; 109: 143–8
- Shimizu E., Hashimoto K., Okamura N., Koike K., Komatsu N., Kumakiri C., et al. Alterations of serum levels of brain‐derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol Psychiatry 2003; 54: 70–5
- Evans S., Choudary P. V., Neal C. R., Li J. Z., Vawter M. P., Tomita H., et al. Dysregulation of the fibroblast growth factor system in major depression. Proc Natl Acad Sci U S A 2004; 101: 15506–11
- Ueyama T., Kawai Y., Nemoto K., Sekimoto M., Tone S., Senba E. Immobilization stress reduced the expression of neurotrophins and their receptors in the rat brain. Neurosci Research 1997; 28: 103–10
- Heine V., Zareno J., Maslam S., Joels M., Lucassen P. J. Chronic stress in the adult dentate gyrus reduces cell proliferation near the vasculature and VEGF and Flk‐1 protein expression. Eur J Neurosci 2005; 21: 1304–14
- Newton S., Collier E., Hunsberger J., Adams D., Salvanayagam E., Duman R. S. Gene profile of electroconvulsive seizures: induction of neurogenic and angiogenic factors. J Neurosci 2003; 23: 10841–51
- Altar C. A., Laeng P., Jurata L. W., Brockman J. A., Lemire A., Bullard J., et al. Electroconvulsive seizures regulate gene expression of distinct neurotrophic signaling pathways. J Neurosci 2004; 24: 2667–77
- Warner‐Schmidt J. L., Duman R. S. VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proc Natl Acad Sci U S A 2007; 104: 4647–52
- Dow A. L., Russell D. S., Duman R. S. Regulation of activin mRNA and Smad2 phosphorylation by antidepressant treatment in the rat brain: effects in behavioral models. J Neurosci 2005; 25: 4908–16
- Khawaja X., Xu J., Liang J. J., Barrett J. E. Proteomic analysis of protein changes developing in rat hippocampus after chronic antidepressant treatment: Implications for depressive disorders and future therapies. J Neurosci Res 2004; 75: 451–60
- Mallei A., Shi B., Mocchetti I. Antidepressant treatments induce the expression of basic fibroblast growth factor in cortical and hippocampal neurons. Mol Pharmacol 2002; 61: 1017–24
- Gonul A. S., Akdeniz F., Taneli F., Donat O., Eker C., Vahip S. Effect of treatment on serum brain‐derived neurotrophic factor levels in depressed patients. Eur Arch Psychiatry Clin Neurosci 2005; 255: 381–6
- Chen B., Dowlatshahi D., MacQueen G. M., Wang J‐F., Young L. T. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry 2001; 50: 260–5
- Aydemir O., Deveci A., Taneli F. The effect of chronic antidepressant treatment on serum brain‐derived neurotrophic factor levels in depressed patients: a preliminary study. Prog Neuropsychopharmacol Biol Psych 2004; 29: 261–5
- Gervasoni N., Aubrey J. M., Bondolfi G., Osiek G., Schwald M., Bertschv G., et al. Partial normalization of serum brain‐derived neurotrophic factor in remitted patients after a major depressive episode. Neuropsychobiology 2005; 51: 234–8
- Fukumoto T., Morinobu S., Okamoto Y., Kagaya A., Yamawaki S. Chronic lithium treatment increases the expression of brain‐derived neurotrophic factor in the rat brain. Psychopharmacology (Berl) 2001; 158: 100–6
- Duman R., Monteggia L. M. A neurotrophic model for stress‐related mood disorders. Biol Psychiatry 2006; 59: 1116–27
- Berton O., McClung C. A., Dileone R. J., Krishnan V., Renthal W., Russo S. J., et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 2006; 311: 864–8
- Eisch A. J., Bolanos C. A., de Wit J., Simonak R. D., Pudiak C. M., Barrot M., et al. Brain‐derived neurotrophic factor in the ventral midbrain‐nucleus accumbens pathway: a role in depression. Biol Psychiatry 2003; 54: 994–1005
- Shirayama Y., Chen A. C., Nakagawa S., Russell D. S., Duman R. S. Brain‐derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci 2002; 22: 3251–61
- Warner J., Duman R. S. Vascular endothelial growth factor mediates antidepressant‐induced neurogenesis. Soc Neurosci Abstract 2005
- Hoshaw B. A., Malberg J. E., Lucki I. Central administration of IGF‐I and BDNF leads to long‐lasting antidepressant‐like effects. Brain Res 2005; 1037: 204–8
- Conti A. C., Cryan J. F., Dalvi A., Lucki I., Blendy J. A. cAMP response element‐binding protein is essential for the upregulation of brain‐derived neurotrophic factor transcription, but not the behavioral or endocrine responses to antidepressant drugs. J Neurosci 2002; 22: 3262–8
- Farooq A., Zhou M. M. Structure and regulation of MAPK phosphatases. Cell Signal 2004; 16: 769–79
- Theodosiou A., Ashworth A. MAP kinase phosphatases. Genome Biol 2002; 3: REVIEWS3009
- Keyse S. M. Protein phosphatases and the regulation of mitogen‐activated protein kinase signalling. Curr Opin Cell Biol 2000; 12: 186–92
- Canettieri G., Morantte I., Guzman E., Asahara H., Herzig S., Anderson S. D., et al. Attenuation of a phosphorylation‐dependent activator by an HDAC‐PP1 complex. Nat Struct Biol 2003; 10: 175–81
- Huang E., Reichardt L. F. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 2003; 72: 609–42
- Rhee S. G. Regulation of phosphoinositide‐specific phospholipase C. Annu Rev Biochem 2001; 70: 281–312
- Battaini F. Protein kinase C isoforms as therapeutic targets in nervous system disease states. Pharmacol Res 2001; 44: 353–61
- Keenan C., Kelleher D. Protein kinase C and the cytoskeleton. Cell Signal 1998; 10: 225–32
- Allison J. H., Stewart M. A. Reduced brain inositol in lithium‐treated rats. Nat New Biol 1971; 233: 267–8
- Allison J. H., Blisner M. E., Holland W. H., Hipps P. P., Sherman W. R. Increased brain myo‐inositol 1‐phosphate in lithium‐treated rats. Biochem Biophys Res Commun 1976; 71: 664–70
- Moore G. J., Bebchuk J. M., Parrish J. K., Faulk M. W., Arfken C. L., Strahl‐Bevacqua J., et al. Temporal dissociation between lithium‐induced changes in frontal lobe myo‐inositol and clinical response in manic‐depressive illness. Am J Psychiatry 1999; 156: 1902–8
- O'Donnell T., Rotzinger S., Nakashima T. T., Hanstock C. C., Ulrich M., Silverstone P. H. Chronic lithium and sodium valproate both decrease the concentration of myo‐inositol and increase the concentration of inositol monophosphates in rat brain. Brain Res 2000; 880: 84–91
- Wolfson M., Hertz E., Belmaker R. H., Hertz L. Chronic treatment with lithium and pretreatment with excess inositol reduce inositol pool size in astrocytes by different mechanisms. Brain Res 1998; 787: 34–40
- Williams R., Cheng L., Mudge A. W., Harwood A. J. A common mechanism of action for three mood‐stabilizing drugs. Nature 2002; 417: 292–5
- Shamir A., Shaltiel G., Greenberg M. L., Belmaker R. H., Agam G. The effect of lithium on expression of genes for inositol biosynthetic enzymes in mouse hippocampus; a comparison with the yeast model. Brain Res Mol Brain Res 2003; 115: 104–10
- Harwood A. J. Lithium and bipolar mood disorder: the inositol‐depletion hypothesis revisited. Mol Psychiatry 2005; 10: 117–26
- Wang H. Y., Johnson G. P., Friedman E. Lithium treatment inhibits protein kinase C translocation in rat brain cortex. Psychopharmacology (Berl) 2001; 158: 80–6
- Wang H., Friedman E. Increased association of brain protein kinase C with the receptor for activated C kinase‐1 (RACK1) in bipolar affective disorder. Biol Psychiatry 2001; 50: 364–70
- Lenox R. H., Watson D. G., Patel J., Ellis J. Chronic lithium administration alters a prominent PKC substrate in rat hippocampus. Brain Res 1992; 570: 333–40
- Manji H. K., Etcheberrigaray R., Chen G., Olds J. L. Lithium decreases membrane‐associated protein kinase C in hippocampus: selectivity for the alpha isozyme. J Neurochem 1993; 61: 2303–10
- Manji H. K., Bersudsky Y., Chen G., Belmaker R. H., Potter W. Z. Modulation of protein kinase C isozymes and substrates by lithium: the role of myo‐inositol. Neuropsychopharmacology 1996; 15: 370–81
- Manji H. K., Lenox R. H. Ziskind‐Somerfeld Research Award. Protein kinase C signaling in the brain: molecular transduction of mood stabilization in the treatment of manic‐depressive illness. Biol Psychiatry 1999; 46: 1328–51
- Gould T., Manji H. K. Glycogen synthase kinase‐3: a putative molecular target for lithium mimetic drugs. Neuropsychopharmacology 2005; 30: 1223–37
- Watcharasit P., Bijur G. N., Zmijewski J. W., Song L., Zmijewska A., Chen X., et al. Direct, activating interaction between glycogen synthase kinase‐3beta and p53 after DNA damage. Proc Natl Acad Sci U S A 2002; 99: 7951–5
- Linseman D. A., Butts B. D., Precht T. A., Phelps R. A., Le S. S., Laessig T. A., et al. Glycogen synthase kinase‐3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci 2004; 24: 9993–10002
- Grimes C. A., Jope R. S. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol 2001; 65: 391–426
- Grimes C. A., Jope R. S. CREB DNA binding activity is inhibited by glycogen synthase kinase‐3 beta and facilitated by lithium. J Neurochem 2001; 78: 1219–32
- Katso R., Okkenhaug K., Ahmadi K., White S., Timms J., Waterfield M. D. Cellular function of phosphoinositide 3‐kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol 2001; 17: 615–75
- Raymond J. R., Mukhin Y. V., Gelasco A., Turner J., Collinsworth G., Gettys T. W., et al. Multiplicity of mechanisms of serotonin receptor signal transduction. Pharmacol Ther 2001; 92: 179–212
- Bennecib M., Gong C. X., Grundke‐Iqbal I., Iqbal K. Role of protein phosphatase‐2A and ‐1 in the regulation of GSK‐3, cdk5 and cdc2 and the phosphorylation of tau in rat forebrain. FEBS Lett 2000; 485: 87–93
- Klein P., Melton D. A. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A 1996; 93: 8455–9
- Stambolic V., Ruel L., Woodgett J. R. Lithium inhibits glycogen synthase kinase‐3 activity and mimics wingless signalling in intact cells. Curr Biol 1996; 6: 1664–8
- De Sarno P., Li X., Jope R. S. Regulation of Akt and glycogen synthase kinase‐3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology 2002; 43: 1158–64
- Kirshenboim N., Plotkin B., Shlomo S. B., Kaidanovich‐Beilin O., Eldar‐Finkelman H. Lithium‐mediated phosphorylation of glycogen synthase kinase‐3beta involves PI3 kinase‐dependent activation of protein kinase C‐alpha. J Mol Neurosci 2004; 24: 237–45
- Chalecka‐Franaszek E., Chuang D. M. Lithium activates the serine/threonine kinase Akt‐1 and suppresses glutamate‐induced inhibition of Akt‐1 activity in neurons. Proc Natl Acad Sci U S A 1999; 96: 8745–50
- Gould T. D., Chen G., Manji H. K. In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase‐3. Neuropsychopharmacology 2004; 29: 32–8
- Chen G., Huang L. D., Jiang Y. M., Manji H. K. The mood‐stabilizing agent valproate inhibits the activity of glycogen synthase kinase‐3. J Neurochem 1999; 72: 1327–30
- Hall A. C., Brennan A., Goold R. G., Cleverley K., Lucas F. R., Gordon‐Weeks P. R., et al. Valproate regulates GSK‐3‐mediated axonal remodeling and synapsin I clustering in developing neurons. Mol Cell Neurosci 2002; 20: 257–70
- Roh M. S., Eom T. Y., Zmijewska A. A., De Sarno P., Roth K. A., Jope R. S. Hypoxia activates glycogen synthase kinase‐3 in mouse brain in vivo: protection by mood stabilizers and imipramine. Biol Psychiatry 2005; 57: 278–86
- Li X., Zhu W., Roh M. S., Friedman A. B., Rosborough K., Jope R. S. In vivo regulation of glycogen synthase kinase‐3beta (GSK3beta) by serotonergic activity in mouse brain. Neuropsychopharmacology 2004; 29: 1426–31
- Madsen T. M., Newton S. S., Eaton M. E., Russell D. S., Duman R. S. Chronic electroconvulsive seizure up‐regulates beta‐catenin expression in rat hippocampus: role in adult neurogenesis. Biol Psychiatry 2003; 54: 1006–14
- Kang U. G., Roh M. S., Jung J. R., Shin S. Y., Lee Y. H., Park J. B., et al. Activation of protein kinase B (Akt) signaling after electroconvulsive shock in the rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry 2004; 28: 41–4
- Roh M. S., Kang U. G., Shin S. Y., Lee Y. H., Jung H. Y., Juhnn Y. S., et al. Biphasic changes in the Ser‐9 phosphorylation of glycogen synthase kinase‐3beta after electroconvulsive shock in the rat brain. Prog Neuropsychopharmacol Biol Psychiatry 2003; 27: 1–5
- Alimohamad H., Rajakumar N., Seah Y. H., Rushlow W. Antipsychotics alter the protein expression levels of beta‐catenin and GSK‐3 in the rat medial prefrontal cortex and striatum. Biol Psychiatry 2005; 57: 533–42
- Emamian E. S., Hall D., Birnbaum M. J., Karayiorgou M., Gogos J. A. Convergent evidence for impaired AKT1‐GSK3beta signaling in schizophrenia. Nat Genet 2004; 36: 131–7
- Kaidanovich‐Beilin O., Milman A., Weizman A., Pick C. G., Eldar‐Finkelman H. Rapid antidepressive‐like activity of specific glycogen synthase kinase‐3 inhibitor and its effect on beta‐catenin in mouse hippocampus. Biol Psychiatry 2004; 55: 781–4
- Gould T. D., Einat H., Bhat R., Manji H. K. AR‐A014418, a selective GSK‐3 inhibitor, produces antidepressant‐like effects in the forced swim test. Int J Neuropsychopharmacol 2004; 7: 387–90
- O'Brien W. T., Harper A. D., Jove F., Woodgett J. R., Maretto S., Piccolo S., et al. Glycogen synthase kinase‐3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci 2004; 24: 6791–8
- Beaulieu J. M., Sotnikova T. D., Yao W. D., Kockeritz L., Woodgett J. R., Gainetdinov R. R., et al. Lithium antagonizes dopamine‐dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A 2004; 101: 5099–104
- Jeon Y. H., Heo Y. S., Kim C. M., Hyun Y. L., Lee T. G., Ro S., et al. Phosphodiesterase: overview of protein structures, potential therapeutic applications and recent progress in drug development. Cell Mol Life Sci 2005; 62: 1198–220
- Tsankova N. M., Berton O., Renthal W., Kumar A., Neve R. L., Nestler E. J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci 2006; 9: 519–25
- Phiel C. J., Zhang F., Huang E. Y., Guenther M. G., Lazar M. A., Klein P. S. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 2001; 276: 36734–41
- Bebchuk J. M., Arfken C. L., Dolan‐Manji S., Murphy J., Hasanat K., Manji H. K. A preliminary investigation of a protein kinase C inhibitor in the treatment of acute mania. Arch Gen Psychiatry 2000; 57: 95–7
- Bhat R. V., Budd Haeberlein S. L., Avila J. Glycogen synthase kinase 3: a drug target for CNS therapies. J Neurochem 2004; 89: 1313–7
- Gould T. D., Zarate C. A., Manji H. K. Glycogen synthase kinase‐3: a target for novel bipolar disorder treatments. J Clin Psychiatry 2004; 65: 10–21
- Tanis K. Q., Newton S. S., Duman R. S. Targeting neurotrophic/growth factor expression and signaling for antidepressant drug development. CNS Neurol Disord Drug Targets 2007; 6: 151–60