Abstract
Alzheimer's disease (AD) is a complex disorder of the central nervous system (CNS). Molecular genetic research has provided a wealth of information regarding the genetic etiology of this devastating disease. Identification and functional characterization of autosomal dominant mutations in the amyloid precursor protein gene (APP) and the presenilin genes 1 and 2 (PSEN1 and PSEN2) have contributed substantially to our understanding of the biological mechanisms leading towards CNS neurodegeneration in AD. Nonetheless, a large part of the genetic etiology remains unresolved, especially that of more common, sporadic forms of AD. While substantial efforts were invested in the identification of genetic risk factors underlying sporadic AD, using carefully designed genetic association studies in large patient-control groups, the only firmly established risk factor remains the ε4 allele of the apolipoprotein E gene (APOE). Nevertheless, one can expect that with the current availability of high-throughput genotyping platforms and dense maps of single-nucleotide polymorphisms (SNPs), large-scale genetic studies will eventually generate additional knowledge about the genetic risk profile for AD. This review provides an overview of the current understanding in the field of AD genetics, covering both the rare monogenic forms as well as recent developments in the search for novel AD susceptibility genes.
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder of the central nervous system (CNS) and is the most common dementia subtype in the elderly (50%–70% of demented patients). Clinically, AD is characterized by progressive deterioration of cognitive functions, ultimately leading to complete dependency and death. Most AD patients present with impairment of recent memory, but during disease progression other symptoms such as changes in personality and behavior become apparent. There is a substantial overlap in clinical symptoms between AD and other CNS degenerative brain diseases involving dementia (e.g. frontotemporal lobar degeneration (FTLD) and Creutzfeldt-Jakob disease). Therefore, a definite diagnosis of AD is obtained only by pathological examination of the autopsied brain. In addition to severe neuronal loss, AD brains show two distinct pathological lesions: extracellular plaques composed of aggregated amyloid β (Aβ) peptides, and intracellular neurofibrillary tangles consisting of filaments of hyperphosphorylated protein tau Citation1.
AD has a complex etiology involving the interplay of both genetic and environmental factors Citation2. Two major risk factors are increased age and a positive family history of dementia. An European population-based study calculated an AD prevalence of 5% in the age group 65 years and older, which increased to 22% among those aged 95 years and older Citation3. Although the majority of patients develop clinical symptoms at later age (>65 years; senile or late-onset AD), 1%–2% of patients have an earlier disease onset (presenile or early-onset AD). Independently of onset age, AD brain pathology is identical, though in young patients the disease progresses more rapidly, and brain pathology is more pronounced. Twin studies identified a substantial genetic component in AD, with an estimated heritability of up to 80% Citation4. Molecular genetic studies performed in the last 20 years have produced important new genetic data, though predominantly in studies of rare monogenic forms of early-onset AD. Highly penetrant mutations were identified in three genes, the amyloid precursor protein gene (APP) and the presenilin genes 1 and 2 (PSEN1 and PSEN2) Citation2. Also, a fourth gene, the apolipoprotein E gene (APOE), a major risk gene was identified in late-onset AD families Citation5. Except for APOE, not much is known about other risk genes contributing to late-onset AD, though several such risk genes must exist with effect sizes equal to or even larger than the APOE ε4 allele Citation6, Citation7.
Key messages
Monogenic forms have contributed significantly to the current understanding of the pathobiology of Alzheimer's disease (AD).
The amyloid precursor protein is a key protein in developing novel therapies for AD.
Novel genetic designs will provide clues for defining risk profiles for sporadic AD.
Monogenic forms of AD
Amyloid precursor protein gene (APP)
APP, located on chromosome 21, was the first gene identified in autosomal dominant early-onset AD families Citation8. Instrumental here were the observation of AD brain pathology in Down's syndrome (DS) or trisomy 21 patients Citation9, the isolation and sequencing of the senile plaque Aβ peptide Citation10, and the mapping of its precursor gene APP Citation11 near the AD-linked chromosomal region on chromosome 21 Citation12, Citation13. Mutation analysis of APP in AD was stimulated by the observation of linkage with APP Citation14 and the identification of a missense mutation in its Aβ sequence in hereditary cerebral hemorrhages with amyloidosis-Dutch type (HCHWA-D) (Dutch E693Q mutation, numbering according to the 770 amino acids isoform) Citation15. HCHWA-D is a rare autosomal dominant disorder characterized by recurrent cerebral hemorrhages due to extensive Aβ congophilic amyloid angiopathy (CAA) affecting small cerebral blood vessel walls Citation16. The first AD mutation in APP was identified near the C-terminal site of the Aβ sequence and was nicknamed London APP mutation (V717I) Citation8. A double-mutation was identified in AD near the N-terminus of the Aβ sequence, the Swedish mutation APP KM670/671NL Citation17. We identified a second mutation within the Aβ sequence, the Flemish APP mutation (A692G) located adjacent to the Dutch APP mutation, which associated AD and CAA Citation18.
Since then, 21 different missense mutations have been identified in APP in 68 families (AD Mutation Database; http://www.molgen.ua.ac.be/ADMutations/). Nonetheless APP mutations explain less than 1% of early-onset AD families Citation19.
Amyloid precursor protein
APP consists of 18 exons within a genomic region of 290 kb with part of exons 16 and 17 coding for the Aβ peptide Citation20 (A). Alternative splicing produces three major isoforms (B), of which APP695 is predominantly expressed in the brain, especially in neurons. APP is a single-pass type I transmembrane glycoprotein with a small cytosolic C-terminal domain and a large luminal N-terminus Citation11.
Figure 1. A: Schematic presentation of Amyloid Precusor Protein (APP) at the genomic, transcript, and protein level. Numbers (genomic and transcript) indicate exons and yellow-colored exons (transcript) designate alternatively spliced exons. At the transcript level, untranslated regions (UTR) are represented as dark gray boxes; coding regions are shown in light gray. Pink boxes indicate the portion of the protein from which the Aβ peptide and the APP intracellular domain (AICD) are formed. B: Different APP isoforms, produced by means of alternative splicing, that have been isolated from human tissue. Each transcript is named according to the protein that can be translated from the transcript, i.e. APP770 encodes an isoform containing 770 amino acids. C: Schematic overview of the two major APP processing pathways, i.e. the constitutive and the amyloidogenic pathway. Arrowheads indicate the respective cleavage sites (α-, β-, and γ-site); arrows indicate the cleavage event by the respective proteases (α-, β-, and γ-secretase).
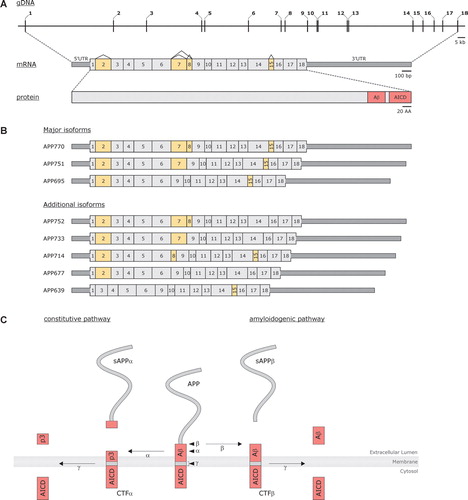
Contrary to what was initially assumed, the Aβ peptide is also formed under normal physiological conditions Citation21, Citation22. APP is proteolyzed by α-, β-, and γ-secretases following one of two mutually exclusive ways: the constitutive or non-amyloidogenic pathway which precludes the formation of intact Aβ peptides and is the major APP processing pathway in most cell types Citation23; or alternatively the amyloidogenic pathway which is particularly enriched in neurons and gives rise to Aβ peptides Citation21, Citation22, Citation24–26 (C). In the latter, APP is cleaved first by β-secretase at N-terminal position 1 of the Aβ sequence (β-site) Citation27. Of note is that β-secretase also cleaves APP at the adjacent β′-site, producing Aβ11–40 and Aβ11–42 Citation27. Next γ-secretase cuts the membrane-bound C-terminal fragment to produce Aβ peptides Citation28, Citation29, a heterogeneous mixture of Aβ peptides with varying C-terminal lengths of 39–43 amino acids. Under physiological conditions two major Aβ species are present. The major form is Aβ40, ending at position 40 (90%), whereas about 10% end at position 42, Aβ42. It is Aβ42 that is predominantly present in amyloid plaques in AD brains Citation30. Studies have shown that this more hydrophobic Aβ42 has a higher aggregation propensity compared to Aβ40.
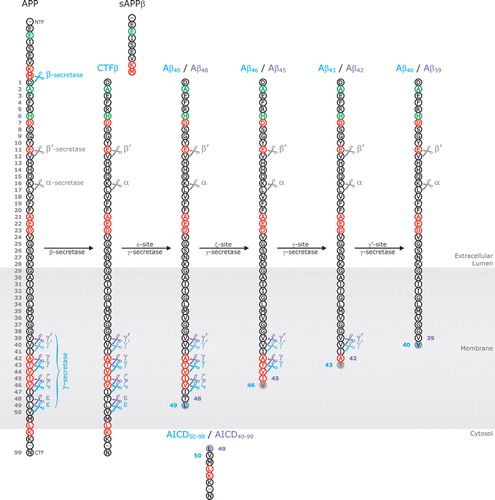
In addition to the regular γ-sites, there are two other cleavage sites located downstream of residue 42, e.g. the γ-like cleavage site or ε-cleavage site Citation31, Citation32 and the ζ-cleavage site Citation33 (). Proteolysis by γ-secretase at these sites generates longer Aβ peptides, e.g. Aβ49 and Aβ48 for ε-cleavage and Aβ46 and Aβ45 for ζ-cleavage, and studies showed that the γ-secretase cleavages of APP C-terminal fragments are likely sequential events. The first cut by γ-secretase occurs at the ε-site releasing the APP intracellular domain (AICD) and producing Aβ49 and Aβ48. These Aβ peptides stay in the enzyme's active site and are subsequently cleaved at the ζ-site (Aβ46/Aβ45) and the γ-site, ultimately releasing Aβ40 and Aβ42 Citation34–38 (). In this manner APP is cleaved every three amino acids which fits well into an α-helical model of APP processing, with Aβ40 and Aβ42 cleavages on opposite sites of the transmembrane domain Citation37.
Figure 2. Schematical illustration of sequential Amyloid Precusor Protein (APP) cleavages producing amyloid Aβ peptide. In a first cleavage event, APP is cut at the Aβ N-terminus (β-secretase action) releasing its large soluble ectodomain (sAPPβ) in the extracellular space. Subsequently, the membrane-bound C-terminal stub (CTFβ) is cleaved downstream of the C-terminal end of Aβ by the γ-secretase complex. The four possible cleavage sites (γ-, γ′-, ζ-, and ε-sites) are indicated by scissors. The first C-terminal cut occurs at the ε-site (ε-site γ-secretase activity) and releases the APP intracellular domain (AICD) into the cytosol. The majority (∼90%) of CTFβ is cleaved between amino acids 49 and 50 (blue scissors) producing Aβ49; however, an alternative cleavage occurs between amino acids 48 and 49 (purple scissors) leading to the formation of Aβ48. Both membrane-bound peptides serve as substrates for subsequent cleavage events (ζ-site, γ- and γ′-site γ-secretase cleavage), cutting the peptides every three amino acids, ultimately producing Aβ40 and Aβ42 (or Aβ39 in case Aβ42 is not yet released into the extracellular lumen before the last cleavage event takes place). Amino acid positions (gray numbers) are numbered starting from the N-terminus of Aβ. Mutated amino acid positions are indicated in red, non-pathogenic variants are shown in green. Major cleavage events, giving rise to the production of Aβ40, are indicated by blue scissors. Minor cleavage events, producing Aβ42, are shown in purple.
APP missense mutations
All pathogenic APP missense mutations are located in or near the Aβ sequence and in the vicinity of protease cleavage sites, exerting their pathogenic effect by influencing APP proteolytic processing (for review see Citation39) (; ). The Swedish APP mutation KM670/671NL increased proteolysis at the β-site, thereby elevating total Aβ levels Citation40, Citation41. Very recently we identified a novel mutation, E682K, in a familial early-onset probable AD patient (onset age 47 years), altering the highly conserved position 11 of the alternate β′-site (Van Broeckhoven et al. unpublished data). At this point, we have not yet completed the genetic studies, neither have we examined the effect of this mutation on Aβ processing. However, a study examining the selectivity of β-secretase cleavage of APP showed that blocking the β′-site by introducing the artificial double-mutation Y681K/E682K shifted β-secretase cleavage entirely to the At this point, we have not yet-site Citation42. Therefore, we can assume that also E682K affects β′-site processing of APP and potentially exerts its pathogenic effect by increasing Aβ1–40 and Aβ1–42 levels as in the Swedish APP mutation. On the other hand, we cannot exclude that this mutation renders Aβ a better substrate for β′-site cleavage, increasing the proportion of N-truncated peptides.
Table I. Overview of the effects of different types of mutations in Alzheimer's disease genes on the processing of APP and the production of Aβ.
Mutations at the γ-site affect the positions where APP C-terminal fragments are cleaved, e.g. position 40 or 42 of the Aβ peptide, with different mutations exerting distinct effects on γ-secretase activity Citation43. Although results differ among studies and cell types used, the overall effect of these C-terminal mutations is an increase in the relative amount of fibrillogenic Aβ42 Citation44, Citation45. Therefore, the best predictor for pathogenicity of an APP mutation in an in vitro cell assay is the Aβ42/Aβ40 ratio. The two most C-terminal APP mutations, Australian L723P and Belgian K724N, also increased the Aβ42/Aβ40 ratio Citation39, Citation46. Since ε- and ζ-cleavages generate intermediates for further γ-processing, it was predicted that alterations in APP processing at these sites would also result in a relative increase of Aβ42 and the Aβ42/Aβ40 ratio. This was confirmed for several C-terminal APP mutations (T714I, V717F, and L723P) Citation35, Citation37, Citation47. For these mutations elevated levels of AICD49–99 (numbering according to Aβ peptide) and its N-terminal counterpart Aβ48 were formed, often combined with a concomitant decrease in the physiologically predominant species (AICD50–99 and Aβ49) Citation35, Citation37, Citation47. Interestingly, the latest model for APP γ-secretase processing proposed that Aβ48 gives rise to the production of Aβ42 while Aβ49 is converted to Aβ40, explaining how differential cleavage at the more C-terminally located cleavage sites would result in an increased Aβ42/Aβ40 ratio Citation36–38. Since these differences in ε-cleavages also generate different AICD fragments, it is plausible that mutations might affect AICD function. The normal function of AICD has not been unambiguously determined yet, but it is presumed to function in neurogenesis Citation48, transcriptional regulation of target genes Citation49, and signal transduction Citation49. Affecting either one or all of these processes could contribute to the mutation's pathogenicity.
Mutations at the α-secretase site interfere with processing of APP at its internal Aβ peptide cleavage site (α-site) Citation50. This was shown for the Flemish APP mutation (A692G) Citation18, which decreased α-cleavage Citation51, Citation52 resulting in more substrate for the amyloidogenic processing into Aβ peptide. However, most α-site mutations do not result in increased Aβ secretion, in fact they are associated with reduced levels of Aβ peptides Citation52–54, suggesting a different mechanistic action. Since α-site mutations are located within the Aβ sequence they alter its amino acid composition and therefore its physicochemical properties. Several studies have shown an enhanced protofibril and/or fibril formation (E693Q, E693G, and D694N) Citation15, Citation53, Citation55–60 or increased fibril stability when formed (A692G) Citation61 for the mutant Aβ peptides. Also the affinity and toxicity towards cerebrovascular cells was altered for some mutant Aβ peptides Citation58, Citation62–67, providing an explanation for the association of several α-site mutations with different clinical/pathological characteristics such as intracerebral hemorrhages due to extensive CAA (E693Q, E693K), intracerebral hemorrhages and dementia (A692G), or dementia only (E693G) Citation50.
APP regulatory and dosage mutations
More recently APP mutations were identified that affect APP copy number or transcriptional activity. An APP locus duplication was identified in five autosomal dominant early-onset AD families Citation68. All five duplications had different chromosomal break points and contained additional genes, implying that the APP locus contained a recombination hot spot. The APP duplication patients had a mixed phenotype of AD and/or intracerebral hemorrhages, caused by extensive CAA Citation68, Citation69. In one Dutch early-onset family, we observed a similar phenotype of AD with CAA caused by a genomic duplication of only APP Citation70. Although there are only few studies, so far APP duplications account for about 8%–10% of autosomal dominant early-onset AD families and 3% of familial AD Citation68–71.
In the 5′ regulatory region of APP we identified mutations in probable early- or late-onset AD that significantly increased APP transcriptional activity in vitro with some mutations increasing expression by a factor of nearly two Citation72, Citation73. Unfortunately, we had no autopsied brain available of AD patients carrying these promoter mutations and thus could not obtain a definite diagnosis of AD, nor could we examine APP expression in vivo. However, very recently one mutation carrier (APP -369C > G) died and neuropathological diagnosis confirmed AD pathology with a strong CAA component similar to AD duplication patients (Brouwers et al. unpublished data). Nonetheless, one genetic study was unable to replicate our findings Citation74, indicating that further studies are needed to define the pathogenic role of APP promoter mutations in risk for AD.
Higher levels of γ-secretase substrate (e.g. APP C-terminal fragments) have been directly correlated with an increased Aβ42/Aβ40 ratio Citation75. This suggests that elevated levels of APP in case of APP duplication or increased APP transcriptional activity (), could ultimately result in an elevated Aβ42/Aβ40 ratio similar to that observed for APP γ-site mutations. Whether this is the case in vivo in patients carrying such a mutation awaits further investigation.
Presenilin 1 and 2 genes (PSEN1 and PSEN2)
Initial linkage studies showed that not all AD families could be explained by a genetic defect located on chromosome 21 Citation13, and mutation analyses showed that the majority of AD families did not segregate an APP mutation Citation19, Citation76–80. Consequently, genetic heterogeneity of familial AD was further investigated, and significant linkage was obtained in multiple AD families supporting a major early-onset AD locus on chromosome 14 (14q24.3) Citation81–84. Eventually, positional cloning identified the chromosome 14 gene, and it was named presenilin 1 (PSEN1) Citation85, based on the observation of missense mutations in several linked pedigrees Citation86, Citation87.
In the extended Volga-German AD family Citation88, a genome-wide study identified linkage to chromosome 1q31–42 Citation89. Following the identification of PSEN1, homology mapping identified a second PSEN gene, PSEN2, in the linked region Citation90, Citation91, in which a missense mutation (N141l) segregated with AD in seven Volga-German kindreds Citation90. A second missense mutation (M239V) in PSEN2 was identified in an Italian family Citation91. To date, 10 PSEN2 mutations have been identified in 18 families, while 164 PSEN1 mutations appear in 361 families Citation92 (AD Mutation Database). In a population-based epidemiological sample of early onset AD, we estimated the overall mutation frequencies of PSEN1 and PSEN2 at, respectively, 6% and 1% Citation93.
Most PSEN mutations cause typical AD, clinically and pathologically indistinguishable from sporadic AD, except for the early-onset age and a more rapid and pronounced disease progression. Nonetheless, PSEN mutations are occasionally associated with AD with onset age >65 years Citation93 (AD Mutation Database). In the Volga-German kindreds some PSEN2 N141l carriers had late-onset AD Citation90, Citation91, and two other PSEN2 mutations (V148I and Q228L) were detected in patients with an onset >65 years Citation94, Citation95. More recently, two PSEN1 mutations (A79V and R269H), previously associated with early-onset AD, were also found in late-onset AD patients Citation96, Citation97. We observed in Belgian AD patients, with onset ≤70 years, missense mutations in PSEN1 (C263F), and PSEN2 (R62C and R71W) in three AD patients with onset age >65 years Citation98. Together these observations suggest that modifying factors influence onset age in PSEN mutation carriers.
Presenilins (PSENs)
PSEN1 covers a genomic region of ±84 kb and comprises 13 exons Citation99, whereas PSEN2 is only ±25 kb in size and has 12 exons Citation100 (A). Apart from differences in genomic size and number of exons, the PSENs have a similar gene structure. Same as APP, PSENs are expressed in a wide variety of tissues including brain, although expression of PSEN2 is remarkably lower in brain Citation85, Citation90, Citation91. In brain, the PSENs are primarily expressed in neurons, with higher levels noted in the cerebellum and the hippocampus Citation101–103. Both genes produce a number of alternatively spliced transcripts.
Figure 3. A: Schematic presentations of the presenilins (PSEN1 and PSEN2) at genomic, transcript, and protein level. Numbers (genomic and transcript) indicate exons. At transcript level, untranslated regions are represented as dark gray boxes; coding regions are shown in light gray. Pink boxes indicate the transmembrane regions (TM) in both proteins, connected by hydrophilic loops (HL). B: Nine-transmembrane topology of PSEN1. Transmembrane regions (TM, pink boxes) are connected by hydrophilic loop structures. In hydrophilic loop VI a portion of the loop is associated with the membrane (MA, gray box).
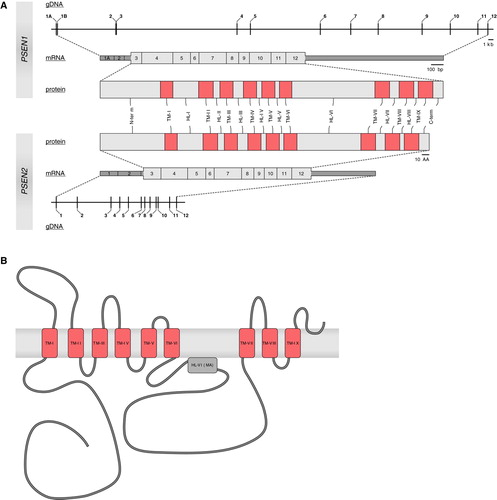
The PSEN proteins share an overall amino acid sequence identity of 67% Citation90, Citation91. Hydrophobicity plots suggested that PSENs represent integral membrane proteins containing at least seven transmembrane domains that are highly conserved between human PSENs and their orthologues Citation85, Citation90, Citation91, but the topology of PSENs remained a matter of debate Citation104–108. Recent evidence indicated that PSENs most likely adopt a nine-transmembrane topology Citation109–111 (B).
Under physiological conditions, the bulk of PSENs are endoproteolytically processed by cleavage within the large hydrophilic loop, yielding N- and C-terminal fragments that are present in a 1:1 stoichiometry, forming stable heterodimeric complexes Citation112–118. Endoproteolytic cleavage is a highly regulated process happening after intramolecular interactions have been established, but fragments derived from the different PSEN molecules do not associate with each other Citation119. Although endoproteolytic cleavage is not necessary to produce functionally active presenilin proteins Citation112, both fragments are essential to ensure presenilin activity Citation118, Citation120, Citation121. PSENs are predominantly located in the endoplasmic reticulum and the intermediate and early-Golgi compartment Citation101, Citation122–124, implying that they are involved in protein processing.
PSENs are γ-secretases
The multiple transmembrane topology of PSENs indicated that they function as cell receptors, channel proteins, or in intracellular trafficking of proteins. Since PSENs show substantial homology with SPE-4, a Caenorhabditis elegans protein that is involved in storage and transport of proteins Citation85, Citation90, it was presumed that the PSENs had a similar function, e.g. regulating intracellular transport of APP Citation85, Citation90. Insights into the function of PSENs resulted from a genetic screen in C. elegans aiming at identifying proteins involved in Notch (LIN-12) signaling. Here a protein (SEL-12) with remarkable sequence similarity to PSENs was identified Citation125. Rescue by human PSENs of the sel-12 phenotype in worms Citation125 and the Notch phenotype in Psen1 null mice indicated a role for PSENs in Notch signaling Citation126, Citation127. Furthermore, PSENs and APP interact with each other to form stable complexes Citation128–130. In neuronal cultures derived from Psen1−/ − null mice, Aβ production was dramatically reduced and accompanied by an accumulation of APP C-terminal fragments Citation131. As α- and β-secretase cleavage of APP was not affected, these results implied that Psen1 is directly involved in γ-secretase cleavage of APP Citation131. Proteolytic cleavage of Notch, producing the Notch intracellular domain (NICD) that is involved in signal transduction, bore a striking resemblance to the γ-secretase processing of APP, and further studies indicated that PSEN1 is also required for the ligand-dependent cleavage of Notch Citation28, Citation132–134. Taken together, these observations suggested that PSENs either function as necessary cofactors or regulators of γ-secretase activity or represent the actual protease activity. Supportive evidence of the latter was obtained using mutagenesis of either one of two highly conserved aspartic acid residues, and using transition state analog inhibitors directed to the active site of aspartyl proteases. Both experiments showed a potent inhibition of γ-secretase activity, similar to that observed in Psen1−/ − cells Citation29, Citation135, Citation136. These data provided evidence that PSENs indeed offer the catalytic activity of γ-secretase, and function as diaspartyl proteases.
Even though it was generally accepted that PSENs are in fact γ-secretases, it was noted that γ-secretases exist as high-molecular-weight complexes in the cell suggesting that other proteins are implicated Citation137–139. Three PSEN interacting proteins, nicastrin (NCSTN) Citation140, presenilin enhancer 2 (PEN2) Citation141, and anterior pharynx-defective 1 (APH1) Citation141, Citation142, were identified as members of the γ-secretase complex, and were shown to be essential components for its activity Citation143–145. APH1 and NCSTN are involved in stabilization of the PSEN holoprotein in the γ-secretase complex, whereas PEN2 is required for endoproteolytic cleavage of the holoprotein Citation146. Thus, the γ-secretase complex consists of four proteins, PSEN1 or 2, NCSTN, APH1 and PEN2, but PSENs are providing the catalytic active site. Also, γ-secretase was shown to process a series of different type I transmembrane proteins through a process nowadays known as ‘regulated intramembrane proteolysis’ (RIP) (for review see Citation147–149).
Gain or loss of PSEN function
The PSEN mutation spectrum comprises primarily missense mutations that are scattered over the protein with some clustering around putative transmembrane domains, e.g. 63% and 67% in PSEN1 and PSEN2, respectively Citation92 (AD Mutation Database). Also most mutations occur at residues conserved among human PSENs (95% in PSEN1 and 100% in PSEN2) and often so among presenilin orthologues, supporting their functional relevance. Some but not all mutations interfere with the endoproteolytic processing Citation113, Citation150–152, like mutations deleting exon 9 (▵9) containing the sequence coding for the endoproteolytic cleavage site Citation112. Deletion of exon 9 inhibits endoproteolytic cleavage and maintains PSEN1 as a stable holoprotein though functionally active as γ-secretase. Therefore, the pathogenic nature of the ▵9 mutation is more likely the result of the introduction of a missense mutation at the junction of exons 8 and 10 (S290C) Citation153.
That PSEN mutations were involved in the pathogenic pathway leading to amyloid deposition was shown by Aβ peptide measurements in plasma and in conditioned medium from fibroblasts of PSEN1 and PSEN2 mutation carriers. PSEN mutations resulted in relatively elevated levels of Aβ42 compared to Aβ40 Citation154, Citation155. Higher levels of Aβ42 were also observed in brains of PSEN1 mutation carriers compared to sporadic AD patients Citation156–158. Further evidence was obtained in transgenic mouse models or transfected cells overexpressing mutant PSENs, where a relatively increased production of longer Aβ species was seen Citation159–163.
Since the vast majority of mutations identified in PSENs were missense mutations that lead to an increase in the Aβ42/Aβ40 ratio, it was hypothesized that PSEN mutations conferred their pathogenic effect by acquiring a toxic gain-of-function. However, it remained unclear how the many different missense mutations scattered over the entire protein were able to cause a similar gain-of-function. Detailed analyses of different mutant PSENs showed that some mutations increased the Aβ42/Aβ40 ratio by decreasing Aβ40 rather than increasing Aβ42 Citation164–168, implying a PSEN loss-of-function as disease causation mechanism. Also, APP processing at the ε-site was reduced for a substantial number of mutant PSENs Citation164–166, Citation169–171, implying a general loss of γ-secretase activity and not only at the γ-site. Also comparable results were obtained for the release of the intracellular domain of other γ-secretase substrates, such as NICD Citation134, Citation165, Citation166, Citation169, Citation170, Citation172–174. In addition to less efficient processing at the ε-site, mutations were shown to alter the preferred cleavage site, since they often increased AICD49–99 and/or decreased AICD50–99 production Citation35, Citation47, similarly to what was observed for APP γ-site mutations. Together with the current sequential APP processing model, this could explain the contradiction of a partial loss-of-function leading to an increased Aβ42/Aβ40 ratio, the apparent toxic gain-of-function. Mutant PSENs that less efficiently process their substrates also cut these substrates more often at the alternative minor ε-cleavage site. The less active mutant γ-secretase releases Aβ42 before it is further processed to Aβ39, resulting in increased amounts of Aβ42 even though there is a general loss of function Citation37, Citation175, Citation176.
The apparent loss-of-function of PSEN mutations corroborates well the partial rescue of the sel-12 phenotype in worms by mutant PSEN compared to the wild type Citation177. Although initial rescue experiments in Psen1 null mice disproved this hypothesis Citation178, Citation179, this was likely due to the rather mild effect on NICD production by the mutant PSEN (A246E) used (only 20% decreased) Citation165, Citation178, Citation179. Also, conditional Psen1 and Psen2 knockout mice displayed memory impairment, synaptic dysfunction, and age-dependent neurodegeneration in the absence of Aβ Citation180, suggesting a role for PSENs in neuronal survival. Thus loss of PSEN function might contribute to neurodegeneration.
Also, previously, several genetic studies reported association of AD with polymorphisms located in the 5′ regulatory region of PSEN1 and PSEN2 Citation181–184. The risk-conferring allele of the associated PSEN1 promoter polymorphism (−22C) leads to a neuron-specific reduction of transcriptional activity of the PSEN1 promoter, due to alterations in transcription factor binding sites Citation185. Moreover, two rare promoter mutations were identified in early-onset AD patients, of which one significantly reduced PSEN1 transcription in neuronal cells Citation183. These data also suggested a pathogenic effect through a loss-of-function mechanism rather than a toxic gain-of-function.
Other monogenic loci/genes for AD
Novel locus on chromosome 7
The identification of mutations in APP and PSENs has substantially contributed to the genetic etiology of familial AD. Nonetheless, in a significant percentage of familial AD patients the genetic cause is still unknown Citation7. In these families, with early- or late-onset AD, the disease is often inherited in an autosomal dominant manner Citation186–189. In a Dutch family with mean onset age 66.8 years (range 47–77 years), we performed a genome-wide screen Citation186, Citation189 and identified a novel locus on chromosome 7q36. Mutation analysis of all 29 known genes in the 5.44 Mb linked region did not unambiguously identify the underlying gene.
Association with non-AD genes
The mutation R406W in the microtubule associated protein tau gene (MAPT), causally related to FTLD Citation190, has been identified in several families diagnosed with clinical AD Citation190–196. A second mutation (▵K281) was found in a patient with a clinical and pathological diagnosis of AD Citation197. Furthermore, the major tau isoform accumulating in neurofibrillary tangles in the patient's brain corresponded to the isoform predominantly produced as a result of the ▵K281 mutation Citation197. While most MAPT mutations are identified in FTLD patients, it is not surprising to find some in AD patients since both dementia subtypes share overlapping clinical symptoms and a differential diagnosis of AD or FTLD is often difficult to establish particularly in the later stages of the disease. Furthermore, it has been suggested that AD and FTLD represent disease expressions in one common spectrum ranging from CAA (amyloid-positive, tau-negative) over AD (amyloid-positive, tau-positive) and tau-positive FTLD, to tau-negative, ubiquitin-positive FTLD (FTLD-U) Citation198.
Another example is the identification in two late-onset AD patients of a null mutation (IVS1 + 5G > C) in the progranulin gene (PGRN) underlying FTLD-U pathology Citation199–201. The intron 1 splice-site mutation IVS1 + 5G > C was originally identified in a large Belgian FTLD-U founder family, consisting of 10 branches with 39 patients. Strikingly, the disease in this large family is characterized by a wide range in onset age (from 45 to 78 years) and association of the PGRN mutation with different clinical phenotypes (FTLD, AD, and Parkinson Disease (PD)) Citation201. A similar effect was observed for another PGRN founder mutation (R493X), with 30% of mutation carriers presenting with memory problems and a clinical diagnosis of AD in three patients Citation202. In both studies postmortem analysis revealed mixed pathologies of FTLD-U and AD (or PD) Citation201, Citation202. As PGRN encodes a growth factor, it is conceivable that the protein could potentially function as a general neuronal survival factor. Null mutations in PGRN lower the threshold for neurodegeneration and formation of pathologic lesions of, for example, AD, occurring because of another disease mechanism or modifying factor Citation201. In this context, we identified several PGRN missense mutations in AD patients that likely disrupt PGRN protein by interfering with the characteristic granulin folds of the protein Citation203. Interestingly, similar missense mutations were observed in FTLD patients Citation204, and the corresponding mutant proteins were shown to be less efficiently secreted and more rapidly degraded Citation205. Taken together, these missense mutations might have a milder effect on the amount of functional protein produced, increasing an individual's risk of developing a neurodegenerative CNS disease at later age.
While these observations are exemplifying the clinical heterogeneity of mutations in dementia genes and their contribution to the complexity of the neurodegeneration process, MAPT or PGRN mutations remain infrequent causes of clinical diagnosed AD. Nevertheless, screening for mutations in these two genes might be warranted in clinically diagnosed AD patients in which mutations in known AD genes are absent.
Searching for AD susceptibility genes
The identification of genes in which mutations are responsible for monogenic early-onset forms of AD has contributed substantially to understanding the molecular mechanisms involved in AD pathogenesis. Nonetheless, the majority of AD patients develop the disease at older age and, although there is a clustering of patients into families, segregation of AD in these families does not follow a Mendelian inheritance pattern.
Apolipoprotein E gene (APOE)
A genome-wide linkage study in late-onset AD families identified a novel locus on chromosome 19 (19q13.1–19q13.3) Citation206. About the same time the apolipoprotein E (APOE) was shown to interact with the Aβ peptide in cerebrospinal fluid, and its gene (APOE) was located near the chromosome 19 linked region Citation207. Moreover, APOE had been associated with senile plaques and neurofibrillary tangles in AD brains Citation208, and APOE transcription was upregulated in brains of AD patients Citation209. Subsequently, it was shown that one of the three major APOE isoforms, APOE ε4, was overrepresented in familial late-onset AD patients compared to aged healthy control individuals Citation207. The APOE ε4 association was extensively confirmed in both familial as well as sporadic late-onset AD patients of different ethnic backgrounds Citation5, Citation210, Citation211, and later also in early-onset AD patients Citation212.
APOE ε4 primarily acts by lowering onset age in a dosage-dependent manner, increasing risk 3 times in heterozygotes and 15 times in homozygotes Citation211. The ε2 isoform, on the other hand, was shown to have a protective effect; however, this could not be consistently replicated Citation213. APOE genotype was also shown to modify onset age in carriers of causal AD mutations. This was extensively shown for APP mutations Citation73, Citation214–217 but also for several PSEN2 mutations Citation218. No effect could be observed for PSEN1 mutations Citation219, with the exception of a large Colombian PSEN1 pedigree Citation220. Apart from the three major protein isoforms, four promoter variants influencing APOE expression levels were shown to affect AD risk in several studies Citation221–226. Although an APOE ε4 independent effect seemed only present for one variant (−491A/T), this finding is noteworthy given that APOE ε4 affects AD risk in a dose-dependent manner.
In vitro studies have indicated that the APOE ε4 isoform binds Aβ peptides with a higher avidity compared to APOE ε3 Citation227. Furthermore, there is a strong correlation between the presence of an APOE ε4 allele and a higher Aβ burden in the brains of AD patients Citation228, Citation229, suggesting that APOE interacts with Aβ in enhancing its deposition in plaques. This is supported by the observation that homozygous Apoe knockout (Apoe−/ − ) mice develop fewer and more diffuse, non-fibrillar Aβ deposits Citation230–232. Some but not all studies assessing the effect of different APOE isoforms on Aβ fibrillization showed that the ε4 isoform leads to increased Aβ aggregation in vitro Citation227, Citation233–236. Similarly, in vivo studies in Apoe−/ − mice indicated that APOE ε4 increased Aβ fibrillization and plaques formation compared to APOE ε3 Citation237, Citation238. Still, it is possible that APOE exerts its effects through different mechanisms, e.g. APOE is a major cholesterol transporter and high cholesterol levels have been associated with an increased Aβ load in animal models Citation239, Citation240 and changes in APP processing Citation241–243. Thus APOE isoform-specific changes in cholesterol binding and transport in brain might also affect plaque formation in AD brains.
Other AD susceptibility loci/genes
Two approaches are being followed to unravel the genetic etiology of late-onset AD, i.e. hypothesis-driven candidate gene studies and hypothesis-free (in terms of biological function or position) genome-wide analyses. Genome-wide linkage and association studies in large samples consisting of late-onset AD families and sib pairs have identified several chromosomal loci harboring potential AD susceptibility genes Citation244. In candidate-gene-based studies researchers have focused on genes encoding functionally relevant proteins, e.g. proteins that belong to the γ-secretase complex (for example PSEN1 Citation185, Citation225, Citation245 or NCSTN Citation225, Citation246), regulate APP trafficking (SORL1 Citation247, Citation248), or are involved in fibrillization or clearance of the Aβ peptide (IDE Citation225, Citation249, ACE Citation225, Citation250, PLAU Citation251, MME Citation252). Also genes that are implicated in other neurodegenerative diseases (PRNP Citation253) or genes located in linked regions have received attention (www.alzgene.org).
Genetic designs
Over the years genetic studies have experienced a marked evolution. Early studies investigated one or few single nucleotide polymorphisms (SNPs) at a time. Unless the functional polymorphism itself was tested, the success of these studies was limited due to a variety of factors, including absence of linkage disequilibrium (LD) between the marker tested and the underlying functional variant. Rapidly advancing technology allowed gene-wide studies utilizing the underlying LD pattern in haplotype-based approaches in order to capture a maximum of genetic information. However, positive results obtained in some of these studies proved difficult to replicate either because the original finding was a false positive, or it was attributable to heterogeneity at the genetic, allelic, mutational, or population level, or it was influenced by differences in study design or statistical power. Recently, geneticists went even further in their search for AD susceptibility genes, analyzing hundreds to thousands of SNPs spread throughout chromosomal regions showing linkage to AD Citation254, Citation255, whole chromosomes Citation256, Citation257, and even the complete genome Citation258–261.
It is not within the scope of this review to go into detail of the numerous studies on the even so numerous candidate AD susceptibility genes that were performed over the last 10–20 years. A regularly updated overview can be found on the Alzgene website (www.alzgene.org) Citation225. Here we will focus on recent developments in identifying novel AD risk genes.
Genome-wide linkage studies
Genome-wide linkage studies in late-onset AD families and sib pairs Citation244 (www.alzgene.org) generally used several hundreds of microsatellite markers spread throughout the genome. These studies identified a substantial number of chromosomal regions implicated in AD. Some of these loci, such as those on chromosomes 9, 10, 12, and 19, were repeatedly linked to AD Citation244, suggesting that these loci contained important AD susceptibility genes. Since multiple genes with small effect sizes were expected to contribute to late-onset AD, several groups performed genome-wide screens in isolated populations Citation188, Citation261–267. Geographically and/or culturally isolated populations are assumed to be less genetically heterogeneous, with disease risk being determined by a smaller number of genes of which some are enriched in the population under study, thus facilitating their identification. These studies confirmed several of the loci identified in outbred populations, for example 1q21 and 1q25 Citation261, and 10q24 Citation261, Citation267. Furthermore, they also extended the genetic spectrum with additional, new genetic loci implicated in AD, for example 8p12–q22 Citation262 and 3q22–q24 Citation261.
Locus- or chromosome-wide association studies
Genetic approaches based on microsatellite markers generally identify extended chromosomal regions harboring a large number of potentially causal genes. Although many candidate gene studies were performed, especially in the linked regions on chromosomes 9, 10, and 12, the proven causal genes have not yet been found (www.alzgene.org). In a renewed attempt researchers resorted to fine-mapping by genotyping large numbers of SNPs across these regions.
On chromosome 12, analysis of a large number of SNPs across the linkage region (12p11–13) showed that the glyceraldehyde-3-phosphate dehydrogenase gene (GAPDH) and some of its paralogues could be implicated in AD pathogenesis Citation254, but decisive arguments for a role of GAPDH as a risk factor for AD are still awaited. For the chromosome 10 AD locus, two independent studies were performed targeting either the locus (10q21.1–10q25.1; 47 cM) Citation255 or the whole chromosome 10 Citation256. Both studies also differed in study design in terms of ethnicity, stratification for APOE genotype, and SNP selection (LD-based versus a gene-centric approach in which SNPs were selected that have a higher chance of having a direct biological effect (e.g. non-synonymous and regulatory variants)). Not surprisingly different genes were identified, dynamin binding protein gene (DNMBP) Citation255 and a gene (LOC439999) encoding a protein similar to ribosomal protein S3a (RPS3A) Citation256. Similarly to chromosome 10, the locus on chromosome 9 was investigated using a chromosome-wide gene-centric approach Citation257, resulting in the identification of several significant SNPs, with the most significantly associated SNP located under the 9q22 linkage peak pointing to the death-associated protein kinase 1 gene (DAPK1).
Although these approaches in identifying risk genes underlying large linkage and association peaks provided some insights into novel pathways potentially involved in AD pathogenesis, replication of these findings in additional independent study populations proved to be hard Citation268–271. Nonetheless, the DNMBP and GAPDH findings could be confirmed in independent studies Citation272, Citation273.
Genome-wide association studies
Locus- and chromosome-wide studies balance between a targeted candidate region approach and a hypothesis-free approach, as to possible biological pathways leading to AD. Candidate genes are usually selected to fit within existing hypotheses, the most prevailing being the amyloid cascade hypothesis. Several other pathways have been suggested, e.g. altered cholesterol metabolism, impaired axonal transport, increased oxidative stress, etc. The number of proteins fitting in existing hypotheses is enormous, making it practically impossible to analyze all the corresponding genes in detail one by one.
Since high-throughput genotyping platforms permit genotyping of large numbers of SNPs and since the completion of the human sequence allows studying common genetic variation across the genome, researchers started to apply genome-wide association studies. One of the promises is that this holistic approach will uncover candidate genes that otherwise would have escaped attention. The approach already proved fruitful in other complex genetic disorders, such as myocardial infarction Citation274, type 2 diabetes Citation275, Citation276, and coronary heart disease Citation277. In AD, however, achievements are still limited. Currently two different approaches are used in AD research, on the one hand there is the gene-centric approach, utilizing genotype data of a large number of, potentially functional, SNPs located in and around genes Citation258, Citation278, while the other consists of an LD-based approach, where SNPs are selected based upon SNP informativeness and the LD structure of the population under study Citation259, Citation260, Citation279. Although these studies already revealed several new genes and pathways potentially implicated in AD, they also generated some questions. It remains to be established whether these genes can be replicated using independent samples, as none of the studies performed to date seem to identify the same genes. Furthermore, genes identified in locus- or chromosome-wide studies are not detected in genome-wide studies on the same samples Citation254, Citation256–258. In fact, the only gene that is clearly associated in different study populations and designs is still APOE.
Conclusions
After more than 20 years of extensive genetic research in the field of AD, we can conclude that AD is a complex and genetically heterogeneous disorder. Most of the insights we have gained so far came from genetic studies in large multigenerational families in which AD is inherited in an autosomal dominant manner. Linkage analyses in these families has led to the identification of three causal genes (APP, PSEN1, and PSEN2) and one susceptibility gene (APOE) that are consistently involved in AD genetic etiology. Yet, there are still additional AD genes to be identified. There is at least one autosomal dominant AD family, in which mutations in the known dementia-causing genes have been excluded, harboring a causal gene at chromosome 7q36 Citation186. For the more common, generally late-onset form of AD, a large number of chromosomal regions linked to or associated with the disease have been discovered, but their underlying genes have not yet been unequivocally identified. As has already been proven in the past with the identification of APP and the PSENs as causal genes, the discovery of novel genes could contribute significantly to our understanding of the disease process by revealing pathways that could provide access points for novel therapeutic strategies. Though much effort has been put into mapping novel AD genes, the search for genes contributing to the risk profile of late-onset AD has been complicated by the many pitfalls of the designs and techniques used in the past, such as candidate-gene-based association studies. However, the field of complex genetics is rapidly evolving, and although strategies currently used to identify risk genes for AD (such as locus-, chromosome-, or genome-wide screens) yielded so far inconsistent data, they are promising since they have already been successfully applied in other complex diseases like myocardial infarction Citation274, type 2 diabetes Citation275, Citation276, and coronary heart disease Citation277. In these studies much larger sample sizes are used compared to what is currently used in genome-wide screens of AD. This implies that in order to be successful, sample sizes used in genome-wide screens for identification of risk alleles with a small effect on the disease, as is expected for a heterogeneous, complex disorder such as late-onset AD, should be increased. Further, as genetic heterogeneity creates a problem in the reliable identification of novel risk factors, more effort should go into minimizing heterogeneity by using populations with fewer founders (isolated populations). Another way of reducing genetic heterogeneity can be established by utilizing endophenotypes in genome-wide studies. Here, phenotypes that are associated with the disease but are closer to the underlying biology and thus less prone to other modifying factors, for example environmental influences, are investigated. This strategy was already proven successful in a genome-wide association study on memory performance Citation280. Also, in the future, in addition to genotyping larger study populations and minimizing genetic heterogeneity, more effort could be put into combining data from different approaches, such as gene expression data, genome-wide association studies, and proteomics, in order to prioritize the number of potentially implicated genes and to discover novel pathways involved in AD pathogenesis.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Acknowledgements
The research in the author's group is funded in part by the Special Research Fund of the University of Antwerp; the Fund for Scientific Research Flanders (FWO-F); the Interuniversity Attraction Poles program P6/43 of the Belgian Science Policy Office (POD); and the ‘Stichting voor Alzheimer Onderzoek-Fondation pour la Recherche sur la Maladie d'Alzheimer’ (SAO-FRMA). KS is a postdoctoral fellow and NB a PhD fellow of the FWO-F.
References
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991; 82: 239–59
- Cruts M, Van Broeckhoven C. Molecular genetics of Alzheimer's disease. Ann Med. 1998; 30: 560–5
- Lobo A, Launer LJ, Fratiglioni L, Andersen K, Di Carlo A, Breteler MM, et al. Prevalence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology. 2000; 54: S4–S9
- Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006; 63: 168–74
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997; 278: 1349–56
- Slooter AJ, Cruts M, Kalmijn S, Hofman A, Breteler MM, Van Broeckhoven C, et al. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: the Rotterdam Study. Arch Neurol. 1998; 55: 964–8
- Warwick DE, Payami H, Nemens EJ, Nochlin D, Bird TD, Schellenberg GD, et al. The number of trait loci in late-onset Alzheimer disease. Am J Hum Genet. 2000; 66: 196–204
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991; 349: 704–6
- Wisniewski KE, Wisniewski HM, Wen GY. Occurrence of neuropathological changes and dementia of Alzheimer's disease in Down's syndrome. Ann Neurol. 1985; 17: 278–82
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985; 82: 4245–9
- Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987; 325: 733–6
- George-Hyslop PH, Tanzi RE, Polinsky RJ, Haines JL, Nee L, Watkins PC, et al. The genetic defect causing familial Alzheimer's disease maps on chromosome 21. Science. 1987; 235: 885–90
- George-Hyslop PH, Haines JL, Farrer LA, Polinsky R, Van Broeckhoven C, Goate A, et al. Genetic linkage studies suggest that Alzheimer's disease is not a single homogeneous disorder. FAD Collaborative Study Group. Nature. 1990; 347: 194–7
- Van Broeckhoven C, Haan J, Bakker E, Hardy JA, Van Hul W, Wehnert A, et al. Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch). Science. 1990; 248: 1120–2
- Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, et al. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990; 248: 1124–6
- Haan J, Hardy JA, Roos RA. Hereditary cerebral hemorrhage with amyloidosis—Dutch type: its importance for Alzheimer research. Trends Neurosci. 1991; 14: 231–4
- Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, et al. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992; 1: 345–7
- Hendriks L, van Duijn CM, Cras P, Cruts M, Van Hul W, van Harskamp F, et al. Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the beta-amyloid precursor protein gene. Nat Genet. 1992; 1: 218–21
- van Duijn CM, Hendriks L, Cruts M, Hardy JA, Hofman A, Van Broeckhoven C. Amyloid precursor protein gene mutation in early-onset Alzheimer's disease. Lancet. 1991; 337: 978
- Yoshikai S, Sasaki H, Doh-ura K, Furuya H, Sakaki Y. Genomic organization of the human amyloid beta-protein precursor gene. Gene. 1990; 87: 257–63
- Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, et al. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992; 359: 322–5
- Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, et al. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science. 1992; 258: 126–9
- Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T, et al. Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science. 1990; 248: 1122–4
- Estus S, Golde TE, Kunishita T, Blades D, Lowery D, Eisen M, et al. Potentially amyloidogenic, carboxyl-terminal derivatives of the amyloid protein precursor. Science. 1992; 255: 726–8
- Golde TE, Estus S, Younkin LH, Selkoe DJ, Younkin SG. Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science. 1992; 255: 728–30
- Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, et al. Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature. 1992; 359: 325–7
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999; 286: 735–41
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999; 398: 518–22
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999; 398: 513–7
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron. 1994; 13: 45–53
- Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, et al. Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2001; 2: 835–41
- Gu Y, Misonou H, Sato T, Dohmae N, Takio K, Ihara Y. Distinct intramembrane cleavage of the beta-amyloid precursor protein family resembling gamma-secretase-like cleavage of Notch. J Biol Chem. 2001; 276: 35235–8
- Zhao G, Mao G, Tan J, Dong Y, Cui MZ, Kim SH, et al. Identification of a new presenilin-dependent zeta-cleavage site within the transmembrane domain of amyloid precursor protein. J Biol Chem. 2004; 279: 50647–50
- Zhao G, Cui MZ, Mao G, Dong Y, Tan J, Sun L, et al. gamma-Cleavage is dependent on zeta-cleavage during the proteolytic processing of amyloid precursor protein within its transmembrane domain. J Biol Chem. 2005; 280: 37689–97
- Kakuda N, Funamoto S, Yagishita S, Takami M, Osawa S, Dohmae N, et al. Equimolar production of amyloid beta-protein and amyloid precursor protein intracellular domain from beta-carboxyl-terminal fragment by gamma-secretase. J Biol Chem. 2006; 281: 14776–86
- Funamoto S, Morishima-Kawashima M, Tanimura Y, Hirotani N, Saido TC, Ihara Y. Truncated carboxyl-terminal fragments of beta-amyloid precursor protein are processed to amyloid beta-proteins 40 and 42. Biochemistry. 2004; 43: 13532–40
- Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, et al. Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J Neurosci. 2005; 25: 436–45
- Yagishita S, Morishima-Kawashima M, Tanimura Y, Ishiura S, Ihara Y. DAPT-induced intracellular accumulations of longer amyloid beta-proteins: further implications for the mechanism of intramembrane cleavage by gamma-secretase. Biochemistry. 2006; 45: 3952–60
- Theuns J, Marjaux E, Vandenbulcke M, Van Laere K, Kumar-Singh S, Bormans G, et al. Alzheimer dementia caused by a novel mutation located in the APP C-terminal intracytosolic fragment. Hum Mutat. 2006; 27: 888–96
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, et al. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature. 1992; 360: 672–4
- Cai XD, Golde TE, Younkin SG. Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science. 1993; 259: 514–6
- Qahwash I, He W, Tomasselli A, Kletzien RF, Yan R. Processing amyloid precursor protein at the beta-site requires proper orientation to be accessed by BACE1. J Biol Chem. 2004; 279: 39010–6
- De Jonghe C, Esselens C, Kumar-Singh S, Craessaerts K, Serneels S, Checler F, et al. Pathogenic APP mutations near the gamma-secretase cleavage site differentially affect Abeta secretion and APP C-terminal fragment stability. Hum Mol Genet. 2001; 10: 1665–71
- Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L, Jr, Eckman C, et al. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science. 1994; 264: 1336–40
- Tamaoka A, Odaka A, Ishibashi Y, Usami M, Sahara N, Suzuki N, et al. APP717 missense mutation affects the ratio of amyloid beta protein species (A beta 1–42/43 and a beta 1–40) in familial Alzheimer's disease brain. J Biol Chem. 1994; 269: 32721–4
- Kwok JB, Li QX, Hallupp M, Whyte S, Ames D, Beyreuther K, et al. Novel Leu723Pro amyloid precursor protein mutation increases amyloid beta42(43) peptide levels and induces apoptosis. Ann Neurol. 2000; 47: 249–53
- Sato T, Dohmae N, Qi Y, Kakuda N, Misonou H, Mitsumori R, et al. Potential link between amyloid beta-protein 42 and C-terminal fragment gamma 49–99 of beta-amyloid precursor protein. J Biol Chem. 2003; 278: 24294–301
- Ma QH, Futagawa T, Yang WL, Jiang XD, Zeng L, Takeda Y, et al. A TAG1-APP signalling pathway through Fe65 negatively modulates neurogenesis. Nat Cell Biol. 2008; 10: 283–94
- Kerr ML, Small DH. Cytoplasmic domain of the beta-amyloid protein precursor of Alzheimer's disease: function, regulation of proteolysis, and implications for drug development. J Neurosci Res. 2005; 80: 151–9
- Van Broeckhoven C, Kumar-Singh S. Genetics and pathology of alpha-secretase site AbetaPP mutations in the understanding of Alzheimer's disease. J Alzheimers Dis. 2006; 9: 389–98
- Haass C, Hung AY, Selkoe DJ, Teplow DB. Mutations associated with a locus for familial Alzheimer's disease result in alternative processing of amyloid beta-protein precursor. J Biol Chem. 1994; 269: 17741–8
- De Jonghe C, Zehr C, Yager D, Prada CM, Younkin S, Hendriks L, et al. Flemish and Dutch mutations in amyloid beta precursor protein have different effects on amyloid beta secretion. Neurobiol Dis. 1998; 5: 281–6
- Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation. Nat Neurosci. 2001; 4: 887–93
- Watson DJ, Selkoe DJ, Teplow DB. Effects of the amyloid precursor protein Glu693→Gln ‘Dutch’ mutation on the production and stability of amyloid beta-protein. Biochem J. 1999; 340 Pt 3: 703–9
- Clements A, Walsh DM, Williams CH, Allsop D. Effects of the mutations Glu22 to Gln and Ala21 to Gly on the aggregation of a synthetic fragment of the Alzheimer's amyloid beta/A4 peptide. Neurosci Lett. 1993; 161: 17–20
- Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem. 1997; 272: 22364–72
- Wisniewski T, Ghiso J, Frangione B. Peptides homologous to the amyloid protein of Alzheimer's disease containing a glutamine for glutamic acid substitution have accelerated amyloid fibril formation. Biochem Biophys Res Commun. 1991; 179: 1247–54
- Van Nostrand WE, Melchor JP, Cho HS, Greenberg SM, Rebeck GW. Pathogenic effects of D23N Iowa mutant amyloid beta-protein. J Biol Chem. 2001; 276: 32860–6
- Kamino K, Orr HT, Payami H, Wijsman EM, Alonso ME, Pulst SM, et al. Linkage and mutational analysis of familial Alzheimer disease kindreds for the APP gene region. Am J Hum Genet. 1992; 51: 998–1014
- Grabowski TJ, Cho HS, Vonsattel JP, Rebeck GW, Greenberg SM. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurol. 2001; 49: 697–705
- Walsh DM, Hartley DM, Condron MM, Selkoe DJ, Teplow DB. In vitro studies of amyloid beta-protein fibril assembly and toxicity provide clues to the aetiology of Flemish variant (Ala692→Gly) Alzheimer's disease. Biochem J. 2001; 355: 869–77
- Davis J, Van Nostrand WE. Enhanced pathologic properties of Dutch-type mutant amyloid beta-protein. Proc Natl Acad Sci U S A. 1996; 93: 2996–3000
- Verbeek MM, de Waal RM, Schipper JJ, Van Nostrand WE. Rapid degeneration of cultured human brain pericytes by amyloid beta protein. J Neurochem. 1997; 68: 1135–41
- Wang Z, Natte R, Berliner JA, van Duinen SG, Vinters HV. Toxicity of Dutch (E22Q) and Flemish (A21G) mutant amyloid beta proteins to human cerebral microvessel and aortic smooth muscle cells. Stroke. 2000; 31: 534–8
- Melchor JP, McVoy L, Van Nostrand WE. Charge alterations of E22 enhance the pathogenic properties of the amyloid beta-protein. J Neurochem. 2000; 74: 2209–12
- Eisenhauer PB, Johnson RJ, Wells JM, Davies TA, Fine RE. Toxicity of various amyloid beta peptide species in cultured human blood-brain barrier endothelial cells: increased toxicity of dutch-type mutant. J Neurosci Res. 2000; 60: 804–10
- Kumar-Singh S, Julliams A, Nuydens R, Ceuterick C, Labeur C, Serneels S, et al. In vitro studies of Flemish, Dutch, and wild-type beta-amyloid provide evidence for two-staged neurotoxicity. Neurobiol Dis. 2002; 11: 330–40
- Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006; 38: 24–6
- Cabrejo L, Guyant-Marechal L, Laquerriere A, Vercelletto M, De la Fourniere F, Thomas-Anterion C, et al. Phenotype associated with APP duplication in five families. Brain. 2006; 129: 2966–76
- Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, et al. APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain. 2006; 129: 2977–83
- Rovelet-Lecrux A, Frebourg T, Tuominen H, Majamaa K, Campion D, Remes AM. APP locus duplication in a Finnish family with dementia and intracerebral haemorrhage. J Neurol Neurosurg Psychiatry. 2007; 78: 1158–9
- Theuns J, Brouwers N, Engelborghs S, Sleegers K, Bogaerts V, Corsmit E, et al. Promoter mutations that increase amyloid precursor-protein expression are associated with Alzheimer disease. Am J Hum Genet. 2006; 78: 936–46
- Brouwers N, Sleegers K, Engelborghs S, Bogaerts V, Serneels S, Kamali K, et al. Genetic risk and transcriptional variability of amyloid precursor protein in Alzheimer's disease. Brain. 2006; 129: 2984–91
- Guyant-Marechal L, Rovelet-Lecrux A, Goumidi L, Cousin E, Hannequin D, Raux G, et al. Variations in the APP gene promoter region and risk of Alzheimer disease. Neurology. 2007; 68: 684–7
- Yin YI, Bassit B, Zhu L, Yang X, Wang C, Li YM. Gamma-secretase substrate concentration modulates the abeta 42/abeta 40 ratio: Implications for Alzheimer's disease. J Biol Chem. 2007; 282: 23639–44
- Tanzi RE, George-Hyslop PH, Haines JL, Polinsky RJ, Nee L, Foncin JF, et al. The genetic defect in familial Alzheimer's disease is not tightly linked to the amyloid beta-protein gene. Nature. 1987; 329: 156–7
- Van Broeckhoven C, Genthe AM, Vandenberghe A, Horsthemke B, Backhovens H, Raeymaekers P, et al. Failure of familial Alzheimer's disease to segregate with the A4-amyloid gene in several European families. Nature. 1987; 329: 153–5
- Crawford F, Hardy J, Mullan M, Goate A, Hughes D, Fidani L, et al. Sequencing of exons 16 and 17 of the beta-amyloid precursor protein gene in 14 families with early onset Alzheimer's disease fails to reveal mutations in the beta-amyloid sequence. Neurosci Lett. 1991; 133: 1–2
- Tanzi RE, Vaula G, Romano DM, Mortilla M, Huang TL, Tupler RG, et al. Assessment of amyloid beta-protein precursor gene mutations in a large set of familial and sporadic Alzheimer disease cases. Am J Hum Genet. 1992; 51: 273–82
- Schellenberg GD, Anderson L, O'dahl S, Wisjman EM, Sadovnick AD, Ball MJ, et al. APP717, APP693, and PRIP gene mutations are rare in Alzheimer disease. Am J Hum Genet. 1991; 49: 511–7
- Schellenberg GD, Bird TD, Wijsman EM, Orr HT, Anderson L, Nemens E, et al. Genetic linkage evidence for a familial Alzheimer's disease locus on chromosome 14. Science. 1992; 258: 668–71
- Van Broeckhoven C, Backhovens H, Cruts M, De Winter G, Bruyland M, Cras P, et al. Mapping of a gene predisposing to early-onset Alzheimer's disease to chromosome 14q24.3. Nat Genet. 1992; 2: 335–9
- George-Hyslop P, Haines J, Rogaev E, Mortilla M, Vaula G, Pericak-Vance M, et al. Genetic evidence for a novel familial Alzheimer's disease locus on chromosome 14. Nat Genet. 1992; 2: 330–4
- Mullan M, Houlden H, Windelspecht M, Fidani L, Lombardi C, Diaz P, et al. A locus for familial early-onset Alzheimer's disease on the long arm of chromosome 14, proximal to the alpha 1-antichymotrypsin gene. Nat Genet. 1992; 2: 340–2
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995; 375: 754–60
- Cruts M, Backhovens H, Wang SY, Van Gassen G, Theuns J, De Jonghe CD, et al. Molecular genetic analysis of familial early-onset Alzheimer's disease linked to chromosome 14q24.3. Hum Mol Genet. 1995; 4: 2363–71
- Clark RF, Hutton M, Fuldner M, Froelich S, Karran E, Talbot C, et al. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Alzheimer's Disease Collaborative Group. Nat Genet. 1995; 11: 219–22
- Bird TD, Lampe TH, Nemens EJ, Sumi SM, Nochlin D, Schellenberg GD, et al. Characteristics of familial Alzheimer's disease in nine kindreds of Volga German ancestry. Prog Clin Biol Res. 1989; 317: 229–34
- Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL, et al. A familial Alzheimer's disease locus on chromosome 1. Science. 1995; 269: 970–3
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995; 269: 973–7
- Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995; 376: 775–8
- Cruts M, Van Broeckhoven C. Presenilin mutations in Alzheimer's disease. Hum Mutat. 1998; 11: 183–90
- Cruts M, van Duijn CM, Backhovens H, Van den BM, Wehnert A, Serneels S, et al. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum Mol Genet. 1998; 7: 43–51
- Lao JI, Beyer K, Fernandez-Novoa L, Cacabelos R. A novel mutation in the predicted TM2 domain of the presenilin 2 gene in a Spanish patient with late-onset Alzheimer's disease. Neurogenetics. 1998; 1: 293–6
- Zekanowski C, Styczynska M, Peplonska B, Gabryelewicz T, Religa D, Ilkowski J, et al. Mutations in presenilin 1, presenilin 2 and amyloid precursor protein genes in patients with early-onset Alzheimer's disease in Poland. Exp Neurol. 2003; 184: 991–6
- Larner AJ, Ray PS, Doran M. The R269H mutation in presenilin-1 presenting as late-onset autosomal dominant Alzheimer's disease. J Neurol Sci. 2007; 252: 173–6
- Kauwe JS, Jacquart S, Chakraverty S, Wang J, Mayo K, Fagan AM, et al. Extreme cerebrospinal fluid amyloid beta levels identify family with late-onset Alzheimer's disease presenilin 1 mutation. Ann Neurol. 2007; 61: 446–53
- Brouwers N, Sleegers K, Theuns J, Engelborghs S, Bogaerts V, Serneels S, et al. Contribution of dementia genes to Alzheimer's disease in Belgium. Alzheimer. s Dement. 2006; 2(Suppl 1)S191
- Rogaev EI, Sherrington R, Wu C, Levesque G, Liang Y, Rogaeva EA, et al. Analysis of the 5′ sequence, genomic structure, and alternative splicing of the presenilin-1 gene (PSEN1) associated with early onset Alzheimer disease. Genomics. 1997; 40: 415–24
- Levy-Lahad E, Poorkaj P, Wang K, Fu YH, Oshima J, Mulligan J, et al. Genomic structure and expression of STM2, the chromosome 1 familial Alzheimer disease gene. Genomics. 1996; 34: 198–204
- Kovacs DM, Fausett HJ, Page KJ, Kim TW, Moir RD, Merriam DE, et al. Alzheimer-associated presenilins 1 and 2: neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat Med. 1996; 2: 224–9
- Lee MK, Slunt HH, Martin LJ, Thinakaran G, Kim G, Gandy SE, et al. Expression of presenilin 1 and 2 (PS1 and PS2) in human and murine tissues. J Neurosci. 1996; 16: 7513–25
- Suzuki T, Nishiyama K, Murayama S, Yamamoto A, Sato S, Kanazawa I, et al. Regional and cellular presenilin 1 gene expression in human and rat tissues. Biochem Biophys Res Commun. 1996; 219: 708–13
- Li X, Greenwald I. Membrane topology of the C. elegans SEL-12 presenilin. Neuron. 1996; 17: 1015–21
- Li X, Greenwald I. Additional evidence for an eight-transmembrane-domain topology for Caenorhabditis elegans and human presenilins. Proc Natl Acad Sci U S A. 1998; 95: 7109–14
- Doan A, Thinakaran G, Borchelt DR, Slunt HH, Ratovitsky T, Podlisny M, et al. Protein topology of presenilin 1. Neuron. 1996; 17: 1023–30
- Dewji NN, Singer SJ. The seven-transmembrane spanning topography of the Alzheimer disease-related presenilin proteins in the plasma membranes of cultured cells. Proc Natl Acad Sci U S A. 1997; 94: 14025–30
- Dewji NN, Valdez D, Singer SJ. The presenilins turned inside out: implications for their structures and functions. Proc Natl Acad Sci U S A. 2004; 101: 1057–62
- Laudon H, Hansson EM, Melen K, Bergman A, Farmery MR, Winblad B, et al. A nine-transmembrane domain topology for presenilin 1. J Biol Chem. 2005; 280: 35352–60
- Spasic D, Tolia A, Dillen K, Baert V, De Strooper B, Vrijens S, et al. Presenilin-1 maintains a nine-transmembrane topology throughout the secretory pathway. J Biol Chem. 2006; 281: 26569–77
- Henricson A, Kall L, Sonnhammer EL. A novel transmembrane topology of presenilin based on reconciling experimental and computational evidence. FEBS J. 2005; 272: 2727–33
- Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996; 17: 181–90
- Mercken M, Takahashi H, Honda T, Sato K, Murayama M, Nakazato Y, et al. Characterization of human presenilin 1 using N-terminal specific monoclonal antibodies: Evidence that Alzheimer mutations affect proteolytic processing. FEBS Lett. 1996; 389: 297–303
- Ward RV, Davis JB, Gray CW, Barton AJ, Bresciani LG, Caivano M, et al. Presenilin-1 is processed into two major cleavage products in neuronal cell lines. Neurodegeneration. 1996; 5: 293–8
- Podlisny MB, Citron M, Amarante P, Sherrington R, Xia W, Zhang J, et al. Presenilin proteins undergo heterogeneous endoproteolysis between Thr291 and Ala299 and occur as stable N- and C-terminal fragments in normal and Alzheimer brain tissue. Neurobiol Dis. 1997; 3: 325–37
- Kim TW, Pettingell WH, Hallmark OG, Moir RD, Wasco W, Tanzi RE. Endoproteolytic cleavage and proteasomal degradation of presenilin 2 in transfected cells. J Biol Chem. 1997; 272: 11006–10
- Shirotani K, Takahashi K, Ozawa K, Kunishita T, Tabira T. Determination of a cleavage site of presenilin 2 protein in stably transfected SH-SY5Y human neuroblastoma cell lines. Biochem Biophys Res Commun. 1997; 240: 728–31
- Steiner H, Capell A, Pesold B, Citron M, Kloetzel PM, Selkoe DJ, et al. Expression of Alzheimer's disease-associated presenilin-1 is controlled by proteolytic degradation and complex formation. J Biol Chem. 1998; 273: 32322–31
- Saura CA, Tomita T, Davenport F, Harris CL, Iwatsubo T, Thinakaran G. Evidence that intramolecular associations between presenilin domains are obligatory for endoproteolytic processing. J Biol Chem. 1999; 274: 13818–23
- Citron M, Eckman CB, Diehl TS, Corcoran C, Ostaszewski BL, Xia W, et al. Additive effects of PS1 and APP mutations on secretion of the 42-residue amyloid beta-protein. Neurobiol Dis. 1998; 5: 107–16
- Tomita T, Tokuhiro S, Hashimoto T, Aiba K, Saido TC, Maruyama K, et al. Molecular dissection of domains in mutant presenilin 2 that mediate overproduction of amyloidogenic forms of amyloid beta peptides. Inability of truncated forms of PS2 with familial Alzheimer's disease mutation to increase secretion of Abeta42. J Biol Chem. 1998; 273: 21153–60
- De Strooper B, Beullens M, Contreras B, Levesque L, Craessaerts K, Cordell B, et al. Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer's disease-associated presenilins. J Biol Chem. 1997; 272: 3590–8
- Annaert WG, Levesque L, Craessaerts K, Dierinck I, Snellings G, Westaway D, et al. Presenilin 1 controls gamma-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J Cell Biol. 1999; 147: 277–94
- Culvenor JG, Maher F, Evin G, Malchiodi-Albedi F, Cappai R, Underwood JR, et al. Alzheimer's disease-associated presenilin 1 in neuronal cells: evidence for localization to the endoplasmic reticulum-Golgi intermediate compartment. J Neurosci Res. 1997; 49: 719–31
- Levitan D, Greenwald I. Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer's disease gene. Nature. 1995; 377: 351–4
- Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, et al. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997; 387: 288–92
- Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997; 89: 629–39
- Weidemann A, Paliga K, Durrwang U, Czech C, Evin G, Masters CL, et al. Formation of stable complexes between two Alzheimer's disease gene products: presenilin-2 and beta-amyloid precursor protein. Nat Med. 1997; 3: 328–32
- Xia W, Zhang J, Perez R, Koo EH, Selkoe DJ. Interaction between amyloid precursor protein and presenilins in mammalian cells: implications for the pathogenesis of Alzheimer disease. Proc Natl Acad Sci U S A. 1997; 94: 8208–13
- Waragai M, Imafuku I, Takeuchi S, Kanazawa I, Oyama F, Udagawa Y, et al. Presenilin 1 binds to amyloid precursor protein directly. Biochem Biophys Res Commun. 1997; 239: 480–2
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998; 391: 387–90
- Ye Y, Lukinova N, Fortini ME. Neurogenic phenotypes and altered Notch processing in Drosophila Presenilin mutants. Nature. 1999; 398: 525–9
- Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 1999; 398: 522–5
- Song W, Nadeau P, Yuan M, Yang X, Shen J, Yankner BA. Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc Natl Acad Sci U S A. 1999; 96: 6959–63
- Steiner H, Duff K, Capell A, Romig H, Grim MG, Lincoln S, et al. A loss of function mutation of presenilin-2 interferes with amyloid beta-peptide production and notch signaling. J Biol Chem. 1999; 274: 28669–73
- Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, et al. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000; 405: 689–94
- Li YM, Lai MT, Xu M, Huang Q, DiMuzio-Mower J, Sardana MK, et al. Presenilin 1 is linked with gamma-secretase activity in the detergent solubilized state. Proc Natl Acad Sci U S A. 2000; 97: 6138–43
- Capell A, Grunberg J, Pesold B, Diehlmann A, Citron M, Nixon R, et al. The proteolytic fragments of the Alzheimer's disease-associated presenilin-1 form heterodimers and occur as a 100-150-kDa molecular mass complex. J Biol Chem. 1998; 273: 3205–11
- Yu G, Chen F, Levesque G, Nishimura M, Zhang DM, Levesque L, et al. The presenilin 1 protein is a component of a high molecular weight intracellular complex that contains beta-catenin. J Biol Chem. 1998; 273: 16470–5
- Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature. 2000; 407: 48–54
- Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, et al. aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell. 2002; 3: 85–97
- Goutte C, Tsunozaki M, Hale VA, Priess JR. APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci U S A. 2002; 99: 775–9
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003; 5: 486–8
- Fraering PC, Ye W, Strub JM, Dolios G, LaVoie MJ, Ostaszewski BL, et al. Purification and characterization of the human gamma-secretase complex. Biochemistry. 2004; 43: 9774–89
- Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc Natl Acad Sci U S A. 2003; 100: 6382–7
- Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, et al. The role of presenilin cofactors in the gamma-secretase complex. Nature. 2003; 422: 438–41
- Xia W, Wolfe MS. Intramembrane proteolysis by presenilin and presenilin-like proteases. J Cell Sci. 2003; 116: 2839–44
- Landman N, Kim TW. Got RIP? Presenilin-dependent intramembrane proteolysis in growth factor receptor signaling. Cytokine Growth Factor Rev. 2004; 15: 337–51
- Annaert W, De SB. A cell biological perspective on Alzheimer's disease. Annu Rev Cell Dev Biol. 2002; 18: 25–51
- Murayama O, Honda T, Mercken M, Murayama M, Yasutake K, Nihonmatsu N, et al. Different effects of Alzheimer-associated mutations of presenilin 1 on its processing. Neurosci Lett. 1997; 229: 61–4
- Murayama O, Tomita T, Nihonmatsu N, Murayama M, Sun X, Honda T, et al. Enhancement of amyloid beta 42 secretion by 28 different presenilin 1 mutations of familial Alzheimer's disease. Neurosci Lett. 1999; 265: 61–3
- Okochi M, Ishii K, Usami M, Sahara N, Kametani F, Tanaka K, et al. Proteolytic processing of presenilin-1 (PS-1) is not associated with Alzheimer's disease with or without PS-1 mutations. FEBS Lett. 1997; 418: 162–6
- Steiner H, Romig H, Grim MG, Philipp U, Pesold B, Citron M, et al. The biological and pathological function of the presenilin-1 Deltaexon 9 mutation is independent of its defect to undergo proteolytic processing. J Biol Chem. 1999; 274: 7615–8
- Martins RN, Turner BA, Carroll RT, Sweeney D, Kim KS, Wisniewski HM, et al. High levels of amyloid-beta protein from S182 (Glu246) familial Alzheimer's cells. Neuroreport. 1995; 7: 217–20
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996; 2: 864–70
- Lemere CA, Lopera F, Kosik KS, Lendon CL, Ossa J, Saido TC, et al. The E280A presenilin 1 Alzheimer mutation produces increased A beta 42 deposition and severe cerebellar pathology. Nat Med. 1996; 2: 1146–50
- Gomez-Isla T, Wasco W, Pettingell WP, Gurubhagavatula S, Schmidt SD, Jondro PD, et al. A novel presenilin-1 mutation: increased beta-amyloid and neurofibrillary changes. Ann Neurol. 1997; 41: 809–13
- Mann DM, Iwatsubo T, Cairns NJ, Lantos PL, Nochlin D, Sumi SM, et al. Amyloid beta protein (Abeta) deposition in chromosome 14-linked Alzheimer's disease: predominance of Abeta42(43). Ann Neurol. 1996; 40: 149–56
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-Tur J, et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996; 383: 710–3
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, et al. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1-42/1–40 ratio in vitro and in vivo. Neuron. 1996; 17: 1005–13
- Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, et al. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997; 3: 67–72
- Tomita T, Maruyama K, Saido TC, Kume H, Shinozaki K, Tokuhiro S, et al. The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid beta protein ending at the 42nd (or 43rd) residue. Proc Natl Acad Sci U S A. 1997; 94: 2025–30
- Larner AJ, Doran M. Clinical phenotypic heterogeneity of Alzheimer's disease associated with mutations of the presenilin-1 gene. J Neurol. 2006; 253: 139–58
- Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsmit E, et al. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat. 2006; 27: 686–95
- Bentahir M, Nyabi O, Verhamme J, Tolia A, Horre K, Wiltfang J, et al. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J Neurochem. 2006; 96: 732–42
- Walker ES, Martinez M, Brunkan AL, Goate A. Presenilin 2 familial Alzheimer's disease mutations result in partial loss of function and dramatic changes in Abeta 42/40 ratios. J Neurochem. 2005; 92: 294–301
- Qi Y, Morishima-Kawashima M, Sato T, Mitsumori R, Ihara Y. Distinct mechanisms by mutant presenilin 1 and 2 leading to increased intracellular levels of amyloid beta-protein 42 in Chinese hamster ovary cells. Biochemistry. 2003; 42: 1042–52
- Mehta ND, Refolo LM, Eckman C, Sanders S, Yager D, Perez-Tur J, et al. Increased Abeta42(43) from cell lines expressing presenilin 1 mutations. Ann Neurol. 1998; 43: 256–8
- Brunkan AL, Martinez M, Wang J, Walker ES, Beher D, Shearman MS, et al. Two domains within the first putative transmembrane domain of presenilin 1 differentially influence presenilinase and gamma-secretase activity. J Neurochem. 2005; 94: 1315–28
- Chen F, Gu Y, Hasegawa H, Ruan X, Arawaka S, Fraser P, et al. Presenilin 1 mutations activate gamma 42-secretase but reciprocally inhibit epsilon-secretase cleavage of amyloid precursor protein (APP) and S3-cleavage of notch. J Biol Chem. 2002; 277: 36521–6
- Wiley JC, Hudson M, Kanning KC, Schecterson LC, Bothwell M. Familial Alzheimer's disease mutations inhibit gamma-secretase-mediated liberation of beta-amyloid precursor protein carboxy-terminal fragment. J Neurochem. 2005; 94: 1189–201
- Moehlmann T, Winkler E, Xia X, Edbauer D, Murrell J, Capell A, et al. Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Abeta 42 production. Proc Natl Acad Sci U S A. 2002; 99: 8025–30
- Zhang DM, Levitan D, Yu G, Nishimura M, Chen F, Tandon A, et al. Mutation of the conserved N-terminal cysteine (Cys92) of human presenilin 1 causes increased A beta42 secretion in mammalian cells but impaired Notch/lin-12 signalling in C. elegans. Neuroreport. 2000; 11: 3227–30
- Steiner H, Revesz T, Neumann M, Romig H, Grim MG, Pesold B, et al. A pathogenic presenilin-1 deletion causes abberrant Abeta 42 production in the absence of congophilic amyloid plaques. J Biol Chem. 2001; 276: 7233–9
- De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007; 8: 141–6
- Wolfe MS. When loss is gain: reduced presenilin proteolytic function leads to increased Abeta42/Abeta40. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007; 8: 136–40
- Levitan D, Doyle TG, Brousseau D, Lee MK, Thinakaran G, Slunt HH, et al. Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1996; 93: 14940–4
- Davis JA, Naruse S, Chen H, Eckman C, Younkin S, Price DL, et al. An Alzheimer's disease-linked PS1 variant rescues the developmental abnormalities of PS1-deficient embryos. Neuron. 1998; 20: 603–9
- Qian S, Jiang P, Guan XM, Singh G, Trumbauer ME, Yu H, et al. Mutant human presenilin 1 protects presenilin 1 null mouse against embryonic lethality and elevates Abeta1-42/43 expression. Neuron. 1998; 20: 611–7
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004; 42: 23–36
- van Duijn CM, Cruts M, Theuns J, Van Gassen G, Backhovens H, Van den Broeck M, et al. Genetic association of the presenilin-1 regulatory region with early-onset Alzheimer's disease in a population-based sample. Eur J Hum Genet. 1999; 7: 801–6
- Lambert JC, Mann DM, Harris JM, Chartier-Harlin MC, Cumming A, Coates J, et al. The (48 C/T polymorphism in the presenilin 1 promoter is associated with an increased risk of developing Alzheimer's disease and an increased Abeta load in brain. J Med Genet. 2001; 38: 353–5
- Theuns J, Del Favero J, Dermaut B, van Duijn CM, Backhovens H, Van den Broeck MV, et al. Genetic variability in the regulatory region of presenilin 1 associated with risk for Alzheimer's disease and variable expression. Hum Mol Genet. 2000; 9: 325–31
- Riazanskaia N, Lukiw WJ, Grigorenko A, Korovaitseva G, Dvoryanchikov G, Moliaka Y, et al. Regulatory region variability in the human presenilin-2 (PSEN2) gene: potential contribution to the gene activity and risk for AD. Mol Psychiatry. 2002; 7: 891–8
- Theuns J, Remacle J, Killick R, Corsmit E, Vennekens K, Huylebroeck D, et al. Alzheimer-associated C allele of the promoter polymorphism (22C > T causes a critical neuron-specific decrease of presenilin 1 expression. Hum Mol Genet. 2003; 12: 869–77
- Rademakers R, Cruts M, Sleegers K, Dermaut B, Theuns J, Aulchenko Y, et al. Linkage and association studies identify a novel locus for Alzheimer disease at 7q36 in a Dutch population-based sample. Am J Hum Genet. 2005; 77: 643–52
- Jimenez-Escrig A, Gomez-Tortosa E, Baron M, Rabano A, Arcos-Burgos M, Palacios LG, et al. A multigenerational pedigree of late-onset Alzheimer's disease implies new genetic causes. Brain. 2005; 128: 1707–15
- Ashley-Koch AE, Shao Y, Rimmler JB, Gaskell PC, Welsh-Bohmer KA, Jackson CE, et al. An autosomal genomic screen for dementia in an extended Amish family. Neurosci Lett. 2005; 379: 199–204
- van Duijn CM, Hendriks L, Farrer LA, Backhovens H, Cruts M, Wehnert A, et al. A population-based study of familial Alzheimer disease: linkage to chromosomes 14, 19, and 21. Am J Hum Genet. 1994; 55: 714–27
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998; 393: 702–5
- Reed LA, Grabowski TJ, Schmidt ML, Morris JC, Goate A, Solodkin A, et al. Autosomal dominant dementia with widespread neurofibrillary tangles. Ann Neurol. 1997; 42: 564–72
- van Swieten JC, Stevens M, Rosso SM, Rizzu P, Joosse M, de Koning I, et al. Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Ann Neurol. 1999; 46: 617–26
- Rademakers R, Dermaut B, Peeters K, Cruts M, Heutink P, Goate A, et al. Tau (MAPT) mutation Arg406Trp presenting clinically with Alzheimer disease does not share a common founder in Western Europe. Hum Mutat. 2003; 22: 409–11
- Ostojic J, Elfgren C, Passant U, Nilsson K, Gustafson L, Lannfelt L, et al. The tau R406W mutation causes progressive presenile dementia with bitemporal atrophy. Dement Geriatr Cogn Disord. 2004; 17: 298–301
- Passant U, Ostojic J, Froelich FS, Gustafson L, Lannfelt L, Larsson EM, et al. Familial presenile dementia with bitemporal atrophy. Dement Geriatr Cogn Disord. 2004; 17: 287–92
- Rosso SM, Donker KL, Baks T, Joosse M, de Koning I, Pijnenburg Y, et al. Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003; 126: 2016–22
- Momeni P Pittman A Lashley T Vandrovcova J Malzer E Luk C , et al Clinical and pathological features of an Alzheimer's disease patient with the MAPT DeltaK280 mutation. Neurobiol Aging. 2007 Aug 25 (Epub ahead of print).
- Dermaut B, Kumar-Singh S, Rademakers R, Theuns J, Cruts M, Van Broeckhoven C. Tau is central in the genetic Alzheimer-frontotemporal dementia spectrum. Trends Genet. 2005; 21: 664–72
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006; 442: 916–9
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006; 442: 920–4
- Brouwers N, Nuytemans K, van der Zee J, Gijselinck I, Engelborghs S, Theuns J, et al. Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch Neurol. 2007; 64: 1436–46
- Rademakers R, Baker M, Gass J, Adamson J, Huey ED, Momeni P, et al. Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C(T (Arg493X) mutation: an international initiative. Lancet Neurol. 2007; 6: 857–68
- Brouwers N Sleegers K Engelborghs S Maurer-Stroh S Gijselinck I van der Zee J , et al Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology. 2008. In press.
- van der Zee J, Le Ber I, Maurer-Stroh S, Engelborghs S, Gijselinck I, Camuzat A, et al. Mutations other than null mutations producing a pathogenic loss of progranulin in frontotemporal dementia. Hum Mutat. 2007; 28: 416
- Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B, et al. FTLD-U linked missense mutations in the progranulin gene reduce progranulin production and secretion. J Biol Chem. 2008; 283: 1744–53
- Pericak-Vance MA, Bebout JL, Gaskell PC, Jr, Yamaoka LH, Hung WY, Alberts MJ, et al. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet. 1991; 48: 1034–50
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993; 90: 1977–81
- Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991; 541: 163–6
- Diedrich JF, Minnigan H, Carp RI, Whitaker JN, Race R, Frey W, et al. Neuropathological changes in scrapie and Alzheimer's disease are associated with increased expression of apolipoprotein E and cathepsin D in astrocytes. J Virol. 1991; 65: 4759–68
- Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993; 43: 1467–72
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993; 261: 921–3
- van Duijn CM, de Knijff P, Cruts M, Wehnert A, Havekes LM, Hofman A, et al. Apolipoprotein E4 allele in a population-based study of early-onset Alzheimer's disease. Nat Genet. 1994; 7: 74–8
- Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, Jr, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994; 7: 180–4
- Houlden H, Collinge J, Kennedy A, Newman S, Rossor M, Lannfelt L, et al. Apolipoprotein E genotype and Alzheimer's disease. Alzheimer's Disease Collaborative Group. Lancet. 1993; 342: 737–8
- George-Hyslop P, McLachlan DC, Tsuda T, Rogaev E, Karlinsky H, Lippa CF, et al. Alzheimer's disease and possible gene interaction. Science. 1994; 263: 537
- Nacmias B, Latorraca S, Piersanti P, Forleo P, Piacentini S, Bracco L, et al. ApoE genotype and familial Alzheimer's disease: a possible influence on age of onset in APP717 Val→Ile mutated families. Neurosci Lett. 1995; 183: 1–3
- Sorbi S, Nacmias B, Forleo P, Piacentini S, Latorraca S, Amaducci L. Epistatic effect of APP717 mutation and apolipoprotein E genotype in familial Alzheimer's disease. Ann Neurol. 1995; 38: 124–7
- Wijsman EM, Daw EW, Yu X, Steinbart EJ, Nochlin D, Bird TD, et al. APOE and other loci affect age-at-onset in Alzheimer's disease families with PS2 mutation. Am J Med Genet B Neuropsychiatr Genet. 2005; 132: 14–20
- Van Broeckhoven C, Backhovens H, Cruts M, Martin JJ, Crook R, Houlden H, et al. APOE genotype does not modulate age of onset in families with chromosome 14 encoded Alzheimer's disease. Neurosci Lett. 1994; 169: 179–80
- Pastor P, Roe CM, Villegas A, Bedoya G, Chakraverty S, Garcia G, et al. Apolipoprotein Eepsilon4 modifies Alzheimer's disease onset in an E280A PS1 kindred. Ann Neurol. 2003; 54: 163–9
- Bullido MJ, Artiga MJ, Recuero M, Sastre I, Garcia MA, Aldudo J, et al. A polymorphism in the regulatory region of APOE associated with risk for Alzheimer's dementia. Nat Genet. 1998; 18: 69–71
- Lambert JC, Pasquier F, Cottel D, Frigard B, Amouyel P, Chartier-Harlin MC. A new polymorphism in the APOE promoter associated with risk of developing Alzheimer's disease. Hum Mol Genet. 1998; 7: 533–40
- Mui S, Briggs M, Chung H, Wallace RB, Gomez-Isla T, Rebeck GW, et al. A newly identified polymorphism in the apolipoprotein E enhancer gene region is associated with Alzheimer's disease and strongly with the epsilon 4 allele. Neurology. 1996; 47: 196–201
- Artiga MJ, Bullido MJ, Frank A, Sastre I, Recuero M, Garcia MA, et al. Risk for Alzheimer's disease correlates with transcriptional activity of the APOE gene. Hum Mol Genet. 1998; 7: 1887–92
- Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007; 39: 17–23
- Laws SM, Hone E, Gandy S, Martins RN. Expanding the association between the APOE gene and the risk of Alzheimer's disease: possible roles for APOE promoter polymorphisms and alterations in APOE transcription. J Neurochem. 2003; 84: 1215–36
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993; 90: 8098–102
- Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993; 90: 9649–53
- Rebeck GW, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer's disease: allelic variation and receptor interactions. Neuron. 1993; 11: 575–80
- Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997; 17: 263–4
- Kindy MS, Rader DJ. Reduction in amyloid A amyloid formation in apolipoprotein-E-deficient mice. Am J Pathol. 1998; 152: 1387–95
- Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, et al. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 1999; 96: 15233–8
- Ma J, Yee A, Brewer HB, Jr, Das S, Potter H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature. 1994; 372: 92–4
- Sanan DA, Weisgraber KH, Russell SJ, Mahley RW, Huang D, Saunders A, et al. Apolipoprotein E associates with beta amyloid peptide of Alzheimer's disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J Clin Invest. 1994; 94: 860–9
- Evans KC, Berger EP, Cho CG, Weisgraber KH, Lansbury PT, Jr. Apolipoprotein E is a kinetic but not a thermodynamic inhibitor of amyloid formation: implications for the pathogenesis and treatment of Alzheimer disease. Proc Natl Acad Sci U S A. 1995; 92: 763–7
- LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem. 1994; 269: 23403–6
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2000; 97: 2892–7
- Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, et al. Expression of human apolipoprotein E reduces amyloid-beta deposition in a mouse model of Alzheimer's disease. J Clin Invest. 1999; 103: R15–R21
- Sparks DL, Scheff SW, Hunsaker JC III, Liu H, Landers T, Gross DR. Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp Neurol. 1994; 126: 88–94
- Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, et al. Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000; 7: 321–31
- Bodovitz S, Klein WL. Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J Biol Chem. 1996; 271: 4436–40
- Howland DS, Trusko SP, Savage MJ, Reaume AG, Lang DM, Hirsch JD, et al. Modulation of secreted beta-amyloid precursor protein and amyloid beta-peptide in brain by cholesterol. J Biol Chem. 1998; 273: 16576–82
- Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci U S A. 1998; 95: 6460–4
- Bertram L Tanzi RE Alzheimer's disease: one disorder, too many genes? Hum Mol Genet. 2004;13 Spec No 1:R135–R41.
- Wragg M, Hutton M, Talbot C. Genetic association between intronic polymorphism in presenilin-1 gene and late-onset Alzheimer's disease. Alzheimer's Disease Collaborative Group. Lancet. 1996; 347: 509–12
- Dermaut B, Theuns J, Sleegers K, Hasegawa H, Van den Broeck M, Vennekens K, et al. The gene encoding nicastrin, a major gamma-secretase component, modifies risk for familial early-onset Alzheimer disease in a Dutch population-based sample. Am J Hum Genet. 2002; 70: 1568–74
- Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007; 39: 168–77
- Bettens K, Brouwers N, Engelborghs S, De Deyn PP, Van Broeckhoven C. SORL1 is genetically associated with increased risk for late-onset Alzheimer disease in the Belgian population. Hum Mutat. 2008; 29: 769–70
- Ertekin-Taner N, Allen M, Fadale D, Scanlin L, Younkin L, Petersen RC, et al. Genetic variants in a haplotype block spanning IDE are significantly associated with plasma Abeta42 levels and risk for Alzheimer disease. Hum Mutat. 2004; 23: 334–42
- Kehoe PG, Russ C, McIlory S, Williams H, Holmans P, Holmes C, et al. Variation in DCP1, encoding ACE, is associated with susceptibility to Alzheimer disease. Nat Genet. 1999; 21: 71–2
- Finckh U, van Hadeln K, Muller-Thomsen T, Alberici A, Binetti G, Hock C, et al. Association of late-onset Alzheimer disease with a genotype of PLAU, the gene encoding urokinase-type plasminogen activator on chromosome 10q22.2. Neurogenetics. 2003; 4: 213–7
- Sodeyama N, Mizusawa H, Yamada M, Itoh Y, Otomo E, Matsushita M. Lack of association of neprilysin polymorphism with Alzheimer's disease and Alzheimer's disease-type neuropathological changes. J Neurol Neurosurg Psychiatry. 2001; 71: 817–8
- Dermaut B, Croes EA, Rademakers R, Van den Broeck M, Cruts M, Hofman A, et al. PRNP Val129 homozygosity increases risk for early-onset Alzheimer's disease. Ann Neurol. 2003; 53: 409–12
- Li Y, Nowotny P, Holmans P, Smemo S, Kauwe JS, Hinrichs AL, et al. Association of late-onset Alzheimer's disease with genetic variation in multiple members of the GAPD gene family. Proc Natl Acad Sci U S A. 2004; 101: 15688–93
- Kuwano R, Miyashita A, Arai H, Asada T, Imagawa M, Shoji M, et al. Dynamin-binding protein gene on chromosome 10q is associated with late-onset Alzheimer's disease. Hum Mol Genet. 2006; 15: 2170–82
- Grupe A, Li Y, Rowland C, Nowotny P, Hinrichs AL, Smemo S, et al. A scan of chromosome 10 identifies a novel locus showing strong association with late-onset Alzheimer disease. Am J Hum Genet. 2006; 78: 78–88
- Li Y, Grupe A, Rowland C, Nowotny P, Kauwe JS, Smemo S, et al. DAPK1 variants are associated with Alzheimer's disease and allele-specific expression. Hum Mol Genet. 2006; 15: 2560–8
- Grupe A, Abraham R, Li Y, Rowland C, Hollingworth P, Morgan A, et al. Evidence for novel susceptibility genes for late-onset Alzheimer's disease from a genome-wide association study of putative functional variants. Hum Mol Genet. 2007; 16: 865–73
- Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, Lince DH, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer's disease. J Clin Psychiatry. 2007; 68: 613–8
- Reiman EM, Webster JA, Myers AJ, Hardy J, Dunckley T, Zismann VL, et al. GAB2 Alleles Modify Alzheimer's Risk in APOE varepsilon4 Carriers. Neuron. 2007; 54: 713–20
- Liu F, Arias-Vasquez A, Sleegers K, Aulchenko YS, Kayser M, Sanchez-Juan P, et al. A genomewide screen for late-onset Alzheimer disease in a genetically isolated Dutch population. Am J Hum Genet. 2007; 81: 17–31
- Giedraitis V, Hedlund M, Skoglund L, Blom E, Ingvast S, Brundin R, et al. New Alzheimer's disease locus on chromosome 8. J Med Genet. 2006; 43: 931–5
- Hahs DW, McCauley JL, Crunk AE, McFarland LL, Gaskell PC, Jiang L, et al. A genome-wide linkage analysis of dementia in the Amish. Am J Med Genet B Neuropsychiatr Genet. 2006; 141: 160–6
- Lee JH, Mayeux R, Mayo D, Mo J, Santana V, Williamson J, et al. Fine mapping of 10q and 18q for familial Alzheimer's disease in Caribbean Hispanics. Mol Psychiatry. 2004; 9: 1042–51
- Lee JH, Cheng R, Santana V, Williamson J, Lantigua R, Medrano M, et al. Expanded genomewide scan implicates a novel locus at 3q28 among Caribbean hispanics with familial Alzheimer disease. Arch Neurol. 2006; 63: 1591–8
- Hiltunen M, Mannermaa A, Thompson D, Easton D, Pirskanen M, Helisalmi S, et al. Genome-wide linkage disequilibrium mapping of late-onset Alzheimer's disease in Finland. Neurology. 2001; 57: 1663–8
- Farrer LA, Bowirrat A, Friedland RP, Waraska K, Korczyn AD, Baldwin CT. Identification of multiple loci for Alzheimer disease in a consanguineous Israeli-Arab community. Hum Mol Genet. 2003; 12: 415–22
- Minster RL, DeKosky ST, Kamboh MI. Lack of association of two chromosome 10q24 SNPs with Alzheimer's disease. Neurosci Lett. 2006; 408: 170–2
- Minster RL DeKosky ST Kamboh MI No association of dynamin binding protein (DNMBP) gene SNPs and Alzheimer's disease. Neurobiol Aging. 2007 Apr 16 (Epub ahead of print).
- Bertram L, Hsiao M, Lange C, Blacker D, Tanzi RE. Single-nucleotide polymorphism rs498055 on chromosome 10q24 is not associated with Alzheimer disease in two independent family samples. Am J Hum Genet. 2006; 79: 180–3
- Liang X, Schnetz-Boutaud N, Bartlett J, Allen MJ, Gwirtsman H, Schmechel DE, et al. No association between SNP rs498055 on chromosome 10 and late-onset Alzheimer disease in multiple datasets. Ann Hum Genet. 2008; 72: 141–4
- Bettens K Brouwers N Engelborghs S De Pooter T De Deyn PP Sleegers K , et al. DNMBP is genetically associated with Alzheimer dementia in the Belgian population. Neurobiol Aging. 2008 Mar 21 (Epub ahead of print).
- Lin PI, Martin ER, Bronson PG, Browning-Large C, Small GW, Schmechel DE, et al. Exploring the association of glyceraldehyde-3-phosphate dehydrogenase gene and Alzheimer disease. Neurology. 2006; 67: 64–8
- Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007; 316: 1491–3
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78.
- Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007; 316: 1336–41
- McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007; 316: 1488–91
- Li Y, Grupe A, Rowland C, Holmans P, Segurado R, Abraham R, et al. Evidence that common variation in NEDD9 is associated with susceptibility to late-onset Alzheimer's and Parkinson's disease. Hum Mol Genet. 2008; 17: 759–67
- Li H, Wetten S, Li L, St Jean PL, Upmanyu R, Surh L, et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol. 2008; 65: 45–53
- Papassotiropoulos A, Stephan DA, Huentelman MJ, Hoerndli FJ, Craig DW, Pearson JV, et al. Common Kibra alleles are associated with human memory performance. Science. 2006; 314: 475–8