I read with interest the paper by Lee et al. Citation[1], in which 3 individuals in an extended family were reported to be affected by two different forms of double heterozygosity for β-thalassemia and Hb New York. Both adults had clinical and hematological features consistent with β-thalassemia trait. The affected child was followed up after birth and manifested the typical course of a thalassemia trait, with no signs of organomegaly or overt hemolysis.
Indeed, Hb New York is relatively common in southern China Citation[2]. In our experience of hemoglobinopathies screening for the past 15 years in the province of Guangdong, Hb New York is the second most frequent Hb variant identified, second only to Hb E and followed by Hb Q Thailand. So it is not an accident to encounter an individual who is a double heterozygote for Hb New York and β-thalassemia. We also had two similar adult subjects identified during our program for prenatal β-thalassemia screening. Their hematological data are summarized in . Both of them were healthy and had never been transfused. They were identified because their partners also were a β-thalassemia carrier, and the fetuses were at risk for homozygous β-thalassemia. In alkaline pH electrophoresis, the samples showed a homozygous Hb New York pattern (). However, the samples manifested as simple β-thalassemia heterozygote on high-performance liquid chromatography (HPLC) study. The two involved pregnancies received prenatal diagnosis at the first trimester of gestation. One fetus was a homozygote for β-thalassemia and was aborted. The other fetus was confirmed to be a heterozygote for β-thalassemia, and the pregnancy continued until term. Effort was not made to reveal Hb New York in this fetus, because we did not believe that this could add any more useful information to the parents. During the follow-up after birth, the child was found to be a double heterozygote for Hb New York and β0-thalassemia. The child grew and developed normally.
TABLE 1 Summary of Hematological Findings and α- and β-Globin Genotypes of the Two Subjects
FIGURE 1 Agarose gel electrophoresis at an alkaline pH: (A) a normal subject; (B) a double heterozygote for Hb New York and β0-thalassemia (codon 17); (C) a double heterozygote for Hb New York and β+-thalassemia (−28); (D) a heterozygote for Hb New York.
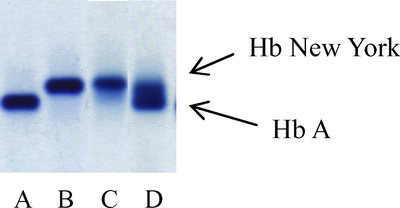
Although Hb New York is a common β-chain variant in our region, a test for it is not usually included in the prenatal screening program for β-thalassemia. The reason is that the method used in our local practice to initially screen for thalassemia trait is mean cell volume (MCV), one of the red cell indices included in a complete blood count generated by automated blood cell counters. Using a cutoff value of MCV < 80 fL, practically all the thalassemic individuals can be identified. However, subjects who are heterozygotes for Hb New York are asymptomatic with normal hemoglobin levels and MCV. Most of these individuals would be omitted, and a few of those with this Hb variant and β-thalassemia were identified only when their partners were also screened positive for β-thalassemia. In the latter situation, Hb New York is usually not tested in the fetus during prenatal diagnosis, considering the mild phenotype in the parents.
REFERENCES
- Lee AC, Ma ES, Chan AY, Szeto SC, Chan LC. Double heterozygosity for Hb New York [beta 113 GTG→GAG; VAL→GLU] and beta degrees-thalassemia mutations manifests as a thalassemia trait. Pediatr Hematol Oncol. 2008; 25: 227–231
- Zeng YT, Huang SZ. Hemoglobin New York (alpha 2 beta 2 113(G15) Val leads to Glu) in China. Hemoglobin 1982; 6: 61–67