
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
After a general discussion of neural networks potential energy functions and their standing within the various approaches of representing the potential energy function of a system, we describe a new interface between the open source atomistic library aenet of Artrith and Urban and the DL_POLY 4 code. As an application example, the training of a neural network for liquid water is described and the network is used in a molecular dynamics simulation. The resulting thermodynamic properties are compared with those from a reference simulation with the same SPC/E model that has been used in the training.
1. Introduction
This chapter attempts to position neural network potential energy functions within the vast area of possible potential energy expressions and to compare them with other approaches.
A suitable representation of the potential energy surface of the system under consideration is needed as input to any classical MD simulation. One can think of this representation to be the intermediate layer between the charges (which cause it) and the nuclei (which are governed by it). It can be expressed as a sum of analytical terms or it can be tabulated in some way. An early and very successful way to approximate the potential energy surface is to treat atoms connected by one up to three bonds separate from other atoms, including those in other molecules. Harmonic terms are applied for bonds and bond angles and angular functions describe dihedral energy contributions. Energy contributions arising from pairs of atoms in the same molecule but not bonded to each other or in a different molecule are described by partial-charge based Coulombic interactions and an appropriate pairwise term, for example but not restricted to the Lennard-Jones expression. The latter one is responsible for a variety of physical effects, like van-der-Waals interactions. Examples of this tremendously successful approach are the various force fields that were developed after 1970 [Citation1], and their applications. It should be pointed out that hydrogen bonds can also be described reasonably well in that way, due to their predominantly electrostatic nature.
This approach has, however, well-known limitations that are often accepted for the sake of simplicity and computational efficiency. The main limitations are the restriction to the pair approximation (except for bond and dihedral angles) and the impossibility of breaking chemical bonds. The latter is obviously due to the harmonic terms for bound atoms. After early attempts to correctly account for bond formation and breaking within the pair approximation it turned out that this is not possible. For example, in Ref. [Citation2] an O-H pair potential function was used that allowed H to dissociate from O (or OH). An H-H pair potential was used to account for the H-O-H angle in intact water. To accomplish this, however, this H-H pair potential included an unphysical ‘kink’ and furthermore, the auto-dissociation constant of H2O was much overestimated. Therefore, this ‘central force’ potential was not really successful for situations where the dissociation of water is important. Therefore, especially in the realm of computational materials science it is important to move beyond this stage.
There are many attempts to create potential energy functions that overcome these limitations. For example, using a ‘valence bond’ – approach [Citation3], bonding and nonbonding energy curves can be combined to a function that incorporates bond breaking. Potential energy functions that account for bond formation or breaking are often called ‘reactive potentials’. On the other side, one way beyond the pairwise approximation is to incorporate atomic dipole polarizability. This is conceptually straightforward and can be done analytically [Citation4] but is more often implemented with the polarisation shell model [Citation5]. Polarisable force fields currently enjoy a renaissance, because the computer power is now available to employ them in large biophysical simulations [Citation6].
Experience has shown that these methods, alone or combined, are often not flexible enough for the simulation of condensed matter. A certain set of analytical functions and parameters may well describe particular situations in terms of stoichiometry, structure, pressure and temperature and pairwise approximations may even include higher order contributions implicitly. However, different situations afford a multitude of different analytical functions, if one finds a good fit at all, and as soon as it comes to mixing of force fields using combining rules, the transferability of the parameters is questionable. Therefore, other approaches prevailed. One way beyond the pairwise approximation is by tuning a pairwise interaction with a function of the coordination number of a particle. This can be called ‘pair functional’ instead of ‘pair potential’. The Sutton-Chen [Citation7] form of the Finnis-Sinclair potential [Citation8] which is often used for metals belongs to this class. The accuracy of this approach is still limited because there is no term modelling directed chemical bonds. To achieve this, a pairwise potential energy function is often augmented with three-body terms, typically giving a contribution only inside a sphere around the middle particle. Combining these two extensions of the pair approximation, one ends up at the stage of the so-called ‘cluster functionals’. The Tersoff-Abell potential [Citation9,Citation10] and other bond order potentials as the ReaxFF force field [Citation11] belong to this class. Recently, approximate density functional calculations based on the tight-binding approximation [Citation12] have made progress as well and it remains to be seen how they compete with other approaches.
The contributions to the potential energy beyond the pair approximation have complicated functional forms and it is by no means clear which mathematical expressions are best suited for them. On the other side, it was known since long that neural networks are about the most flexible constructions that can be used to fit arbitrary functions. This has naturally led to attempts to use neural networks in this respect, although their main application was always more towards classification and not the reproduction of exact numbers. Early attempts to use neural networks as potential energy functions worked well [Citation13] but had no big impact because there were more efficient ways to achieve the same goal.
The present renaissance of neural network potentials for atomistic simulations can be deducted from their use in the spirit of the ‘cluster functionals’ mentioned above. A central atom interacts with its surroundings in a complicated way governed by the radial and angular density of its neighbours. The modern implementations of neural networks [Citation14] start at this level. Each type of atom is assigned its own neural network that calculates the energy contribution from this atom. The input are the coordinates of the neighbours. In case of the ‘cluster functionals’ (bond order potentials) one must convert their Cartesian coordinates into distances and angles. In case of a neural network, a similar step must be performed. The energy contribution of an atom must be invariant with respect to permutations of the same neighbour atoms, to rotations and translations and, possibly, to other symmetry operations. Therefor the Cartesian coordinates are converted to symmetry coordinates which are the actual input to the neural network. The symmetry functions used are somewhat more general than in case of the cluster potentials, but they are also constructed from distances and angles. There are several other approaches as well [Citation15,Citation16]. It is important to realise that the neural network of an atom contains all information of the specific atom type (normally the specific element with nuclear charge Z) and its ‘fingerprint’. This guarantees transferability of the potential between systems of different size and, ideally, chemical composition. Together with the symmetry functions, this also makes up for a conceptionally different viewpoint towards the potential energy of a system, compared with other force fields.
Since the NN potential is analytic, and the atomic contributions sum up, the forces acting on each atom can be calculated without problems by repeated application of the chain rule. The fact that the total energy is the sum of the neural network energy of each atom facilitates also the fitting process, since in quantum chemical calculations the total energy is calculated and individual interaction energies are normally not directly accessible.
Neural network potential energy functions seem to have warranted their existence by being potentially more accurate (but slower) than other force fields and by being faster (but not as accurate) as all-electron (ab-initio) type simulations. Currently, the number of publications in which they are used or developed increases drastically (2016–17: 17, 2014–15: 8, before 2014: 10). Neural network hardware is already built into some processors (for example, Apple’s A11) and dedicated neural network coprocessors like Intel’s Nervana chip are currently entering the market so that the computational aspect looks promising. Altogether it seems to be of interest that such potential energy functions become available in a general-purpose molecular dynamics code like DL_POLY, where they can be combined with the multitude of other interaction types implemented.
2. Implementation details
In the scheme of Behler and Parrinello [Citation14], the total energy is calculated as a sum over individual atomic energies Ei. Each contribution Ei depends on the central atom i and its neighbours within a sphere defined by a cutoff distance. The neighbourhood of atom i is described in terms of symmetry functions which map both the relative atomic positions and the atom types into a set of input nodes for the neural network. The number of input nodes is constant and the input node values are independent from exchange of like atoms, rotation and translation. Since the way how energy and forces are constructed varies a bit from other potential energy functions, we have not implemented them as DL_POLY subroutines, but have interfaced the DL_POLY 4.08 code with the neural network library aenet. For a detailed description of the open source atomistic library aenet by Nongnuch and Urban, and the Behler approach in general, we refer to Refs. [Citation14,Citation17–19]. The interface allows to read ANN-parameter files and network structures generated with the aenet-library and to call the aenet energy prediction functions with simple keywords from the DL_POLY input files. It thus adds further functionality to the code, while other functionalities are maintained and can be used in conjunction with the new neural network interface. In particular, it is possible to use the standard analytical functions together with neural network files, which can be useful for repulsive short-range interactions or for long-range interactions that are difficult to cover with the neural network. Also, the virial and the stress tensor are calculated within the new interface, so that the instantaneous pressure is available for simulations performed in a constant pressure ensemble.
When the domain decomposition MPI parallelisation of DL_POLY 4 is used, the total space is divided into spatial cells which are mapped onto single cores. Each cell stores the information for the atoms within the cell and for the cells neighbourhood within the cutoff including for periodic boundary conditions, the so-called halo. Thus, all relevant information for the call to the neural network library is available for each atom on each node, if the DL_POLY cutoff is chosen larger than any of the cutoffs specified in the neural network. In the current implementation, we forgo a linked-cell algorithm, since extremely large simulations are unlikely to be performed with feedforward neural networks due to their higher computational demand compared to conventional force fields. Instead, a simple neighbour list is updated for every timestep by calculating distances to all neighbours within the domain. Profiling shows that this does not affect the total performance of the interface since the execution time is dominated by the call to the aenet library. A code that supports linked-cell lists is in preparation for the next version of the interface. Having identified all coordinates and types of the neighbours of an atom, its energy contribution Ei and the forces resulting from the derivative of this energy contribution w.r.t. the positions of atom i and its neighbours are calculated by the library function aenet_atomic_energy_and_forces. This is done for every atom and the energies, forces and virial contributions are summed up, corresponding to the standard implementation for energy prediction with Behler’s method, which we denote with nnets.
In a second alternative approach, denoted by nnpairs, the total energy is calculated as a sum over molecular pair potentials. The code identifies all molecule pairs within the cutoff and applies Behler’s method to each pair in turn:(1)
(1) where Eij is the pair energy contribution between molecule i and j calculated from the sum over atomic energies
(2)
(2)
n goes over all atoms in molecule i and m over all atoms in molecule j. For each atomic energy, only the neighbours within the molecular pair are considered and are fed via the symmetry functions into the feedforward neural network. nnpairs is slower than nnets and loses the ability to describe reactive processes, but allows for more flexibility in terms of combinations of different neural network potentials. Hereby, the fitting is restricted to pairwise interactions between two molecules and each type of molecule-pair is assigned its own neural network. The energies from different molecule-pairwise neural networks are summed up neglecting three-molecule contributions in a similar way that three-atom contributions are neglected in pairwise force fields. For example, it is possible to combine a neural network for water pair interactions with a neural network for sodium-water and one for chloride-water interaction, or for liquid mixtures such as ethylene glycol – water mixtures that are of interest in the framework of solvent dynamics [Citation20]. nnpairs may also be an alternative for simulations with larger biomolecules in solution, with neural networks fitting the potential energy surface for each type of molecular pair. It can only be used to describe inter-molecular interactions and flexible molecules need a different treatment for their intra-molecular interactions. Although intra-molecular forces could, in principle, also be treated with a similar neural network, a combination of nnpairs with an intra-molecular neural network has not yet been implemented and tested.
3. Liquid water as an example
Given the importance and complexity of systems involving water, it is no surprise that a number of works on molecular dynamics with neural network potentials have already been performed. Small water clusters have been investigated in [Citation21–23]. A number of studies deal with metal or metal oxide surfaces in contact with water [Citation24–26] and with nanosystems and water [Citation27]. If the (auto)dissociation of water in aqueous solutions is to be modelled, reactive potentials are required and such simulations with the neural network approach are reported in [Citation28,Citation29].
In the present work, we use our new neural network implementation to compare two molecular dynamics simulations on liquid water, one with a neural network potential trained from the SPC/E [Citation30] water model, and one using the SPC/E model itself, with otherwise identical conditions. We describe our procedure and compare the results. In other words, we tried to imitate the SPC/E model with the neural network and could directly compare its results with the SPC/E reference. In this example we stay within the pair approximation and do not take advantage of chemical bond breaking/bond formation but strife to verify our implementation which can be readily extended to simulate more complex cases.
At first, a training set has to be created and the network architecture must be specified. The training data was obtained by simulating 1501 collisions of two water molecules with collision energies from 17.3 kcal/mol to 86.48 kcal/mol. Since the SPC/E model consists of pairwise additive terms only, it is not necessary to include data for three or more interacting molecules in the training set. Initial conditions with random relative orientation and impact parameters between 0.0 and 3.0 Å were generated using the venus direct dynamics code [Citation31]. The classical trajectories for non-reactive H2O-H2O collisions were then obtained with DL_POLY using the standard set of parameters and the water geometry of the SPC/E force field. The collision trajectories were integrated with a time step of 0.5 fs, a total simulation time of 0.75 ps, and a recording stride of 3 for each trajectory. From the whole set, we discarded many of the low-energy configurations with large intermolecular distances and then randomly extracted 10,000 configurations from which 90% were chosen for the training set and 10% for the testing set.
A shallow feedforward neural network with 2 hidden layers and 10 nodes per layer was chosen, together with a hyperbolic tangent activation function. We used a set of Behler type-2 and type-4 symmetry functions [Citation32] for H and for O with a cutoff of 6 Å. The ANN binary parameter files are given in the supplementary information and also contain the chosen parameters of the symmetry functions. The network was trained using the limited-memory Broyden-Fletcher-Goldfarb-Shanno optimisation algorithm [Citation33] up to a final mean average error of 0.4 meV per atom for the testing set. The ANN training was performed with the aenet programme [Citation18].
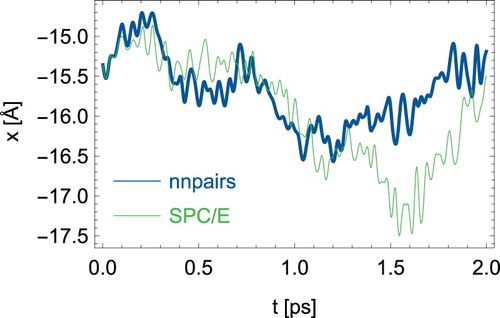
Having thus established a neural network representation of the SPC/E force field, a cubic periodic box of edge length 39.15 Å with 2000 H2O molecules was prepared using packmol [Citation34] and equilibrated with the analytical SPC/E force field for 1 ns. The mass to volume ratio corresponds to a density of 9971 kg/m3. A timestep of 0.2 fs was chosen. The temperature of 298 K was controlled with the Nosé-Hoover thermostat using a relaxation constant of 0.5 ps [Citation35,Citation36]. Two typical trajectories of the x-coordinates of a hydrogen atom are compared in for the full 2 ps simulation time. The trajectories start out at the same point with the same velocity, resemble each other in the first 70 fs and deviate strongly thereafter. This is not unexpected, of course, and it remains to be seen how static and dynamic quantities compare with each other.
Figure 1. (Colour online) Comparison of two trajectories between nnpairs and the analytical SPC/E model.
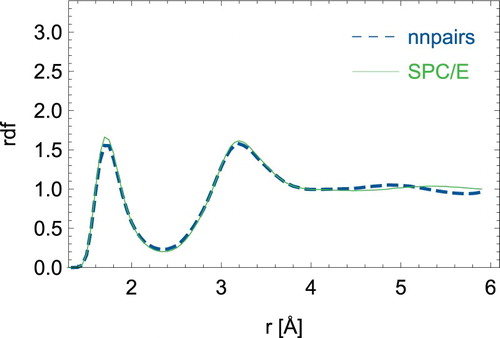
First, we discuss static quantities derived from averaging over 2 ps simulations. In and , we show the radial distribution functions for oxygen-oxygen distances and oxygen–hydrogen distances respectively. The neural network model can mimic the SPC/E model especially for the short-ranged structure. However, significant differences can be seen for the second coordination shell. On average, a water molecule is involved in nHB = 3.4 number of hydrogen-bonds in the SPC/E model, while nHB = 3.29 for nnpairs. The hydrogen-bond connectivity was obtained in analogy to Ref. [Citation37] with the distance criteria ROO ≤ 3.3 Å and ROH ≤ 2.35 Å and the angle criterion . Note that we employed a smaller cutoff of 6 Å for the neural network, while we used 10 Å for the original SPC/E model.
Figure 2. (Colour online) Radial distribution function between pairs of oxygen atoms for the neural network nnpairs model and the analytical SPC/E water model.
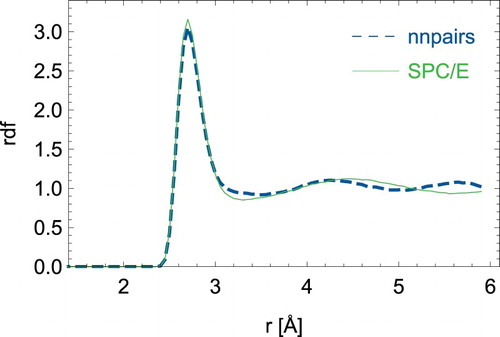
Figure 3. (Colour online) Radial distribution function between oxygen and hydrogen atoms for the neural network nnpairs model and the analytical SPC/E water model.
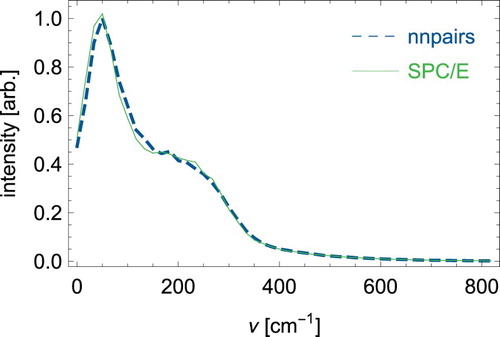
Even more interesting is a direct comparison of dynamical quantities. For the rather short 2ps simulation, we obtain an Einstein diffusion coefficient of 2.95*10−9 m2/s for SPC/E from the mean square displacements of the water centre of mass, while nnpairs reaches a very close value of 2.97*10−9 m2/s. Both compare well with literature data [Citation38]. As an example of dynamic properties, we calculated the autocorrelation function of the centre of mass velocities of the H2O molecules, see . The nnpairs correlation function comes very close to the SPC/E curve and both lead to very similar power spectra as shown in . A discussion on the power spectrum of SPC/E water can be found for example in Ref. [Citation20]. It is interesting to note that nnpairs performs quite good for dynamic quantities although the SPC/E radial distribution in and could not be accurately reproduced for distances larger than the position of the first maximum. A possible reason could be that we used dynamic trajectories of colliding water molecules for the training set generation together with a cutoff of 6 Å which seems to capture the parts of the force field that are needed for dynamics ().
Figure 4. (Colour online) Velocity autocorrelation function of the H2O centre of mass velocities.
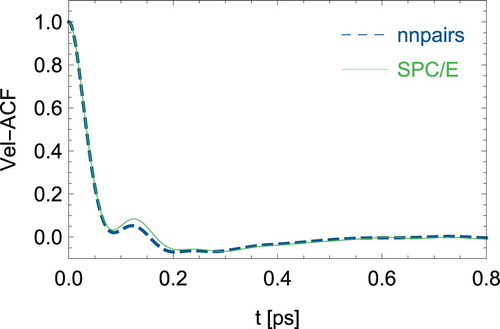
Figure 5. (Colour online) Power spectrum obtained from the velocity autocorrelation function of the H2O centre of mass velocities.
In our present case the analytical SPC/E model is faster by a factor of about 100 than the implemented nnpairs model on the same high-performance computing architecture using 4 cores.
4. Conclusions and outlook
An interface between the feedforward neural network library aenet and the general purpose molecular dynamics simulation code DL_POLY has been implemented. The standard implementation nnets follows the Behler approach by considering for each atom all neighbor atoms within a spherical cutoff distance and also allows for reactive simulations. The second approach, nnpairs, uses a pairwise approximation, where energy and forces of molecule-pairs are predicted by the neural network framework. The pair-approximation was tested in comparison to the SPC/E water model reference force field and yielded reasonably accurate results for static and dynamic quantities. Both nnets and nnpairs can be combined with the usual functionality of DL_POLY and in particular also with analytical force fields, with rigid body structures, and with thermo- and barostats. It will be interesting to see for example, how well standard analytical intermolecular potentials can be improved by correction of intrinsic inaccuracies with feedforward neural networks, especially in the intermediate distances in between steep repulsive parts of the potential and well known long-range interactions. Our intention now is to make this interface available for the scientific community free of charge.
Supplemental Material
Download PDF (271.3 KB)Acknowledgments
We thank Nongnuch Artrith and Alexander Urban for many fruitful discussions and for sharing their aenet library. The computational results presented have been achieved using the HPC infrastructure LEO of the University of Innsbruck.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Allinger NL. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J Am Chem Soc. 1977;99:8127–8134. doi: 10.1021/ja00467a001
- Rahman A, Stillinger FH, Lemberg HL. Study of a central force model for liquid water by molecular dynamics. J Chem Phys. 1975;63:5223–5230. doi: 10.1063/1.431307
- Schlegel HB, Sonnenberg JL. Empirical valence-bond models for reactive potential energy surfaces using distributed gaussians. J Chem Theory Comput. 2006;2:905–911. doi: 10.1021/ct600084p
- Masia M, Probst M, Rey R. On the performance of molecular polarization methods. I. water and carbon tetrachloride close to a point charge. J Chem Phys. 2004;121:7362–7378. doi: 10.1063/1.1791637
- Straatsma TP, McCammon JA. Free energy thermodynamic integrations in molecular dynamics simulations using a noniterative method to include electronic polarization. Chem Phys Lett. 1990;167:252–254. doi: 10.1016/0009-2614(90)85014-4
- Baker CM. Polarizable force fields for molecular dynamics simulations of biomolecules. WIREs Comput Mol Sci. 2015;5:241–254. doi: 10.1002/wcms.1215
- Sutton AP, Chen J. Long-range finnis–sinclair potentials. Philos Mag Lett. 1990;61:139–146. doi: 10.1080/09500839008206493
- Finnis MW, Sinclair JE. A simple empirical N-body potential for transition metals. Philos Mag A. 1984;50:45–55. doi: 10.1080/01418618408244210
- Abell G. Empirical chemical pseudopotential theory of molecular and metallic bonding. Phys Rev B. 1985;31:6184–6196. doi: 10.1103/PhysRevB.31.6184
- Tersoff J. New empirical approach for the structure and energy of covalent systems. Phys Rev B. 1988;37:6991–7000. doi: 10.1103/PhysRevB.37.6991
- van Duin ACT, Dasgupta S, Lorant F, et al. ReaxFF: A reactive force field for hydrocarbons. J Phys Chem A. 2001;105:9396–9409. doi: 10.1021/jp004368u
- Goringe CM, Bowler DR, Hernández E. Tight-binding modelling of materials. Rep Prog Phys. 1997;60:1447–1512. doi: 10.1088/0034-4885/60/12/001
- Gassner H, Probst M, Lauenstein A, et al. Representation of intermolecular potential functions by neural networks. J Phys Chem A. 1998;102:4596–4605. doi: 10.1021/jp972209d
- Behler J, Parrinello M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys Rev Lett. 2007;98:146401. doi: 10.1103/PhysRevLett.98.146401
- Bartók AP, Kondor R, Csányi G. On representing chemical environments. Phys Rev B. 2013;87:184115. doi: 10.1103/PhysRevB.87.184115
- Shao K, Chen J, Zhao Z, et al. Communication: fitting potential energy surfaces with fundamental invariant neural network. J Chem Phys. 2016;145:071101. doi: 10.1063/1.4961454
- Behler J. Representing potential energy surfaces by high-dimensional neural network potentials. J Phys: Condens Matter. 2014;26:183001.
- Artrith N, Urban A. An implementation of artificial neural-network potentials for atomistic materials simulations: performance for TiO2. Comp Mat Sci. 2016;114:135–150. doi: 10.1016/j.commatsci.2015.11.047
- Artrith N, Urban A, Ceder G. Efficient and accurate machine-learning interpolation of atomic energies in compositions with many species. Phys Rev B. 2017;96:014112. doi: 10.1103/PhysRevB.96.014112
- Kaiser A, Ritter M, Nazmutdinov R, et al. Hydrogen bonding and dielectric spectra of ethylene glycol–water mixtures from molecular dynamics simulations. J Phys Chem B. 2016;120:10515–10523. doi: 10.1021/acs.jpcb.6b05236
- Morawietz T, Sharma V, Behler J. A neural network potential-energy surface for the water dimer based on environment-dependent atomic energies and charges. J Chem Phys. 2012;136:064103. doi: 10.1063/1.3682557
- Morawietz T, Behler J. A density-functional theory-based neural network potential for water clusters including van der Waals corrections. J Phys Chem A. 2013;117:7356–7366. doi: 10.1021/jp401225b
- Morawietz T, Behler J. A full-dimensional neural network potential-energy surface for water clusters up to the hexamer. Zeitschrift für Physikalische Chemie. 2013;227:1559–1581. doi: 10.1524/zpch.2013.0384
- Kondati Natarajan S, Behler J. Self-Diffusion of surface defects at copper–water interfaces. J Phys Chem C. 2017;121:4368–4383. doi: 10.1021/acs.jpcc.6b12657
- Natarajan SK, Behler J. Neural network molecular dynamics simulations of solid–liquid interfaces: water at low-index copper surfaces. Phys Chem Chem Phys. 2016;18:28704–28725. doi: 10.1039/C6CP05711J
- Quaranta V, Hellström M, Behler J. Proton-transfer mechanisms at the water–ZnO interface: The role of presolvation. J Phys Chem Lett. 2017;8:1476–1483. doi: 10.1021/acs.jpclett.7b00358
- Artrith N, Kolpak AM. Understanding the composition and activity of electrocatalytic nanoalloys in aqueous solvents: A combination of DFT and accurate neural network potentials. Nano Lett. 2014;14:2670–2676. doi: 10.1021/nl5005674
- Hellström M, Behler J. Concentration-dependent proton transfer mechanisms in aqueous NaOH solutions: from acceptor-driven to donor-driven and back. J Phys Chem Lett. 2016;7:3302–3306. doi: 10.1021/acs.jpclett.6b01448
- Hellström M, Behler J. Structure of aqueous NaOH solutions: insights from neural-network-based molecular dynamics simulations. Phys Chem Chem Phys. 2017;19:82–96. doi: 10.1039/C6CP06547C
- Berendsen HJC, Grigera JR, Straatsma TP. The missing term in effective pair potentials. J Phys Chem. 1987;91:6269–6271. doi: 10.1021/j100308a038
- Lourderaj U, Sun R, Kohale SC, et al. The VENUS/NWChem software package. Tight coupling between chemical dynamics simulations and electronic structure theory. Comput Phys Commun. 2014;185:1074–1080. doi: 10.1016/j.cpc.2013.11.011
- Behler J. Constructing high-dimensional neural network potentials: A tutorial review. Int J Quantum Chem. 2015;115:1032–1050. doi: 10.1002/qua.24890
- Byrd R, Lu P, Nocedal J, et al. A limited memory algorithm for bound constrained optimization. SIAM Journal on Scientific Computing. 1995;16:1190–1208. doi: 10.1137/0916069
- Martínez L, Andrade R, Birgin EG, et al. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J Comput Chem. 2009;30:2157–2164. doi: 10.1002/jcc.21224
- Hoover WG. Canonical dynamics: Equilibrium phase-space distributions. Phys Rev A. 1985;31:1695–1697. doi: 10.1103/PhysRevA.31.1695
- Nosé S. A molecular dynamics method for simulations in the canonical ensemble. Mol Phys. 1984;52:255–268. doi: 10.1080/00268978400101201
- Kaiser A, Ismailova O, Koskela A, et al. Ethylene glycol revisited: molecular dynamics simulations and visualization of the liquid and its hydrogen-bond network. J Mol Liqu. 2014;189:20–29. doi: 10.1016/j.molliq.2013.05.033
- Mark P, Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J Phys Chem A. 2001;105:9954–9960. doi: 10.1021/jp003020w