
Abstract
We attempted to increase the thermostability of Moloney murine leukemia virus (MMLV) reverse transcriptase (RT). The eight-site saturation mutagenesis libraries corresponding to Ala70−Arg469 in the whole MMLV RT (Thr24−Leu671), in each of which 1 out of 50 amino acid residues was replaced with other amino acid residue, were constructed. Seven-hundred and sixty eight MMLV RT clones were expressed using a cell-free protein expression system, and their thermostabilities were assessed by the temperature of thermal treatment at which they retained cDNA synthesis activity. One clone D200C was selected as the most thermostable variant. The highest temperature of thermal treatment at which D200C exhibited cDNA synthesis activity was 57ºC, which was higher than for WT (53ºC). Our results suggest that a combination of site saturation mutagenesis library and cell-free protein expression system might be useful for generation of thermostable MMLV RT in a short period of time for expression and selection.
Site saturation mutagenesis library of MMLV RT was constructed and expressed using cell-free protein expression system.
Reverse transcriptase (RT) [EC 2.7.7.49] is the enzyme responsible for RNA viral genome replication. It possesses RNA- and DNA-dependent DNA polymerase and RNase H activities. RT from Moloney murine leukemia virus (MMLV) is widely used in cDNA synthesis.Citation1,2) MMLV RT is a 75-kDa monomer, consisting of fingers, palm, thumb, connection, and RNase H domains.Citation3–7) It has two active sites, one in the fingers/palm/thumb domain for the DNA polymerase reaction and the other in the RNase H domain for the RNase H reaction.
High reaction temperature reduces RNA secondary structures and nonspecific primer binding. However, MMLV RT is thermolabile.Citation8) Thus, improvement of the thermostability of MMLV RT has been an important research target. The thermostability of MMLV RT was first improved by eliminating the RNase H activity.Citation9–11) We improved the thermostability of MMLV RT by site-directed mutagenesis and generated MM4 (E286R/E302K/L435R/D524A) in which negatively charged (Glu286, Glu302) or hydrophobic (Leu435) residues thought to interact with a template-primer were replaced with positively charged ones and the catalytic residue for RNase H activity (Asp524) was replaced with Ala.Citation12) We also generated V433R in which Val433 at the molecular surface was replaced with Arg.Citation13) Recently, we designed 29 mutations, focusing on the number of surface charge and stabilization of hydrophobic core, and finally generated a sextuple thermostable variant MM3.14 (A32V/L72R/E286R/E302K/W388R/L435R).Citation14) Others improved thermostability of MMLV RT by random mutagenesis, in which error-prone PCR was used for the construction of MMLV RT gene library, and filter assayCitation15) or emulsion PCRCitation16) was used for the screening of MMLV RT with higher thermostability.
Site saturation mutagenesis is a technique, whereby a set of codons is randomized to produce a library of variants with every possible amino acid at each randomized position in the target region.Citation17–19) This method can be contrasted with error-prone PCR, in which many mutations that do not correspond to amino acid substitution are generated. Recently, it has become possible to create site saturation mutagenesis libraries using commercially available custom oligonucleotide libraries.
Cell-free protein expression system has been developed to produce a target protein, in which cellular components were used, instead of living cells, to translate DNA to RNA and translate RNA to protein.Citation20,21) Such components have been obtained from cells of various species, including Escherichia coli, wheat germ, yeast, rabbit reticulocytes, and insects. We previously expressed the wild-type MMLV RT (WT) and MM4 in an insect cell-free protein expression system, showing that the WT and MM4 expressed in that system had the same activity and thermostability as the WT and MM4, respectively, expressed in E. coli.Citation22) Unlike an expression in the living cells, a cell-free protein expression enables the completion of whole process including DNA template preparation, expression, and selection within a few days.Citation23) Thus, our previous resultsCitation22) suggest that a cell-free protein expression system might be useful for further stabilization of MMLV RT.
In this study, we constructed a site saturation mutagenesis library of MMLV RT, expressed MMLV RT variants using cell-free protein expression system, and screened for thermostable ones. Our results have shown that this novel strategy might be useful to increase the thermostability of MMLV RT.
Materials and methods
Materials
The concentration of the MMLV RT expressed in E. coli was determined using Protein Assay CBB Solution (Nacalai Tesque, Kyoto, Japan) with bovine serum albumin (Nacalai Tesque) as standard. Standard RNA, which is an RNA of 1014-nucleotides corresponding to DNA sequence 8353-9366 of the cesA gene of Bacillus cereus (GenBank accession number DQ360825), was prepared by in vitro transcription.Citation24) p(dT)15 was purchased from Fasmac (Tokyo, Japan). [methyl-3H]dTTP (1.52 TBq/mmol) and poly(rA) was from GE Healthcare (Buckinghamshire, UK).
Construction of a site saturation mutagenesis library
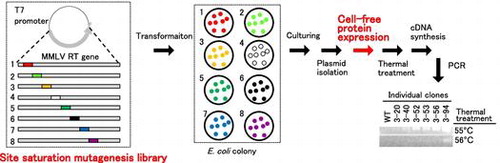
We designed eight site saturation mutagenesis libraries, in which one of all 50 amino acids in region 1 (Ala70-Asn119), 2 (Lys120-Ala169), 3 (Phe170-Leu219), 4 (Leu220-Leu269), 5 (Gly270-Arg319), 6 (Met320-Phe369), 7 (Val370-Leu419), or 8 (Thr420-Arg469), respectively, was replaced with all 19 other amino acids. Each library was constructed one by one using the QuikChange method with the expression plasmid for the wild-type MMLV RT (WT), pET-MRTHis (Fig. S1), as a template and the corresponding 150-bp oligonucleotide set as primers.
pET-MRTHis was constructed as described previously.Citation25) Briefly, the 2002-bp DNA fragment encoding MMLV RT was amplified from the pUC19 plasmid that contains the complete MMLV gene (8332bp) (GenBank accession No. J02255), using the primers 5′-AAGGAGATATACATATGACATGGCTGTCTG-3′ and 5′-CTGAATTCTAGTGGTGATGGTGGTGGTGGAGGAGGGTAGAGGTGTCT-3′ (NdeI and EcoRI sites are underlined), and the amplified DNA was digested with NdeI and EcoRI, and inserted in pET-22b(+).
Eight 150-bp oligonucleotide mixture sets, each corresponding to one of region 1−8, were designed and synthesized by Agilent Technologies using a novel microarray technology the company developed. QuikChange HT Protein Engineering System (Agilent Technologies, Santa Clara, CA) was used in the following steps. Each of the eight oligonucleotide sets was amplified by PCR (50 μL) in the presence of the oligonucleotide library and the corresponding forward and reverse custom primers (0.5 μM each) (Table S1). Each of the eight site saturation mutagenesis libraries was constructed by a thermal cycling reaction (25 μL) in the presence of 1 ng/μL pET-MRTHis and the corresponding oligonucleotide set, and transfected into E. coli SoloPack Gold supercompetent cells.
Expression of a site saturation mutagenesis library
Five hundred microlitre of L broth containing 50 μg/mL ampicillin in the 96-well pate was inoculated with a colony of the transformed E. coli cells and incubated for 12‒16 h with shaking at 37 °C. Then, 100 μL of each eight culture mediums were mixed, from which plasmid DNAs were purified. To amplify the DNA fragments containing the T7 promoter and the entire MMLV RT gene, the reaction mixture for PCR (25 μL) was prepared by mixing the plasmid preparation (2 μL), water (38 μL), 10 × PCR buffer (2.5 μL), 2 mM dNTP (1.5 μL), 10 μM T7 promoter primer 5′-CCCGCGAAATTAATACGAC-3′ (1 μL) and 10 μM termination primer 5′-ATGCTAGTTATTGCTCAGCG-3′, and 1 U/μL recombinant Taq polymerase (0.3 μL) (Toyobo, Osaka, Japan) under 30 cycles of 30 s at 95 °C, 30 s at 55 °C, and 180 s at 72 °C. The cell-free protein expression reaction (6 μL) was performed using Cell-free expression kit Purefrex 1.0 (Genefrontier, Kashiwa, Japan) in the presence of the PCR product (2 μL) at 37 °C for 3 h. After the reaction, 10 μL of water was added to the reaction solution.
Screening of a site saturation mutagenesis library
Aliquots (3 μL) of the diluted reaction solution above mentioned were incubated at different temperatures (55 or 56 °C) for 7 min followed by the incubation on ice for 30–60 min. Then, the reaction mixture for cDNA synthesis (20 μL) was prepared by mixing water (11 μL), 10 × RT buffer (250 mM Tris-HCl (pH 8.3) buffer, 500 mM KCl, 20 mM DTT, 50 mM MgCl2) (2 μL), 2.0 mM dNTP (2 μL), 160 pg/μL cesA RNA (2 μL), 10 μM RV-R26 primer 5′-TGTGGAATTGTGAGCGGTGTCGCAATCACCGTAACACGACGTAG-3′ (1 μL) and the thermally treated product (2 out of 3 μL). The reaction was run at 45 °C for 30 min and 65 °C for 5 min. The reaction mixture for PCR (25 μL) was prepared by mixing the reaction product of cDNA synthesis (2 μL), water (17.7 μL), 10 × PCR buffer (2.5 μL), 2 mM dNTP (1.5 μL), 10 μM F5 primer 5′-TGCGCGCAAAATGGGTATCAC-3′ (0.5 μL) and 10 μM RV primer 5′-TGTGGAATTGTGAGCGG-3′ (0.5 μL), and 1 U/μL recombinant Taq polymerase (0.3 μL). The reaction was run under 30 cycles of 30 s at 95 °C, 30 s at 55 °C, and 60 s at 72 °C. The amplified products were separated on 1.0% w/v agarose gel and stained with ethidium bromide (1 μg/mL).
Expression in E. coli and purification of MMLV RT
Expression of WT and its thermostable variant D200C in E. coli and purification were performed as described in the previous reports.Citation8,12) Briefly, E. coli strain BL21(DE3) was transformed with pET-MRTHis or pET-D200CHis. WT and D200C were expressed in the soluble fractions of the transformants, from which active enzymes were purified by ion-exchange chromatography followed by ammonium sulfate fractionation and Ni2+ affinity chromatography.
Western blot
The solution containing MMLV RT expressed in the cell-free protein expression system was applied to 10% w/v polyacrylamide gel. After separation, the proteins were transferred by electroblotting onto a polyvinylidene difluoride (PVDF) membrane Sequi-BlotTM PVDF (BIO-RAD, Hercules, CA) in 25 mM Tris-HCl (pH 8.3) buffer, 192 mM glycine, 20% (v/v) methanol at 25 V for 45 min. After blotting, the membrane was washed with 50 mM Tris-HCl (pH 8.3) buffer, 138 mM NaCl, 2.7 mM KCl, 0.05% Tween 20 (TBS-T), blocked with TBS-T containing 5% (w/v) skim milk, and incubated with Anti-His-tag mAb-HRP DirectT (MBL life Science, Nagoya, Japan) diluted by 5000-fold in TBS-T containing skim milk. After washing with TBS-T for three times and 50 mM Tris-HCl (pH 8.3) buffer, 138 mM NaCl, 2.7 mM KCl (TBS) for one time, the protein bands were visualized using a Peroxidase Stain Kit for Immuno-blotting (Nacalai Tesque).
DNA polymerase activity to incorporate dTTP into poly(rA)-p(dT)15
The reaction was carried out in 25 mM Tris-HCl (pH 8.3), 50 mM KCl, 2 mM DTT, 5 mM MgCl2, 24 μM poly(rA)-p(dT)15 (this concentration is expressed based on p(dT)15), 0.2 mM [3H]dTTP (1.85 Bq/pmol), and 5 nM MMLV RT at 37 °C. An aliquot (20 μL) was taken from the reaction mixture at specified times and immediately spotted onto the glass filter. Unincorporated [3H]dTTP was removed by three times of wash with chilled 5% (w/v) trichloroacetic acid for 10 min each, followed by one wash with chilled 95% ethanol. The radioactivity retained on the dried filter was counted in 2.5 mL Ecoscint H (National Diagnostics, Yorkshire, UK). The initial reaction rate was estimated from the time-course for incorporation of [3H]dTTP. One unit is defined as the amount which incorporates 1 nmol of dTTP into poly(rA)-p(dT)15 in 10 min.
Results and discussion
Construction of the site saturation mutagenesis library
Currently, rational design and random mutation are both widely used for protein engineering. In the stabilization of MMLV RT, the drawback of rational design is that detailed structural knowledge of MMLV RT is unavailable. Crystal structure of the full length molecule has not been determined yet although that of the fingers, palm, thumb, and connection domains (Thr24-Pro474)Citation5) and that of the RNase H domain (His503-Ser668)Citation6) have been independently determined. In the alteration of MMLV RT’s function, the drawback of random mutation is that preparation of MMLV RT is time-consuming: MMLV RT is expressed inside E. coli cells, from which active enzyme should be purified. Therefore, high throughput screening of thermostable MMLV RT has not been realized. To overcome these obstacles, in this study, we constructed site saturation mutagenesis libraries of MMLV RT and expressed MMLV RT variants using cell-free protein expression system. Unlike error-prone PCR, site saturation mutagenesis is expected to introduce all possible amino acid substitutions into a target region.
In MMLV RT, the DNA polymerase activity is located in the fingers (Thr24-Asp124 and Phe156-Ser195), palm (Ile125-Phe155 and Pro196-Glu275), thumb (Gly276-Leu338), and connection (Pro339-Asp468) subdomains while the RNase H activity is in the RNase H domain (Arg469-Leu671) (Fig. (A)). Since we intended to increase the thermostability of the DNA polymerase domain rather than the RNase H domain, we selected Ala70-Arg469 as the target region for mutagenesis. As shown in Fig. , eight site saturation libraries, each corresponding to one of regions 1‒8, were constructed.
Fig. 1. Structure of MMLV RT.
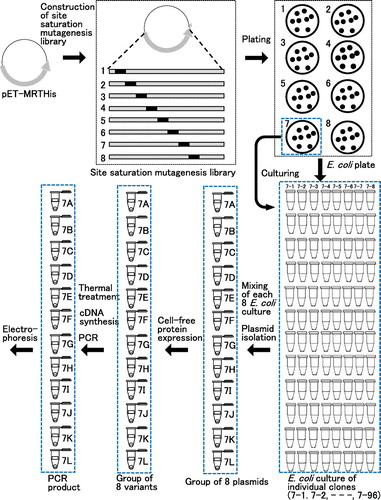
Fig. 2. Workflow to construct a site saturation mutagenesis library and to screen a thermostable MMLV RT variant.
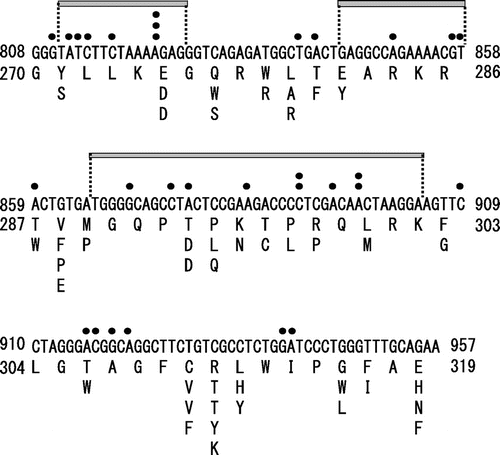
We first investigated the quality of the libraries. The nucleotide sequences of the MMLV RT genes of 73 clones in the library for region 5 were analyzed as one example. Twenty-seven clones (37%) had expected mutations corresponding to one amino acid substitution, while 42 clones (58%) had unexpected mutations (one nucleotide deletion for 32 clones; two amino acid substation for 7 clones; and 15‒39 nucleotides deletion for 3 clones), and 4 clones (5%) had no mutation. These results indicate that the libraries had substantial numbers of unexpected mutations. This may have been caused by errors in oligonucleotides synthesis in the microarray at a fixed frequency, such that the library contained a certain proportion of oligonucleotides with unexpected sequences. Similar results were obtained in the analysis of the quality of site saturation mutagenesis libraries constructed by using oligonucleotides synthesized in the standard solid-phase method.Citation26,27) It is difficult to quantitatively evaluate the success rate for the expected mutation because the success of protein engineering study depends on various factors including the success rate for the expected mutation. However, we considered that the success rate (37%) to be high enough for subsequent screening. Indeed, random mutagenesis methods such as error-prone PCR theoretically create greater numbers of mutations that do not correspond to amino acid substitution than the site saturation mutagenesis method.
The mutation spectrum of region 5, shown in Fig. , exhibited five hot spots (deletion of A822 for three clones and deletion of C890, deletion of A897, substitutions corresponding to E275D/R311T, and substitutions corresponding to T293D/C310 V for two clones each), indicating that the library was biased toward certain unexpected mutations. On the other hand, there was no such bias for the expected mutations.
Fig. 3. Sequence analysis of the site saturation mutagenesis library of MMLV RT.
Expression of the site saturation mutagenesis library using cell-free protein expression system and screening of thermostable MMLV RT variants
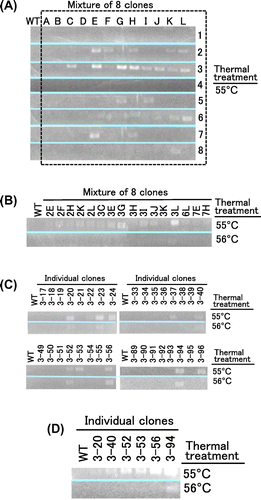
Considering the amounts of experiment, we decided to examine 768 clones (96 clones/one region). Fig. also shows the working flow for the expression and screening. Each 768 clones were cultured, then equal volumes of each eight cultures were mixed. We thought that this equal-volume mixing made the screening less time-consuming but did not make the expression levels different among clones. These groups were named according to the numbers of target region and individual clones (for example, 1A represents the mixture of 1-1 to 1-8). Plasmid DNA was prepared, and the MMLV RT (Thr24-Leu671) was expressed using a cell-free protein expression system for each group. The thermostabilities of MMLV RT variants were examined as the following. (i) The reaction solutions containing MMLV RT variants were thermally treated; (ii) A cDNA synthesis reaction was carried out at 45 °C for 30 min with the reaction solution exposed to 55 or 56 °C for 7 min; (iii) PCR was carried out from the reaction product for the cDNA synthesis reaction; and (iv) Agarose gel electrophoresis was performed for the detection of 601-bp band in the PCR products.
Fig. shows the results of screening of 96 groups of eight clones. In the first round of screening, 16 groups (2E, 2F, 2H, 2K, 2L, 3C, 3E, 3G, 3H, 3I, 3J, 3K, 3L, 6L, 7E, and 7H) that exhibited cDNA synthesis activity after the thermal treatment at 55 °C were selected out of the 96 groups (Fig. (A)). In the second round of screening, four groups (3C, 3E, 3G, and 3L) that exhibited relatively high cDNA synthesis activity after the thermal treatment at 55 or 56 °C were selected out of these 16 groups (Fig. (B)). In the third round of screening, six clones (3-20, 3-40, 3-52, 3-53, 3-56, and 3-94) were selected from the 32 clones in these four groups (Fig. (C)). Finally, 3-94 was selected as the most thermostable variant out of these six clones (Fig. (D)). These results indicated that the library for region 3 contained the largest number of thermostable variants (Fig. (B)), suggesting the region 3 is the most highly involved in stability. Such finding has not been obtained by site-directed mutagenesis study based on the crystal structure of MMLV RT.
Fig. 4. Screening of the site saturation mutagenesis library of MMLV RT.
We selected 19 clones which exhibited higher thermostability than WT (3-1, 3-2, 3-73, 3-74, 3-75, and 3-76 (data not shown) and other 13 clones (Fig. )) and analyzed their sequences (Table ). The most thermostable 3-94 clone had the D200C mutation. As shown in Fig. S2, Asp200 belongs to the palm subdomain and located closely in the catalytically important residues Asp225 and Asp226. Thirteen mutational sites were identified: nine (Asp200, Glu201, Leu202, Leu207, Ala208, Phe210, Gln213, Ile218, and Gly248) were in the palm subdomain and four (Gly178, Thr184, Thr186, Leu188) were in the fingers domain. The mutation at Ile218 was detected in three clones, and these at Thr186, Asp200, Glu201, and Leu202 were detected in two clones, respectively. These results suggest that the priority of the subdomains as the site to be mutated for the thermostabilization of MMLV RT is in the order of palm > fingers > thumb, connection, and that Thr186, Asp200, Glu201, Leu202, and Ile218 are highly involved in thermostability.
Table 1. Mutations which increase thermostability of MMLV RT.
Fig. S2 shows the comparison of the amino acid sequences of MMLV RT and avian myeloblastosis virus reverse transcriptase (AMV RT). Like MMLV RT, AMV RT is widely used in cDNA synthesis. AMV RT is a heterodimer consisting of a 63-kDa α subunit and a 95-kDa β subunit. The β subunit comprises the fingers, palm, thumb, connection, RNase H, and integrase domains. The α subunit is a proteolytic cleavage product of the β subunit, and lacks the integrase domain. The sequence homology of fingers, palm, and thumb are 24, 35, and 21%, respectively.Citation28) Except for Leu188 and Leu207, the 12 amino acid residues that were mutated in the thermostable variants identified in this study were not conserved in both RTs. This result suggests that these residues are not necessary for activity and accords with the observation that the variation of these residues do not abolish activity.
Preparation of the thermostable MMLV RT variant using an E. coli expression system and evaluation of its thermostability
To compare the thermostability of D200C with that of WT, WT and D200C were expressed in E. coli and purified from the cells. Fig. S3(A) shows the results of SDS-PAGE analysis under reducing conditions at various purification stages. Purified WT (lane 5) and D200C (lane 10) yielded a single band with a molecular mass of 75 kDa, corresponding to C-terminally (His)6-tagged MMLV RT. The yields of the purified enzyme preparations from 500 mL of culture were 2.6 mg for WT and 3.3 mg for D200C. Fig. S3(B) shows the results of western blot analysis. Unpurified WT and D200C preparations which had been expressed in the cell-free protein expression system and purified WT and D200C preparations which had been expressed in E. coli exhibited a 75-kDa protein band.
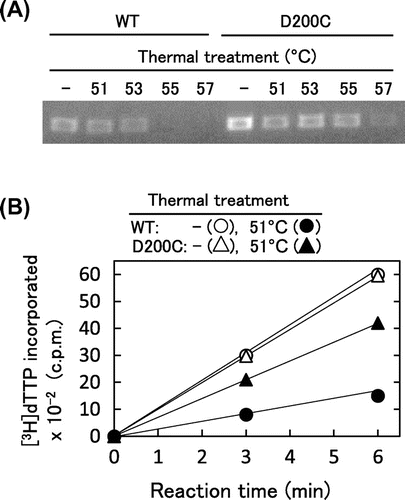
Fig. shows the comparison of thermostabilities between WT and D200C. In the cDNA synthesis assay after thermal treatment, the highest temperature of heat treatment at which D200C exhibited cDNA synthesis activity was 57 °C, which was higher than for WT (53 °C) (Fig. (A)). In the DNA polymerase assay for the incorporation of dTTP into poly(rA)-p(dT)15 after the thermal incubation at 51 °C for 7 min, D200C retained 70% of the initial activity, while WT retained 25% of the initial activity (Fig. (B)). These results indicated that D200C was more thermostable than WT. However, D200C was less thermostable than the thermostable quadruple variant we generated, MM4 (E286R/E302 K/L435R/D524A), which retained activity after thermal treatment at 60 °C.Citation23) Mutational combination is the next strategy for further stabilization, which was reported to be effective.Citation12,14–16)
Fig. 5. Effects of thermal treatment on the activities of MMLV RT.
In conclusion, thermostable MMLV RT variant D200C was generated using the combination of site saturation mutagenesis library and cell-free protein expression system. Our results suggest that this combination might be valuable for engineering various enzymes that are difficult to manipulate, such as MMLV RT, whose expression, purification, and activity measurements are problematic and time-consuming. We propose that site saturation mutagenesis alone might also be valuable for engineering enzymes that are easier to manipulate. Our results also suggest that increasing the quality of site saturation mutagenesis libraries is an important target for future research.
Authors contribution
K.Y. designed research; Y.K., T.I. M.B., and M.N. performed research; Y.K., T.I., M.I., K.K., T.T., and K.Y. analyzed data; Y.K., M.B., and K.Y. wrote the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by Japan Society for the Promotion of Science; Grants-in-Aid for Scientific Research [grant number 21580110]; Japan Science and Technology Agency (SENTAN).
Supplementary material
The supplementary material for this paper is available online at https://doi.org/10.1080/09168451.2017.1394790.
Supplementary Figure
Download PDF (100.1 KB)Supplementary Table
Download MS Word (39.3 KB)Acknowledgments
We appreciate Ms. Tomomi Yamasaki of Kyoto University for her contribution to this work.
Notes
Abbreviations: MMLV, Moloney murine leukemia virus; RT, reverse transcriptase; T/P, template primer.
Related Research Data
References
- Kimmel AR, Berger SL. Preparation of cDNA and the generation of cDNA libraries: overview. Methods Enzymol. 1987;152:307–316.10.1016/0076-6879(87)52035-3
- Roberts JD, Bebenek K, Kunkel TA. The accuracy of reverse transcriptase from HIV-1. Science. 1988;242:1171–1173.10.1126/science.2460925
- Patel PH, Jacobo-Molina A, Ding J, et al. Insights into DNA polymerization mechanisms from structure and function analysis of HIV-1 reverse transcriptase. Biochemistry. 1995;34:5351–5363.10.1021/bi00016a006
- Georgiadis MM, Jessen SM, Ogata CM, et al. Mechanistic implications from the structure of a catalytic fragment of Moloney murine leukemia virus reverse transcriptase. Structure. 1995;3:879–892.10.1016/S0969-2126(01)00223-4
- Das D, Georgiadis MM. The crystal structure of the monomeric reverse transcriptase from Moloney murine leukemia virus. Structure. 2004;12:819–829.10.1016/j.str.2004.02.032
- Lim D, Gregorio GG, Bingman C, et al. Crystal structure of the Moloney murine leukemia virus RNaseH domain. J Virol. 2006;80:8379–8389.10.1128/JVI.00750-06
- Cote ML, Roth MJ. Murine leukemia virus reverse transcriptase: structural comparison with HIV-1 reverse transcriptase. Virus Res. 2008;134:186–202.10.1016/j.virusres.2008.01.001
- Yasukawa K, Nemoto D, Inouye K. Comparison of the thermal stabilities of reverse transcriptases from avian myeloblastosis virus and Moloney murine leukaemia virus. J Biochem. 2008;143:261–268.10.1093/jb/mvm217
- Kotewicz ML, D’Alessio JM, Driftmier KM, et al. Cloning and overexpression of Moloney murine leukemia virus reverse transcriptase in Escherichia coli. Gene. 1985;35:249–258.10.1016/0378-1119(85)90003-4
- Gerard GF, Potter RJ, Smith MD, et al. The role of template-primer in protection of reverse transcriptase from thermal inactivation. Nucleic Acids Res. 2002;30:3118–3129.10.1093/nar/gkf417
- Mizuno M, Yasukawa K, Inouye K. Insight into the mechanism of the stabilization of Moloney murine leukaemia virus reverse transcriptase by eliminating RNase H activity. Biosci Biotechnol Biochem. 2010;74:440–442.10.1271/bbb.90777
- Yasukawa K, Mizuno M, Konishi A, et al. Increase in thermal stability of Moloney murine leukaemia virus reverse transcriptase by site-directed mutagenesis. J Biotechnol. 2010;150:299–306.10.1016/j.jbiotec.2010.09.961
- Konishi A, Ma X, Yasukawa K. Stabilization of Moloney murine leukemia virus reverse transcriptase by site-directed mutagenesis of the surface residue Val433. Biosci Biotechnol Biochem. 2014;78:147–150.
- Baba M, Kakue R, Leucht C, et al. Further increase in thermostability of Moloney murine leukemia virus reverse transcriptase by mutational combination. Protein Eng Des Sel. 2017;30:551–557.
- Arezi B, Hogrefe H. Novel mutations in Moloney murine leukemia virus reverse transcriptase increase thermostability through tighter binding to template-primer. Nucleic Acids Res. 2009;37:473–481.10.1093/nar/gkn952
- Baranauskas A, Paliksa S, Alzbutas G, et al. Generation and characterization of new highly thermostable and processive M-MuLV reverse transcriptase variants. Protein Eng Des Sel. 2012;25:657–668.10.1093/protein/gzs034
- Reetz MT, Carballeira JD. Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat Protoc. 2007;2:891–903.10.1038/nprot.2007.72
- Reetz MT, Prasad S, Carballeira JD, et al. Iterative saturation mutagenesis accelerates laboratory evolution of enzyme stereoselectivity: rigorous comparison with traditional methods. J Am Chem Soc. 2010;132:9144–9152.10.1021/ja1030479
- Cheng F, Xu J-M, Xiang C, et al. Simple-MSSM: a simple and efficient method for simultaneous multi-site saturation mutagenesis. Biotechnol Lett. 2017;39:567–575.10.1007/s10529-016-2278-x
- Hodgman CE, Jewett MC. Cell-free synthetic biology: thinking outside the cell. Metab Eng. 2012;14:261–269.10.1016/j.ymben.2011.09.002
- Whittaker JW. Cell-free protein synthesis: the state of the art. Biotechnol Lett. 2013;35:143–152.10.1007/s10529-012-1075-4
- Katano Y, Hisayoshi T, Kuze I, et al. Expression of Moloney murine leukemia virus reverse transcriptase in a cell-free protein expression system. Biotechnol Lett. 2016;38:1203–1211.10.1007/s10529-016-2097-0
- Kanno T, Tozawa Y. Protein engineering accelerated by cell-free technology. Methods Mol Biol. 2010;607:85–99.10.1007/978-1-60327-331-2
- Yasukawa K, Agata N, Inouye K. Detection of cesA mRNA from Bacillus cereus by RNA-specific amplification. Enzyme Microbiol Technol. 2010;46:391–396.10.1016/j.enzmictec.2009.12.009
- Konishi A, Shinomura M, Yasukawa K. Enzymatic characterization of human immunodeficiency virus type 1 reverse transcriptase for use in cDNA synthesis. Appl Biochem Biotechnol. 2013;169:77–87.10.1007/s12010-012-9953-8
- Acevedo-Rocha CG, Reetz MT, Nov Y. Economical analysis of saturation mutagenesis experiments. Sci Rep. 2015;5:10654.10.1038/srep10654
- Zhao J, Frauenkron-Machedjou VJ, Kardashliev T, et al. Amino acid substitutions in random mutagenesis libraries: lessons from analyzing 3000 mutations. Appl Microbiol Biotechnol. 2017;101:3177–3187.10.1007/s00253-016-8035-1
- Yasukawa K, Mizuno M, Inouye K. Characterization of Moloney murine leukaemia virus/avian myeloblastosis virus chimeric reverse transcriptases. J Biochem. 2009;145:315–324.10.1093/jb/mvn166