
ABSTRACT
Glycoengineered yeast cells, which express human-compatible glycan structures, are particularly attractive host cells to produce therapeutic glycoproteins. Disruption of OCH1 gene, which encodes an α-1,6-mannosyltransferase required for mannan-type N-glycan formation, is essential for the elimination of yeast-specific N-glycan structures. However, the gene disruption causes cell wall defects leading to growth defects. Here, we tried to identify factors to rescue the growth defects of och1Δ cells by in vivo mutagenesis using piggyBac (PB)-based transposon. We isolated a mutant strain, named 121, which could grow faster than parental och1Δ cells. The PB element was introduced into the promoter region of BEM4 gene and upregulated the BEM4 expression. Overexpression of BEM4 suppressed growth defects in och1Δ cells. The slow grow phenotypes were partially rescued by expression of Rho1p, whose function is regulated by Bem4p. Our results indicate that BEM4 would be useful to produce therapeutic proteins in glycoengineered yeast without the growth defects.
Graphical Abstract
PB transposon in yeast for in vivo mutagenesis. The PB-mutagenized cells that grew faster than parental och1Δ cells were isolated. BEM4 was identified in the screening.
Biopharmaceuticals such as monoclonal antibodies have high specificity and high affinity against target molecules and have low side effects, compared to traditional small molecular compounds. Recombinant therapeutic proteins are becoming essential pharmaceutical agents for treating intractable diseases such as cancer and autoimmune diseases [Citation1]. Many of the therapeutic proteins receive post-translational modifications such as glycosylation, disulfide bond formation and proteolytic cleavage, which influence the protein structure and proper function [Citation2]. Approximately 70% of therapeutic proteins are modified with asparagine (N)-linked glycans [Citation3]. N-glycans on proteins play critical roles in protein quality control at the endoplasmic reticulum (ER) to ensure the correct protein folding [Citation4,Citation5]. In addition, the N-glycans contribute to the protein activity, stability in the serum, and targeting to the cells [Citation6]. Most therapeutic proteins are currently produced in mammalian cells such as Chinese hamster ovary (CHO) cells because these cell types improve the yields of correctly folded proteins and facilitate appropriate post-translational modifications [Citation7,Citation8]. Production of biopharmaceuticals in mammalian cells, however, has several drawbacks, including high cost and the potential for infections such as viruses and prions [Citation6].
There is the development of alternative host cells and organisms for the production of pharmaceutical proteins [Citation6]. Yeasts for that matter, provide several advantages in the host cell expression system; high growth rate and large-scale culturing to produce recombinant proteins [Citation9,Citation10]. However, the use of the recombinant proteins from yeast in human causes immunogenicity, as a result of different N-glycan structures in yeasts from those in mammalian cells. To overcome the issue, there has been the development of glycoengineered yeast strains (budding yeast, fission yeast, and methylotrophic yeasts) that produce recombinant proteins having human compatible N-glycans [Citation9,Citation11–Citation16]. Mannosyltransferases form yeast-specific mannan-type N-glycans at the Golgi apparatus [Citation17]. Och1p is a cis-Golgi-localized α-1,6-mannosyltransferase required for the initiation of mannan-type N-glycan formation [Citation18]. It is essential to disrupt OCH1 genes to establish yeast strain producing human compatible N-glycans [Citation9,Citation15,Citation19]. Since proteins having mannan-type N-glycans (mannoproteins) are components of mannan layers in the yeast cell wall, disruption of OCH1 causes defects in the cell wall structures, leading to growth defects and temperature sensitivity [Citation18,Citation20]. The phenotypes cause adverse effects on the recombinant protein production [Citation21].
So far, several trials have been performed to overcome the issue. One of the ways is to express some specific genes. Overexpression of RHO1, which encodes the Rho1p small GTPase, partly recovered the growth defects in och1Δ alg3Δ double mutant cells [Citation22]. Rho1p is a master regulator of the cell wall integrity [Citation23], hence the overexpression of RHO1 increased the glucan layer in the cell wall. Also, the overexpression of FKS2 encoding a β-1,3-glucan synthase partially rescued the defects in och1Δ alg3Δ double mutant cells [Citation22]. Alternatively, genetic screens were used to isolate cells that alleviate the growth defects. The glycoengineered yeast strains were mutagenized in vivo by the introduction of proofreading-deficient DNA polymerase variant [Citation21]. Then, the glycoengineered mutant cells that rescued the growth phenotypes were successfully obtained. The isolated, glycoengineered mutant cells upregulate genes involved in gluconeogenesis and glyoxylate cycle [Citation24]. Although in vivo mutagenesis by mutant DNA polymerase and chemical mutagenesis are useful to isolate mutant cells of interests, the identification of the gene responsible for a mutant phenotype is difficult and time-consuming.
In this study, we used transposon-based in vivo mutagenesis to obtain the mutant och1Δ cells that rescue the growth defects. The piggyBac (PB) transposon is a TTAA-specific transposon isolated from the cabbage looper moth, Trichoplusia ni [Citation25,Citation26]. Compared with other transposon-based methods, the PB transposon system is active in various organisms and the PB insertion sites are distributed uniformly with little bias in the genome [Citation27]. Therefore, this makes it useful as a mutagenesis tool in many organisms including fission yeast, protozoa, plants, insects, and vertebrates [Citation28]. We hereby constructed a system to screen mutant cells using PB transposon in budding yeast. Using the system, a mutant och1Δ strain named 121, which restored the cell growth at 37°C, was isolated. In the 121 cells, the PB element was incorporated into the promoter region of BEM4, and upregulated the expression of BEM4. Again, overexpression of BEM4 rescued the growth defects of och1Δ cells. Expression of Rho1p, whose function is regulated by Bem4p, partially rescued the phenotypes, consistent with our previous results [Citation22]. Our results suggest that activation of the BEM4-RHO1 pathway could be useful to overcome the growth defects in the glycoengineered yeast strains.
Materials and methods
Media and strains
Yeast strains were grown on YPAD (1% yeast extract, 2% peptone, 0.003% adenine, 2% glucose), SD (0.67% yeast nitrogen base, 2% glucose, and required amino acids), and SG (0.67% yeast nitrogen base, 2% galactose and required amino acids) media. The yeast strains used in this study were BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0; EUROSCARF), BY4741PB (MATa ade2::PB-HIS3, BY4741; this study), och1Δ (och1Δ::hphNT1, BY4741PB; this study), 121 (mutant och1Δ strain in which PB element was inserted at the promoter region of the BEM4 gene; this study), and 121_PBr (121 strain, in which PB transposon was removed; this study). Gene disruption in yeast was performed using a one-step method as described previously [Citation29]. To construct the strain BY4741PB, a 3ʹPB-PTEF-HIS3–5ʹPB element, which was amplified using primers (ADE2-PBdel-F: 5ʹ-tggtatatttggtgtggaaatgttctatttagaaacaggggaattgcttattaaccctagaaagatagtctgcgtaaaattg-3ʹ and ADE2-PBdel-R: 5ʹ-aagcatcaatggtataatgtccagagttgtgaggccttggggcaatttcgttaaccctagaaagataatcatattgtgacgt-3ʹ) from plasmid pPB-PTEF-HIS3, was inserted into the ADE2 gene. The OCH1 gene was deleted by the hygromycin B resistance cassette (hphNT1), amplified from the p303H through homologous recombination [Citation30]. 121_PBr was obtained from the mutant strain 121 by removing the PB element using PBase induction as described below.
Plasmids
A plasmid pRS316-GALp was constructed by cutting the GAL1 promoter fragment from plasmid pRS424-GALp [Citation31] with SpeI and SacI sites. The PBase DNA fragment was amplified from pCMV-hyPBase [Citation32], which was kindly provided by Kosuke Yusa (Sanger Institute). The fragment was ligated into the EcoRI and XhoI sites of the plasmid pRS316-GALp, generating pRS316-GALp-PBase. To construct pPB-PTEF-HIS3, the PTEF-HIS3 fragment was amplified from plasmid pFA6a-HIS3MX6, which contains TEF promoter and HIS3 gene from Saccharomyces kluyveri. The fragment was ligated into a plasmid pPB-R1R2-NP [Citation33], which was kindly provided by Kosuke Yusa, generating pPB-PTEF-HIS3. The BEM4 gene fragment was amplified from genomic DNA extracted from the strain BY4741, and inserted into the plasmid YEp352GAPII [Citation34] between the EcoRI and SalI sites, to construct plasmid YEp352GAPII-BEM4. The construction of the plasmid YEp352GAPII-CDC42 was achieved by amplifying the CDC42 gene fragment, which was inserted into the plasmid YEp352GAPII between the EcoRI and SalI sites. The construction of pY26-RHO1 was described previously [Citation22].
PB transposon-based in vivo mutagenesis and screening
For screening, och1Δ cells harboring pRS316-GALp-PBase were cultured in liquid SD −uracil (Ura) medium for 1 day. Yeast cells of 0.625 OD660 units were transferred to 25 mL SG −Ura medium, (OD660 = 0.025) and were cultured for 27 h to allow the expression of PBase. Subsequently, 1 mL (OD660 = 0.8) of cell suspension was transferred to 25 mL SD −Ade −His medium and cultured at 30°C for 24 h. Then, 1 mL (OD660 = 0.5) of cells were further cultured in SD −Ade −His medium at 37°C for 36 h. Cells were spread on SD −Ade −His selective plates (about 5–6 × 103 cells/plate × 2) after the cultivation at 37°C. Colonies with faster growth rate on the plates were picked up and streaked on SD −Ade −His plates.
Removal of the PB insertion from mutant cells
For the recovery experiment, the PB insertion was required to be removed from mutant cells. The PBase was induced in galactose medium to excise the PB element from the insertion site. The cells were spread on SD −Ade plates after the induction. The colonies were picked up and streaked on SD −Ade −His plates. Yeast strains that grew on SD −Ade plates, but did not grow on SD −Ade −His plates were isolated as strains with the PB transposon removed.
Sequence analysis of PB insertion sites in single colonies
Genomic DNA was extracted from 5 OD660 units of cells with an Ezup Column Yeast Genomic DNA Purification Kit (Sangon Biotech), and 2 μg of DNA was digested with HaeIII (NEB) for 12–16 h. A splinkerette adaptor composed of the oligonucleotides (Spl-top: 5ʹ-cgaatcgtaaccgttcgtacgagaattcgtacgagaatcgctgtcctctccaacgagccaagg-3ʹ and SplB-BLT: 5ʹ-ccttggctcgtttttttttgcaaaaa-3ʹ) was ligated to the digested DNA fragments with T4 DNA ligase (NEB) as described previously [Citation35–Citation37]. After purification, the fragments were used as templates for splinkerette PCR with the 1st primer set (Spl-P1: 5ʹ-cgaatcgtaaccgttcgtacgagaa-3ʹ and 3PB-1st: 5ʹ-tatacagaccgataaaacacatgcgt-3ʹ), followed by nested PCR with the 2nd primer set (Spl-P2: 5ʹ-tcgtacgagaatcgctgtcctctcc-3ʹ and 3PB-2nd: 5ʹ-cgcatgattatctttaacgtacgtcacaa-3ʹ). After purification, The resulting DNA fragments after purification were phosphorylated by T4 polynucleotide kinase (NEB), and then ligated into the EcoRV sites of the plasmid pBluescriptII (Agilent) and sequenced.
Growth curve
Cells were precultured in SD media, and the cells (1.25 OD595) were later inoculated in 25 mL of SD or YPAD media to begin the measurement. The cell suspensions were taken at indicated times, and absorbance (OD595) was measured.
Spot test
Cells were cultivated in the corresponding selective medium until the OD660 was more than 1.0. The cell concentration was adjusted to OD600 = 0.5. The samples were serially diluted ten times. Four μL of each sample was spotted on the plates.
Quantitative RT-PCR analysis
Cells were cultured in YPAD medium at 30°C overnight, then 1 mL of the cell suspension was transferred to fresh 10 mL of YPAD medium and cultured at 30°C until the absorbance of the cell suspension (OD660) reached to 0.6. Cells (1.5 OD660) were harvested, and RNA was extracted according to the manufacturer’s protocol (Promega), with the total RNA resuspended in 50 μL RNase-free water. To synthesize cDNA, the RNA concentration was measured by Nanodrop 2000 (Thermo Fisher Scientific). In the 50 μL system, 500 ng of RNA and 10 μL of 5×PrimeScript RT Master Mix for RT-qPCR (Takara, Shiga, Japan) was added. The reaction was incubated for 15 min at 37°C, for 5 s at 85°C afterward, and was later maintained at 4°C. For Real-Time PCR, 2 μL cDNA was mixed with 10 μL SYBR Premix Ex Taq II (Tli RNaseH Plus, Takara), 0.4 μL ROX Reference Dye II, 6 μL RNase-free water, 0.8 μL forward and reverse primers (10 μM). For BEM4, we designed the primer set (forward: 5ʹ-ggaaattctcttggattctggagg-3ʹ; reverse: 5ʹ-gaccgcatctgtaatttatccactaag-3ʹ). The expression level was normalized by expression of TDH1 gene (primer set: forward: 5ʹ-tctaacgattatgctgcttacatgg-3ʹ; reverse: 5ʹ-ctttcttggtaggtagcgatcttgaca-3ʹ) as the control. Triplicate reactions for each cDNA and primer pair were run using standard parameters in an Applied Biosystems StepOnePlus Real-Time PCR system (Thermo Fisher Scientific).
Quantitative measurements of β-1,3-glucan
The amount of β-1,3-glucan was measured using aniline blue as described previously [Citation22,Citation38]. Cells were precultured in YPAD or SD – Ura media, then cultured in SD media. Cells (2.5 × 106) were harvested, washed twice with TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8), and resuspended to 250 μL in TE. Then, 6 N NaOH was added to a final concentration of 1 N. The suspension was incubated in a water bath at 80°C for 30 min. After incubation, 1.05 mL of AB mix (0.03% aniline blue (Sinopharm Chemical Reagent), 0.18 M HCl, and 0.49 M glycine-NaOH, pH 9.5) was added, vortexed briefly, incubated at 50°C for 30 min and an additional 30 min at room temperature. Fluorescence of β-1,3-glucan was quantified using a fluorescence spectrophotometer (F-7000, Hitachi). Excitation wavelength was 400 nm and emission wavelength was 460 nm.
Results
Disruption of OCH1 in S. cerevisiae showed growth defects
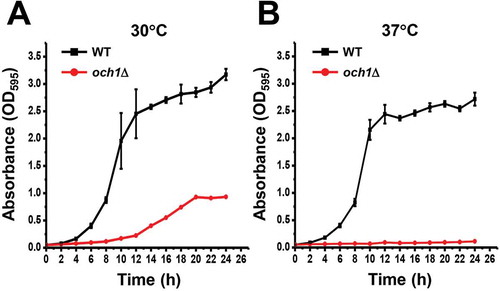
The secretory proteins expressed in S. cerevisiae are modified with mannan-type N-glycans, which cause immunogenicity in human. To overcome this hurdle, genes required for formation of yeast-specific N-glycan structures need to be disrupted for the expression of therapeutic proteins [Citation9,Citation15,Citation19]. A strain S. cerevisiae with disrupted OCH1 could eliminate outer glycan chains consisting of the α-1,6-linked poly-mannose outer chain from the core N-glycan structures. However, the disruption of OCH1 causes slow growth at permissive temperature and lethality at a higher temperature, resulting in the negative effects on the recombinant protein production. To overcome this problem, we tried to identify factors to restore the slow growth phenotypes in och1Δ cells by genetic screens. First, we disrupted OCH1 gene in our yeast strain background. Consistent with previous reports [Citation18,Citation20], the och1Δ cells showed slow growth at 30°C and 37°C ().
Figure 1. Disruption of OCH1 caused slow growth.
(A and B) Wild-type (BY4741PB; WT) and och1Δ cells were pre-cultured in SD media. Cells of 1.25 OD600 were inoculated in 25 mL of SD media and cultured at 30°C (A) or 37°C (B). The culture media were taken at indicated time and absorbance at 595 nm (A595) was measured. The values shown are means ± SE of triplicate determinants.
Construction of a PB-based mutagenesis system in budding yeast
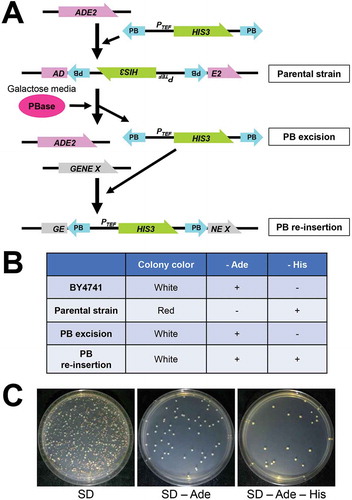
It has been reported that, the PB element can transpose in budding yeast [Citation39]. In previous studies, simple model constructs such as a plasmid carrying the URA3 gene, split with an actin intron, in which the PB element was inserted, were used to analyze transposition activity [Citation39]. Using such model systems, the hyperactive PB transposase (hyPBase) was isolated [Citation32,Citation40]. To use the PB transposon system in budding yeast, we first constructed a plasmid with a 3ʹPB-PTEF-HIS3–5ʹPB element, which contained the HIS3 gene under the TEF promoter flanked by PB inverted terminal repeats. The PB element was inserted into a TTAA sequence of the ADE2 gene in yeast (). The yeast colonies appeared as red colonies on SD plates upon disruption of the ADE2 gene. (). Also, a plasmid for PBase expression, controlled by the GAL promoter was constructed. After the expression of the PBase in galactose medium, we observed white and red colonies on SD plates ( and ). On SD −Ade plates, white colonies grew but not red ones, indicating that the 3ʹPB-PTEF-HIS3–5ʹPB element was excised from the ADE2 gene by PBase activity, and ADE2 expression was restored in the white yeast colonies. Some of the white yeast colonies grew on SD −Ade −His plates when the excised 3ʹPB-PTEF-HIS3–5ʹPB element was re-inserted into the genome (). These results indicate that, the constructed PB system worked well in budding yeast.
Figure 2. Transposition of PB element in S. cerevisiae.
(A) A schematic view of the PB transposon system. The ADE2 gene is intact in BY4741 background strains. A PB element containing the HIS3 gene under the TEF promoter (PTEF) is inserted into a TTAA sequence in ADE2 in the parental strain. By inducing PBase, whose expression is regulated by the GAL promoter, the PB element is excised from the ADE2 gene. Then, the PB is randomly re-inserted into another gene.(B) Colony color on SD plates and growth on SD −Ade and SD −His of BY4741, the parental strain, the strain after PB excision and the strain after PB re-insertion.(C) After PBase was expressed in galactose medium, about 500 cells were spread on plates. White and red colonies could be observed on SD plates. White colonies but not red ones grew on SD −Ade plates. Some of the white yeast colonies grew on SD −Ade −His plates.
A PB-based screening to isolate mutant och1Δ cells that restore the growth
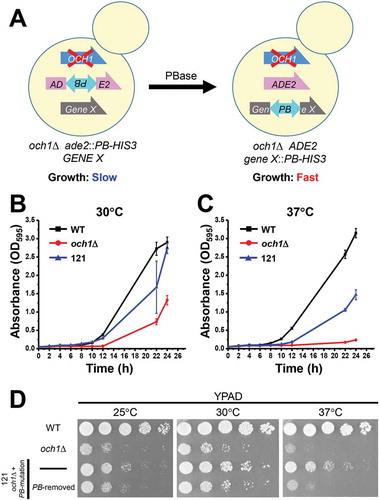
Genetic screens were performed to isolate mutant cells that rescue the slow growth phenotype in och1Δ cells (). In the preliminary experiment, the optimal time for the excision and insertion of the PB element was determined as 27 h after induction of PBase (excision rate: about 10.6%; Re-insertion rate: about 2.6%). After PBase was expressed in galactose medium for 27 h at 30°C, the yeast cells were further cultured in glucose medium at 30°C for 24 h, followed by cultured in glucose medium at 37°C for 36 h. Then, the yeast cells were plated on SD −Ade −His plates and cultured at 37°C. Three days later, a large number of colonies were observed and picked up. One mutant strain, named 121 (och1Δ + PB mutation), grew faster than parental och1Δ strain at both 30°C and 37°C ( and ). To check whether the rescue of phenotype was dependent upon PB insertion or not, the PB element was removed from the cells by the re-induction of PBase. After removal of PB insertion, the growth rate of the 121 on YPAD plates became similar to that of the parental cells (), suggesting that the phenotype of 121 was dependent upon the PB insertion.
Figure 3. Isolation of 121 mutant cells from a PB-based screen.
(A) A schematic representation of the screening. The parental strain (och1Δ ade2::PB-HIS3) was mutagenized by induction of PBase. The PB-mutagenized cells were cultured at 37°C and mutant cells that grew faster than parental cells were isolated.(B and C) Wild-type (WT), och1Δ parental, and 121 cells were precultured in SD media for 24 h. Cells (1.25 OD600) were inoculated in 25 mL of YPAD media and cultured at 30°C (B) or 37°C (C). The culture media were taken at indicated times, and the absorbance at 595 nm (A595) was measured. The values shown are means ± SE of triplicate determinants.(D) The WT, och1Δ parental, 121 cells and 121 cells with removed PB insertion site were spotted onto YPAD plates and incubated at the indicated temperatures for 2 days.
PB was inserted in the promoter region of BEM4 gene
Genomic DNA was extracted from the 121 mutant cells, and the DNA region containing the inserted PB element was amplified by PCR. Further, the PB insertion site was determined by sequencing. The PB element was integrated into the promoter region (−178 bp upstream) of the BEM4 gene (). We designed a primer set in the promoter region of BEM4 and in the PB region respectively and analyzed the PB insertion by PCR. The expected band was observed from 121, but not from the parental och1Δ cells (), indicating that PB element was inserted in the position.
Figure 4. The BEM4 gene was identified in PB-based genetic screening.
(A) A schematic view of the PB insertion site in the 121 mutant strain. The dark grey box is the BEM4 coding sequence. The black line shows the promoter and terminator regions of the BEM4 gene. The PB element was inserted at the TTAA site in the promoter region of the BEM4 gene (178 bp upstream from the start codon). A forward and a reverse primer were designed at the promoter region of BEM4 and at the PB element, respectively. The expected size of the PCR product using the primers is 442 bp.(B) Genomic DNA was extracted from och1Δ parental and 121 mutant cells. The PB inserted region was amplified by a primer set in (A) and analyzed by agarose gel electrophoresis.(C) Quantitative PCR analysis of BEM4 mRNA levels in och1Δ parental and 121 mutant cells. The TDH1 values were used to normalize the data. The relative expression was calculated by the – ΔΔCT method. The bars represent RQ (relative quantification) values ± RQmax and RQmin (error bars) of triplicate samples. A P-value was calculated from ΔCt values of each sample by two-sided Student’s t-test. *, P < 0.01.
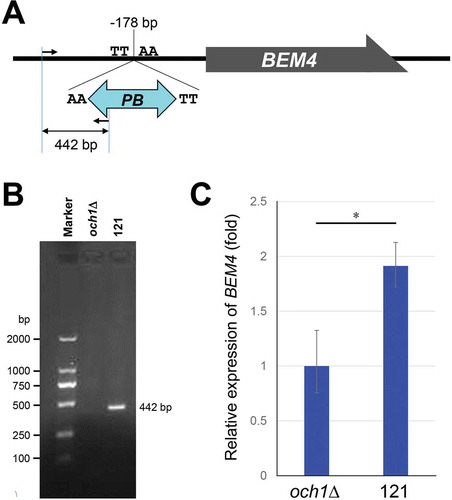
Overexpression of BEM4 rescued growth phenotypes in och1δ cells
Since the PB element was inserted into the promoter region of the BEM4 gene, it was then checked whether the phenotype observed in 121 was due to inactivation or overexpression of BEM4. We analyzed the expression level of BEM4 mRNA using quantitative PCR. The BEM4 expression was upregulated (1.9 fold) in 121 mutant cells (). The result suggests that the growth restoration in 121 cells were due to the overexpression of BEM4. To confirm the former,, we overexpressed the BEM4 gene in och1Δ cells and checked growth on the YPAD plates. Overexpression of BEM4 rescued the slow growth at 30°C, and temperature sensitivity at 37°C in och1Δ cells (). These results indicate that overexpression of BEM4 could bypass the growth defects of och1Δ cells.
Figure 5. Overexpression of BEM4 and RHO1 recovered growth defects in och1Δ cells.
(a) The WT, och1Δ parental, 121 cells, 121 cells with removed PB insertion site, and och1Δ cells carrying YEp352GAPII-BEM4 were spotted onto YPAD plates and incubated at the indicated temperatures for 2–3 days. (b) The och1Δ cells carrying YEp352GAPII, YEp352GAPII-BEM4, YEp352-CDC42 or Y26-RHO1 were spotted onto YPAD plates and incubated at the indicated temperatures for 2–3 days. (C) Colorimetric quantification of the total amount of β-1,3-glucan. The och1Δ and 121 cells were pre-cultured in YPAD media, and och1Δ carrying YEp352GAPII (+ Vector), YEp352GAPII-BEM4 (+ BEM4) or Y26-RHO1 (+ RHO1) were pre-cultured in SD – Ura, followed by culturing in SD media. 2.5 × 106 cells were harvested. The amount of β-1,3-glucan was measured with aniline blue staining and is expressed as the relative fluorescence intensity. The average fluorescence intensities of in och1Δ cells were set as 1. Mean of the relative fluorescence intensity with S.D. of triplicate measurements are plotted. The P-values was calculated by two-sided Student’s t-test. *, P < 0.01; * *, P < 0.02.
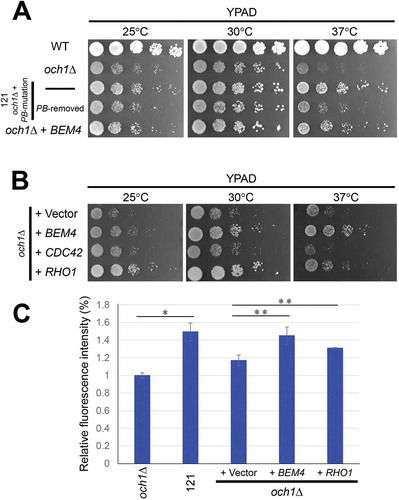
Overexpression of RHO1, a downstream gene of BEM4, partially recovered the growth defects of och1Δ cells
It has been reported that Bem4p interacts with Rho-type GTPases such as Rho1p and Cdc42p, and functions in cell wall integrity and cell polarity. BEM4 was originally isolated as multicopy suppressors of cells expressing a dominant-negative form of Rho1p and cdc42 mutant cells [Citation41,Citation42]. Cdc42p regulates cell polarity and cellular signaling [Citation43,Citation44]. Rho1p is a sensor of the cell wall integrity and is involved in cell wall biogenesis, actin organization, and polarized secretion [Citation23]. Based on the results that och1Δ cell growth was recovered by expressing of BEM4 gene, we analyzed next, the downstream genes that are regulated by Bem4p. Overexpression of RHO1, but not CDC42, partly rescued the growth phenotype in och1Δ cells ().
It has been reported that overexpression of Rho1p upregulates the β-1,3-glucan content in the yeast cell wall. To further analyze the relationship between Bem4p and Rho1p, we quantified the total amount of β-1,3-glucan by using a fluorescence spectrophotometer with aniline blue staining. Compared to och1Δ cells, the glucan contents were increased in 121 mutant cells (). The β-1,3-glucan levels were also increased in both och1Δ cells overexpressing BEM4 or RHO1, suggesting that Bem4p has a positive effect on the glucan synthesis probably through activating Rho1p. Considering both our results and previous report [Citation22], overexpression of BEM4 contributes to the upregulation of cell wall components through activation of Rho1p dependent pathway.
Discussion
Compared with classical mutagenesis, insertional mutagenesis is advantageous for identifying the gene responsible for a mutation, because common sequences in the insertional module can be used to determine the insertion site [Citation45,Citation46]. The PB derived from T. ni can transpose not only into insect cells but also in a variety of living organisms including yeasts, protozoa, plants, and vertebrates [Citation28]. Therefore, the PB transposon is widely used for genetic manipulations such as transgenesis and mutagenesis [Citation47–Citation50]. In budding yeast, PB and related piggyBat elements from the brown bat Myotis lucifugus can transpose in vitro and in vivo [Citation39,Citation51]. PB transpositions such as excision and integration have been analyzed using a simple assay system. However, there have been no reports of the use of the PB transposon for practical genetic screens in vivo in budding yeasts. To our knowledge, this is the first report of a genetic screen using the PB transposon in budding yeast. PB-based gene transfer is carried out through a “cut and paste” mechanism [Citation39]. By activation of PBase, a PB transposon is excised precisely from a donor DNA, which leaves no footprint [Citation26,Citation39]. The transposon is then inserted into another genomic DNA at the TTAA site, leading to duplication of the TTAA sequence flanking the new insertion site. In vivo mutagenesis by the PB transposon is useful in the following respects. Firstly, it is easy to mutagenize the genome just by inducing PBase, which is not harmful compared with chemical mutagens. Secondly, the gene that PB is inserted into can be easily identified by PCR. Moreover, the mutant cells can be restored to the parental strain by re-excision of the PB element.
Here, screening methods using the PB transposon in budding yeast was established. We constructed a yeast strain with a 3ʹPB-PTEF-HIS3–5ʹPB element, which was inserted in the ADE2 gene coding sequence. The PBase was expressed under a galactose-inducible promoter. To check whether our PB system worked in budding yeast, we screened for mutant cells that restore the growth defects of och1Δ cells. From the screen, the BEM4 gene was identified as a suppressor. In the 121 mutant cells, BEM4 expression was upregulated. BEM4 overexpression overcame the slow growth phenotype in och1Δ cells. This observation suggests our PB-based screening is useful for isolating genes required for phenotypes of interest in budding yeast.
Although the precise function of Bem4p is not well known, Bem4p is involved in multiple cellular signaling pathways mediated by multiple Rho-type GTPase members [Citation42,Citation52]. Rho-type GTPases including Cdc42p and Rho1p function in the establishment and maintenance of cell polarity [Citation52]. Bem4p seems to bring Rho-type GTPases and other proteins to the bud neck in the cell polarity development. It has been reported that Bem4p associates with Cdc42p and its GDP/GTP exchange factor (GEF) Cdc24p to regulate the mitogen-activated protein kinase (MAPK) pathway that controls filamentous growth [Citation53]. Overexpression of BEM4 restored the temperature sensitivity of cdc42 mutant cells [Citation42]. However, overexpression of CDC42 could not rescue the growth defects of och1Δ cells (), suggesting that Cdc42p-mediated pathway is not involved in the phenotype rescue by BEM4 expression in this study. On the other hand, overexpression of RHO1 recovered the growth defects of och1Δ cells, similar to BEM4 expression. BEM4 was originally isolated as a multicopy suppressor of cells expressing the dominant-negative Rho1p form [Citation41]. Bem4p directly interacts with Rho1p and regulates Rho1p-mediated signaling pathway. The disruption of BEM4 in yeast shows a temperature-sensitive growth phenotype. The temperature sensitivity of bem4Δ cells was partly rescued by overexpression of RHO1, ROM2 or PKC1 [Citation41]. ROM2 encodes a GEF for Rho1p, and PKC1 encodes a protein kinase C that is a downstream target of Rho1p [Citation23]. These results suggest that BEM4 is functionally relevant to RHO1. Furthermore, the glucan synthase activity, which is activated by Rho1p, was significantly reduced in bem4Δ cells [Citation41]. In addition, overexpression of RHO1 and FKS2 encoding β-1,3-glucan synthase partially restored the slow growth phenotypes in och1Δ alg3Δ double mutant cells [Citation22]. Our results suggest that Bem4p positively regulates Rho1p signaling and compensates cell wall weakness due to the OCH1-disruption probably through increasing β-glucan synthesis.
Construction of a suitable expression system is critical for the production of a therapeutic protein having human-compatible glycans in yeast. In this study, we provide evidence that enhanced expression of the Bem4p-Rho1p pathway would be useful to facilitate its future industrial applications for the production of therapeutic glycoproteins in the glycoengineered yeast cells.
Author contribution
D. M., S. B. Z., Y. S. L., and E. O. M. performed experiments and analyzed data. X. D. G. and M. F. proposed and supervised the project. D. M., E. O. M., and M. F. wrote the manuscript. All authors confirmed and edited the manuscript.
Acknowledgments
We thank Dr. Kosuke Yusa and Sanger Institute for plasmids, Ms. Lin-Xi Zhou (Jiangnan University) for the initial construction of the yeast strains, Drs. Toshihiko Kitajima, Hideki Nakanishi, Zi-Jie Li and Yun Hu (Jiangnan University) for their helpful discussion. This work was supported by grants-in-aid from the National Natural Science Foundation of China (31770853), the Young Thousand Program (to M. Fujita), the Program of Introducing Talents of Discipline to Universities (No. 111–2–06), National first-class discipline program of Light Industry Technology and Engineering (LITE2018–015), Collaborative Innovation Center of Jiangsu Modern Industrial Fermentation, and the International Joint Research Laboratory for Production of Therapeutic Glycoproteins at Jiangnan University.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Lagasse HA, Alexaki A, Simhadri VL, et al. Recent advances in (therapeutic protein) drug development. F1000Res. 2017;6:113.
- Jenkins N, Murphy L, Tyther R. Post-translational modifications of recombinant proteins: significance for biopharmaceuticals. Mol Biotechnol. 2008 Jun;39:113–118.
- Pandhal J, PC W. N-linked glycoengineering for human therapeutic proteins in bacteria. Biotechnol Lett. 2010 Sep;32:1189–1198.
- Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049.
- Tannous A, Pisoni GB, Hebert DN, et al. N-Linked Sugar-Regulated Protein Folding and Quality Control in the ER Semin Cell Dev Biol. 2015 May;41:79–89.
- Sethuraman N, Stadheim TA. Challenges in therapeutic glycoprotein production . Curr Opin Biotechnol. 2006 Aug;17:341–346.
- Kim JY, Kim Y-G, Lee GM. CHO cells in biotechnology for production of recombinant proteins: current state and further potential. Appl Microbiol Biotechnol. 2012 Feb;93:917–930.
- Walsh G. Biopharmaceutical benchmarks 2014. Nat Biotechnol. 2014 Oct;32:992–1000.
- Chiba Y, Suzuki M, Yoshida S, et al. Production of human compatible high mannose-type (Man5GlcNAc2) sugar chains in Saccharomyces cerevisiae. J Biol Chem. 1998 Oct;9(273):26298–26304.
- Cregg JM, Cereghino JL, Shi J, et al. Recombinant protein expression in Pichia pastoris . Mol Biotechnol. 2000;16:23–52.
- Chiba Y, Sakuraba H, Kotani M, et al Production in yeast of alpha-galactosidase A, a lysosomal enzyme applicable to enzyme replacement therapy for Fabry disease. Glycobiology. 2002;12:821–828.
- Hamilton SR, Bobrowicz P, Bobrowicz B, et al Production of complex human glycoproteins in yeast. Science. 2003 Aug;301(5637):1244–1246.
- Kuroda K, Kobayashi K, Tsumura H, et al. Production of Man5GlcNAc2-type sugar chain by the methylotrophic yeast Ogataea minuta. FEMS Yeast Res. 2006;6:1052–1062.
- Ohashi T, Nakakita S, Sumiyoshi W, et al. Production of heterologous glycoproteins by a glycosylation-defective alg3och1 mutant of Schizosaccharomyces pombe. J Biotechnol. 2010;150:348–356.
- Wildt S, Gerngross TU. The humanization of N-glycosylation pathways in yeast. Nat Rev Microbiol. 2005 Feb;3:119–128.
- Jacobs PP, Geysens S, Vervecken W, et al Engineering complex-type N-glycosylation in Pichia pastoris using GlycoSwitch technology. Nat Protoc. 2009;4:58–70.
- Jigami Y. Yeast glycobiology and its application. Biosci Biotechnol Biochem. 2008 Mar;72:637–648.
- Nakayama K, Nagasu T, Shimma Y, et al. OCH1 encodes a novel membrane bound mannosyltransferase: outer chain elongation of asparagine-linked oligosaccharides. EMBO J. 1992;11:2511–2519.
- Jacobs PP, Callewaert N. N-glycosylation engineering of biopharmaceutical expression systems. Curr Mol Med. 2009 Sep;9:774–800.
- Nagasu T, Shimma Y, Nakanishi Y, et al. Isolation of new temperature-sensitive mutants of Saccharomyces cerevisiae deficient in mannose outer chain elongation. Yeast. 1992;8:535–547.
- Abe H, Takaoka Y, Chiba Y, et al. Development of valuable yeast strains using a novel mutagenesis technique for the effective production of therapeutic glycoproteins. Glycobiology. 2009;19:428–436.
- Xu S, Zhang G-Y, Zhang H, et al. Effects of Rho1, a small GTPase on the production of recombinant glycoproteins in Saccharomyces cerevisiae. Microb Cell Fact. 2016 Oct 21;15:179.
- Levin DE. Regulation of cell wall biogenesis in Saccharomyces cerevisiae: the cell wall integrity signaling pathway. Genetics. 2011 Dec;189:1145–1175.
- Abe H, Fujita Y, Chiba Y, et al. Upregulation of genes involved in gluconeogenesis and the glyoxylate cycle suppressed the drug sensitivity of an N-glycan-deficient Saccharomyces cerevisiae . Mutant Biosci Biotechnol Biochem. 2009;73:1398–1403.
- Cary LC, Goebel M, Corsaro BG, et al. Transposon mutagenesis of baculoviruses: analysis of Trichoplusia ni transposon IFP2 insertions within the FP-locus of nuclear polyhedrosis viruses. Virology. 1989;172:156–169.
- Fraser MJ, Cary L, Boonvisudhi K, et al Assay for movement of lepidopteran transposon IFP2 in insect cells using a baculovirus genome as a target DNA. Virology. 1995 Aug;20(2):397–407.
- Thibault ST, Singer MA, Miyazaki WY, et al. A complementary transposon tool kit for Drosophila melanogaster using P and piggyBac. Nat Genet. 2004;36:283–287.
- Yusa K. piggyBac Transposon. Microbiol Spectr. Apr 32015;3. MDNA3–0028–2014.
- Longtine MS, McKenzie A 3rd, Demarini DJ, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in saccharomyces cerevisiae. Yeast. 1998;14:953–961.
- Taxis C, Knop M. System of centromeric, episomal, and integrative vectors based on drug resistance markers for Saccharomyces cerevisiae. Biotechniques. 2006 Jan;40:73–78.
- Mohamed LA, Tachikawa H, Gao XD, et al. Yeast cell-based analysis of human lactate dehydrogenase isoforms. J Biochem. Dec 2015;158:467–476.
- Yusa K, Zhou L, Li MA, et al A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci. 2011 Jan;25(108):1531–1536.
- Yusa K, Rashid ST, Strick-Marchand H, et al Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent. Stem Cells Nature. 2011 Oct;12(478):391–394.
- Fujita M, Yoko OT, Jigami Y. Inositol deacylation by Bst1p is required for the quality control of glycosylphosphatidylinositol-anchored proteins. Mol Biol Cell. 2006 Feb;17:834–850.
- Liu YS, Guo XY, Hirata T, et al. N-Glycan-dependent protein folding and endoplasmic reticulum retention regulate GPI-anchor processing. J Cell Biol. 2018 Feb;5(217):585–599.
- Matabaro E, He Z, Liu YS, et al Molecular switching system using glycosylphosphatidylinositol to select cells highly expressing recombinant proteins. Sci Rep. 2017 Jun;22(7):4033.
- Rong Y, Nakamura S, Hirata T, et al Genome-wide screening of genes required for glycosylphosphatidylinositol biosynthesis. PLoS One. 2015;10(e0138553).
- Sekiya-Kawasaki M, Abe M, Saka A, et al. Dissection of upstream regulatory components of the Rho1p effector, 1,3-beta-glucan synthase, in Saccharomyces cerevisiae Genetics.Oct 2002 ;162:663–676.
- Mitra R, Fain-Thornton J, Craig NL. piggyBac can bypass DNA synthesis during cut and paste transposition. EMBO J. 2008 Apr;09(27):1097–1109.
- Li X, Burnight ER, Cooney AL, et al piggyBac transposase tools for genome engineering. Proc Natl Acad Sci. 2013 Jun;18(25):E2279–2287.
- Hirano H, Tanaka K, Ozaki K, et al ROM7/BEM4 encodes a novel protein that interacts with the Rho1p small GTP-binding protein in Saccharomyces cerevisiae . Mol Cell Biol. 1996 Aug;16(8):4396–4403.
- Mack D, Nishimura K, Dennehey BK, et al. Identification of the bud emergence gene BEM4 and its interactions with rho-type GTPases in Saccharomyces cerevisiae. Mol Cell Biol.. 1996;16:4387–4395.
- Bendezú FO, Martin SG. Cdc42 explores the cell periphery for mate selection in fission yeast. Curr Biol.. 2013 Jan;23(1):42–47.
- Melendez J, Grogg M, Zheng Y. Signaling role of Cdc42 in regulating mammalian physiology. J Biol Chem. 2011 Jan;286(286):2375–2381.
- Horecka J, Jigami Y. Identifying tagged transposon insertion sites in yeast by direct genomic sequencing. Yeast. 2000 Jul;16:967–970.
- Ross-Macdonald P, Sheehan A, Friddle C, et al Transposon mutagenesis for the analysis of protein production, function, and localization . Methods Enzymol. 1999;303:512–532.
- Ding S, Wu X, Li Get al Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice . Cell. Aug 2005;122(3):473–483.
- Handler AM, Harrell RA 2nd. Germline transformation of Drosophila melanogaster with the piggyBac transposon vector. Insect Mol Biol. 1999 Nov;8:449–457.
- Rad R, Rad L, Wang W, et al PiggyBac transposon mutagenesis: a tool for cancer gene discovery in mice. Science. 2010 Nov;330(6007):1104–1107.
- Li J, Zhang J-M, Li X, et al. A piggyBac transposon-based mutagenesis system for the fission yeast Schizosaccharomyces pombe. Nucleic Acids Res. 2011;39:e40.
- Mitra R, Li X, Kapusta A, et al Functional characterization of piggyBat from the bat Myotis lucifugus unveils an active mammalian DNA transposon. Proc Natl Acad Sci. 2013 Jan;110(1):234–239.
- Drees BL, Sundin B, Brazeau E, et al. A protein interaction map for cell polarity development. J Cell Biol. 2001 Aug;154(3):549–576.
- Pitoniak A, Chavel CA, Chow J, et al Cdc42p-interacting protein Bem4p regulates the filamentous-growth mitogen-activated protein kinase pathway. Mol Cell Biol. 2015 Jan;35(2):417–436.