
ABSTRACT
To achieve rhamnose-inducible efficient protein expression in Bacillus subtilis, we assembled the strong promoters of B. subtilis cdd and ylbP genes and the regulatory region (PrhaEW) of B. subtilis rhaEWRBMA operon, whose transcription is induced by rhamnose and repressed by glucose, to produce various hybrid constructs. These constructs were evaluated using B. subtilis strains carrying a fusion of each construct to the gene encoding a mutated green fluorescent protein in the chromosome. When these strains were cultivated in the presence of glucose or rhamnose, the strain carrying a fusion of a partial PrhaEW region, lacking the intrinsic Shine-Dalgarno (SD) sequence, and the ylbP SD sequence most strictly controlled the promoter activity depending on sugar species. Moreover, the strain carrying a fusion of the cdd core promoter and the ylbP SD sequence showed the highest promoter activity when it was cultivated in the presence of glucose until the late stationary phase.
Abbreviations: RNAP: RNA polymerase; cre: catabolite-responsive element; SD: Shine-Dalgarno; PAGE: polyacrylamide gel electrophoresis; GFP: green fluorescent protein; OD600: optical density at 600 nm; LB: Luria-Bertani; a.u.: arbitrary unit; SDS: sodium dodecyl sulfate.
Graphical Abstract
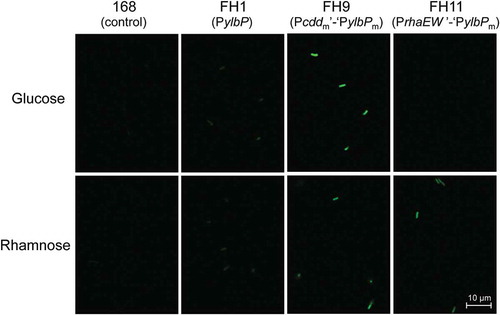
Fluorescence micrographs of B. subtilis reporter strains carrying each egfp fusion and strain 168 (control), which had been cultivated in the presence of glucose or rhamnose.
Many of the commercially available enzymes are produced by the genus Bacillus and related genera, and most of these enzymes are intrinsic and naturally secreted in the culture medium, e.g., alkaline proteases and amylases [Citation1,Citation2]. Some species of these bacteria are also used as hosts for producing recombinant proteins such as enzymes for sugar degradation [Citation3] and antibiotic biosynthesis [Citation4]. Bacillus subtilis is a soil-dwelling bacterium that has been extensively and closely studied as a Gram-positive model bacterium. Next to Escherichia coli, B. subtilis is one of the most frequently used bacterial hosts to produce recombinant proteins, because it grows quickly in a simple and inexpensive medium to achieve high cell density, and its genetic engineering techniques are well established [Citation5,Citation6]. While the Gram-negative bacterium E. coli contains lipopolysaccharides referred to as endotoxins, which cause fever in humans and other mammals, B. subtilis does not possess such endotoxins and is regarded as a GRAS (generally recognized as safe) organism by the U.S. Food and Drug Administration. Therefore, from the aspect of excluding toxic substances, B. subtilis is more favorable than E. coli for protein production in the food, feed, and pharmaceutical industries. Moreover, in contrast to E. coli, B. subtilis natively has high secretory systems, and thus, if the target recombinant protein can be secreted through any of these systems by adducting a signal peptide, it will simplify the protein recovery and purification steps [Citation2,Citation7].
Protein production in bacterial cells is mainly controlled at the transcriptional level, in which promoters are an important factor that determines the transcript amount. Depending on the processes of cellular differentiation and the circumstances around the cells, RNA polymerase (RNAP) associated with a certain sigma (σ) factor binds to a specific promoter sequence to initiate transcription of the gene whose expression is required. Each σ factor recognizes a unique consensus sequence (core promoter), thereby directing the RNAP complex to the transcription initiation of a specific set of genes. In B. subtilis, σA participates in the transcription initiation of most of the housekeeping genes, and the consensus sequence recognized by σA (5ʹ-TTGACA-17 nt-TATAAT-3ʹ) is usually located 10 and 35 bp upstream of the transcription start site [Citation8]. On the other hand, σB is activated in response to multiple physical stress stimuli as well as energy starvation conditions, generally during the late logarithmic and stationary phases, and the target genes of σB involve the general stress regulon comprised of approximately 200 genes. The consensus sequence of σB-dependent promoters (5ʹ-DGWKTND-12 ~ 15 nt-GGRWAW-3ʹ; D, W, K, R, and N stand for A/G/T, A/T, G/T, A/G, and A/C/G/T, respectively) is also located around 10 and 35 bp upstream of the transcription start site [Citation9].
Transcriptional factors bind to a specific site(s) around the promoter of their target genes, by which they promote (as an activator) or inhibit (as a repressor) the RNAP complex binding to the promoter, alternatively interrupting (as a repressor) the transcript elongation by RNAP. The promoters that have been used for heterologous protein expression in bacterial cells are largely classified into three types (constitutive, inducer-specific, and auto-inducible ones), and most inducible promoters are controlled by transcriptional factors [Citation5,Citation7]. To date, several inducer-specific promoters, such as the IPTG (isopropyl β-D-1-thiogalactopyranoside)-inducible spac promoter and the xylose-inducible xyl promoter, have been used in the B. subtilis host cells [Citation5,Citation7]; however, the variety of such inducer compounds is limited, and thus a novel inducible promoter system with a different inducer compound in B. subtilis is being requested as well as higher induced promoter activity than ever.
The B. subtilis cdd promoter, also known as the P43 promoter, is for the gene encoding cytidine deaminase, and this promoter has been representatively used as a constitutive promoter in B. subtilis cells [Citation10,Citation11]. The auto-inducible promoter is regarded to increase its activity as the culture time proceeds via the accumulation of signaling molecules [Citation5,Citation7,Citation12]. It was recently reported that the B. subtilis ylbP promoter of the gene encoding a putative N-acetyltransferase was apparently an auto-inducible promoter and exhibited remarkably high activity during the stationary phase, although its induction mechanism remained obscure [Citation11]. Both cdd and ylbP promoter regions contain the core promoter sequence recognized by σA. In addition, the cdd promoter region contains another core promoter sequence that σB recognizes [Citation10]. Since the σA- and σB-recognized core promoter sequences of the cdd gene overlap, we handled them together and used the term “the cdd core promoter” in this study ().
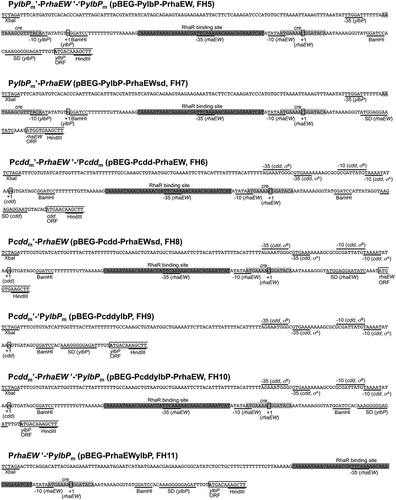
Figure 1. Organization of the hybrid protein expression systems designed in this study.
The −35 and −10 sequences of core promoters of the ylbP, cdd, and rhaEW genes are indicated by solid lines, and the transcription start sites (+ 1) from each promoter are enclosed in rectangles. The Shine-Dalgarno (SD) sequences and the restriction sites are underlined. The partial coding region of each gene is indicated by lines. The direct repeat in the RhaR binding site and the cre sequence for CcpA/P-Ser-HPr binding are indicated by dark gray and light gray shades, respectively. The names of plasmids and B. subtilis reporter strains carrying each construct are indicated in parentheses after the construct’s compositions ().
In our previous study, the transcriptional regulatory mechanism of the B. subtilis rhaEWRBMA operon, which is involved in the catabolism of L-rhamnose (hereafter referred to as rhamnose), was closely analyzed [Citation13]. B. subtilis catabolizes rhamnose via phosphorylation, and the relevant enzymes and a DeoR-type transcriptional factor (RhaR) are encoded in this operon. RhaR regulates the rhaEW promoter in concert with CcpA, a global transcriptional factor for carbon catabolite control. The core promoter sequence in the rhaEW regulatory region (PrhaEW region) is also recognized by σA. In the absence of rhamnose, RhaR binds to a specific site containing a unique direct repeat in the PrhaEW region to repress the operon, and RhaR binding is inhibited by rhamnulose-1-phosphate, an intermediate of the rhamnose catabolism, leading to derepression of the operon () [Citation13].
The CcpA-mediated carbon catabolite control is conserved in many low-GC Gram-positive bacteria. When certain carbohydrates, such as glucose, are transported into the cells through the phosphoenolpyruvate-dependent phosphotransferase system, the intracellular concentration of fructose-1,6-bisphosphate increases, by which HPr kinase/phosphorylase is activated and phosphorylates the specific serine residue of the HPr or Crh protein. CcpA is complexed with either of these seryl-phosphorylated proteins (P-Ser-HPr and P-Ser-Crh) and binds to catabolite-responsive elements (cres) of the target genes, leading to their repression or activation [Citation14]. In the case of the rhaEWRBMA operon, the CcpA complex binds to the cre in the PrhaEW region, leading to catabolite repression of the operon () [Citation13].
Notably, the regulatory mechanism of this B. subtilis operon through the two transcriptional factors is totally different from that of the rhamnose-catabolic gene cluster in E. coli [Citation15]. Although the E. coli regulatory mechanism is utilized for the protein expression system strictly inducible by rhamnose and repressible by glucose in E. coli cells, this expression system cannot be applicable to the B. subtilis host cells due to difference in the regulatory mechanism [Citation16]. Because rhamnose is less expensive and nontoxic to the bacterial host cells as wells as humans, it is expected to be very useful as an inducer compound for the B. subtilis protein expression system, especially in the case of producing the proteins for food processing and as pharmaceutical agents.
In this study, in order to develop a novel protein expression system in B. subtilis cells that is highly efficient and controllable by glucose and rhamnose, we combined the cdd and ylbP promoter regions including the highly efficient promoters, with the PrhaEW region covering the binding sites of the two transcriptional factors (RhaR and CcpA) to produce various types of hybrid protein expression constructs. These constructs were fused to the gene encoding a mutated green fluorescent protein (EGFP) as a reporter, and each egfp fusion was introduced into the B. subtilis chromosome. By culturing the resultant reporter strains in the presence of glucose or rhamnose, followed by quantification of their fluorescence intensities, the expression strength and induction specificity of these hybrid constructs were evaluated. As a result, the strain carrying a fusion composed of a partial PrhaEW region, which contains the intrinsic core promoter and the RhaR and CcpA binding sites but lacks the intrinsic Shine-Dalgarno (SD) sequence, and the SD sequence of the ylbP gene most strictly controlled the promoter activity depending on the sugar species (repression by glucose and induction by rhamnose) and showed significantly high rhamnose-induced promoter activity. Unexpectedly, the strain carrying a fusion composed of the cdd core promoter and the ylbP SD sequence showed the highest promoter activity when it was cultivated in the glucose-containing medium until the late stationary phase, among all of the reporter strains grown under either culture condition. Therefore, we consider that the former construct is suitable for strictly controllable protein expression if B. subtilis host cells are cultivated in the presence of glucose or rhamnose and that the latter construct fits for a high level of protein expression when the host cells are cultivated in the glucose-containing medium until the stationary phase.
Materials and methods
DNase I footprinting analysis
The recombinant B. subtilis CcpA and HPr proteins were prepared and purified, and then the Ser-46 of HPr was phosphorylated to form P-Ser-HPr using the recombinant B. subtilis HPr kinase/phosphorylase as reported previously [Citation17]. The phosphorylation efficiency was estimated to be ca. 80%.
DNase I footprinting analysis was performed according to the procedure previously reported [Citation18]. The DNA probe for the footprinting was prepared by PCR with the genomic DNA of B. subtilis strain 168 () as the template and primer pair PylbP_XF/PylbP_BR (). Prior to PCR amplification, the 5ʹ terminus of only one of the primers was labeled with [γ-32P]ATP (PerkinElmer, Waltham, MA, USA), using a MEGALABEL kit (TaKaRa Bio, Shiga, Japan). The DNA probe (0.04 pmol), labeled at the 5ʹ end, was mixed with CcpA and P-Ser-HPr to obtain a DNA-protein complex, which was then partially digested with DNase I (TaKaRa Bio) in 50 µL of a reaction mixture and subjected to urea-polyacrylamide gel electrophoresis (urea-PAGE) with sequencing ladders prepared using each of the 5ʹ-end-labeled primers and a non-labeled template DNA corresponding to the DNA probe. The autoradiograms were obtained using a Typhoon 9400 Variable Mode Imager (GE Healthcare UK Ltd., Little Chalfont, Buckinghamshire, England).
Table 1. B. subtilis strains used in this study.
Table 2. Oligonucleotide primers used in this study.
Construction of plasmids and B. subtilis strains
A plasmid pGFP, which carries the coding sequence of the wild-type green fluorescent protein (GFP) from Aequorea victoria, was a product of Clontech purchased from TaKaRa Bio. Site-directed mutagenesis of the GFP coding sequence was performed to introduce two amino acid substitutions (F64L and S65T) into the GFP protein, thereby substantially enhancing its fluorescence intensity [Citation19]. A two-step overlapping PCR method was employed for the mutagenesis, in which primer pairs of GFP_HF/GFP_F64L_S65T_R and GFP_F64L_S65T_F/GFP_SR () with the pGFP plasmid as the template and ExTaq DNA polymerase (TaKaRa Bio) were used to amplify the 5ʹ- and 3ʹ-parts of the mutated GFP (EGFP) coding sequence (egfp), respectively. These two fragments were combined in a reaction mixture containing ExTaq DNA polymerase and deoxynucleoside triphosphates without any primer oligonucleotide. The resultant full-length egfp was amplified with a primer pair of GFP_HF/GFP_SR and then cloned into a pCR2.1 TOPO vector (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) to produce a plasmid pCR-EGFP.
A plasmid pCRE-PdhbA was constructed by integrating the promoter region of the B. subtilis dhbA gene into the pCRE-test2 vector [Citation20] that had been treated with XbaI and BamHI (K. Hirooka, unpublished). pCRE-PdhbA was treated with HindIII and SacI, by which the partial lacZ gene was removed, and then ligated with the egfp fragment that had been excised from pCR-EGFP by HindIII and SacI digestion, resulting in a plasmid pBEG-PdhbA.
The promoter regions of the B. subtilis ylbP and cdd genes were amplified by PCR with the genomic DNA of strain 168 and primer pairs of PylbP_XF/PylbP_HR and Pcdd_XF/Pcdd_HR (). To introduce a BamHI site into each of the ylbP and cdd promoter regions, PCR was performed with the genomic DNA of strain 168 and primer pairs of PylbP_XF/PylbP_HBR and Pcdd_XF/Pcdd_HBR (). The four obtained fragments were trimmed with XbaI and HindIII digestion and respectively cloned into the pBEG-PdhbA vector that had been treated with the same restriction enzymes, resulting in plasmids pBEG-PylbP_if, pBEG-Pcdd_if, pBEG-PylbP_m, and pBEG-Pcdd_m.
Two DNA fragments, which cover the intrinsic promoter and the binding sites of transcriptional regulators (RhaR and CcpA), without/with the SD sequence of the B. subtilis rhaEWRBMA operon, were amplified by PCR with the genomic DNA of strain 168 and primer pairs of PrhaEWi_BF/PrhaEWi_BR and PrhaEWi_BF/PrhaEWi_HR (). The obtained fragments were trimmed by BamHI or BamHI/HindIII and cloned into the pBEG-PylbP_m and pBEG-Pcdd_m vectors that had been treated with the same restriction enzyme(s), resulting in plasmids pBEG-PylbP-PrhaEW, pBEG-Pcdd-PrhaEW, pBEG-PylbP-PrhaEWsd, and pBEG-Pcdd-PrhaEWsd ().
The DNA fragment corresponding to the cdd promoter region with the additional BamHI site was prepared by PCR as mentioned above, digested by XbaI and BamHI, and then ligated with the pBEG-PylbP_m vector that had been treated with the same restriction enzymes to produce a plasmid pBEG-PcddylbP. In this plasmid, the cdd core promoter (−35 and −10 sequences) and the ylbP SD sequence were arranged in sequence. The DNA fragment corresponding to the rhaEW regulatory region without the SD sequence was prepared by PCR as mentioned above, trimmed with BamHI digestion, and then inserted into the pBEG-PcddylbP vector at the same restriction site to produce a plasmid pBEG-PcddylbP-PrhaEW ().
A DNA fragment that corresponds to the rhaEW regulatory region without the SD sequence and carries XbaI and BamHI sites at the 5ʹ and 3ʹ ends, respectively, was amplified by PCR with the genomic DNA of strain 168 and primer pair of PrhaEW_XF/PrhaEWi_BR (), followed by trimming with XbaI and BamHI digestion and cloning into the pBEG-PylbP_m vector that had been treated with the same restriction enzymes, resulting in a plasmid pBEG-PrhaEWylbP ().
The 11 plasmids (pBEG-PylbP_if, pBEG-Pcdd_if, pBEG-PylbP_m, pBEG-Pcdd_m, pBEG-PylbP-Prha-EW, pBEG-Pcdd-PrhaEW, pBEG-PylbP-PrhaEWsd, pBEG-Pcdd-PrhaEWsd, pBEG-PcddylbP, pBEG-PcddylbP-PrhaEW, and pBEG-PrhaEWylbP) were linearized by PstI digestion, and each of them was integrated into the amyE locus of B. subtilis strain 168 through double-crossover transformation to obtain chloramphenicol resistance, which resulted in strains FH1–FH11 (, ).
Cultivation of B. subtilis strains and measurement of their fluorescence intensity
B. subtilis strains carrying each egfp fusion and B. subtilis strain 168 as a control were pregrown at 30°C overnight on tryptose blood agar base (Difco, Becton, Dickinson and Company, Franklin Lakes, NJ, USA) plates supplemented with 0.18% glucose, which contained chloramphenicol (5 μg/mL) or not according to the drug resistance. The cells of each strain were inoculated into 25 mL of a minimal medium [Citation21] supplemented with a mixture of 16 amino acids (glutamine, histidine, tyrosine, and asparagine were omitted) [Citation22] (MM+16aa medium) in a 200 mL Erlenmeyer flask to give an optical density at 600 nm (OD600) of 0.05 and then incubated at 37°C with reciprocal shaking (120 rpm). This medium contained glucose at 25 mM as a carbon source. The cells were cultivated in a similar medium, in which the 25 mM glucose was replaced with 25 mM rhamnose. As for strain FH11, the cells were also cultivated in the minimal medium containing 25 mM glucose or 25 mM rhamnose, in the coexistence of 5 mM IPTG or 25 mM xylose, as well as in a minimal medium containing 25 mM xylose as a sole carbon source. Moreover, the cells of strains FH1–FH4 were cultivated in a Luria-Bertani (LB) medium [Citation23].
When the OD600 reached 1.0, which corresponds to the late logarithmic phase, and after the cell growth reached the late stationary phase (18 h after the inoculation), the cells in a 2 mL aliquot of each culture were collected by centrifugation (17,400 x g, 4°C, 5 min), and the supernatant was removed. The cell pellets were resuspended in Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, and 1 mM dithiothreitol, pH 7.0) so that the OD600 of each suspension was adjusted to be nearly 2.0. Subsequently, 200 µL aliquots of the suspensions were transferred to wells of a flat-bottomed microtiter plate, and the fluorescence intensity was measured using the Typhoon 9400 Variable Mode Imager (λex = 488 nm and λem = 520 nm). The fluorescence intensity values (a.u., arbitrary unit) were normalized by dividing them by the OD600 values of the corresponding suspensions.
Sodium dodecyl sulfate (SDS)-PAGE analysis
The cells of each B. subtilis strain, which had been cultivated in 25 mL of the minimal medium containing glucose or rhamnose until the late stationary phase, were harvested by centrifugation (3,000 x g, 4°C, 10 min) and washed with Z buffer. Each of the cell pellets was resuspended in 5 mL of Z buffer containing 10 µg/mL of lysozyme, followed by incubation at 37°C for 20 min. Afterward, the cells were completely disrupted by sonication, and then the supernatant was recovered as a crude lysate by centrifugation (17,400 x g, 4°C, 30 min). The protein concentration of each lysate was determined by the Bradford method with bovine serum albumin as the standard, using a protein assay kit (Bio-Rad, Hercules, CA, USA). Equal amounts (10 µg) of the total proteins in the lysates were subjected to SDS-PAGE with Coomassie Brilliant Blue staining.
Confocal fluorescence microscopy
The cells of each B. subtilis strain, which had been cultivated and collected as those used in the SDS-PAGE analysis, were resuspended in Z buffer so that the OD600 of the suspension was adjusted to be nearly 1.0. Fluorescence images of the cells in each suspension were taken using a Leica TCS-SPE DMI4000B system (Leica Microsystems, Wetzlar, Germany). Wavelengths of 488 nm were used for the fluorescence excitation of EGFP, and its fluorescence emission was collected from 500–600 nm.
Results
Verification of CcpA/P-Ser-HPr binding to the cre in the ylbP regulatory region
To achieve high-level and rhamnose-inducible protein expression in B. subtilis, we attempted to construct a hybrid protein expression system in which a highly efficient promoter and the regulatory region of the B. subtilis rhamnose-catabolic operon (rhaEWRBMA operon) are combined in sequence. The rhaEWRBMA operon is regulated through two transcriptional factors (RhaR and CcpA), and its regulatory region (PrhaEW) contains the binding sites of both transcriptional factors as well as the core promoter (−35 and −10 sequences). In the absence of rhamnose, RhaR binds to the specific site in the PrhaEW region to repress the operon, and RhaR binding is inhibited by rhamnulose-1-phosphate, an intermediate of rhamnose catabolism, leading to derepression of the operon. On the other hand, in the presence of certain energetically favorable carbohydrates such as glucose, CcpA complexed with P-Ser-HPr binds to the cre in the PrhaEW region, leading to catabolite repression of the operon [Citation13].
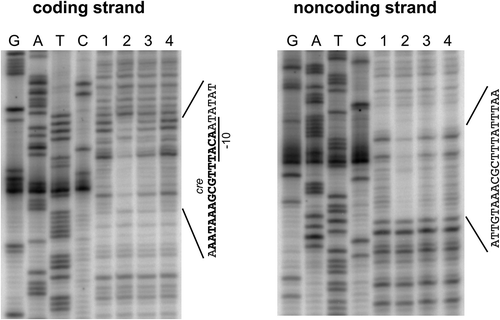
For a highly efficient promoter component, we selected promoters of the B. subtilis cdd and ylbP genes because the cdd promoter is commonly used as a constitutive promoter in B. subtilis [Citation10,Citation11], and the ylbP promoter has been reported to be highly efficient in the stationary phase of B. subtilis cell growth [Citation11]. It has also been reported that the ylbP expression is under catabolite repression [Citation24], and a putative cre (AATAAAGCGTTTACA) was found in the ylbP promoter region (), which corresponds to the consensus sequence of one subgroup of cres (WTGAAARCGYTTWNN; W, R, Y, and N stand for A/T, A/G, C/T, and A/C/G/T, respectively) [Citation14]. Since the binding of the CcpA/P-Ser-HPr complex to this candidate cre had not been experimentally confirmed, we performed the DNase I footprinting analysis by using the DNA probe corresponding to the ylbP promoter region with the 5ʹ-short part of the ylbP coding sequence (bases −140 to 105; base 1 is the transcription start base) and recombinant CcpA and P-Ser-HPr proteins, which were prepared as reported previously [Citation17]. The CcpA/P-Ser-HPr complex, composed of two copies of each protein, was formed upon their addition at an equimolar ratio to the reaction mixture; the proportion of phosphorylated HPr was 80% of the total HPr protein. As a result, a region covering the candidate cre was protected against DNase I upon the CcpA/P-Ser-HPr addition (bases -24 to −3 of the coding strand and bases −26 to −7 of the noncoding strand), indicating that the catabolite repression of the ylbP promoter is exerted through the CcpA/P-Ser-HPr binding to the true cre ( and ).
Figure 2. DNase I footprinting analysis to confirm binding of the CcpA/P-Ser-HPr complex to the cre in the ylbP promoter region.
A 5ʹ-end-32P-labeled DNA probe (0.04 pmol), corresponding to the region around the ylbP promoter, was incubated with/without the combination of CcpA and P-Ser-HPr proteins in the reaction mixture (50 µL), followed by partial digestion with DNase I and urea-PAGE. The products of the mixture containing CcpA at 8.0 µM and HPr (phosphorylated and non-phosphorylated forms) at 10 µM and of the mixture containing CcpA at 4.0 µM and HPr at 5.0 µM, each as a monomer, were applied to lanes 2 and 3; the mixtures were assembled to contain the CcpA/P-Ser-HPr heterotetramer at 4.0 µM and 2.0 µM, respectively. The product of the mixture containing no recombinant protein was applied to lanes 1 and 4. To lanes G, A, T, and C, the products of the dideoxy sequencing reactions with the same 5ʹ-labeled primers were applied. The regions protected by the CcpA/P-Ser-HPr complex are indicated at the right of each panel. The −10 sequence of the ylbP core promoter is underlined, and the cre sequence is shown in boldface type.
Construction of hybrid expression systems controllable by glucose and rhamnose
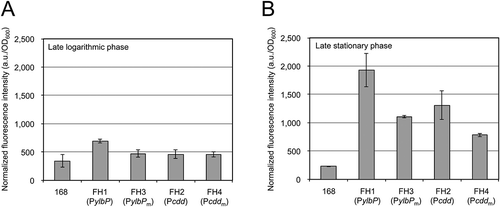
As the first step in construction of the hybrid protein expression systems, we fused each of the ylbP promoter region (PylbP, −140 to 34) and the cdd promoter region (Pcdd, −123 to 48) to the gene encoding the GFP mutant carrying the F64L and S65T substitutions (EGFP) so that the egfp coding sequence was fused in-frame to the 5ʹ-short fragment of the ylbP or cdd coding sequence. We also prepared PylbP and Pcdd derivatives (PylbPm and Pcddm), in which a BamHI site was introduced between the core promoter and SD sequence of each gene, and the PylbPm and Pcddm fragments were similarly fused to the egfp gene. Each egfp fusion was integrated into the amyE locus on the chromosome of B. subtilis strain 168 to produce four reporter strains (FH1–FH4) (). These strains were cultivated in LB medium, and then their fluorescence intensities were quantified. In the late logarithmic phase (OD600 = 1), strain FH1 (PylbP-egfp) showed a fluorescence intensity slightly higher than that of strain 168 (control), although those of the other three reporter strains (FH2, FH3, and FH4) remained as low as the control’s (). In the late stationary phase (18 h after inoculation), all four reporter strains showed distinctly higher fluorescence intensities than the control strain did, and the fluorescence intensities of strains FH3 (PylbPm-egfp) and FH4 (Pcddm-egfp) were somewhat lower than those of strains FH1 (PylbP-egfp) and FH2 (Pcdd-egfp), respectively, indicating that introduction of the BamHI site affected each promoter activity to some extent ().
Figure 3. Fluorescence intensities of B. subtilis reporter strains carrying each egfp fusion.
Each reporter strain was cultivated in LB medium at 37°C with shaking until the OD600 reached 1.0 (A) or for 18 h after inoculation (B), corresponding to the late logarithmic and the late stationary phases, respectively. The cells of each strain were harvested and resuspended in the buffer, and then the fluorescence intensity of each suspension was measured as described in “Materials and methods.” Strain 168 was used as a negative control. The experiments of cell cultivation and fluorescence measurement were repeated at least two times. The fluorescence intensity values (a.u.) were normalized by dividing them by the OD600 values of the corresponding cell suspensions, and bars and error bars in the graphs indicate the mean values and standard deviations (n = 2), respectively.
Next, the PrhaEW (−70 to 44) fragment and its truncated form lacking the SD sequence (PrhaEW’, −70 to 24) were respectively inserted into the PylbPm-egfp and Pcddm-egfp fragments to produce four egfp fusions, in which the rhaEW regulatory region is placed downstream of the ylbP or cdd core promoter and the egfp transcript is translated from the SD sequence of the ylbP (PylbPm’-PrhaEW’-‘PylbPm-egfp), cdd (Pcddm’-PrhaEW’-‘Pcddm-egfp), or rhaEW (PylbPm’-PrhaEW-egfp and Pcddm’-PrhaEW-egfp) gene. Each egfp fusion was integrated into the chromosome of strain 168, which resulted in four reporter strains (FH5–FH8) ( and ).
To evaluate glucose’s repressing and rhamnose’s inducing effects on each construct, we cultivated the reporter strains in a minimal medium containing 25 mM glucose or rhamnose as a carbon source, and their fluorescence intensities were quantified. As shown in , in the late logarithmic phase, the fluorescence intensity of strain FH1 (PylbP-egfp, without the BamHI-site introduction) grown in the rhamnose-containing medium was higher than that of the same strain grown in the glucose-containing medium; the rhamnose-induced ratio (the value obtained by rhamnose/that by glucose) was 7.1. When strain FH1 was cultivated until the late stationary phase in either medium, comparable fluorescence intensities were obtained, and these values were 1.3–1.5 times higher than that of the same strain grown until the late logarithmic phase in the rhamnose-containing medium (). Probably, the ylbP promoter was repressed by the activated CcpA in the presence of glucose, and when glucose was exhausted in the medium, the promoter was relieved from the CcpA repression; this is unlikely to be a rhamnose-specific induction of the ylbP promoter.
Figure 4. Fluorescence intensities of B. subtilis reporter strains carrying each egfp fusion.
Each reporter strain was cultivated in the minimal medium containing 25 mM glucose (white bar) or 25 mM rhamnose (gray bar) at 37°C with shaking until the OD600 reached 1.0 (A, B, and C) or for 18 h after inoculation (D, E, and F), corresponding to the late logarithmic and the late stationary phases, respectively. The cells of each strain were harvested and resuspended in the buffer, and then the fluorescence intensity of each suspension was measured as described in “Materials and methods.” Strain 168 was used as a negative control. The experiments of cell cultivation and fluorescence measurement were repeated at least three times. The fluorescence intensity values (a.u.) were normalized by dividing them by the OD600 values of the corresponding cell suspensions, and bars and error bars in the graphs indicate the mean values and standard deviations (n = 3), respectively. The p values from the t test are presented when the differences are small but statistically significant.
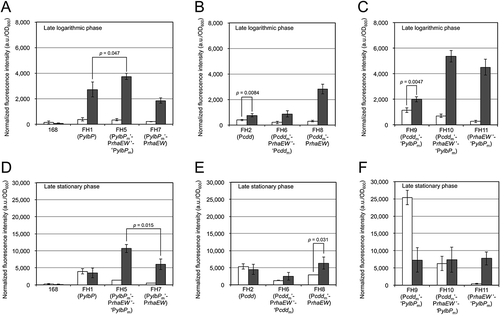
In the late logarithmic phase, the rhamnose-induced ratios of strains FH5 (PylbPm’-PrhaEW’-‘PylbPm-egfp) and FH7 (PylbPm’-PrhaEW-egfp) were more remarkable (11 and 9.1, respectively) than that obtained by strain FH1 in the same growth phase (), and the inducing effect on these strains was still observed when they were cultivated until the late stationary phase (7.7 for FH5 and 11 for FH7) (). These results indicate that introduction of the rhaEW regulatory region into the downstream of the ylbP promoter enables the protein expression to be rhamnose-dependently inducible even in the late stationary phase. Moreover, the rhamnose-induced fluorescence intensity of strain FH5 was higher than those of strains FH1 and FH7 in both growth phases (). Strain FH5’s superiority over strain FH7 suggested that translation from the ylbP SD sequence is more efficient than that from the rhaEW SD sequence.
When strain FH2 (Pcdd-egfp, without the BamHI-site introduction) was cultivated in the minimal medium containing glucose or rhamnose, the fluorescence intensity was slightly induced by rhamnose in the late logarithmic phase; the rhamnose-induced ratio was 1.9 (p = 0.0084 according to the t test) (). In the late stationary phase, comparable fluorescence intensities were obtained from both medium conditions (). In a comparison of the fluorescence data between strains FH1 and FH2, in the late logarithmic phase, strain FH1 grown in the rhamnose-containing medium showed the highest fluorescence intensity (), whereas the fluorescence intensities of strains FH1 and FH2 grown in each medium were similar in the late stationary phase; a statistical difference was not obtained in the t test (p > 0.05) ().
The fluorescence intensity of strain FH8 (Pcddm’-PrhaEW-egfp) was distinctly induced by rhamnose in the late logarithmic phase; however, the rhamnose-induced ratio declined as the culture time proceeded (9.4 in the logarithmic phase and 2.2 in the stationary phase). Meanwhile, strain FH6 (Pcddm’-PrhaEW’-‘Pcddm-egfp) did not show a clear induction by rhamnose in either growth phase, and its fluorescence intensities were consistently lower than those of strain FH8 grown in the same medium until the same growth phase (). This suggested that the translation efficiency of the cdd SD sequence is lower than that of the rhaEW SD sequence (i.e., ylbP SD > rhaEW SD > cdd SD).
Among the four hybrid expression constructs including the rhaEW regulatory region, strain FH5’s seemed to be the most promising as a highly efficient and conditional protein expression system in B. subtilis cultivated to either growth phase.
Further improvement of the hybrid expression systems by replacing components
As described in the previous section, the translation from the SD sequence of the ylbP gene was considered to be the most efficient among the three SD sequences tested, at least in the case that the cells were cultivated in the rhamnose-containing medium. Moreover, by comparing the fluorescence intensities of strains FH7 and FH8 grown in the presence of rhamnose until the late logarithmic phase, considering that the egfp transcript is translated from the rhaEW SD sequence in both strains, it was suggested that the transcriptional efficiency of the cdd core promoter is higher than that of the ylbP core promoter for such a medium condition and growth stage (). Hence, we assumed that the combination of the cdd core promoter, the rhaEW regulatory region without the SD sequence, and the ylbP SD sequence in this order could make the rhamnose-induced protein expression level more enhanced in the logarithmic phase while keeping this high level of expression during the stationary phase. To test this possibility, we first combined the cdd core promoter and the ylbP SD sequence to produce a hybrid expression unit and then fused it to the egfp gene (Pcddm’-‘PylbPm-egfp). Subsequently, the rhaEW regulatory region without its own SD sequence (PrhaEW’, −70 to 24) was inserted into Pcddm’-‘PylbPm-egfp to produce a rhamnose-inducible hybrid expression system (Pcddm’-PrhaEW’-‘PylbPm-egfp). These two egfp fusions were respectively integrated into the chromosome of B. subtilis strain 168 to produce two reporter strains (FH9 and FH10) ( and ).
On the other hand, as shown in , strains FH7 and FH8 exerted similar fluorescence intensities when they were grown in the rhamnose-containing medium until the late stationary phase, although the fluorescence intensity of strain FH8 grown in the presence of glucose until the late stationary phase was higher than that of strain FH7 for the same medium condition and growth stage. Therefore, we speculated that the ylbP and cdd core promoters placed upstream of the rhaEW regulatory region do not contribute to the protein expression level induced by rhamnose in the stationary phase, while the cdd core promoter appeared to strengthen the protein expression in the presence of glucose in the stationary phase. Thus, we further assumed that for the highly efficient and rhamnose-inducible protein expression in B. subtilis cells, it is sufficient to combine the PrhaEW’ (including the RhaR and CcpA/P-Ser-HPr binding regions and the intrinsic core promoter but lacking the SD sequence) region and the ylbP SD sequence without placing the ylbP or cdd core promoter upstream of the PrhaEW’ region. To examine this assumption, we prepared the PrhaEW’ (−144 to 24) fragment and fused it to the ‘ylbPm (corresponding to the ylbP SD sequence)-egfp fragment to produce the PrhaEW’-‘ylbPm-egfp construct, which was then introduced into the chromosome of strain 168, resulting in strain FH11 ( and ). This strain, together with strains FH9 and FH10, was subjected to similar characterization.
In the late logarithmic phase, the fluorescence intensity of strain FH9 (Pcddm’-‘PylbPm-egfp) was slightly induced by rhamnose; the rhamnose-induced ratio was 1.7 (p = 0.0047), and the fluorescence intensities of this strain were much higher than those of strain FH2 (Pcdd-egfp) under the corresponding medium condition (). Also, the fluorescence intensities of strain FH9 were higher than and comparable (p = 0.11) to those of strain FH1 (PylbP-egfp) when the reporter strains were grown in the glucose-containing medium and the rhamnose-containing medium, respectively (). To our surprise, in the late stationary phase, strain FH9 showed a strikingly (3.5-fold) higher fluorescence intensity in the glucose-containing medium than did the same strain grown in the rhamnose-containing medium (). This fluorescence intensity is the highest among those of all the reporter strains tested, including strains FH1 and FH2, under both medium conditions in both growth phases. This result indicates that the maximal protein (EGFP) expression level obtained by the Pcddm’-‘PylbPm construct was significantly (4.7–6.4-fold) higher than those by the ylbP and cdd promoters, which have been reported to be highly efficient in the stationary phase and constitutively highly efficient, respectively [Citation11], at least under the culture conditions that we tested ().
When strains FH10 (Pcddm’-PrhaEW’-‘PylbPm-egfp) and FH11 (PrhaEW’-‘PylbPm-egfp) were cultivated in the rhamnose-containing medium until the late logarithmic phase, they exhibited comparable fluorescence intensities (p = 0.13), which were much higher than that obtained by strain FH9 for the same medium condition and growth stage (). A comparison of the rhamnose-induced ratios of strains FH10 (7.6) and FH11 (17) in this growth phase revealed that the construct of strain FH11 more strictly regulates the protein expression in response to sugar species (glucose and rhamnose) than does that of strain FH10. In the late stationary phase, the fluorescence intensity of strain FH11 was specifically induced by rhamnose (the rhamnose-induced ratio was 19), whereas comparable fluorescence intensities were obtained from strain FH10 grown in the medium containing either glucose or rhamnose (). Moreover, while the rhamnose-induced fluorescence intensities of strains FH5 and FH11 were comparable (p > 0.05), the rhamnose-induced ratios of strain FH11 were higher than those of strain FH5 (11 and 7.7) in the late logarithmic and late stationary phases. These comparative data ensure both the high efficiency and strict sugar-selective inducibility of strain FH11’s construct.
In addition, the fluorescence intensity of strain FH11 was distinctly higher than those of strains FH1 and FH2 when each strain was cultivated in the rhamnose-containing medium until the late logarithmic phase (), and strain FH11 showed the fluorescence intensity higher than that of strain FH1 (p = 0.033) but comparable to that of strain FH2 (p = 0.080) when they were grown in the presence of rhamnose until the late stationary phase (). These comparisons indicate that, under those culture conditions, the PrhaEW’-‘ylbP construct exerted the protein expression higher than or comparable to those from the ylbP and cdd promoters.
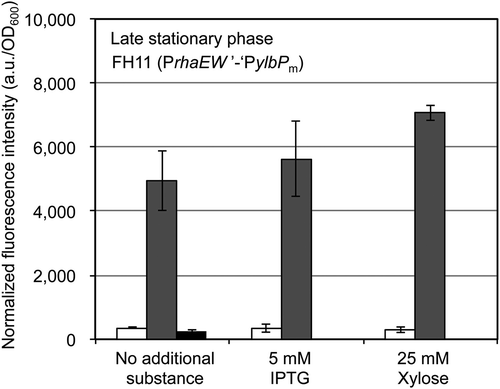
Furthermore, we examined whether glucose’s repressing and rhamnose’s inducing effects on the protein expression by strain FH11’s construct were affected by 5 mM IPTG or 25 mM xylose, which had been individually added in the glucose- or rhamnose-containing minimal medium, and strain FH11 in each medium was cultivated until the late stationary phase. As a result, IPTG did not affect both glucose’s repressing and rhamnose’s inducing effects, and xylose did not inhibit the repression by glucose. However, rhamnose’s inducing effect was slightly enhanced by the xylose addition; in the absence of rhamnose, xylose did not induce the protein (EGFP) expression in strain FH11 (). In addition, when strain FH11 was cultivated in each medium until the late logarithmic phase, the xylose addition did not enhance the rhamnose-induced protein expression (data not shown). Thus, we speculate that, when the B. subtilis cells were cultivated until the late stationary phase, a considerable amount of rhamnose was consumed as a carbon source, which was able to be alleviated by the addition of xylose used as another carbon source, resulting in persistence of rhamnose as the inducer for the PrhaEW’-‘ylbP construct in strain FH11. Since IPTG and xylose did not intrinsically impair both glucose’s repressing and rhamnose’s inducing effects on the PrhaEW’-‘ylbP construct, this construct has great promise as a component of the conditional coexpression system, combined with a construct for protein expression inducible by a compound such as IPTG and xylose, but other than rhamnose and glucose.
Figure 5. Effects of IPTG and xylose on the glucose-dependent repression and the rhamnose-dependent induction of the EGFP expression controlled by the PrhaEW’-‘ylbP construct.
B. subtilis strain FH11, carrying the PrhaEW’-‘ylbP-egfp, was cultivated in the minimal medium containing 25 mM glucose (white bar), 25 mM rhamnose (gray bar), or 25 mM xylose (black bar), with no additional substance (left), and in the coexistence of 5 mM IPTG (center) or 25 mM xylose (right), at 37°C with shaking for 18 h after inoculation, corresponding to the late stationary phase. The cells were harvested and resuspended in the buffer, and then the fluorescence intensity of each suspension was measured as described in “Materials and methods.” The experiments of cell cultivation and fluorescence measurement were repeated at least two times, independently of those performed to obtain the data in . The fluorescence intensity values (a.u.) were normalized by dividing them by the OD600 values of the corresponding cell suspensions, and bars and error bars in the graphs indicate the mean values and standard deviations (n = 2), respectively.
Detection of EGFP production by SDS-PAGE and confocal fluorescence microscopy
To confirm that the two B. subtilis reporter strains (FH9 and FH11) carrying each of the promising hybrid expression constructs actually produce a high level of EGFP on the appropriate growth conditions, these reporter strains as well as strains 168 and FH1 as controls were cultivated in the minimal medium containing glucose or rhamnose until the late stationary phase, and then the cells of each strain were harvested for preparation of the crude lysate, which was subjected to the SDS-PAGE analysis. Despite our expectation, the band corresponding to EGFP (27.6 kDa), which was translated from the ylbP initiation codon, was not clearly detected on either crude lysate (data not shown). This is probably because only one copy of each construct has been integrated into the chromosome of each reporter strain. The band of EGFP could be detected if each construct was mounted on a multicopy plasmid as reported by the other groups [Citation5,Citation7,Citation11], although the chromosome-integrated type that we employed in this study appears to be more stably retained in the B. subtilis cells [Citation25].
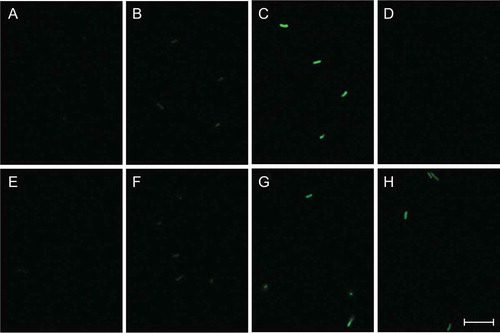
We also performed confocal fluorescence microscopy using the cells of each B. subtilis strain, which had been cultivated and collected as those used in the SDS-PAGE analysis. The fluorescence micrographs in were obtained under the same detection condition. Clear fluorescence images were observed from the cells of strain FH9 grown in the medium containing either sugar () and strain FH11 grown in the rhamnose-containing medium (). The fluorescence intensity of strain FH9 grown in the glucose-containing medium was the highest, and a comparable level of fluorescence was observed when strains FH9 and FH11 were grown in the rhamnose-containing medium. A blurry fluorescence image was observed when strain FH1 was cultivated in the medium containing either sugar, and no fluorescence was detected from the cells of strain FH11 grown in the presence of glucose () or strain 168 grown in either medium (). These observations were in good agreement with the data of fluorescence quantification presented in . Moreover, the fluorescence intensities emitted from the cells of the same reporter strain appeared almost homogeneous, suggesting that the protein (EGFP) expression level did not significantly vary among the individual cells of the same strain.
Figure 6. Micrographs of confocal fluorescence microscopy of B. subtilis reporter strains carrying each egfp fusion.
Strains FH1 (B and F), FH9 (C and G), and FH11 (D and H) were cultivated in the minimal medium containing 25 mM glucose (A–D) or 25 mM rhamnose (E–H) at 37°C with shaking for 18 h after inoculation, corresponding to the late stationary phase. The cells of each strain were harvested and resuspended in the buffer, and then the fluorescence images were observed via confocal fluorescence microscope as described in “Materials and methods.” Strain 168 was used as a negative control (A and E). All fluorescence micrographs were obtained under the same detection condition. Scale bar = 10 μm.
Discussion
To achieve highly efficient and rhamnose-inducible protein expression in B. subtilis cells, we assembled the B. subtilis-derived highly efficient promoters of the two genes (cdd and ylbP) and the regulatory region (PrhaEW) of the B. subtilis rhaEWRBMA operon, the transcription of which is induced by rhamnose and repressed by glucose, to produce various types of hybrid protein expression constructs. The protein expression level and rhamnose-induced ratio of these constructs were evaluated via fluorescence quantification of the B. subtilis reporter strains carrying each of the hybrid construct-egfp fusions.
The cdd and ylbP promoter regions (Pcdd and PylbP) used in this study contained the SD sequences of the respective genes as well as the intrinsic core promoters (−35 and −10 sequences). A comparison of the fluorescence intensities between strains FH5 and FH7 (), between strains FH6 and FH8 (), and between strains FH7 and FH8 () indicated that the SD sequence of the ylbP gene is more efficient than those of the cdd and rhaEW genes and suggested that, at least in the late logarithmic phase, the core promoter strength of the cdd gene is higher than that of the ylbP gene. The high efficiency of the ylbP SD sequence might be attributed to its close resemblance to the canonical SD sequence for B. subtilis (5ʹ-AAAGGAGGTGATC-3ʹ) [Citation26].
These pieces of information gave us the idea that the sequential combination of the cdd core promoter, the partial PrhaEW fragment (PrhaEW’, including the core promoter and the binding sites of the two transcriptional factors but lacking the SD sequence), and the ylbP SD sequence could be the optimum construct to provide highly potent and rhamnose-specific induced promoter activity. However, the strain carrying this construct (FH10) did not show rhamnose-specific induction in the late stationary phase; comparable fluorescence intensities were obtained by this strain grown in the medium containing glucose or rhamnose (), whereas the rhamnose-specific induction was observed when this strain was cultivated until the late logarithmic phase (). Based on comparisons between strains FH7 and FH8, between strains FH5 and FH10, and between strains FH1 and FH9, we assume that the cdd core promoter is likely to be activated when the cells are grown in the presence of glucose until the late stationary phase, although the activation mechanism is currently unknown. On the other hand, the rhaEW core promoter in the PrhaEW region is repressed and induced in the presence of glucose and rhamnose, respectively. Such conflicting features of these core promoters may have caused the functional failure of the strain FH10 construct.
The fluorescence data of strains FH9, FH10, and FH11 were nearly consistent with our aforementioned assumption; the efficiency of the ylbP SD sequence is better than those of the cdd and rhaEW SD sequences, and the activity of the cdd core promoter is higher than that of the ylbP core promoter in the late logarithmic phase. The only exception is that the fluorescence intensity of strain FH9 was not higher than that of strain FH1 when they were grown in the rhamnose-containing medium until the late logarithmic phase, which means that replacement with the cdd core promoter in the PylbP region did not enhance the rhamnose-induced fluorescence intensity under this culture condition ().
Strain FH11, which carries a combination of the PrhaEW’ fragment and the ylbP SD sequence (PrhaEW’-‘PylbPm), showed a rhamnose-induced fluorescence intensity comparable to that of strain FH5 (PylbPm’-PrhaEW’-‘PylbPm), and the rhamnose-induced ratio of strain FH11 was higher than that of strain FH5 (). Thus, we concluded that among the constructs tested, strain FH11’s is the best for protein expression with both high yield and strict sugar specificity, which is controllable by glucose and rhamnose. Placement of the PylbPm’ fragment, which includes the ylbP core promoter and the CcpA/P-Ser-HPr binding site (cre), upstream of the PrhaEW’-‘ylbPm fragment did not significantly elevate the rhamnose-induced fluorescence intensity; rather, it raised the fluorescence intensity in the presence of glucose in the late stationary phase, indicating that the ylbP core promoter was activated, and it surpassed the repression by RhaR even under this culture condition. The binding of two molecules of the CcpA/P-Ser-HPr complexes to the respective cres in the PylbPm’ and PrhaEW’ regions was unlikely to be effective for the glucose-specific repression.
Judging from comparisons of the fluorescence intensities mentioned above, we estimate that replacement of the rhaEW SD sequence with the ylbP’s improved the translational efficiency, although we did not test an egfp fusion with the PrhaEW fragment covering the core promoter, the two transcriptional factors’ binding sites, and the intrinsic SD sequence.
Strain FH9, which carries the combination of the cdd core promoter and the ylbP SD sequence (Pcddm’-‘ylbPm), unexpectedly showed a remarkable degree of fluorescence intensity when it was cultivated in the glucose-containing medium until the late stationary phase (). This is probably due to the combination of the highly efficient ylbP SD sequence (involving translation) and the cdd core promoter (involving transcription) that is activated when the cells are grown in the presence of glucose until the late stationary phase. Since the protein expression level by the construct of strain FH9 under this culture condition was the highest among those of all reporter strains tested under both medium conditions in both growth phases, we propose that this construct is appropriate for the overexpression of a target protein in B. subtilis cells grown in the medium containing glucose, which is a favorable carbon source for the cell growth, until the late stationary phase.
Author contributions
AT conducted most of the experiments and analyzed the results under the supervision of KH. KH drafted the research plan and wrote the manuscript with AT.
Acknowledgements
We are grateful to Dr. Jun Kamishikiryo for assistance with confocal fluorescence microscopy. We also thank Saki Shioda, Rikiya Yanai, and Mizuki Handa for their help with the experiments.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Westers L, Westers H, Quax WJ. Bacillus subtilis as cell factory for pharmaceutical proteins: a biotechnological approach to optimize the host organism. Biochim Biophys Acta. 2004;1694:299–310.
- Degering C, Eggert T, Puls M, et al Optimization of protease secretion in Bacillus subtilis and Bacillus licheniformis by screening of homologous and heterologous signal peptides. Appl Environ Microbiol. 2010;76:6370–6376.
- Sak-Ubol S, Namvijitr P, Pechsrichuang P, et al Secretory production of a beta-mannanase and a chitosanase using a Lactobacillus plantarum expression system. Microb Cell Fact. 2016;15:81.
- Zobel S, Kumpfmüller J, Süssmuth RD, et al Bacillus subtilis as heterologous host for the secretory production of the non-ribosomal cyclodepsipeptide enniatin. Appl Microbiol Biotechnol. 2015;99:681–691.
- Schumann W. Production of recombinant proteins in Bacillus subtilis. Adv Appl Microbiol. 2007;62:137–189.
- Dong H, Zhang D. Current development in genetic engineering strategies of Bacillus species. Microb Cell Fact. 2014;13:63.
- Song Y, Nikoloff JM, Zhang D. Improving protein production on the level of regulation of both expression and secretion pathways in Bacillus subtilis. J Microbiol Biotechnol. 2015;25:963–977.
- Jarmer H, Larsen TS, Krogh A, et al Sigma A recognition sites in the Bacillus subtilis genome. Microbiology. 2001;147:2417–2424.
- Petersohn A, Bernhardt J, Gerth U, et al Identification of σB-dependent genes in Bacillus subtilis using a promoter consensus-directed search and oligonucleotide hybridization. J Bacteriol. 1999;181:5718–5724.
- Wang PZ, Doi RH. Overlapping promoters transcribed by Bacillus subtilis σ55 and σ37 RNA polymerase holoenzymes during growth and stationary phases. J Biol Chem. 1984;259:8619–8625.
- Yu X, Xu J, Liu X, et al Identification of a highly efficient stationary phase promoter in Bacillus subtilis. Sci Rep. 2015;5:18405.
- Guan C, Cui W, Cheng J, et al Construction and development of an auto-regulatory gene expression system in Bacillus subtilis. Microb Cell Fact. 2015;14:150.
- K, Kodoi Y, Satomura T, et al. Regulation of the rhaEWRBMA operon involved in L-rhamnose catabolism through two transcriptional factors, RhaR and CcpA, in Bacillus subtilis. J Bacteriol. 2016;198:830–845.
- Fujita Y. Carbon catabolite control of the metabolic network in Bacillus subtilis. Biosci Biotechnol Biochem. 2009;73:245–259.
- Wickstrum JR, Skredenske JM, Balasubramaniam V, et al The AraC/XylS family activator RhaS negatively autoregulates rhaSR expression by preventing cyclic AMP receptor protein activation. J Bacteriol. 2010;192:225–232.
- Terpe K. Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2006;72:211–222.
- Tojo S, Satomura T, Matsuoka H, et al Catabolite repression of the Bacillus subtilis FadR regulon, which is involved in fatty acid catabolism. J Bacteriol. 2011;193:2388–2395.
- Fujita Y, Miwa Y. Identification of an operator sequence for the Bacillus subtilis gnt operon. J Biol Chem. 1989;264:4201–4206.
- Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544.
- Miwa Y, Fujita Y. Involvement of two distinct catabolite-responsive elements in catabolite repression of the Bacillus subtilis myo-inositol (iol) operon. J Bacteriol. 2001;183:5877–5884.
- Yoshida K, Ishio I, Nagakawa E, et al Systematic study of gene expression and transcription organization in the gntZ-ywaA region of the Bacillus subtilis genome. Microbiology. 2000;146:573–579.
- Atkinson MR, Wray LV Jr, Fisher SH. Regulation of histidine and proline degradation enzymes by amino acid availability in Bacillus subtilis. J Bacteriol. 1990;172:4758–4765.
- Sambrook J, Russell DW. Molecular cloning: a laboratory manual. 3rd ed. Cold Spring Harbor, New York (NY): Cold Spring Harbor Laboratory Press; 2001.
- Blencke HM, Homuth G, Ludwig H, et al Transcriptional profiling of gene expression in response to glucose in Bacillus subtilis: regulation of the central metabolic pathways. Metab Eng. 2003;5:133–149.
- Bron S, Luxen E, Swart P. Instability of recombinant pUB110 plasmids in Bacillus subtilis: plasmid-encoded stability function and effects of DNA inserts. Plasmid. 1988;19:231–241.
- Band L, Henner DJ. Bacillus subtilis requires a “stringent” Shine-Dalgarno region for gene expression. DNA. 1984;3:17–21.