
ABSTRACT
Apoptosis-linked gene 2 (ALG-2) is a Ca2+-binding protein with five repetitive EF-hand motifs, named penta-EF-hand (PEF) domain. It interacts with various target proteins and functions as a Ca2+-dependent adaptor in diverse cellular activities. In the cytoplasm, ALG-2 is predominantly localized to a specialized region of the endoplasmic reticulum (ER), called the ER exit site (ERES), through its interaction with Sec31A. Sec31A is an outer coat protein of coat protein complex II (COPII) and is recruited from the cytosol to the ERES to form COPII-coated transport vesicles. I will overview current knowledge of the physiological significance of ALG-2 in regulating ERES localization of Sec31A and the following adaptor functions of ALG-2, including bridging Sec31A and annexin A11 to stabilize Sec31A at the ERES, polymerizing the Trk-fused gene (TFG) product, and linking MAPK1-interacting and spindle stabilizing (MISS)-like (MISSL) and microtubule-associated protein 1B (MAP1B) to promote anterograde transport from the ER.
Graphical Abstract
ALG-2 localizes at the Sec31A-positive endoplasmic reticulum exit site (ERES).
Apoptosis-linked gene 2 (ALG-2), a product of the PDCD6 gene, is a 22-kDa penta-EF-hand (PEF) type Ca2+-binding protein (UniProt ID: O75340) [Citation1,Citation2]. There is confusion caused by the use of the same or similar name for different proteins and genes; for example, the product of the ALG2 gene (without hyphen between “ALG” and “2”) is termed ALG2 as an α-1,3/1,6-mannosyltransferase (UniProt ID: Q9H553) and the gene alg-2 (Argonaute-like gene 2) in C. elegans encodes a member of the highly conserved eukaryotic RDE-1/AGO1/PIWI family of proteins (UniProt ID: O16720). Here, the term “ALG-2” is used to refer to a product of the PDCD6 gene.
A cDNA encoding murine Pdcd6 was first isolated by the death-trap method, in which a cDNA library constructed in a mammalian expression vector was transiently transfected into mouse T cell hybridoma 3DO cells, apoptosis was then induced with T cell receptor stimulation, and the plasmids were recovered from surviving cells [Citation3]. The obtained plasmids containing the Pdcd6 insert all encoded anti-sense transcripts. The resultant reduced expression of ALG-2 protein conferred resistance to cell death induced by several stimuli, including glucocorticoids, T cell receptors and Fas triggering. Thus, it is conceivable that ALG-2 functions as a proapoptotic protein [Citation4]. However, an unexpected finding is that Pdcd6-deficient mice generated by gene targeting exhibit no abnormality in the immune system and that both immature thymocytes and mature T cells from the Pdcd6-deficient mouse retain their susceptibility to apoptotic stimuli [Citation5]. Nonetheless, potential roles of ALG-2 in promoting cell death induced by ER stress [Citation6], tumor necrosis factor receptor 1 [Citation7], and DNA damage [Citation8] have been reported. On the other hand, ALG-2 has been reported to be up-regulated in a variety of tumors [Citation9–Citation12]. ALG-2 overexpression is an independent poor prognostic factor in early stage lung adenocarcinoma [Citation11], whereas lower expression levels of PDCD6 mRNA are correlated significantly with poor overall survival of gastric cancer patients who have received chemotherapy [Citation12]. Thus, ALG-2 may be an important modulator involved in the cellular decision between cell proliferation and cell death [Citation13]. However, the molecular mechanisms of proapoptotic and survival functions of ALG-2 are not yet fully understood.
To elucidate the molecular functions of ALG-2 and its role in Ca2+ signaling, searching for interacting proteins of ALG-2 has been conducted by several groups using yeast two-hybrid screening and mass-spectrometric analyses of immunoprecipitates of endogenous or tagged proteins or of pulldown products of recombinant ALG-2 expressed in E. coli. Evaluation of different in vitro and/or in vivo methods has been carried out to validate the direct interactions of ALG-2 with candidate proteins and their Ca2+-dependency [Citation14,Citation15]. Until now, more than 20 proteins have been identified as Ca2+-dependent interacting proteins for ALG-2 [Citation15]. The target proteins of ALG-2 are distributed in various intracellular compartments such as the endoplasmic reticulum [Sec31A [Citation16–Citation18], Scotin [Citation19], Trk-fused gene (TFG) [Citation20] and MAPK1-interacting and spindle-stabilizing (MISS)-like (MISSL) [Citation21]], endocytic organelles [ALG-2 interacting protein X (ALIX) [Citation22,Citation23], tumor susceptibility gene 101 (TSG101) [Citation24], vacuolar protein sorting 37C (VPS37C) [Citation25], IST1 [Citation26], and Mucolipin-1/TRPML1 [Citation27]] and nucleus [RBM22 [Citation28] and calcium homeostasis endoplasmic reticulum protein (CHERP) [Citation29]]. Multifaceted roles of ALG-2 in these compartments were reviewed recently [Citation15]. Here, some of the more recent advances in understanding adaptor functions of ALG-2 in protein transport from the endoplasmic reticulum (ER) are reviewed.
ALG-2 is predominantly localized to the Sec31A-positive ERES
Approximately one-third of all proteins in eukaryotic cells are estimated to translocate into the ER during their synthesis on membrane-bound ribosomes. Only properly folded proteins are packaged into coat protein complex II (COPII)-coated transport vesicles to be dispatched to their final destinations via the Golgi apparatus. Five evolutionally conserved proteins, namely a small GTPase Sar1, inner coat subunits Sec23 and Sec24, and outer coat subunits Sec13 and Sec31, constitute the minimal machinery for generating COPII vesicles [Citation30] (). They are sequentially recruited from the cytosol to a discrete sub-region of the ER called the ER exit site (ERES) or transitional ER (tER) as follows: 1) exchange of GDP for GTP in Sar1 by the guanine nucleotide exchange factor Sec12 leads to exposure of N-terminal amphipathic α-helix of Sar1, which is inserted into the cytoplasmic leaflet of the ERES membrane, 2) the active form of Sar1 recruits the Sec23-Sec24 heterodimer from the cytosol to the ERES, where Sec24 recognizes and binds exit motifs in the cytosolic region of cargo proteins or cargo receptors to form a pre-budding complex, and 3) the pre-budding complex in turn recruits the Sec13-Sec31 heterotetramer, which forms the outer layer of the COPII coat [Citation31–Citation34].
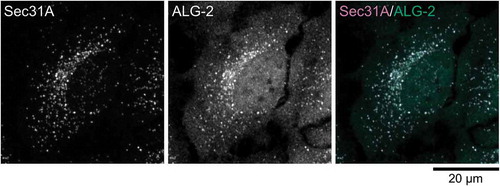
Figure 1. ERES localization of ALG-2. (a) Schematic representation of COPII vesicle formation in the ERES. (b) Representative micrograph showing the ERES localization of ALG-2. HeLa cells were fixed with 4% paraformaldehyde in phosphate buffer, permeabilized with 0.1 % Triton X-100, and then triple stained with antibodies against an ER marker, calnexin (a rabbit polyclonal antibody, Enzo Life Sciences), an ERES marker, Sec31A (a mouse monoclonal antibody, BD Biosciences) and ALG-2 (a goat polyclonal antibody) [Citation98]. The images are superimpositions of serial optical sections taken by a confocal laser-scanning microscope through the whole thickness of the cell. The right panel shows a merged image with the pseudocolors as follows: green (ALG-2) and magenta (Sec31A).
![Figure 1. ERES localization of ALG-2. (a) Schematic representation of COPII vesicle formation in the ERES. (b) Representative micrograph showing the ERES localization of ALG-2. HeLa cells were fixed with 4% paraformaldehyde in phosphate buffer, permeabilized with 0.1 % Triton X-100, and then triple stained with antibodies against an ER marker, calnexin (a rabbit polyclonal antibody, Enzo Life Sciences), an ERES marker, Sec31A (a mouse monoclonal antibody, BD Biosciences) and ALG-2 (a goat polyclonal antibody) [Citation98]. The images are superimpositions of serial optical sections taken by a confocal laser-scanning microscope through the whole thickness of the cell. The right panel shows a merged image with the pseudocolors as follows: green (ALG-2) and magenta (Sec31A).](/cms/asset/2189ad18-be30-48db-ab6f-db349c3cc347/tbbb_a_1525274_f0001_oc.jpg)
In contrast to the reticular pattern of the ER, the ERES in mammalian cells shows a small punctate structure distributed throughout the cytoplasm but usually concentrated in the juxtanuclear region detected by the indirect immunofluorescent method (). Three-dimensional immuno-electron tomography of the ERES revealed that COPII components exist on ER-associated buds as well as on vesicles free from the ER and tubules adjacent to the ER [Citation35]. Therefore, in images obtained by the indirect immunofluorescence method using conventional confocal microscopy, each fluorescence spot labeled with an antibody against the COPII component should correspond to one ERES containing a cluster of nascent buds and vesicles and tubules after budding off.
Several independent groups including ours reported that ALG-2 interacts Ca2+-dependently with Sec31A and distributes predominantly to the Sec31A-positive ERES in the cytoplasm [Citation16,Citation17] (). Live-cell imaging of fluorescent protein-fused ALG-2 revealed that ALG-2 translocates from the cytosol to the Sec31A-positive ERES following exposure of cells to Ca2+-mobilizing agents [Citation18,Citation36]. A mutant with glutamate residues substituted for alanine in Ca2+-coordinating positions of EF1 and EF3 (ALG-2E47A/E114A) does not interact with Sec31A, and overexpressed ALG-2E47A/E114A is seen distributed diffusely to the cytoplasm and nucleus with no co-localization with ERES marker proteins [Citation16,Citation17]. In addition, the cytoplasmic punctate localization pattern of ALG-2 is lost to a great extent in Sec31A knockdown cells [Citation16]. Therefore, Ca2+-dependent binding of ALG-2 with Sec31A is essential for the ERES localization of ALG-2.
ALG-2 binding site in Sec31A
ALG-2 has five repetitive EF-hand motifs in its C-terminus (), but, like calmodulin, it lacks catalytic activities and is thought to perform its biological functions by interacting with target proteins [Citation15]. Two short peptide motifs that are recognized by ALG-2 have been identified and named ABM-1 (ALG-2 binding motif type-1) of PPYP(X)nYP (X, variable amino acids, n = 3–6) [Citation37] and ABM-2 of [PΦ]PX[PΦ]G[FW]Ω ([PΦ], Pro or hydrophobic; [FW], Phe or Trp; Ω, large side chain; X, variable) [Citation38].
Figure 2. Structures of ALG-2. (a) Schematic representation of primary structure of ALG-2. ALG-2 has an N-terminal extension rich in Ala, Gly and Pro, and a C-terminal penta-EF-hand (PEF) domain. Calcium ions (Ca2+) bind to EF1 and EF3 under physiological conditions. ALG-2ΔGF122 is an alternatively spliced variant of ALG-2 lacking two amino acids, Gly121 and Phe122. (b) Co-crystal structure of the complex between ALG-2 and Sec31A peptide (PDB code, 3WXA). Chain A and B (ALG-2 molecule A and B) are shown by cartoon (green, EF1 and EF3; dark green, EF5, dark grey, EF2 and EF4) and surface representation (blue, pocket 1; light blue, pocket 2; green, pocket 3), respectively, using PyMOL software. Chain C and D (Sec31A peptides) are shown by stick in magenta. Grey spheres indicate zinc ions (Zn2+). (c) Schematic representation of the ALG-2 dimer-mediated physical association between two target proteins. The ALG-2 dimer couples two target proteins in a Ca2+-dependent manner.
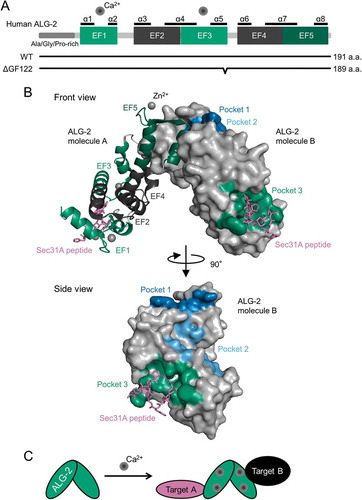
Murine Pdcd6 encodes two alternatively spliced transcripts, and the short isoform lacks 6 base pairs; therefore, the corresponding protein lacks Gly121 and Phe122 [Citation39] (). Here, the long and short isoforms are denoted by ALG-2WT and ALG-2ΔGF122, respectively (). ALG-2WT binds both target proteins having ABM-1 or ABM-2. In contrast, ALG-2ΔGF122 has no binding avidity to proteins with ABM-1 or ABM-1-like motifs, such as ALIX [Citation39], TSG101 [Citation24], Scotin [Citation19], annexin A7 and annexin A11 [Citation40]. On the other hand, ALG-2ΔGF122 shows binding to proteins with ABM-2, including Sec31A [Citation17], PLSCR3 [Citation40] and TFG [Citation20]. An RNase protection assay revealed that both the long and short RNAs are transcribed in a molar ratio of 2:1 in the brain, ovary and kidney in mice [Citation39]. In addition to mice, both isoforms of ALG-2 are deposited in human, rat, chicken and frog NCBI protein databases under the following accession numbers: Homo sapiens, NP_037364.1 and NP_001254485.1; Rattus norvegicus, NP_001100922.1 and XP_006227846.1; Gallus gallus, XP_004935237.1 and XP_419075.2; Xenopus tropicalis, NP_001008004.1 and XP_012820018.1. While ALG-2ΔGF122 has a lower Ca2+ affinity than that of ALG-2WT [Citation39], they have comparable Mg2+-binding signatures [Citation41], which contribute to dimer stability of ALG-2 proteins [Citation42]. However, it has remained unclear how the expression ratio between isoforms is regulated and whether there are differences between cells in the expression ratio of the two isoforms at protein levels.
To elucidate the biological significance of the interaction between ALG-2 and Sec31A, we first identified the ALG-2 binding site in human Sec31A. An in vitro overlay assay using biotin-labeled ALG-2 revealed that a sequence (839-PPPGFIMHGNVNP-851) in the Pro-rich region of Sec31A is necessary and sufficient for binding to ALG-2 [Citation36]. In vertebrates, there are two paralogous genes of Sec31: SEC31A and SEC31B. Sec31A is expressed abundantly and ubiquitously, whereas Sec31B is enriched particularly in the testis [Citation43–Citation45]. The two proteins share a high degree of amino acid similarity (92% in humans) at their N-terminal WD40 repeat domain. Both proteins have a Pro-rich region at their C-terminus with 70% amino acid similarity. Sec31B, however, has no sequence similar to the ALG-2 binding site of Sec31A. Although there is the possibility that ALG-2 could bind Sec31B through an unidentified motif sequence, the two Sec31 proteins might play distinct roles in the COPII-mediated secretory pathway by interacting with different proteins through their Pro-rich regions. The ALG-2 binding site of human Sec31A is only conserved in homologues of mammals and avians, not in other vertebrates. It is interesting, however, that Saccharomyces cerevisiae Pef1p, a unique PEF protein, binds Sec13p-Sec31p in the absence of Ca2+, not in the presence of Ca2+ [Citation46]. The opposite dependency of Ca2+ in interaction between Pef1p and Sec13p-Sec31p may be related to differences in Ca2+ requirement for the ER-to-Golgi transport between mammals and yeast.
We have co-crystallized a complex of an N-terminally deleted ALG-2 (des3-20ALG-2) with a Sec31A peptide (837-NPPPPGFIMHGN-848) in the presence of Zn2+ and refined the crystal structure at 2.4 Å resolution [Citation38] (). Like the crystal structure of des3-20ALG-2/the ALIX peptide [Citation37], two ALG-2 molecules are contained as a dimer in an asymmetric unit. The ALIX peptide binds hydrophobic pockets, named Pocket 1 and Pocket 2, whereas the Sec31A peptide is accommodated in Pocket 3 of each ALG-2 molecule, in which the N-terminal Pro residues (838-PPPP-841) form left-handed type II polyproline helices (). Pocket 3 consists of 19 residues that are present in EF1, EF2, EF3, EF4 and the loop connecting EF1 and EF2 [Citation38]. Thus, two distinct ALG-2 binding motifs bind different hydrophobic pockets of ALG-2; therefore, the ALG-2 dimer has at least four binding surfaces and is proposed to function as a Ca2+-dependent adaptor bridging two (or more) target proteins ().
ALG-2 stabilizes Sec31A to the ERES
Biochemical reconstitution studies of purified yeast Sec13p-Sec31p complex demonstrated that this heterotetramer is recruited to the budding site by the active form of Sar1p and the Sec23p-Sec24p complex [Citation30]. Structurally, Sec23p and Sec24p form a bow-tie-shaped dimer with a positively charged concave surface that can make contact with acidic membrane phospholipids to drive curvature [Citation47]. The Sec13p-Sec31p heterotetramer forms an elongated rod-like structure [Citation48] that assembles in vitro into polyhedral cages with flexibility in size to accommodate cargoes of various shapes and sizes [Citation49]. Enzymatically, Sec23p functions as a GTPase-activating protein (GAP) for Sar1p [Citation50], and the outer complex further stimulates GAP activity [Citation51]. Hydrolysis of GTP by Sar1p is required for scission of vesicles [Citation31]. It also promotes disassembly of coat components from the membrane [Citation51]; however, loading of cargoes has been shown to delay the uncoating process [Citation52,Citation53]. A peptide sequence corresponding to residues 899–947 located in the middle of the Pro-rich region of yeast Sec31p was identified as an active fragment responsible for the GAP-stimulating activity [Citation54]. The active fragment binds as an extended polypeptide across the composite surface of Sec23p and Sar1p in a crystal structure of the ternary complex. Four critical residues in the active fragment to exert the activity are conserved in vertebrate homologs of Sec31p, while the ALG-2-binding site in human Sec31A is not found in yeast Sec31p as mentioned above. Since the ALG-2-binding site is separated from the active fragment by about 140 amino acids [Citation36], it was thought that Sec31A could bind simultaneously Sec23 and ALG-2 in the presence of Ca2+.
Several independent studies demonstrated that stable localization of mammalian Sec31A to the ERES is required for Ca2+ and ALG-2. First, treatment of cells with a membrane-permeable Ca2+-chelator, BAMTA-AM, resulted in removal of Sec31A from the ERES [Citation16,Citation17]. In cells suppressing ALG-2 expression, Sec31A proteins are mainly present in the cytosolic fraction [Citation16]. Second, by using purified recombinant proteins, la Cour et al. found that recruitment of both an inner coat, Sec23-Sec24, and an outer coat, Sec13-Sec31A, to artificial liposomes is increased by the presence of ALG-2 and Ca2+ [Citation55]. In addition, they demonstrated that Sec23A is sufficient for the ALG-2/Ca2+-mediated recruitment of Sec13-Sec31A. Mechanistically, it is postulated that binding of ALG-2 to Sec31A endows Sec31A with the ability to robustly bind to Sec23A. Finally, the molecular dynamics of Sec31A on and off the ERES membrane in live cells has been demonstrated by fluorescence recovery after photobleaching (FRAP) analysis in cells expressing green fluorescent protein (GFP)-fused Sec31A. In this analysis, deletion of the ALG-2 binding region in Sec31A reduced the high-affinity population of Sec31A to the ERES to nearly half in HeLa cells, indicating that the region constitutes a high affinity binding site for the ERES [Citation36]. In human fibrosarcoma HT1080 cells, there is a population of Sec31A stably associated with the ERES membrane [Citation56]. ALG-2 knockdown results in a reduction in the stable immobile population and an increase in the fast mobile population. Thus, ALG-2 stabilizes Sec31A to the ERES in HT1080 cells. It is interesting that expression of ALG-2WT, but not expression of ALG-2ΔGF122, rescues the stable population of Sec31A, even though both isoforms of ALG-2 can interact with Sec31A [Citation56]. The reason why only ALG-2WT is related to stabilization of Sec31A to the ERES is described below.
Effects of ALG-2 knockdown on ER-to-Golgi transport
To monitor transport of newly synthesized proteins from the ER, a temperature-sensitive variant of vesicular stomatitis virus glycoprotein (tsO45 VSV-G) is routinely used as a cargo protein. This variant is a type I transmembrane protein having an F204S substitution in the lumenal/extracellular domain [Citation57]. At a restrictive temperature, the mutant protein is synthesized but misfolded, which allows it to remain in the ER. After shifting from a restrictive temperature to a permissive temperature, the protein is folded correctly to form a trimer and then the proteins are synchronously transported from the ER in a COPII-dependent fashion [Citation58]. To investigate the role of ALG-2 in the ER-to-Golgi transport, the effects of ALG-2 knockdown on the tsO45 VSV-G transport have been monitored by three independent laboratories and inconsistent data were reported. In the first report by Yamasaki et al. [Citation16], it was stated that there was no detectable difference in the rate of ER-to-Golgi transport of tsO45 VSV-G in the ALG-2-depleted HeLa cells. We established human fibrosarcoma HT1080 cells stably expressing GFP-fused tsO45 VSV-G and demonstrated that ALG-2 knockdown enhanced the rate of ER-to-Golgi transport of the GFP-fused protein, suggesting that ALG-2 functions as a negative regulator [Citation56]. This is consistent with the results of an in vitro study by la Cour et al. [Citation55] showing that Ca2+-dependent binding of ALG-2 to Sec31A attenuates budding of COPII. Recently, Sec31A has been reported to be modified by O-linked β-N-acetylglucosamine (O-GlcNAc) on Ser964 in the Pro-rich region. Although the modification occurs outside the ALG-2 binding sequence, the O-GlcNAc-modified Sec31A has reduced ability to bind ALG-2 and therefore accelerates the ER-to-Golgi transport [Citation59]. In contrast, Helm et al. [Citation60] showed delay of the transport of tsO45 VSV-G by ALG-2 knockdown in normal rat kidney (NRK) cells. A later study from the same laboratory showed that knockdown of peflin, a heterodimer partner of ALG-2, enhances the rate of transport of tsO45 VSV-G by increased localization of ALG-2 to the ERES in NRK cells, supporting the role of ALG-2 as a positive regulator [Citation61]. A possible explanation for the discrepancies in these results of ALG-2 knockdown could be the different cell lines used. ALG-2 may play both roles – a positive regulator and a negative regulator – in the ER-to-Golgi transport by selectively interacting with its various target proteins. Different expression levels of peflin and ERES-located target proteins of ALG-2 from cell to cell might provide an enormous combinatorial potential for generating apparent differences in the functional role of ALG-2 in ER-to-Golgi transport.
Large cargoes, such as procollagen fibers and chylomicron particles, are newly synthesized in the ER with sizes that are too large to be incorporated into typical COPII vesicles (60 – 90 nm), but their export from the ER is essentially dependent on COPII components. Missense mutations in the SAR1B gene have been shown to be associated with chylomicron retention disease (CMRD) [Citation62], whereas an F382L mutation in the SEC23A gene causes cranio-lenticulo-sutural dysplasia (CLSD), an autosomal recessive syndrome characterized by facial dysmorphism, late-closing fontanels, cataracts, and skeletal defects [Citation63,Citation64]. Fibroblasts isolated from patients exhibit diffuse cytoplasmic mislocalization of Sec31 proteins and accumulation of procollagen I within the ER [Citation63]. Structurally, F382 in Sec23A is located on the binding surface for the active fragment of Sec31A in its Pro-rich region as described above [Citation54]. The F382L mutation causes failure to recruit the outer coat Sec13-Sec31A, thereby inhibiting vesicle formation [Citation65]. Recently, molecular machineries required for export of large cargoes from the ER have been identified [Citation66,Citation67] (). Transport and Golgi organization protein 1 (TANGO1, a product of the MIA3 gene) and cutaneous T-Cell lymphoma-associated antigen 5 (cTAGE5) are transmembrane proteins localized to the ERES, where they form a complex [Citation68]. The SH3 domain of TANGO1 faces into the lumen of the ER and directly binds procollagen VII [Citation69] or indirectly binds other procollagens through interaction with HSP47, a collagen-specific molecular chaperone [Citation70], to function as a procollagen receptor. cTAGE5 binds and concentrates Sec12 at the ERES, which is required for efficient export of procollagen VII [Citation71]. Furthermore, recruitment of Sedlin, also known as TRAPPC2, by TANGO1 facilitates efficient GDP-GTP cycling of Sar1 [Citation72]. Both TANGO1 and cTAGE5 bind Sec23-Sec24 via their Pro-rich regions in the cytoplasm, and this binding is thought to stall recruitment of the outer Sec13-Sec31 complex, thereby enabling the COPII vesicle to grow [Citation69,Citation73,Citation74]. Another key molecule is Kelch-like protein 12 (KLHL12), a KLHL family protein that forms a CUL3-based ubiquitin ligase complex, referred to as CUL3KHLH12 hereafter. Although KLHL12 interacts directly with Sec31A [Citation75], McGourty et al. [Citation76] reported that the ALG-2-peflin heterodimer functions as a Ca2+-dependent adaptor to bridge CUL3KLHL12 and Sec31A for facilitating mono (or multi)-ubiquitylation of Sec31A (). Recently, Gorur et al. [Citation77] reported large Sec31A-positive structures that encapsulate procollagen I in human fibrosarcoma K16 cells, in which pro-α1(I) collagen and KLHL12 are overexpressed, as well as in human osteosarcoma SaOS-2 cells. In SaOS-2 cells, knockdown of either ALG-2 or peflin resulted in retention of procollagen I within the ER [Citation76]. Although such large Sec31A-positive structures have not been observed in human IMR-90 fibroblasts, which secrete high levels of procollagen I, we presented evidence showing that ALG-2 knockdown leads to a delayed exit of procollagen I from the ER in IMR-90 cells [Citation21]. Thus, both ALG-2 and Sec31A contribute to efficient export of procollagen from the ER, but it remains uncertain whether large Sec31A-positive-structures mediated by KLHL12 are essential for procollagen secretion.
Table 1. Combinations of proteins bridged by ALG-2 homodimer and ALG-2-peflin heterodimer.
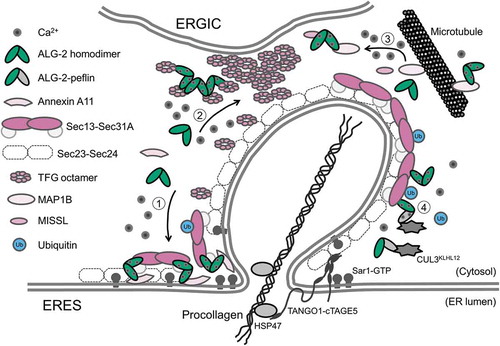
Figure 3. Schematic representation of a potential model for adaptor functions of ALG-2 in the ERES. The ALG-2 homodimer couples following combinations of target proteins: 1) Sec31A and annexin A11, 2) two TFG octamers, and 3) MISSL and MAP1B, whereas 4) the ALG-2-peflin heterodimer bridges between Sec31A and CUL3KLHL12 in a Ca2+-dependent manner.
Adaptor functions of ALG-2 in the ERES
The initial evidence for ALG-2 as a Ca2+-dependent adaptor protein came from biochemical studies on endosomal complex required for transport (ESCRT) machineries and their associated proteins in mammalian cells. ALIX is the first identified protein that interacts Ca2+-dependently with ALG-2 [Citation22,Citation23]. In the ESCRT-mediated membrane remodelling, ALIX binds and recruits ESCRT-III protein CHMP4 through its N-terminal BRO1 domain [Citation78–Citation80], which promotes the formation of a membrane-attached ESCRT-III filament [Citation81,Citation82]. The ESCRT-I protein TSG101 was reported to bind its binding sequence (717-PSAP-720 in human ALIX) in the C-terminal Pro-rich region [Citation79,Citation80]. Therefore, ALIX has been believed to link between ESCRT-I and ESCRT-III. However, in our biochemical analysis using HEK293T cells, the interaction between ALIX and TSG101 was detected only in the presence of Ca2+. Then, we found that ALG-2 homodimer mediates the Ca2+-dependent interaction between ALIX and TSG101 [Citation83]. In the ESCRT-I components, VPS37B and VPS37C have shown to be stronger interacting proteins for ALG-2 than TSG101 [Citation25]. The interaction of ALG-2 and these VPS37 isoforms contributes to stabilization of the Ca2+-sensitive complex containing ALG-2, ALIX and ESCRT-I ().
Since ALG-2 predominantly distributes to the ERES in the cytoplasm, it was conceivable that ALG-2 could function as an adaptor, not only in the ESCRT system, but also in the early secretory pathway. Screening of a bridging partner for Sec31A mediated by ALG-2 and searching for novel ALG-2-interacting proteins have unveiled pleiotropic roles of ALG-2 in the ER-to-Golgi transport through its adaptor functions to bridge between different combinations of target proteins as described below () ().
Sec31A and annexin A11
Annexins are a family of Ca2+-dependent membrane lipid-binding proteins with a highly conserved C-terminal core domain comprising four or eight annexin repeats, each of which is about 70 amino acids in length [Citation84]. In contrast, N-terminal head regions are variable in length and sequence. Annexin A11 has the longest N-terminal head of the 12 human annexins and provides binding sites for three EF-hand proteins, including calcyclin/S100A6 [Citation85], sorcin [Citation86], and ALG-2 [Citation87,Citation88]. The ALG-2-binding property of anneixn A11 is not similar to that of Sec31A: ALG-2WT binds both proteins, but ALG-2ΔGF122 fails to bind annexin A11. As described above, a reduced stable population of Sec31A at the ERES in ALG-2 knockdown cells was rescued by overexpression of ALG-2WT, but not by overexpression of ALG-2ΔGF122, raising the possibility that ALGWT recruits a specific ALG-2-interacting protein(s). In pulldown and immunoprecipitation analyses, we identified annexin A11 as a bridging partner for Sec31A mediated by ALG-2 [Citation56]. Although only a subpopulation of annexin A11 localizes to the Sec31A-positive ERES, knockdown of annexin A11 in HT1080 cells causes phenotypes similar to those of ALG-2: a decrease in the Sec31A population that is stably associated with the ERES, a scattering of juxtanuclear ERES to the cell periphery, and an acceleration of transport of VSV-G tsO45 from the ER to the Golgi apparatus. From these observations, we propose that ALG-2 and annexin A11 function in the same process by forming a ternary complex with Sec31A [Citation56].
One important question is how annexin A11 regulates Sec31A dynamics at the ERES. Annexins bind negatively charged phospholipids in a calcium-dependent manner and can organize membrane domains that function as platforms for many membrane-related events including membrane trafficking and membrane-cytoskeleton linkages [Citation84,Citation89]. The specific protein-lipid interaction in combination with protein-protein interactions might enable annexin A11 to form a structural prerequisite for stable localization of the COPII coat component at the ERES. In an in vitro reconstitution experiment, it was found that acidic phospholipids, particularly phosphatidylinositol-4,5-bisphosphate and phosphatidylinositol-4-phosphate, are actually required for efficient recruitment of the COPII component to lipid bilayers in an active Sar1-dependent manner [Citation30]. Annexin A11 assembly at the Sec31A-positive ERES might induce segregation of membrane lipids, with certain acidic phospholipids accumulating underneath annexin A11 clusters. Annexin A11 induces the formation of lens structures in the lipid bilayer in vitro [Citation90]. Such an activity could be responsible for organization of membrane domains at the ERES, which may influence the post-translational modification status of Sec31A, including ubiquitylation [Citation75], phosphorylation [Citation91], and O-GlcNAcylation [Citation59], and provide a link between calcium signaling and COPII function. Recently, ALS-associated annexin A11 mutations have been found to affect binding to S100A6/calcyclin [Citation92]. Although there is no effect of annexin A11 mutations on binding to ALG-2, annexin A11 mutations have been shown to sequester the wild-type annexin A11 into aggregates with the mutant protein. Therefore, ALS-associated annexin A11 could behave in a dominant negative-manner, which may in turn cause perturbation in protein transport from the ER.
Polymerization of TFG
Proteins that interact Ca2+-dependently with the ALG-2 homodimer and the ALG-2-peflin heterodimer continue to be identified. Although a protein that interacts Ca2+-dependently with the heterodimer has not been found, we recently identified the TRK-fused gene (TFG) product as an uncharacterized protein interacting with the ALG-2 homodimer [Citation20]. Since TFG had been shown to localize to the ERES [Citation93], it was predicted that ALG-2 could bridge between Sec31A and TFG, similar to Sec31A and annexin A11. However, Sec31A was not detected in the immunoprecipitate of TFG in the presence of Ca2+, even though overexpressed ALG-2 was efficiently precipitated with TFG [Citation20]. In addition, TFG has been shown to bind Sec23A, resulting in outcompeting Sec31A for binding to Sec23A [Citation94]. Thus, it is less likely that TFG associates and cooperates with Sec31A in regulation of transport from the ER. It is possible that ALG-2 has diverse functions through its mutually exclusive interaction with different proteins, including Sec31A, TFG and MISSL (as described below).
TFG is a ubiquitously expressed gene [Citation95] that encodes a protein with a Phox and Bem1p (PB1) domain at the N-terminus, followed by a coiled-coil domain and a C-terminal Pro and Gln (P/Q)-rich region. The protein oligomerizes to form octamers via its N-terminal region including the PB1 and coiled-coil domains in vitro. The oligomerization capability is necessary for transforming activity of the TRK-T3 gene product, which is expressed as a fusion protein of an N-terminal region of TFG and a tyrosine-kinase domain of NTRK1, a receptor for nerve growth factor [Citation96]. A mutant protein of TFG in autosomal recessive hereditary spastic paraplegia (SPG57), in which Arg106 in the coiled-coil region is replaced by Cys, p.R106C), causes a defect in normal oligomerization [Citation97]. Interestingly, the R106C mutant is defective in ability to bind to ALG-2 [Citation20], suggesting that the incapability of TFG for self-assembly and for binding to ALG-2 can lead to the development of neurodegenerative disorders. Furthermore, TFG has an ABM2-like sequence in its C-terminal P/Q-rich region that is necessary for efficient binding to ALG-2 [Citation20,Citation98].
Hanna et al. [Citation94,Citation99] recently proposed a phase separation model for regulation of TFG in the anterograde transport of COPII-coated vesicles, in which TFG at an elevated concentration potentially undergoes liquid-liquid phase separation at the interface between the ERES and ER-Golgi intermediate compartment (ERGIC). Actually, they showed that the C-terminal P/Q-rich region of TFG tethers COPII vesicles via interaction of the region with an inner coat component, Sec23A or Sec23B [Citation94]. Double depletion of Sec23A and Sec23B causes TFG to become spread diffusely throughout the cytoplasm and to fail to accumulate near the Sec16A-positive ERES [Citation94]. Although depletion of ALG-2 has no effect on the distribution pattern of TFG, overexpression of ALG-2 and Ca2+-mobilization lead to elevated levels of TFG protein at the ERES [Citation20]. TFG oligomers have been shown to self-associate into larger polymers in vitro [Citation100]. The polymerization of TFG oligomers is promoted by ALG-2 in the presence of Ca2+ [Citation20]. Therefore, in close juxtaposition of ERGIC, ALG-2 may function as a Ca2+ sensor to concentrate TFG, leading to its phase separation (). The ALG-2-TFG complex might regulate dissociation of the outer coat and restriction of diffusion of COPII vesicles coated with the inner coat, leading to disassembly of the inner coat to fuse homotypically or heterotypically with ERGIC membranes.
MISSL and MAP1B
MAPK1-interacting and spindle-stabilizing (MISS)-like (MISSL) is a Pro-rich protein of 245 amino acids containing 83 prolines (33.8%) and two PYP(X)nYP motifs that are similar to the ALG-2 binding region in ALIX. Results of our binding analysis suggested that ALG-2 recognizes multiple regions in MISSL [Citation21]. MISSL was named after the homology to mouse MISS protein [Citation101], but the C-terminal functional region in MISS, including the MAPK-docking site, is lacking in MISSL. When MISSL was identified as an ALG-2-interacting protein with ABM-1 motifs [Citation98], it was initially an uncharacterized protein with no known function. Live-cell imaging of GFP-fused MISSL (GFP-MISSL) and indirect immunofluorescence analyses revealed translocation of MISSL from the cytosol to the ERES in response to Ca2+-mobilization [Citation21]. ALG-2 is necessary for the ERES translocation of MISSL. As in the case of TFG, MISSL did not associate with Sec31A in our binding experiments. Knockdown of MISSL resulted in dispersed localization of Sec31A and Sec16A (ERES markers), ERGIC-53 (an ERGIC marker) and GM130 (a cis-Golgi marker). These results indicate that MISSL is recruited to the ERES and has a role in proper positioning of organelles of the early secretory pathway [Citation21].
To understand the molecular mechanism underlying MISSL function in the early secretory pathway, we searched for MISSL-interacting proteins using HeLa cells stably expressing GFP-MISSL and we identified microtubule-associated protein 1B (MAP1B) in the immunoprecipitate of GFP-MISSL from the cell lysate [Citation21]. Further analyses revealed that ALG-2 directly interacts with MAP1B and that ALG-2 mediates indirect association between MISSL and MAP1B in a Ca2+-dependent fashion [Citation21,Citation102]. When secretion of secreted alkaline phosphatase (SEAP) as a model protein from HeLa cells stably expressing SEAP [Citation91] was monitored, it was found that knockdown of either MISSL or ALG-2 resulted in a reduction of the secretion of SEAP [Citation21]. Although knockdown of MAP1B has little effect on secretion of SEAP, it reverts the reduced secretion of SEAP by knockdown of ALG-2 or MISSL. Taken together with previous findings that MAP1B outcompetes dynein for binding to microtubules [Citation103] and that association of inner coat components of COPII with dynactin, an essential cofactor for dynein, appears to be required for directional transport from the ER [Citation104], it is possible that MAP1B is sequestered by MISSL and ALG-2 away from microtubules (), thereby allowing dynein/dynactin to bind to microtubules to promote protein transport from the ER to the Golgi. Since the expression level of MAP1B in cells affects the amount of ALG-2 in the ERES [Citation102], it would be interesting to investigate the involvement of MISSL, ALG-2 and MAP1B in protein transport in neurons, in which MAP1B is highly expressed.
Although ALG-2 directly binds to MAP1B, no sequence similar to ABM-1 and ABM-2 was found in MAP1B. We recently narrowed down the region of mouse MAP1B that is responsible for ALG-2 binding to 36 amino acids, in which the 1825-PYGPR-1829 motif is necessary [Citation102]. In the Catalogue of Somatic Mutations in Cancer (COSMIC) database, there are two mutations, P1829L (mouse P1825L) and R1833G (mouse R1829G), in human MAP1B. These cancer-associated mutations result in an impairment of binding to ALG-2 [Citation102]. Since MAP1B knockout HeLa cells display enhanced recruitment of ALG-2 to the Sec31A-positive ERES [Citation102], the loss of binding of MAP1B to ALG-2 might cause accumulation of ALG-2 at the ERES, which might contribute to a high incidence as well as rapid progression and poor survival in cancer patients.
Conclusions and future directions
ALG-2 binds multiple target proteins in response to Ca2+-mobilization and acts as a multifunctional adaptor by utilizing distinct combinations of these targets to deliver different outputs in the early secretory pathway, including intracellular distribution of the ERES, budding of COPII vesicles, and directional transport along microtubules. It will be important to explore in more detail the extent to which ALG-2 bridges distinct combinations of target proteins in different cells. Further studies will also be needed to elucidate structural features in ALG-2 that govern the specificity of bridging partners. Recently, ALG-2 has been reported to work in repair of the plasma membrane and lysosomal membrane by sensing Ca2+ leaked from the site of injury [Citation105,Citation106]. If multiple membrane sites are damaged in severe cases, most of ALG-2 will be recruited to injury sites and the amount of ALG-2 in the ERES will be reduced, thereby affecting protein transport from the ER. Thus, it will be intriguing to see whether ALG-2 acts as a signalling molecule in response to local elevation of intracellular Ca2+, which contributes to communication between the ERES and other spatially separated organelles.
Acknowledgments
I would like to express my deepest and most sincere gratitude to Professor Masatoshi Maki (Nagoya University) for providing me the opportunity to work on this project and for his continued support, guidance and encouragement. I am grateful to Professor Kiyotaka Hitomi (Nagoya University) and Lecturer Terunao Takahara (Nagoya University) for their conscientious advice and fruitful discussions. I would also like to heartily thank Professor Stephen E. Moss (University College London) for his kind support during my stay in his laboratory. I would further like to express my appreciation to current and past members of the Laboratory of Molecular and Cellular Regulation (Nagoya University) for their wonderful collaborations in this study.
Disclosure statement
No potential conflict of interest was reported by the author.
Additional information
Funding
References
- Maki M, Kitaura Y, Satoh H, et al Structures, functions and molecular evolution of the penta-EF-hand Ca2+-binding proteins. Biochim Biophys Acta. 2002;1600:51–60.
- Maki M, Maemoto Y, Osako Y, et al Evolutionary and physical linkage between calpains and penta-EF-hand Ca2+-binding proteins. FEBS J. 2012;279:1414–1421.
- Vito P, Lacanà E, D’Adamio L. Interfering with apoptosis: Ca2+-binding protein ALG-2 and Alzheimer’s disease gene ALG-3. Science. 1996;271:521–525.
- Lacanà E, Ganjei JK, Vito P, et al Dissociation of apoptosis and activation of IL-1beta-converting enzyme/Ced-3 proteases by ALG-2 and the truncated Alzheimer’s gene ALG-3. J Immunol. 1997;158:5129–5135.
- Jang IK, Hu R, Lacaná E, et al Apoptosis-linked gene 2-deficient mice exhibit normal T-cell development and function. Mol Cell Biol. 2002;22:4094–4100.
- Rao RV, Poksay KS, Castro-Obregon S, et al Molecular components of a cell death pathway activated by endoplasmic reticulum stress. J Biol Chem. 2004;279:177–187.
- Mahul-Mellier AL, Strappazzon F, Petiot A, et al Alix and ALG-2 are involved in tumor necrosis factor receptor 1-induced cell death. J Biol Chem. 2008;283:34954–34965.
- Suzuki K, Dashzeveg N, Lu ZG, et al Programmed cell death 6, a novel p53-responsive gene, targets to the nucleus in the apoptotic response to DNA damage. Cancer Sci. 2012;103:1788–1794.
- la Cour JM, Mollerup J, Winding P, et al Up-regulation of ALG-2 in hepatomas and lung cancer tissue. Am J Pathol. 2003;163:81–89.
- la Cour JM, Høj BR, Mollerup J, et al The apoptosis linked gene ALG-2 is dysregulated in tumors of various origin and contributes to cancer cell viability. Mol Oncol. 2008;1:431–439.
- Aviel-Ronen S, Coe BP, Lau SK, et al Genomic markers for malignant progression in pulmonary adenocarcinoma with bronchioloalveolar features. Proc Natl Acad Sci USA. 2008;105:10155–10160.
- Yamada Y, Arao T, Gotoda T, et al Identification of prognostic biomarkers in gastric cancer using endoscopic biopsy samples. Cancer Sci. 2008;99:2193–2199.
- Krebs J, Saremaslani P, Caduff R. ALG-2: a Ca2+-binding modulator protein involved in cell proliferation and in cell death. Biochim Biophys Acta. 2002;1600:68–73.
- Osugi K, Shibata H, Maki M. Biochemical and immunological detection of physical interactions between penta-EF-hand protein ALG-2 and its binding partners. Methods Mol Biol. 2013;963:187–200.
- Maki M, Takahara T, Shibata H. Multifaceted roles of ALG-2 in Ca2+-Regulated membrane trafficking. Int J Mol Sci. 2016;17:E1401.
- Yamasaki A, Tani K, Yamamoto A, et al The Ca2+-binding protein ALG-2 is recruited to endoplasmic reticulum exit sites by Sec31A and stabilizes the localization of Sec31A. Mol Biol Cell. 2006;17:4876–4887.
- Shibata H, Suzuki H, Yoshida H, et al ALG-2 directly binds Sec31A and localizes at endoplasmic reticulum exit sites in a Ca2+-dependent manner. Biochem Biophys Res Commun. 2007;353:756–763.
- la Cour JM, Mollerup J, Berchtold MW. ALG-2 oscillates in subcellular localization, unitemporally with calcium oscillations. Biochem Biophys Res Commun. 2007;353:1063–1067.
- Draeby I, Woods YL, la Cour JM, et al The calcium binding protein ALG-2 binds and stabilizes Scotin, a p53-inducible gene product localized at the endoplasmic reticulum membrane. Arch Biochem Biophys. 2007;467:87–94.
- Kanadome T, Shibata H, Kuwata K, et al The calcium-binding protein ALG-2 promotes endoplasmic reticulum exit site localization and polymerization of Trk-fused gene (TFG) protein. FEBS J. 2017;284:56–76.
- Takahara T, Inoue K, Arai Y, et al The calcium-binding protein ALG-2 regulates protein secretion and trafficking via interactions with MISSL and MAP1B proteins. J Biol Chem. 2017;292:17057–17072.
- Missotten M, Nichols A, Rieger K, et al Alix, a novel mouse protein undergoing calcium-dependent interaction with the apoptosis-linked-gene 2 (ALG-2) protein. Cell Death Differ. 1999;6:124–129.
- Vito P, Pellegrini L, Guiet C, et al Cloning of AIP1, a novel protein that associates with the apoptosis-linked gene ALG-2 in a Ca2+-dependent reaction. J Biol Chem. 1999;274:1533–1540.
- Katoh K, Suzuki H, Terasawa Y, et al The penta-EF-hand protein ALG-2 interacts directly with the ESCRT-I component TSG101, and Ca2+-dependently co-localizes to aberrant endosomes with dominant-negative AAA ATPase SKD1/Vps4B. Biochem J. 2005;391:677–685.
- Okumura M, Katsuyama AM, Shibata H, et al VPS37 isoforms differentially modulate the ternary complex formation of ALIX, ALG-2, and ESCRT-I. Biosci Biotechnol Biochem. 2013;77:1715–1721.
- Okumura M, Takahashi T, Shibata H, et al Mammalian ESCRT-III-related protein IST1 has a distinctive met-pro repeat sequence that is essential for interaction with ALG-2 in the presence of Ca2+. Biosci Biotechnol Biochem. 2013;77:1049–1054.
- Vergarajauregui S, Martina JA, Puertollano R. Identification of the penta-EF-hand protein ALG-2 as a Ca2+-dependent interactor of mucolipin-1. J Biol Chem. 2009;284:36357–36366.
- Montaville P, Dai Y, Cheung CY, et al Nuclear translocation of the calcium-binding protein ALG-2 induced by the RNA-binding protein RBM22. Biochim Biophys Acta. 2006;1763:1335–1343.
- Sasaki-Osugi K, Imoto C, Takahara T, et al Nuclear ALG-2 protein interacts with Ca2+ homeostasis endoplasmic reticulum protein (CHERP) Ca2+-dependently and participates in regulation of alternative splicing of inositol trisphosphate receptor type 1 (IP3R1) pre-mRNA. J Biol Chem. 2013;288:33361–33375.
- Matsuoka K, Orci L, Amherdt M, et al COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell. 1998;93:263–275.
- Bannykh SI, Rowe T, Balch WE. The organization of endoplasmic reticulum export complexes. J Cell Biol. 1996;135:19–35.
- Budnik A, Stephens DJ. ER exit sites - localization and control of COPII vesicle formation. FEBS Lett. 2009;583:3796–3803.
- Zanetti G, Pahuja KB, Studer S, et al COPII and the regulation of protein sorting in mammals. Nat Cell Biol. 2011;14:20–28.
- D’Arcangelo JG, Stahmer KR, Miller EA. Vesicle-mediated export from the ER: COPII coat function and regulation. Biochim Biophys Acta. 2013;1833:2464–2472.
- Zeuschner D, Geerts WJ, van Donselaar E, et al Immuno-electron tomography of ER exit sites reveals the existence of free COPII-coated transport carriers. Nat Cell Biol. 2006;8:377–383.
- Shibata H, Inuzuka T, Yoshida H, et al The ALG-2 binding site in Sec31A influences the retention kinetics of Sec31A at the endoplasmic reticulum exit sites as revealed by live-cell time-lapse imaging. Biosci Biotechnol Biochem. 2010;74:1819–1826.
- Suzuki H, Kawasaki M, Inuzuka T, et al Structural basis for Ca2+-dependent formation of ALG-2/Alix peptide complex: Ca2+/EF3-driven arginine switch mechanism. Structure. 2008;16:1562–1573.
- Takahashi T, Kojima K, Zhang W, et al Structural analysis of the complex between penta-EF-hand ALG-2 protein and Sec31A peptide reveals a novel target recognition mechanism of ALG-2. Int J Mol Sci. 2015;16:3677–3699.
- Tarabykina S, Møller AL, Durussel I, et al Two forms of the apoptosis-linked protein ALG-2 with different Ca2+ affinities and target recognition. J Biol Chem. 2000;275:10514–10518.
- Shibata H, Suzuki H, Kakiuchi T, et al Identification of Alix-type and Non-Alix-type ALG-2-binding sites in human phospholipid scramblase 3: differential binding to an alternatively spliced isoform and amino acid-substituted mutants. J Biol Chem. 2008;283:9623–9632.
- Tanner JJ, Frey BB, Pemberton T, et al EF5 is the high-affinity Mg2+ site in ALG-2. Biochemistry. 2016;55:5128–5141.
- Henzl MT. Ligation events influence ALG-2 dimerization. Biophys Chem. 2018;239:16–28.
- Shugrue CA, Kolen ER, Peters H, et al Identification of the putative mammalian orthologue of Sec31P, a component of the COPII coat. J Cell Sci. 1999;112:4547–4556.
- Tang BL, Zhang T, Low DY, et al Mammalian homologues of yeast sec31p. An ubiquitously expressed form is localized to endoplasmic reticulum (ER) exit sites and is essential for ER-Golgi transport. J Biol Chem. 2000;275:13597–13604.
- Stankewich MC, Stabach PR, Morrow JS. Human Sec31B: a family of new mammalian orthologues of yeast Sec31p that associate with the COPII coat. J Cell Sci. 2006;119:958–969.
- Yoshibori M, Yorimitsu T, Sato K. Involvement of the penta-EF-hand protein Pef1p in the Ca2+-dependent regulation of COPII subunit assembly in Saccharomyces cerevisiae. PLoS One. 2012;7:e40765.
- Bi X, Corpina RA, Goldberg J. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 2002;419:271–277.
- Fath S, Mancias JD, Bi X, et al Structure and organization of coat proteins in the COPII cage. Cell. 2007;129:1325–1336.
- Noble AJ, Zhang Q, O’Donnell J, et al A pseudoatomic model of the COPII cage obtained from cryo-electron microscopy and mass spectrometry. Nat Struct Mol Biol. 2013;20:167–173.
- Yoshihisa T, Barlowe C, Schekman R. Requirement for a GTPase-activating protein in vesicle budding from the endoplasmic reticulum. Science. 1993;259:1466–1468.
- Antonny B, Madden D, Hamamoto S, et al Dynamics of the COPII coat with GTP and stable analogues. Nat Cell Biol. 2001;3:531–537.
- Sato K, Nakano A. Dissection of COPII subunit-cargo assembly and disassembly kinetics during Sar1p-GTP hydrolysis. Nat Struct Mol Biol. 2005;12:167–174.
- Forster R, Weiss M, Zimmermann T, et al Secretory cargo regulates the turnover of COPII subunits at single ER exit sites. Curr Biol. 2006;16:173–179.
- Bi X, Mancias JD, Goldberg J. Insights into COPII coat nucleation from the structure of Sec23.Sar1 complexed with the active fragment of Sec31. Dev Cell. 2007;13:635–645.
- la Cour JM, Schindler AJ, Berchtold MW, et al ALG-2 attenuates COPII budding in vitro and stabilizes the Sec23/Sec31A complex. PLoS One. 2013;8:e75309.
- Shibata H, Kanadome T, Sugiura H, et al A new role for annexin A11 in the early secretory pathway via stabilizing Sec31A protein at the endoplasmic reticulum exit sites (ERES). J Biol Chem. 2015;290:4981–4993.
- Gallione CJ, Rose JK. A single amino acid substitution in a hydrophobic domain causes temperature-sensitive cell-surface transport of a mutant viral glycoprotein. J Virol. 1985;54:374–382.
- Doms RW, Keller DS, Helenius A, et al Role for adenosine triphosphate in regulating the assembly and transport of vesicular stomatitis virus G protein trimers. J Cell Biol. 1987;105:1957–1969.
- Cho HJ, Mook-Jung I. O-GlcNAcylation regulates endoplasmic reticulum exit sites through Sec31A modification in conventional secretory pathway. FASEB J. 2018 Apr 17. DOI:10.1096/fj.201701523R
- Helm JR, Bentley M, Thorsen KD, et al Apoptosis-linked gene-2 (ALG-2)/Sec31 interactions regulate endoplasmic reticulum (ER)-to-Golgi transport: a potential effector pathway for luminal calcium. J Biol Chem. 2014;289:23609–23628.
- Rayl M, Truitt M, Held A, et al Penta-EF-hand protein peflin is a negative regulator of ER-to-golgi transport. PLoS One. 2016;11:e0157227.
- Jones B, Jones EL, Bonney SA, et al Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat Genet. 2003;34:29–31.
- Boyadjiev SA, Fromme JC, Ben J, et al Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat Genet. 2006;38:1192–1197.
- Boyadjiev SA, Kim SD, Hata A, et al Cranio-lenticulo-sutural dysplasia associated with defects in collagen secretion. Clin Genet. 2011;80:169–176.
- Fromme JC, Ravazzola M, Hamamoto S, et al The genetic basis of a craniofacial disease provides insight into COPII coat assembly. Dev Cell. 2007;13:623–634.
- McCaughey J, Stephens DJ. COPII-dependent ER export in animal cells: adaptation and control for diverse cargo. Histochem Cell Biol. 2018 Jun;18. DOI:10.1007/s00418-018-1689-2
- Aridor M. COPII gets in shape: lessons derived from morphological aspects of early secretion. Traffic. 2018 Jul;6. DOI:10.1111/tra.12603
- Saito K, Katada T. Mechanisms for exporting large-sized cargoes from the endoplasmic reticulum. Cell Mol Life Sci. 2015;72:3709–3720.
- Saito K, Chen M, Bard F, et al TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell. 2009;136:891–902.
- Ishikawa Y, Ito S, Nagata K, et al Intracellular mechanisms of molecular recognition and sorting for transport of large extracellular matrix molecules. Proc Natl Acad Sci USA. 2016;113:E6036–E6044.
- Saito K, Yamashiro K, Shimazu N, et al Concentration of Sec12 at ER exit sites via interaction with cTAGE5 is required for collagen export. J Cell Biol. 2014;206:751–762.
- Venditti R, Scanu T, Santoro M, et al Sedlin controls the ER export of procollagen by regulating the Sar1 cycle. Science. 2012;337:1668–1672.
- Saito K, Yamashiro K, Ichikawa Y, et al cTAGE5 mediates collagen secretion through interaction with TANGO1 at endoplasmic reticulum exit sites. Mol Biol Cell. 2011;22:2301–2308.
- Malhotra V, Erlmann P. Protein export at the ER: loading big collagens into COPII carriers. Embo J. 2011;30:3475–3480.
- Jin L, Pahuja KB, Wickliffe KE, et al Ubiquitin-dependent regulation of COPII coat size and function. Nature. 2012;482:495–500.
- McGourty CA, Akopian D, Walsh C, et al Regulation of the CUL3 ubiquitin ligase by a calcium-dependent co-adaptor. Cell. 2016;167:525–538.
- Gorur A, Yuan L, Kenny SJ, et al COPII-coated membranes function as transport carriers of intracellular procollagen I. J Cell Biol. 2017;216:1745–1759.
- Katoh K, Shibata H, Suzuki H, et al The ALG-2-interacting protein Alix associates with CHMP4b, a human homologue of yeast Snf7 that is involved in multivesicular body sorting. J Biol Chem. 2003;278:39104–39113.
- Strack B, Calistri A, Craig S, et al AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell. 2003;114:689–699.
- von Schwedler UK, Stuchell M, Müller B, et al The protein network of HIV budding. Cell. 2003;114:701–713.
- Hanson PI, Roth R, Lin Y, et al Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J Cell Biol. 2008;180:389–402.
- Pires R, Hartlieb B, Signor L, et al A crescent-shaped ALIX dimer targets ESCRT-III CHMP4 filaments. Structure. 2009;17:843–856.
- Okumura M, Ichioka F, Kobayashi R, et al Penta-EF-hand protein ALG-2 functions as a Ca2+-dependent adaptor that bridges Alix and TSG101. Biochem Biophys Res Commun. 2009;386:237–241.
- Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6:449–461.
- Sudo T, Hidaka H. Regulation of calcyclin (S100A6) binding by alternative splicing in the N-terminal regulatory domain of annexin XI isoforms. J Biol Chem. 1998;273:6351–6357.
- Brownawell AM, Creutz CE. Calcium-dependent binding of sorcin to the N-terminal domain of synexin (annexin VII). J Biol Chem. 1997;272:22182–22190.
- Satoh H, Shibata H, Nakano Y, et al ALG-2 interacts with the amino-terminal domain of annexin XI in a Ca2+-dependent manner. Biochem Biophys Res Commun. 2002;291:1166–1172.
- Satoh H, Nakano Y, Shibata H, et al The penta-EF-hand domain of ALG-2 interacts with amino-terminal domains of both annexin VII and annexin XI in a Ca2+-dependent manner. Biochim Biophys Acta. 2002;1600:61–67.
- Lizarbe MA, Barrasa JI, Olmo N, et al Annexin-phospholipid interactions. Functional implications. Int J Mol Sci. 2013;14:2652–2683.
- Boye TL, Jeppesen JC, Maeda K, et al Annexins induce curvature on free-edge membranes displaying distinct morphologies. Sci Rep. 2018;8:10309.
- Koreishi M, Yu S, Oda M, et al CK2 phosphorylates Sec31 and regulates ER-To-Golgi trafficking. PLoS One. 2013;8:e54382.
- Smith BN, Topp SD, Fallini C, et al Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis. Sci Transl Med. 2017;9:eaad9157.
- Witte K, Schuh AL, Hegermann J, et al TFG-1 function in protein secretion and oncogenesis. Nat Cell Biol. 2011;13:550–558.
- Hanna MG 4th, Block S, Frankel EB, et al TFG facilitates outer coat disassembly on COPII transport carriers to promote tethering and fusion with ER-Golgi intermediate compartments. Proc Natl Acad Sci USA. 2017;114:E7707–E7716.
- Greco A, Mariani C, Miranda C, et al The DNA rearrangement that generates the TRK-T3 oncogene involves a novel gene on chromosome 3 whose product has a potential coiled-coil domain. Mol Cell Biol. 1995;15:6118–6127.
- Greco A, Fusetti L, Miranda C, et al Role of the TFG N-terminus and coiled-coil domain in the transforming activity of the thyroid TRK-T3 oncogene. Oncogene. 1998;16:809–816.
- Beetz C, Johnson A, Schuh AL, et al Inhibition of TFG function causes hereditary axon degeneration by impairing endoplasmic reticulum structure. Proc Natl Acad Sci USA. 2013;110:5091–5096.
- Osugi K, Suzuki H, Nomura T, et al Identification of the P-body component PATL1 as a novel ALG-2-interacting protein by in silico and far-Western screening of proline-rich proteins. J Biochem. 2012;151:657–666.
- Hanna MG, Peotter JL, Frankel EB, et al Membrane transport at an organelle interface in the early secretory pathway: take your coat off and stay a while: evolution of the metazoan early secretory pathway. Bioessays. 2018;40:e1800004.
- Johnson A, Bhattacharya N, Hanna M, et al TFG clusters COPII-coated transport carriers and promotes early secretory pathway organization. EMBO J. 2015;34:811–827.
- Lefebvre C, Terret ME, Djiane A, et al Meiotic spindle stability depends on MAPK-interacting and spindle-stabilizing protein (MISS), a new MAPK substrate. J Cell Biol. 2002;157:603–613.
- Takahara T, Arai Y, Kono Y, et al A microtubule-associated protein MAP1B binds to and regulates localization of a calcium-binding protein ALG-2. Biochem Biophys Res Commun. 2018;497:492–498.
- Koonce MP, Tikhonenko I. Functional elements within the dynein microtubule-binding domain. Mol Biol Cell. 2000;11:523–529.
- Watson P, Forster R, Palmer KJ, et al Coupling of ER exit to microtubules through direct interaction of COPII with dynactin. Nat Cell Biol. 2005;7:48–55.
- Scheffer LL, Sreetama SC, Sharma N, et al Mechanism of Ca2⁺-triggered ESCRT assembly and regulation of cell membrane repair. Nat Commun. 2014;5:5646.
- Skowyra ML, Schlesinger PH, Naismith TV, et al Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science. 2018;360:eaar5078.