
Abstract
Thrombocytopenia or platelet dysfunction is a risk factor for severe infection. Staphylococcus aureus (S. aureus) releases a variety of virulence factors especially toxic shock syndrome toxin 1 (TSST-1), which may cause toxic shock syndrome. S. aureus, when carrying the tst gene, is more prone to cause toxic shock syndrome and is responsible for an especially high rate of mortality. However, the effect of TSST-1 protein on platelets is unknown. Patients with the tst gene positive S. aureus bacteremia showed more serious infection, higher mortality and lower platelet count. The tst gene positive S. aureus strains induce more platelet apoptosis and activation and corresponding up-regulation of Bak and down-regulation of Bcl-XL in addition to the activation of Caspase-3. C57BL/6 mice infected with the tst gene positive strains resulted in both a decrease in platelet count and an increase in platelet apoptosis and/or activation events and mortality. Moreover, TSST-1 protein, encoded by tst gene, caused the decrease of platelet count, the increase of platelet apoptosis and activation events and the level of inflammatory cytokines in vivo. However, TSST-1 protein was unable to induce traditional activation and apoptosis on human platelets in vitro. These results suggested that TSST-1 protein may exert indirect effects on platelet activation and apoptosis in vivo.
Introduction
Staphylococcus aureus (S. aureus), one of the deadliest bacterial pathogens, causes a wide spectrum of clinical disorders, including skin and soft tissue infections, bacteremia, sepsis, and toxic shock syndrome (TSS) [Citation1]. It produces a variety of virulence factors, such as staphylococcal enterotoxins (SE), exfoliative toxins (ET) A and B, as well as toxic shock syndrome toxin-1 (TSST-1). The 29.1-kDa TSST-1 protein is encoded by the tst gene and is present in 37–47% of the S. aureus strains isolated from TSS patients [Citation2,Citation3]. TSS is an acute and potentially fatal illness characterized by high fever, erythematous rash, hypotension, and respiratory distress, which may lead to multiple organ dysfunction syndrome (MODS). Patients may develop lethal shock within 24 h after onset of symptoms without appropriate treatment [Citation4]. Studies have shown that excessive inflammatory response and coagulation dysfunction in sepsis patients are pathophysiologically associated with the occurrence and development of MODS [Citation5]. Activated platelets, serving as the bridge between thrombosis and inflammation, play important roles in the pathogenesis of sepsis [Citation6,Citation7].
S. aureus pathogenicity Islands (SaPIs) are 15–17 kb mobile genetic elements that play an important role in the pathogenesis and virulence of staphylococci. The SaPIs usually carry two or three superantigen toxins and are responsible for most superantigen-related human diseases, especially TSS. The SaPIs are inserted at specific chromosomal sites and each is always in the same orientation. They are found to be highly mobile, owing to their relationship with certain phages [Citation8]. A previous study showed that several kinds of SaPIs harbor tst and other virulence gene such as seb, sec, selk, sell, and selq [Citation9]. Furthermore, the tst gene is found together with selk and selq in SaPI1, with sec3 and sell in SaPIm1 and SaPIn1, with sell and sec in SaPIbov1, with seb, selq and selk in SaPI3 [Citation8]. SaPIs induction is prone to increase the copy number of the toxin genes and therefore increase the toxin production [Citation10].
Platelet apoptosis contributes to the pathogenesis of thrombocytopenia [Citation11–14]. To the best of our knowledge, several aspects have been reported to be associated with platelet apoptosis, including mitochondrial inner transmembrane potential (Δψm) depolarization, alternation of Bcl-2 protein expression [Citation15], caspases activation, exposure of acidic antiphospholipid phosphatidylserine (PS) on the platelet plasma membrane [Citation16], as well as blebbing and filopodia formation on platelet membrane [Citation17]. Accumulating evidence indicates that Bcl-2 family proteins regulate the mitochondrial apoptosis pathway in platelets. Among these pro-survival Bcl-2 proteins, only Bcl-XL has been confirmed as interacting with Bak, one of the fated killer of platelets, causing platelet apoptosis [Citation18]. The mutations in Bcl-XL would reduce platelet survival in a dose-dependent manner, which can be corrected by deletion of Bax and Bak [Citation19]. Exposure to cytotoxic stimuli often triggers Bax/Bak activation, ultimately activating the caspase proteases. Caspase-3 is known as an executioner caspase in apoptosis, because it cleaves hundreds of intracellular substrates, leading to DNA damage, repression of transcription and protein translation, and the inhibition of other essential cellular processes involved in the process of inflammation and tissue repair [Citation20]. These studies reveal how crucial apoptotic proteins are in the regulation of platelet apoptosis.
Thrombocytopenia poses a big risk to ICU patients [Citation21]. Additionally, a close relationship was reported between thrombocytopenia, multiple-organ failure and mortality in ICU patients. In those patients, thrombocytopenia usually leads to severe sepsis and a poor prognosis [Citation21]. Many studies have confirmed that S. aureus could obviously induce the platelet activation, aggregation and apoptosis in vitro [Citation22] and in vivo [Citation23]. Platelet integrin GPIIb/IIIa can directly bind to S. aureus adhesion factor SdrG or aggregation factor ClfA through fibrinogen or directly to induce platelet activation [Citation24,Citation25]. Platelets can also interact with S. aureus surface SpA, SdrG or ClfA through Fc receptor FcγRIIa or complement receptor C1qR Combination leads to aggregation [Citation26,Citation27]. Besides, S. aureus could induce apoptotic events in platelets that include the degradation of Bcl-XL, an essential regulator of platelet survival [Citation28]. However, little is known about the activation and apoptosis of platelets induced by S. aureus with or without tst gene. In this study, we aim to explore the effects of purified TSST-1 protein and S. aureus with or without tst gene on the viability of platelets.
Materials and Methods
Patients with S. Aureus Bacteremia
Ninety-three patients with blood cultures positive for MRSA (Methicillin-resistant Staphylococcus aureus) were involved from the Second Affiliated Hospital of Soochow University between August 2018 and August 2021 retrospectively. Species identification was performed using the Phoenix 100 Automated Microbiology System (Becton-Dickinson, United States), and confirmed by 16S rRNA sequencing. This study was approved by the institutional review board (IRB) of the Second Affiliated Hospital of Soochow University. The details about the patients are found in .
Table 1. Baseline characteristics in patients with S. aureus bacteremia.
Antibodies and Reagents
Fluorescein isothiocyanate (FITC)-conjugated PAC-1 was purchased from BD Bioscience (CA, USA). The 5,5’,6,6’-tetrachloro-1,1’,3,3’- tetraethylbenzimidazolcarbocyanine iodide (JC-1) and E64 were purchased from Roche (Indianapolis, IN, USA). FITC-conjugated annexin V was purchased from Jiamay Biotech (Beijing, China). FITC-conjugated anti-mouse CD62P antibody and anti-human CD62P antibody were from Biolegend (San Diego, CA, USA). Antibodies against Caspase-3, Bax, Bak, and Bcl-XL were purchased from Santa Cruz (Santa Cruz, CA, USA). Anti-β-actin was purchased from Sigma-Aldrich (LA, USA). The purified TSST-1 protein was purchased from Toxin Technology Inc (Sarasota, USA) with an over 98% purity.
Bacterial Culture
The tst gene positive and negative strains were obtained from the Clinical Laboratory of The Second Affiliated Hospital of Soochow University (Soochow, China). All strains were investigated with pulsed field gel electrophoresis (PFGE) combined with multilocus sequence typing (MLST) to identify the genetic background, as described previously [Citation29,Citation30]. Subsequently, whole genome sequencing technology was used to select sixteen strains which belong to the same ST 5 clone. Among those strains, three isolates characterized with the Staphylococcus aureus Pathogenicity Island (SaPI) where tst was found together with sec3 and sel gene were chosen. Correspondingly, three isolates that without SaPI containing tst gene used as control. The assemblies were annotated using RAST (Rapid Annotation using Subsystem Technology) server, and the SaPI-containing contigs were extracted, followed by manual curation using NCBI BLAST (https://blast.ncbi.nlm.nih.gov/Blast) against other S. aureus SaPIs. Other virulent genes including hla, lukD, lukE, seg, sei, and sem were present in six selected isolates and they all belonged to SCCmec type II characterized with spa type t002.
All strains were cultured in a Luria-Bertani (LB) medium at 37°C for 12 h. Then the solution was centrifuged at 2,000 g for 5 min. The pellet was resuspended in fresh LB media with 30% glycerin, while the culture supernatant was stored at −80°C until use. The number of bacteria was determined by a petri dish colony-counting method. Before experiments, S. aureus stains with or without SaPI containing tst gene (2.2 × 1010 CFU/ml) were centrifuged at 2,000 g for 5 min and resuspended in modified Tyrode’s buffer (MTB, 12.1 mM NaHCO3, 136.9 mM NaCl, 5.6 mM d-glucose, 2.6 mM KCl, 2.4 mM HEPES, 1 mM CaCl2, 1 mM MgCl2, and 0.1% BSA, pH 7.4). Then these strains were resuscitated for 2 hours at 25°C before usage.
Platelet and PBMC Preparation
Blood specimens were obtained with consent from healthy volunteers (aged between 20 and 50 years). For platelet preparation, the whole blood was anti-coagulated with acid-citrate dextrose (ACD) containing 2.5% trisodium citrate, 2.0% D-glucose and 1.5% citric acid. Platelet rich plasma (PRP) was collected after centrifugation at 200 g for 11 min. The generated PRP was washed twice with CGS buffer, and then the platelets were resuspended in modified Tyrode’s buffer (MTB) to reach a final concentration of 3 × 108 /ml. Finally, the platelets were incubated at room temperature for 1.5 h before usage. For PBMC preparation, heparinized whole blood was diluted with RPMI 1640 medium and PBMC were isolated by discontinuous density gradient centrifugation on Percoll at 450 g for 30 min. PBMC harvested from the ‘buffy’ layers were washed twice by centrifugation at 600 g for 6 min with RPMI 1640 medium. The cells were counted and resuspended in complete cell culture medium. This study was approved by the Ethics Committee of the Second Affiliated Hospital of Soochow University, and informed consent was approved according to the Declaration of Helsinki.
Mitochondrial Δψm Depolarization Assay
The apoptotic inner membrane (IM) permeabilization was assessed by determining a reduction in the Δψm. PRP or MTB washed platelets (3 × 108/ml) were incubated with the tst gene positive or negative S. aureus cells or vehicle control at 37°C for 1.5 h. Lipophilic cationic probe JC-1 was added to the pretreated platelets to a final concentration of 2 μg/ml, followed by incubation at 37°C in the dark for 5 min. Mitochondrial Δψm depolarization was detected by flow cytometry according to the previous description. The JC-1 monomers (i.e. lex 514 nm and lem 529 nm) and aggregates (lex 585 nm and lem 590 nm) were calculated as the fluorescence ratio of red to green. Red fluorescence represented potential-dependent aggregation in the mitochondria, and green fluorescence reflected the monomeric form of JC-1 in the cytosol after mitochondrial membrane potential depolarization.
PS Externalization Assay
Pretreated platelets were stained with annexin V-FITC (10:1:50) in annexin V-binding buffer. Samples were incubated at room temperature for 15 min in the dark with gentle mixture; then flow cytometry was used to analyzed.
Platelet Activation
Platelet activation was assessed by P-selectin (CD62P) surface exposure and integrin αIIbβ3 activation. PRP or washed platelets in MTB (3 × 108/ml) were incubated with the tst gene positive or negative S. aureus cells or vehicle control at 37°C for 1.5 h. The treated platelets were incubated with FITC-labeled PAC-1 or FITC-labeled anti-CD62P antibody at room temperature for 20 min in the dark and then subjected to flow cytometry analysis.
Western blot Analysis
Washed platelets (3 × 108/ml) were incubated with the tst gene positive or negative S. aureus cells (2.2 × 1010 CFU/ml) or vehicle control at 37°C for 1.5 h and lysed with an equal volume of lysis buffer on ice for 30 min. Proteins were separated by SDS-PAGE. After blocking, membranes were incubated with different primary antibodies (anti-Bax, anti-Bcl-XL, anti-Bak and anti-Caspase-3) and protein bands were visualized by the ECL Chemiluminescence System on Kodak film. Quantification was performed with ImageJ software.
In Vitro Platelet Experiments
To determine the effects of bacteria on platelets, the washed platelets (3 × 108/ml) were incubated with the tst gene associated SaPI positive or negative S. aureus cells (2.2 × 1010 CFU/ml) and MTB serving as the vehicle at 37°C for 1.5 h. The bacterial suspension was revitalized for four hours at room temperature before usage. The ratio of platelets to S. aureus cells was 1: 20 in this study. To investigate the effects of the metabolites of bacteria on platelets, the platelets (3 × 108/ml) were incubated with culture supernatant of S. aureus stains and LB medium serving as the vehicle at 37°C for 1.5 h.
S. Aureus Infection and TSST-1 Treatment in Mice
Male C57BL/6 mice (6–7 weeks) were purchased from Shanghai Institute for Biological Sciences (Shanghai, China). Mice were housed individually under routine conditions at animal facilities of Soochow University at 22–24°C in a cycle of 12 h/12 h. All interventions were carried out in strict compliance with the stipulations of Regulations for the Administration of Affairs Concerning Experimental Animals (China). Experimental mice were injected with 100 μl normal saline containing the tst gene positive or negative S. aureus cells (2.2 × 108 or 2.2 × 109 CFU/mouse) or purified TSST-1 protein (0, 1, 10, 100 μg/mouse) via the posterior ophthalmic vein. Whole blood was collected at 0, 1, 2, 6, 24 and 48 h after treatment from the orbital veins of the mice. Then platelet, white blood cell and red blood cell counts were performed using KX-21 N Blood Cell Analyzer (Sysmex Corporation, Kobe, Japan). PRP was collected from whole blood after resting for 30 min and antibody labeled platelets from PRP were analyzed by flow cytometry (FACSCanto II, BD). For the evaluation of CFU determination in vivo, whole blood or aseptic saline diluted blood (1: 10 or 1: 100) was evenly coated on LB solid medium and incubated overnight in an incubator at 37°C. The number of bacteria in the whole blood was calculated by averaging the number of colonies on three plates. For the determination of inflammatory factors, serum was collected from whole blood by centrifugation at 1,000 g for 5 min. Serum inflammatory cytokines (MPC-1, TNF-α, IL-10, IFN-γ, IL-12p70, and IL-6) were tested by using a BD™ Cytometric Bead Array (CBA) Mouse Inflammation Kit.
Statistical Analysis
The significance of data was analyzed using GraphPad Prism 9 software. The normal distribution data was expressed as mean ± SEM, otherwise was expressed as median with range. Differences are considered as significant at P< .05.
RESULTS
Patients with the Tst Gene Positive S. Aureus Bacteremia Showed More Serious Infection, Higher Mortality and Lower Platelet Count
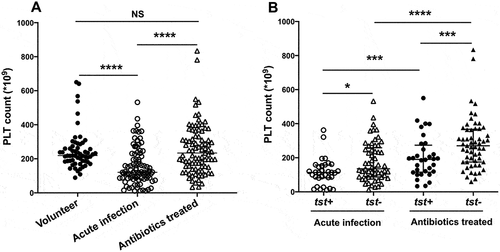
Ninety-three patients with blood cultures positive for MRSA were involved. Thrombocytopenia (<50 × 109/L) at bacteremia onset was present in 11.8% (11/93) patients. As shown in , compared with healthy volunteers, patients with acute S. aureus infection had lower platelet count (p < .0001) when S. aureus was isolated from their peripheral blood. After antibiotic treatment, platelet count was obviously elevated (p < .0001). Then, 32 strains with tst gene positive and 61 strains with tst gene negative from peripheral blood of patients were identified by PCR. As shown in , tst gene positive S. aureus infected patients had lower platelet count in the stage of acute infection (p < .05). Then, two weeks antibiotics treatment was conducted. Patients infected with tst gene positive S. aureus still had less platelets (p = .007), compared to those patients infected with tst gene negative S. aureus. The clinical data of involved 93 patients and 60 volunteers were present in the . As shown, the tst gene positive S. aureus infected patients had higher level of serum PCT concentration at the initial stage of infection. After antibiotic treatment, those patients had higher mortality with elevated CRP concentration and positive blood culture rate (p < .05). There was no difference of body temperature, breath, heart rate and blood pressure between those two group patients infected with tst gene positive or negative S. aureus.
Figure 1. Ninety-three patients with S. aureus bacteremia and 60 healthy volunteers were involved. All those patients received antibacterial treatment. Peripheral blood samples were collected when S. aureus was isolated at acute infection or after antibiotic treatment for 2 weeks. Platelets were counted using a Hemocytometer. (A) Platelet count of volunteers and patients with S. aureus bacteremia in the period of acute infection and after antibiotic treatment. Ninety-three patients were then divided into two groups, 32 patients infected with tst gene positive (tst+) isolates and 61 patients infected with tst gene negative (tst-) isolates. (B) Platelet count of tst+ and tst- in the period of acute infection and after antibiotic treatment. ****P<0.0001; **P<0.01; *P<0.05, Tukey test.
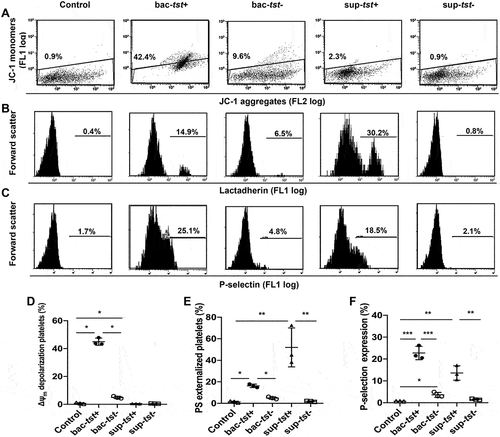
The Tst Gene Positive S. Aureus Strains Induced More Platelet Apoptosis and Activation in Vitro
To investigate the different effects of S. aureus isolates on platelet apoptosis and activation, three tst gene positive stains and three tst gene negative strains were chosen according to the M&M. The TSST-1 protein was continuously expressed on the S. aureus cells or secreted into the culture supernatant. Therefore, S. aureus cells or their supernatant were incubated with washed platelets from healthy volunteers for 1.5 h at 37°C, and then flow cytometry was used to determine the conditions of those platelets. All S. aureus cells induced significant apoptosis events in platelets with increased Δψm depolarization ( a nd D) and PS exposure ( a nd E). Meanwhile, those S. aureus cells also induced obvious activation of platelets by P-selectin expression ( a nd F) but not PAC-1 expression (data not shown). Expectedly, the tst gene positive strains had stronger effects on the platelets by inducing more apoptosis and activation than the negative strains. Notably, culture supernatant of the tst gene positive isolates can also cause PS exposure and P-selectin expression on the platelets ( a nd F). On the contrary, the culture supernatant of the tst gene negative isolates had no effect on the platelets.
Figure 2. The bacterial cells and culture supernatant of S. aureus induced platelet apoptosis and activation. Washed platelets (3×108/ml) were incubated with the bacterial pellet or culture supernatant of the tst gene associated SaPI positive (bac-tst+) or negative strains (bac-tst-) (1:20) (sup-tst+ or sup-tst-) (10 μl) or vehicle control (MTB buffer) at 37°C for 1.5 h. Representative flow cytometric figures (left) and quantification (right) of Δψm depolarization (A), PS exposure (B) and P-selectin expression (C) were shown. Data were expressed as mean ± SEM from 3 independent experiments. ****P<0.0001; ***P<0.001; *P<0.05, Tukey test.
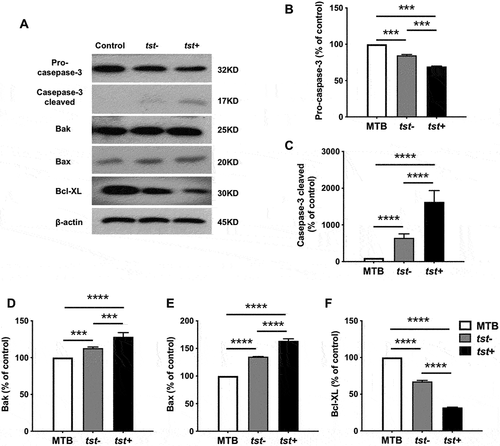
The Tst Gene Positive S. Aureus Strains Induced Higher Expression of Apoptotic Protein in Platelets
As reported, platelet apoptosis is a kind of programmed cell death mediated by mitochondria dysfunction. Upon apoptotic stimulation, Bcl-2 family proteins interact with the mitochondrial outer membrane, leading to transmembrane potential (Δψm) depolarization as well as activation of caspases [Citation17]. To clarify whether the tst gene positive S. aureus trigger more platelet apoptosis through the mitochondrial-related signal pathway, the levels of Bcl-2 family members and activated Caspase-3 were examined. Immunoblots showed that the tst gene positive S. aureus induced about 2fold increase in the caspase-3 activity, which was reflected with the elevated cleaved caspase-3 (). Meanwhile, it induced higher levels of apoptotic executors Bak () and Bax (), along with subsequent decline of pro-survival protein Bcl-XL (). Consistent with the flow cytometry data, the tst gene positive S. aureus caused more apoptotic cascades in platelets compared with the tst gene negative S. aureus.
Figure 3. S. aureus strains induced up-regulation of Bak and Bax, down-regulation of Bcl-XL, and activation of caspase-3 in platelets. Washed platelets (3×108/ml) were incubated with the tst gene associated SaPI positive (tst+) or negative isolates (tst-) (1:20) or vehicle (MTB buffer) at 37°C for 1.5 h. Representative immunoblots (A) and quantification for 32 kDa pro-caspase-3 (B), and 17 kDa cleaved caspase-3 (C), Bak (D), Bax (E) and Bcl-XL(F) are shown. Data were expressed as mean ± SEM from 3 independent experiments. ****P<0.0001; ***P<0.001, Tukey test.
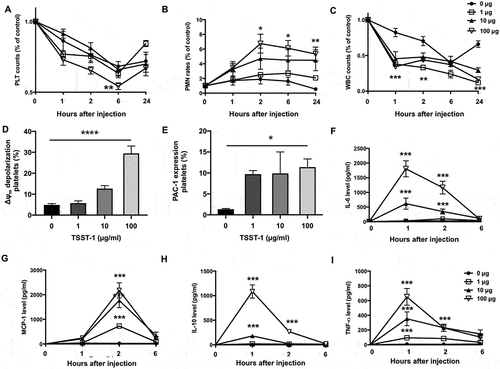
The Tst Gene Positive S. Aureus Strains Triggered Severe Sepsis in Mice by Reducing Platelets Counts, Promoting Cytokine Production and Increasing Mortality
As the tst gene-positive isolates potently influenced platelets function in vitro, whether those strains could trigger apoptosis and activation of circulating platelets in vivo need to be examined. Mice were intravenously injected with S. aureus strains and whole blood samples were collected at a series of time points. As reported, bacterial infection causes red blood cells to rupture, reducing the number of RBC in the peripheral blood [Citation31] whereas WBC usually experience a short period of decreased and a reversely increase from onset of infection to recovery [Citation32]. Herein, mice received the tst gene positive strain injection showed marked decline in platelet and RBC count at 6 h after injection, compared with those injected with the negative isolate (). There was no big difference in WBC count between those two groups (). To count the live S. aureus in blood, diluted blood was culture onto an agar medium and the colony forming units (CFUs) were counted. The average CFUs of the tst gene positive isolates-infected mice were 5fold or 10fold more, respectively, than those infected with the negative isolates when measured at 2 h or 6 h (). Accordingly, mice infected with the tst gene positive isolates showed higher mortality (). To explore the causes of thrombocytopenia, we studied platelet apoptosis and activation in the mice infected with S. aureus. As shown in , platelets from the tst gene positive isolates-infected mice presented more PS exposure, compared with those infected with the tst gene negative isolates. No significant difference in Δψm depolarization () and p-selectin expression () of platelets was observed between the tst gene positive and negative isolates-infected mice. To determine the level of inflammatory cytokines in the blood, we measured the concentration of MCP-1, TNF-α, IL-10, IFN-γ, IL-12p70, and IL-6 at 2 h after infection, the time point at which most cytokines were produced at high mounts in the preliminary experiment. S. aureus infection could cause a large release of IL-6, TNF-α, and MCP-1 into serum (); The tst gene positive induced higher level of serum MCP-1 (P < .05).
Figure 4. Peripheral blood cell counts, CFU counts, and life span of S. aureus-infected mice were shown. For A-E: The tst gene associated SaPI positive (tst+) or negative strains (tst-) (1×106 CFU/mouse) or vehicle (MTB buffer) were injected into mice through the subarachnoid vein. (A) platelet, (B) blood cell count (RBC) and (C) white blood cell (WBC) count were determined at the indicated time points. Data were expressed as mean ± SEM (n=5). *P<0.05, compared with tst-, Mann-Whitney U test. Representative image of CFU in 10 μl blood (D). CFU counts were determined at the indicated time points (E). Data were expressed as median with range (n=5). For F: Mice were injected with the tst gene associated SaPI positive (tst+) (2.2×108 or 2.2×109 CFU/mouse) or negative isolates (tst-) (2.2×108 or 2.2×109 CFU/mouse) or MTB buffer via the posterior ophthalmic vein, and the survival rate of the mice were observed for 3 days (n=5). The survival rate of mice that were treated with S. aureus infection or vehicle was determined (F).
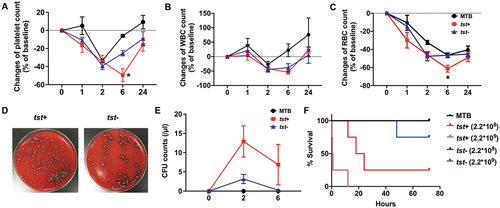
Figure 5. S. aureus strains induced circulating platelet PS exposure and inflammatory cytokine release in mice. C57BL/6 mice were injected with the tst gene associated SaPI positive (tst+) or negative isolates (tst-) (1×106 CFU/mouse) or vehicle (MTB buffer) through the subarachnoid vein. PS externalization (A), normal Δψm depolarization (B) and p-selectin (CD62p) expression (C) of platelets were determined by flow cytometry at the indicated time points. The concentration of IL-6 (D), TNF-α (E) and MCP-1 (F) in the serum were detected by CBA at 2h after injection. Quantitative data from 5 mice per group were illustrated as mean ±SEM. *P<0.05, compared with tst-, Mann-Whitney U test.
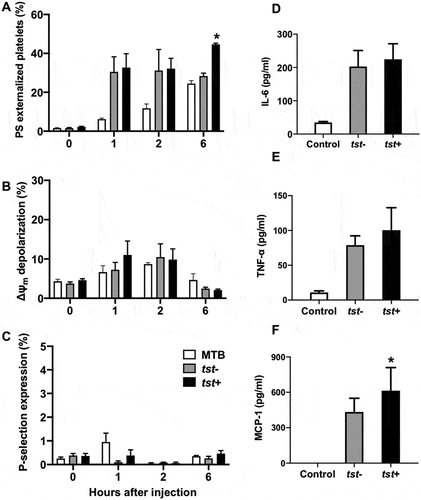
TSST-1 Protein Reduced Platelet Count and Induced Inflammatory Cytokines Production in C57BL/6 Mice
Mice were intravenously injected with purified TSST-1 and whole blood samples were collected at a series of time points. As shown, a significant decline of platelet counts was present in those mice injected with 100 μg/mouse TSST-1 after 6 h (). Meanwhile, 100 μg/mouse TSST-1 injection caused more decline in WBC counts and an increase in PMN rate ( a nd C). The circulating platelets showed increased Δψm depolarization () and PS exposure (). Purified TSST-1 could cause a high level of IL-6, MCP-1, IL-10, and TNF-α in serum at 1 h and 2 h after injection (). Those data illustrated that purified TSST-1 injection could cause platelets reduce and cytokines production in vivo.
Figure 6. TSST-1 injection could cause platelets reduce and cytokines production in vivo. Purified TSST-1(0-100 μg/mouse) was intravenously injected and whole blood samples were collected at 0, 1, 2, 6, 24 h. The platelet count (A), WBC count (B) and PMN rate (C) were determined by a Hemocytometer. Δψm depolarization (D) and PS exposure (E) were detected by a flow cytometry. ****P<0.0001, *P<0.05, Linear regression. The levels of several cytokines, including IL-6 (F), MCP-1 (G), IL-10 (H) and TNF-α (I) in serum were detected by CBA. ***P < 0.001, **P < 0.01, *P < 0.05 compared with 0 μg, Mann-Whitney U test.
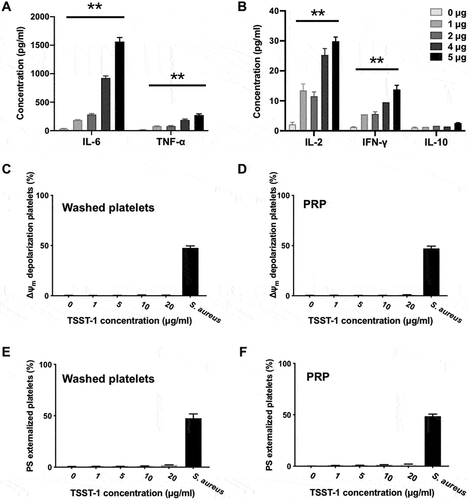
TSST-1 Protein Could Not Induce Platelet Activation and Apoptosis in Vitro
To investigate the effect of TSST-1, encoded by the tst gene, on platelets, purified TSST-1 was co-cultured with human PRP or washed platelets. We tested the activity of purified TSST-1 on human PBMC before usage. PBMC were co-cultured with TSST-1 and the level of several cytokines in the supernatant was detected by CBA. The results showed that TSST-1 could induce high level of IL-6, TNF-α, IL-2 and IFN-γ in a dose-dependent manner (). The conditions of platelets were determined by flow cytometry and the results showed that no obvious change of Δψm depolarization and PS exposure on platelets was observed even after 20 μg/ml TSST-1 stimulation (). Those data illustrated that TSST-1 was unable to induce platelet activation and apoptosis in vitro.
Figure 7. TSST-1 induced cytokine production by PBMC but was unable to stimulate platelet activation and apoptosis. PBMC (6.8×106/ml) separated from healthy volunteers were co-cultured with TSST-1(1-5 μg/ml) at 37°C for 24 h. The level of several cytokines, including IL-6, TNF-α (A), IL-2, IFN-γ, IL-10 (B) in the supernatant was detected by CBA. Human PRP and washed platelets were stimulated with TSST-1 (1-20 μg/ml) at 37°C for 1.5 h. Live S. aureus were used as a positive control. The normal Δψm depolarization (C, D) and PS externalization (E, F) of platelets were determined by flow cytometry. **P<0.01, Linear regression.
Discussion
Sepsis is a complex syndrome characterized by immune and metabolic disorders, which results in multiple organ failure. Increasing evidence suggests that there might be a close relationship between thrombocytopenia and multiple-organ failure in patients [Citation21]. In clinical settings, TSST-1 is a cause of toxic shock syndrome and multi-organ failure due to a cytokine storm triggered by T-cell activation. However, there was not much know about the influence of purified TSST-1 on platelets and sepsis. Our results showed that TSST-1 injection cause platelet reduction and inflammatory cytokine secretion in mice. However, up to 20 μg/ml of TSST-1 was inert to induce activation or apoptosis on washed human platelets or PRP in vitro. One of the potential reasons may lie in the different signaling pathways between nucleated cells and platelets. TSST-1 could bind class II major histocompatibility antigen, HLA-DR and HLA-DQ on T cells and activate T cells [Citation33]. Until now, there was no information about the effects of TSST-1 or its receptor on human platelets. Therefore, TSST-1 may exert an indirect effect on platelet through inflammatory cytokines, such as TNF-α, IL-6 and MCP-1. In our study, TSST-1 could induce several kinds of cytokine secretion, such as TNF-α, IL-6, MCP-1 and IL-10. TNF-α is a strong inductor of apoptosis through the induction of caspases. The injection of TNF-α in mice induced profound thrombocytopenia, due to an increase activation of integrin αIIbβ3 on platelet and the quick consumption of platelets. The levels of TNF-a were dependently associated with decrease of platelet count [Citation34,Citation35]. In human, TNF-α behaved as a trigger of platelet activation through the stimulation of the arachidonic acid pathway in patients with heart failure and SLE [Citation36,Citation37]. IL-6 is a proinflammatory cytokine and exerts its biological activities through the membrane-bound β-receptor glycoprotein 130, which is expressed on platelets. In vitro, IL-6 directly caused platelet hyper-activation and spreading by using scanning electron microscopy [Citation38]. Besides, other cytokines such as MCP-1, IL-1b, IL-8, and IL-10 were reported to associate with severe thrombocytopenia in dengue infection, due to the formation of platelet–monocyte aggregates [Citation39,Citation40].
Several staphylococcal virulence factors, including the cytolytic a-toxin (AT), peptidoglycan (PNG), clumping factor A (ClfA) and extracellular adherence protein (Eap) have been shown to activate platelets. AT is expressed by most clinical staphylococcal isolates and AT expression level correlates with disease severity [Citation41]. It became the first recognized bacterial cytolysin to activate human platelets and promoted blood coagulation in subcytolytic concentrations [Citation42]. It could also impair the function of platelets through glycoprotein VI modification in vitro, resulting in reduced platelet adhesion and aggregation to fibrinogen and von Willebrand factor (vWF) [Citation43]. Intravenous AT injection induced rapid platelet aggregation, dynamic micro-thrombi formation in the microcirculation and associated lethality in mice. It also was essential for S. aureus intracellular survival in liver. Neutralizing AT were completely protected against AT-induced mortality [Citation44]. Furthermore, AT mediated heterotypic platelet-neutrophil aggregation by inducing P-selectin expression on platelets [Citation45]. PNG is the major structural polymer in most bacterial cell walls. PGN induced platelet aggregation, expression of the activated form of αIIbβ3 integrin, and exposure of phosphatidylserine (PS) in vitro. In vivo, PNG might mediate S. aureus induced platelet aggregation and DIC. Pretreated with PNG, the mice had a prolong survival [Citation46]. Besides, PGN also triggered apoptosis of platelets in activation-dependent manner, characterized by mitochondrial depolarization, caspase-3 activation and cell membrane scrambling [Citation47]. ClfA is the archetype of a family of surface proteins expressed by staphylococci. It is expressed on the surface of nearly all S. aureus strains and mediates bacterial adherence to immobilized fibrinogen and cell clumping in soluble fibrinogen. ClfA binding to fibrinogen contributed to platelet aggregation via a fibrinogen- or complement-dependent mechanism or fibrinogen-independent processes [Citation48]. Lacking the bacterial ClfA brought the survival benefits in mice [Citation49]. Eap of S. aureus is a secreted protein that exerts a number of adhesive and immunomodulatory properties. Eap binding to platelets induced the activation of αIIbβ3 integrin, enhanced binding of fibrinogen and other adhesive proteins, platelet aggregation, and granule secretion [Citation50]. In this study, the lack of tst gene was associated with less CFUs in peripheral blood and lower mortality in mice.
Previous study showed that the toxin genes tst was found more frequently among MRSA isolates compared with MSSA (Methicillin-sensitive Staphylococcus aureus) isolates. ST5 was the most common ST and SCCmec type II was the most frequent SCCmec type among MRSA isolates in China, Shanghai [Citation51]. In this study, we chose MRSA strains belongs to ST5 and SCCmec type II. The tst gene positive MRSA strains induced showed more serious infection, higher mortality and more severe thrombocytopenia in this study. This conclusion is consistent with precious research. A prospective, multicenter, observational study of South Korea showed that the tst positivity of the causative S. aureus isolates was associated with an increased 2-week mortality rate in Patients with bloodstream staphylococcal infections [Citation52]. Besides, A case–control study reported that tst gene was an independent predictor of the 4-week mortality in S. aureus bacteremia patients [Citation53]. Experimental data using isogenic tst deletion mutants showed that tst+/sea+ strain induced significantly higher morbidity and mortality in tampon-associated vaginal infection model in NZW rabbits, compared with tst-/sea+ strain [Citation54]. Until now, there are very few reports on the relationship between tst gene positive MRSA isolates or TSST-1 protein and platelets. Therefore, this study is necessary and innovative. To clarify the optimal ratio and time for coculture of platelets and S. aureus, we conducted some preliminary experiments. We found that 200 million bacteria per mouse could induce a significant decrease of platelets in 24 h after bacterial infection. The count of platelets would restore in the next 24 h. Most of the experimental mice are still alive, and live S. aureus (about 100 colony forming unit [CFU] in 10 μl blood) can be detected in the peripheral blood (data not shown). 20 million bacteria per mouse could not induce obviously decrease of platelets. Meanwhile, live S. aureus in bloodstream would be cleared up within 2 h. Finally, 200 million bacteria per mouse was chosen in our study.
In summary, the tst gene positive S. aureus infection caused lower platelet count and higher mortality in human and in mice. Purified TSST-1 protein injection cause platelet reduction and inflammatory cytokine secretion in mice. However, it was inert to induce activation or apoptosis on washed human platelets or PRP in vitro. Therefore, TSST-1 protein exerted an indirect effect on platelet activation and apoptosis.
Author Contributions
MG, TY, KD, and CH conceived and designed the experiments. MG and TY performed the experiments. QW, DW, and PF analyzed the data. TY and CH wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
Disclosure Statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- SMcCaig LF, McDonald LC, Mandal S, Jernigan DB. Staphylococcus aureus-associated skin and soft tissue infections in ambulatory care. Emerg Infect Dis 2006; 12: 1715–1723. doi:10.3201/eid1211.060190.
- Jaulhac B, De Buyser ML, Dilasser F, Prevost G, Piedmont Y. Screening of staphylococci for the toxic shock syndrome toxin-1 (TSST-1) gene. Lett Appl Microbiol 1991; 13: 90–92. doi:10.1111/j.1472-765x.1991.tb00578.x.
- Weckbach LS, Thompson MR, Staneck JL, Bonventre PF. Rapid screening assay for toxic shock syndrome toxin production by Staphylococcus aureus. J Clin Microbiol 1984; 20: 18–22. doi:10.1128/JCM.20.1.18-22.1984.
- Bukowski M, Wladyka B, Dubin B. Exfoliative toxins of staphylococcus aureus. Toxins (Basel) 2010; 2: 1148–1165. doi:10.3390/toxins2051148.
- Anas AA, Wiersinga WJ, de Vos AF, van der Poll T. Recent insights into the pathogenesis of bacterial sepsis. Neth J Med 2010; 68: 147–152. PMID: 20421654.
- Dewitte A, Lepreux S, Villeneuve J, Rigothier C, Combe C, Ouattara A, Ripoche J. Blood platelets and sepsis pathophysiology: a new therapeutic prospect in critically ill patients? Ann Intensive Care 2017; 7: 115. doi:10.1186/s13613-017-0337-7.
- van der Meijden P, Heemskerk J. Platelet biology and functions: new concepts and clinical perspectives. Nat Rev Cardiol 2019; 16: 166–179. doi:10.1038/s41569-018-0110-0.
- Novick RP, Subedi A. The SaPIs: mobile pathogenicity Islands of Staphylococcus. Chem Immunol Allergy 2007; 93: 42–57. doi:10.1159/000100857.
- Sato’o Y, Omoe K, Ono HK, Nakane A, Hu DL. A novel comprehensive analysis method for Staphylococcus aureus pathogenicity Islands. Microbiol Immunol 2013; 57: 91–99. doi:10.1111/1348-0421.12007.
- Sumby P, Waldor MK. Transcription of the toxin genes present within the Staphylococcal phage phiSa3ms is intimately linked with the phage’s life cycle. J Bacteriol 2003; 185: 6841–6851. doi:10.1128/JB.185.23.6841-6851.2003.
- Leytin V, Allen DJ, Mykhaylov S, Lyubimov E, Freedman J. Thrombin-triggered platelet apoptosis. J Thromb Haemost 2006; 4: 2656–2663. doi:10.1111/j.1538-7836.2006.02200.x.
- Wang Z, Li S, Shi Q, Yan R, Liu G, Dai K. Calmodulin antagonists induce platelet apoptosis. Thromb Res 2010; 125: 340–350. doi:10.1016/j.thromres.2010.02.001.
- Zhang W, Zhao LL, Liu J, Du J, Wang Z, Ruan C, Dai K. Cisplatin induces platelet apoptosis through the ERK signaling pathway. Thromb Res 2012; 130: 81–91. doi:10.1016/j.thromres.2012.02.013.
- Zhang J, Chen M, Zhang Y, Zhao L, Yan R, Dai K. Carmustine induces platelet apoptosis. Platelets 2015; 26: 437–442. doi:10.3109/09537104.2014.928676.
- Rong Y, Distelhorst CW. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol 2008; 70: 73–91. doi:10.1146/annurev.physiol.70.021507.105852.
- Danial NN, Korsmeyer S. Cell death: critical control points. Cell 2004; 116: 205–219. doi:10.1016/s0092-8674(04)00046-7.
- Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, et al. Programmed anuclear cell death delimits platelet life span. Cell 2007; 128: 1173–1186. doi:10.1016/j.cell.2007.01.037.
- Kodama T, Takehara T, Hikita H, Shimizu S, Shigekawa M, Li W, Miyagi T, Hosui A, Tatsumi T, Ishida H, et al. BH3-only activator proteins Bid and Bim are dispensable for Bak/Bax-dependent thrombocyte apoptosis induced by Bcl-xL deficiency: molecular requisites for the mitochondrial pathway to apoptosis in platelets. J Biol Chem 2011; 286: 13905–13913. doi:10.1074/jbc.M110.195370.
- Kelly PN, White MJ, Goschnick MW, Fairfax KA, Tarlinton DM, Kinkel SA, Bouillet P, Adams JM, Kile BT, Strasser A. Individual and overlapping roles of BH3-only proteins Bim and Bad in apoptosis of lymphocytes and platelets and in suppression of thymic lymphoma development. Cell Death Differ 2010; 17: 1655–1664. doi:10.1038/cdd.2010.43.
- Josefsson EC, Burnett DL, Lebois M, Debrincat MA, White MJ, Henley KJ, Lane RM, Moujalled D, Preston SP, O’Reilly LA, et al. Platelet production proceeds independently of the intrinsic and extrinsic apoptosis pathways. Nat Commun 2014; 5: 3455. doi:10.1038/ncomms4455.
- Vanderschueren S, De Weerdt A, Malbrain M, Vankersschaever D, Frans E, Wilmer A, Bobbaers H. Thrombocytopenia and prognosis in intensive care. Crit Care Med 2000; 28: 1871–1876. doi:10.1097/00003246-200006000-00031.
- Speth C, Rambach G, Würzner R, Lass-Flörl C, Kozarcanin H, Hamad OA, Nilsson B, Ekdahl KN. Complement and platelets: mutual interference in the immune network. Mol Immunol 2015; 67: 108–118. doi:10.1016/j.molimm.2015.03.244.
- Jenne CN, Urrutia R, Kubes P. Platelets: bridging hemostasis, inflammation, and immunity. Int J Lab Hematol 2013; 35: 254–261. doi:10.1111/ijlh.12084.
- Miajlovic H, Zapotoczna M, Geoghegan JA, Kerrigan SW, Speziale P, Foster TJ. Direct interaction of iron-regulated surface determinant IsdB of Staphylococcus aureus with the GPIIb/IIIa receptor on platelets. Microbiology (Reading, England) 2010; 156: 920–928. doi:10.1099/mic.0.036673-0.
- Fitzgerald JR, Loughman A, Keane F, Brennan M, Knobel M, Higgins J, Visai L, Speziale P, Cox D, Foster TJ. Fibronectin-binding proteins of Staphylococcus aureus mediate activation of human platelets via fibrinogen and fibronectin bridges to integrin GPIIb/IIIa and IgG binding to the FcgammaRIIa receptor. Mol Microbiol 2006; 59: 212–230. doi:10.1111/j.1365-2958.2005.04922.x.
- Loughman A, Fitzgerald JR, Brennan MP, Higgins J, Downer R, Cox D, Foster TJ. Roles for fibrinogen, immunoglobulin and complement in platelet activation promoted by Staphylococcus aureus clumping factor A. Mol Microbiol 2005; 57: 804–818. doi:10.1111/j.1365-2958.2005.04731.x.
- Kerrigan SW. The expanding field of platelet-bacterial interconnections. Platelets 2015; 26: 293–301. doi:10.3109/09537104.2014.997690.
- Zhao L, Liu J, He C, Yan R, Zhou K, Cui Q, Meng X, Li X, Zhang Y, Nie Y, et al. Protein kinase A determines platelet life span and survival by regulating apoptosis. J Clin Invest 2017; 127: 4338–4351. doi:10.1172/JCI95109.
- Wang M, Zheng Y, Mediavilla JR, Chen L, Kreiswirth BN, Song Y, Yang R, Du H. Hospital dissemination of tst-1-positive clonal complex 5 (CC5) methicillin-resistant Staphylococcus aureus. Front Cell Infect Microbiol 2017; 7: 101. doi:10.3389/fcimb.2017.00101.
- Zheng Y, Qin C, Zhang X, Zhu Y, Li A, Wang M, Tang Y, Kreiswirth BN, Chen L, Zhang H, et al. The tst gene associated Staphylococcus aureus pathogenicity Island facilitates its pathogenesis by promoting the secretion of inflammatory cytokines and inducing immune suppression. Microb Pathog 2020; 138: 103797. doi:10.1016/j.micpath.2019.103797.
- Bateman RM, Sharpe MD, Singer M, Ellis CG. The effect of sepsis on the erythrocyte. Int J Mol Sci 2017; 18: 1932. doi:10.3390/ijms18091932.
- Honda T, Uehara T, Matsumoto G, Arai S, Sugano M. Neutrophil left shift and white blood cell count as markers of bacterial infection. Clin Chim Acta 2016; 457: 46–53. doi:10.1016/j.cca.2016.03.017.
- Krogman A, Tilahun A, David CS, Chowdhary VR, Alexander MP, Rajagopalan G. HLA-DR polymorphisms influence in vivo responses to staphylococcal toxic shock syndrome toxin-1 in a transgenic mouse model. HLA 2017; 89: 20–28. doi:10.1111/tan.12930.
- Davizon-Castillo P, McMahon B, Aguila S, Bark D, Ashworth K, Allawzi A, Campbell RA, Montenont E, Nemkov T, D’Alessandro A, et al. TNF-α-driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood 2019; 134: 727–740. doi:10.1182/blood.2019000200.
- Tacchini-Cottier F, Vesin C, Redard M, Buurman W, Piguet PF. Role of TNFR1 and TNFR2 in TNF-induced platelet consumption in mice. J Immunol 1998; 160: 6182–6186.
- Nagahama M, Nomura S, Ozaki Y, Yoshimura C, Kagawa H, Fukuhara S. Platelet activation markers and soluble adhesion molecules in patients with systemic lupus erythematosus. Autoimmunity 2001; 33: 85–94. doi:10.3109/08916930108995993.
- Pignatelli P, De Biase L, Lenti L, Tocci G, Brunelli A, Cangemi R, Riondino S, Grego S, Volpe M, Violi F. Tumor necrosis factor-alpha as trigger of platelet activation in patients with heart failure. Blood 2005; 106: 1992–1994. doi:10.1182/blood-2005-03-1247.
- Bester J, Pretorius E. Effects of IL-1β, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci Rep 2016; 6: 32188. doi:10.1038/srep32188.
- Azeredo EL, Zagne SM, Santiago MA, Gouvea AS, Santana AA, Neves-Souza PC, Nogueira RM, Miagostovich MP, Kubelka CF. Characterisation of lymphocyte response and cytokine patterns in patients with dengue fever. Immunobiology 2001; 204: 494–507. doi:10.1078/0171-2985-00058.
- Hottz ED, Medeiros-de-moraes IM, Vieira-de-abreu A, de Assis EF, Vals-de-souza R, Castro-Faria-Neto HC, Weyrich AS, Zimmerman GA, Bozza FA, Bozza PT. Platelet activation and apoptosis modulate monocyte inflammatory responses in dengue. J Immunol 2014; 193: 1864–1872. doi:10.4049/jimmunol.1400091.
- Jenkins A, Diep BA, Mai TT, Vo NH, Warrener P, Suzich J, Stover CK, Sellman BR. Differential expression and roles of Staphylococcus aureus virulence determinants during colonization and disease. mBio 2015; 6: e02272–14. doi:10.1128/mBio.02272-14.
- Bhakdi S, Muhly M, Mannhardt U, Hugo F, Klapettek K, Mueller-Eckhardt C, Roka L. Staphylococcal alpha toxin promotes blood coagulation via attack on human platelets. J Exp Med 1988; 168: 527–542. doi:10.1084/jem.168.2.527.
- Powers ME, Becker RE, Sailer A, Turner JR, Bubeck Wardenburg J. Synergistic action of Staphylococcus aureus alpha-toxin on platelets and myeloid lineage cells contributes to lethal sepsis. Cell Host Microbe 2015; 17: 775–787. doi:10.1016/j.chom.2015.05.011.
- Surewaard B, Thanabalasuriar A, Zeng Z, Tkaczyk C, Cohen TS, Bardoel BW, Jorch SK, Deppermann C, Bubeck Wardenburg J, Davis RP, et al. α-toxin induces platelet aggregation and liver injury during Staphylococcus aureus sepsis. Cell Host Microbe 2018; 24: 271–284.e3. doi:10.1016/j.chom.2018.06.017.
- Parimon T, Li Z, Bolz DD, McIndoo ER, Bayer CR, Stevens DL, Bryant AE. Staphylococcus aureus α-hemolysin promotes platelet-neutrophil aggregate formation. J Infect Dis 2013; 208: 761–770. doi:10.1093/infdis/jit235.
- Murphey ED, Fang G, Sherwood ER. Pretreatment with the Gram-positive bacterial cell wall molecule peptidoglycan improves bacterial clearance and decreases inflammation and mortality in mice challenged with Staphylococcus aureus. Crit Care Med 2008; 36: 3067–3073. doi:10.1097/CCM.0b013e31818c6fb7.
- Towhid ST, Nega M, Schmidt EM, Schmid E, Albrecht T, Münzer P, Borst O, Götz F, Lang F. Stimulation of platelet apoptosis by peptidoglycan from Staphylococcus aureus 113. Apoptosis 2012; 17: 998–1008. doi:10.1007/s10495-012-0718-1.
- Loughman A, Fitzgerald JR, Brennan MP, Higgins J, Downer R, Cox D, Foster TJ. Roles for fibrinogen, immunoglobulin and complement in platelet activation promoted by Staphylococcus aureus clumping factor A. Mol Microbiol 2005; 57: 804–818. doi:10.1111/j.1365-2958.2005.04731.x.
- Flick MJ, Du X, Prasad JM, Raghu H, Palumbo JS, Smeds E, Höök M, Degen JL. Genetic elimination of the binding motif on fibrinogen for the S. aureus virulence factor ClfA improves host survival in septicemia. Blood 2013; 121: 1783–1794. doi:10.1182/blood-2012-09-453894.
- Bertling A, Niemann S, Hussain M, Holbrook L, Stanley RG, Brodde MF, Pohl S, Schifferdecker T, Roth J, Jurk K, et al. Staphylococcal extracellular adherence protein induces platelet activation by stimulation of thiol isomerases. Arterioscler Thromb Vasc Biol 2012; 32: 1979–1990. doi:10.1161/ATVBAHA.112.246249.
- Gu F, He W, Xiao S, Wang S, Li X, Zeng Q, Ni Y, Han L. Antimicrobial resistance and molecular epidemiology of Staphylococcus aureus causing bloodstream infections at ruijin hospital in Shanghai from 2013 to 2018. Sci Rep 2020; 10: 6019. doi:10.1038/s41598-020-63248-5.
- Kim D, Hong JS, Yoon EJ, Lee H, Kim YA, Shin KS, Shin JH, Uh Y, Shin JH, Park YS, et al. Toxic shock syndrome toxin 1-producing methicillin-resistant Staphylococcus aureus of clonal complex 5, the New York/Japan epidemic clone, causing a high early-mortality rate in patients with bloodstream infections. Antimicrob Agents Chemother 2019; 63: e01362–19. doi:10.1128/AAC.01362-19.
- Yi J, Park JS, Hong KH, Lee SH, Kim EC. Serum capacity to neutralize superantigens does not affect the outcome of Staphylococcus aureus bacteremia. Eur J Clin Microbiol Infect Dis 2012. doi:10.1007/s10096-011-1541-2.
- De Boer ML, Kum WW, Chow AW. Staphylococcus aureus isogenic mutant, deficient in toxic shock syndrome toxin-1 but not staphylococcal enterotoxin A production, exhibits attenuated virulence in a tampon-associated vaginal infection model of toxic shock syndrome. Can J Microbiol 1999; 45: 250–256. PMID: 10408098 doi:10.1139/w99-015.