
Abstract
Alantolactone (ALT), a sesquiterpene lactone compound isolated from Inula helenium L., has recently attracted much attention for its anti-tumor properties. ALT reportedly functions by regulating the Akt pathway, which has been shown to be involved in programmed platelet death (apoptosis) and platelet activation. However, the precise effect of ALT on platelets remains unclear. In this study, washed platelets were treated with ALT in vitro, and apoptotic events and platelet activation were detected. In vivo, platelet transfusion experiments were employed to detect the effect of ALT on platelet clearance. Platelet counts were examined after intravenous injection of ALT. We found that ALT treatment induced Akt activation and Akt-mediated apoptosis in platelets. ALT-activated Akt elicited platelet apoptosis by activating phosphodiesterase (PDE3A) and PDE3A-mediated protein kinase A (PKA) inhibition. Pharmacological inhibition of the PI3K/Akt/PDE3A signaling pathway or PKA activation was found to protect platelets from apoptosis induced by ALT. Moreover, ALT-induced apoptotic platelets were removed faster in vivo, and ALT injection resulted in the platelet count decline. Either PI3K/Akt/PDE3A inhibitors or a PKA activator could protect platelets from clearance, ultimately ameliorating the ALT-induced decline in platelet count in the animal model. These results reveal the effects of ALT on platelets and their related mechanisms, suggesting potential therapeutic targets for the prevention and alleviation of possible side effects resulting from ALT treatments.
Plain Language Summary
What is the context?
In the past several decades, natural products, including traditional Chinese medicine (TCM), have been developed for the treatment of a variety of diseases.
Alantolactone (ALT), a natural herb compound mainly extracted from the root of Inula helenium L., is the essential active component in many TCM formulas. ALT has attracted extensive attention because of its anti-cancer capacity recently.
However, adverse events (AEs) induced by drugs are common in chemotherapy, and the side effects of ALT treatment remain unclear.
What is new?
In this study, experiments were conducted to clarify the precise effect of ALT on platelets. We demonstrated for the first time that ALT induces platelet apoptosis and platelet count decline, suggesting possible side effects of ALT treatment.
ALT-activated Akt elicited platelet apoptosis by activating phosphodiesterase (PDE3A) and PDE3A-mediated protein kinase A (PKA) inhibition.
Our work provides experimental evidence supporting the hypothesis that the effects of ALT on Akt may vary depending on cell types. Therefore. More research is needed to explore the side effects of ALT on other cells before clinical application.
What is the impact?
This study reveals possible side effects of ALT treatment, providing the reference for clinic drug administrate and estimation of medicine safety. Significantly, our findings demonstrated relevant molecular mechanisms, providing strategies for controlling or alleviating these side effects in the future.
Introduction
ALT is a natural herb compound mainly extracted from the root of Inula helenium L., which is also a well-known plant in traditional Chinese medicine and is officially listed in some European Pharmacopoeias as elecampane [Citation1]. In traditional medicine, ALT is widely used to treat diseases such as asthma, bronchitis, chronic enterogastritis, and tuberculosis [Citation2,Citation3]. Recently, ALT has received attention owing to its anti-tumor efficacy [Citation4], which has been demonstrated in various types of cancer, such as lung cancer [Citation5], liver cancer [Citation6], hematological malignancy [Citation7], osteosarcoma [Citation8] and gastric cancer [Citation9,Citation10]. Previously, ALT was shown to inhibit proliferation and trigger apoptosis in multiple myeloma (MM) cells, partly damage the bone marrow (BM) microenvironment, and overcome proteasome inhibitor resistance [Citation11]. Compared to conventional systemic cytotoxic chemotherapies, antineoplastic drugs targeting specific molecules cause lower levels of toxicity. Recent studies have investigated the molecular mechanisms by which ALT affects cancer cells. Results suggest that ALT exerts its anti-tumor effects by regulating signaling pathways and abnormal regulatory factors that affect the progression of cancer cells [Citation12]. However, adverse events can also occur as drug-targeted molecules may be expressed in normal cells, the modulation of which can affect important cellular functions. The side effects of ALT need to be clearly identified to determine whether ALT could be utilized clinically.
Platelets, anucleate cytoplasmic discs derived from megakaryocytes (MKs), play a pivotal role in hemostasis and arterial thrombogenesis. At sites of vascular injury, platelet adhesion, activation, and aggregation and the elaboration of procoagulant surface activity are critical events to stop bleeding [Citation13,Citation14]. Approximately one trillion platelets circulate in the blood of an adult human, and their average lifespan is approximately 810 days. The steady-state number of mature platelets results from the balance between platelet production and destruction, which is strictly regulated under physiological conditions [Citation15,Citation16]. Apoptosis or programmed cell death, exclusively attributed to nucleated cells, is a crucial physiologic mechanism regulating the platelet lifespan, and serves as the controlled deletion of senescent platelets [Citation17]. However, accelerated platelet apoptosis triggered by multiple pathological stimuli and chemical compounds are associated with a low platelet count (thrombocytopenia), leading to bleeding disorders [Citation18,Citation19]. Platelet apoptosis and thrombocytopenia induced by chemotherapy are common, potentially profound adverse effects that severely limit the application of numerous drugs [Citation20,Citation21]. Our team has demonstrated that several drugs such as ethanol, carmustine, aspirin, and carbamazepine as well as antiplatelet antibodies lead to thrombocytopenia and hemorrhage by facilitating platelet apoptosis [Citation22–26].
Platelets have highly specialized intracellular signaling systems. The intrinsic mitochondria-dependent pathway of apoptosis in platelets has been well-documented and is defined by an imbalance between the pro-survival Bcl-2 proteins (Bcl-2, Bcl-xL, Mcl-1, Bcl-W and A1) and pro-apoptotic proteins Bak and Bax, which leads to mitochondrial transmembrane potential (ΔΨm) depolarization and activation of caspases [Citation17]. Platelets undergoing apoptosis also exhibit increased phosphatidylserine (PS) exposure on the membrane surface [Citation19]. Moreover, the involvement of functional proteins and related signaling pathways, such as the apoptosis signal-regulating kinase (ASK1) [Citation27], p38 mitogen-activated protein (MAP) kinase/cytosolic phospholipase A2 (cPLA2) pathway [Citation28], extracellular signal-regulated protein kinase (ERK) [Citation29], and c-Jun NH2-terminal kinase (JNK) [Citation30,Citation31], were identified in apoptotic platelets. We previously revealed that Akt, the downstream effector of PI3K, was activated after anti-GPIbα antibody binding. Akt activation plays an important role in anti-GPIbα antibody-induced platelet apoptosis [Citation26]. Importantly, the PI3K/Akt signaling pathway plays an essential role in the pharmacological activity of ALT. ALT was demonstrated to promote Akt activation during the treatment of chronic skeletal muscle inflammation [Citation2]. On the contrary, the suppression of Akt activity after ALT treatment was detected in squamous cell lung cancer [Citation32], liver cancer [Citation6], osteosarcoma [Citation8], and breast cancer [Citation33]. The effects of ALT on Akt may vary depending on cell types; therefore, the impact of ALT on platelets is unpredictable.
In this study, to clarify the effects of ALT on platelets, human and murine platelets were incubated with ALT in vitro, and platelet apoptosis and activation were assessed. The activity of the Akt pathway and its downstream effectors were also evaluated. The clearance rate of ALT-treated platelets was examined and the platelet count in circulation was measured after ALT injection to investigate the effects of ALT on the lifespan of platelets in vivo. Our study might be valuable in developing strategies to prevent and treat the possible side effects resulting from the use of ALT.
Materials and methods
Healthy volunteers
Age- and sex-matched healthy volunteers were recruited for this study. To obtain whole-blood samples, approval was obtained from the Ethics Committee of the First Affiliated Hospital of Soochow University. Each participant provided written informed consent and the study was conducted in accordance with the Helsinki Declaration.
Animals
Bad-/- mice were generated using a 129/SvJ background [Citation19], and the Akt-/- mice were generated using a 129/Ola and C57BL/6J mixed background as described previously [Citation18]. C57BL/6J WT mice were purchased from JOINN Laboratories (Suzhou, China). All mutations had been backcrossed onto the C57BL/6J background for at least 10 generations prior to use in this study. Mice were 612 weeks old, weighing 20-25 g. Each experiment included groups with balanced numbers of males and females unless otherwise stated. Animals were maintained in the SPF-class Laboratory Animal Center of Soochow University. All animal experiments followed the standard biosafety procedures and were approved by the Ethics Committee of the First Affiliated Hospital of Soochow University.
Antibodies and reagents
Alantolactone (ALT) (M44233) was purchased from Abmole BioScience (Houston, Texas, United States). FITC-conjugated PAC-1 (3405007) was purchased from BD Biosciences (San Jose, CA, United States). FITC-conjugated anti-human P-selectin antibody (304904) was purchased from Biolegend (San Diego, CA, United States). FITC-conjugated lactadherin (BLAC-FITC) was purchased from Hematologic Technologies (Essex Junction, VT, United States). Antibodies against caspase-3 (9662), phospho-Akt (Ser473) (4060), phospho-Akt (Thr308) (13038), Akt (9272), Bcl-xL (2764), Bax (2772), phospho-p53 (Ser15) (9284), phospho-VASP (Ser157) (84519), and GAPDH (5174) were purchased from Cell Signaling Technology (Beverly, MA, United States). Phospho-Bad (Ser155) (sc101641) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, United States). JC-1 (C2005) and forskolin (1612) were purchased from the Beyotime Institute of Biotechnology (Beyotime, Shanghai, China). PE-conjugated anti-mouse JON/A antibody was purchased from Emfret analytics (Eibelstadt, Germany). Akt inhibitor Akti VIII (HY-10355), MK 2206 (HY10358) and PDE3A inhibitor Milirone (HY-14252) were purchased from MedChem Express (Princeton, NJ, United States). PI3K inhibitor LY294002 (M1925) and Wortmannin (M2053) were purchased from Abmole BioScience (Houston, TX, United States). PDE3A inhibitor Cilostazol (S1294) was purchased from Selleck (Houston, TX, United States). Protease inhibitor cocktail (K1007) was purchased from APExBIO (Houston, TX, United States).
Platelet preparation and counting
Whole-blood samples were taken from the cubital veins of the healthy human volunteers and anticoagulated with a 1/7 volume of acidcitratedextrose (ACD: 2.5% trisodium citrate, 2.0% D-glucose, 1.5% citric acid). For mice, blood samples were collected from the inferior vena cava and anticoagulated with a 1/9 volume of ACD. Platelets were washed with CGS buffer (0.123 M NaCl, 0.033 M D-glucose, and 0.013 M trisodium citrate, pH 6.5), resuspended in modified Tyrode’s buffer (MTB) (2.5 mM HEPES, 150 mM NaCl, 2.5 mM KCl, 12 mM NaHCO3, 5.5 mM D-glucose, 1 mM CaCl2, 1 mM MgCl2, pH 7.4) to a final concentration of 3 × 108/mL, and then allowed to stay at 22°C for 1-2 h. Platelet counting was performed with a Sysmex XP-100 Hematologic Analyzer (Sysmex Corporation, Kobe, Japan).
Treatment of platelets with ALT in vitro
Human or murine washed platelets were incubated with 5 μM ALT for 4 h, 8 h, and 12 h or incubated with different concentrations of ALT for 8 h. All platelet samples were gently stirred continuously under sterile conditions and stored at 37°C. In inhibition experiments, platelets were pre-incubated with diverse inhibitors or vehicle control dimethyl sulfoxide (DMSO) at 37°C for 10 min before ALT treatment.
Flow cytometry
Platelet apoptosis indexes, mitochondrial inner transmembrane potential (ΔΨm) depolarization, and PS exposure of the human or murine platelets were detected by JC-1 (2 μg/mL) and FITC-labeled lactadherin (10 μg/mL), respectively. Platelet activation was detected by P-selectin (CD62P) expression and integrin αIIbβ3 activation. P-selectin expression was detected with FITC-labeled anti-human (10 μg/mL) or anti-mouse P-selectin (1:5) antibodies. αIIbβ3 activation was detected by FITC-labeled PAC-1 (25 μg/mL) binding to human platelets or PE-labeled JON/A (1:5) binding to murine platelets.
Western blot
Washed human or murine platelets (3 × 108/mL) were lysed with a 1/9 volume of 10 × lysis buffer containing phenylmethylsulfonyl fluoride (1 mM) and protease inhibitor cocktail on ice for 30 min and boiled for 10 min. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the membranes were incubated with primary antibodies and HRP-conjugated secondary antibodies. Blots were developed using an ECL chemiluminescence system. Quantification was performed with ImageJ software (https://imagej.net/Fiji).
Effects of ALT on the counts for platelets in circulation
C57BL/6J WT mice were intravenously injected ALT (2 mg/kg) dissolved in 100 μL DMSO or vehicle control. In the PKA activation experiment, C57BL/6J WT mice were pre-treated with forskolin (5 mg/kg) dissolved in 100 μL phosphate-buffered saline (PBS) or vehicle control through intraperitoneal injection for 30 min, before the addition of ALT (2 mg/kg). Peripheral blood samples were collected via a capillary tube from the postorbital veins at various time points and anticoagulated with ACD. Platelet counts were determined by the Sysmex XP-100 hematology analyzer.
Platelet lifespan analysis after ALT treatment
Washed murine platelets labeled with calcein were incubated with ALT (10 μM) at 37°C for 4 h and then injected intravenously into C57BL/6J receptor mice (1 × 108 platelets in 100 μL MTB). In inhibition experiments, calcein-labeled platelets were pre-treated with milrinone (10 μM), cilostazol (10 μM), forskolin (5 μM), or vehicle control at 37°C for 10 min before incubation with ALT (5 μM) in vitro, and they were then transfused to receptor mice. Whole blood was collected via a capillary tube from the post-glomus venous plexus at 0 h, 0.25 h, and 0.5 h after transfusion and anticoagulated with ACD. Whole blood (5 μL) was diluted with MTB (45 μL) and incubated with PE-conjugated anti-mouse CD41 antibody at 22°C for 30 min. The percentage of calcein-labeled platelets was measured by flow cytometry (FC 500, Beckman-Coulter, Miami, FL, USA).
Statistical analysis
All experiments were repeated at least three times and data were presented as the mean ± SD. The significance of the data was analyzed using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA) software. The variables were conformed to a normal distribution using the ShapiroWilk’s test. Differences between groups were analyzed using one-way (for a single variable) or two-way (for multiple variables) analysis of variance (ANOVA) when the samples were homoscedastic through Levene’s test. The means of two groups were compared using the two-tailed Student’s t-test. For all analyses, P < .05 was considered to be statistical signficant. All animal experiments were subject to randomization based on litter. No animals or samples were excluded from the study. The sample size was predetermined based on the variability observed in prior experiments and on preliminary data. Investigators were not blinded to outcome assessment.
Results
ALT induces platelet apoptosis in vitro
Experiments were performed on human and murine platelets to assess whether ALT modulates platelet apoptosis. The lipophilic cation JC-1 was used to detect ΔΨm depolarization by flow cytometry. We found that ALT time-dependently triggered ΔΨm depolarization in human platelets, with an efficacy of~90% within 12 h (). On the other hand, ALT markedly promoted caspase-3 activation in human platelets, indicated by the appearance of 17-kDa fragments and a reduction in total caspase-3 (). Furthermore, activated caspases combined with other apoptogenic enzymes disrupted plasma membrane integrity, leading to PS externalization during apoptosis; ALT was shown to induce PS exposure of human platelets in a time-dependent manner (). In addition, the results from murine platelets followed that in human platelets. (Supplemental Figure S1).
Figure 1. ALT time-dependently induces human platelet apoptosis. Washed human platelets were incubated with ALT (5 μM) or vehicle control (DMSO) at 37°C for 4 h, 8 h or 12 h, and platelet apoptosis was detected at different time points. (a) ΔΨm depolarization was detected and quantitatively analyzed by flow cytometry. (b-d) Caspase-3 activity was evaluated using western blot analysis (b), and the densitometric analyses of the immunoblots for full caspase-3 (c) and cleaved caspase-3 (d) were performed. (e) PS exposure was measured by flow cytometry. Data presented are from three independent experiments and expressed as the mean ± SD; * p < .05, ** p < .01, and *** p < .001 versus the control with a two-tailed Student’s t-test.
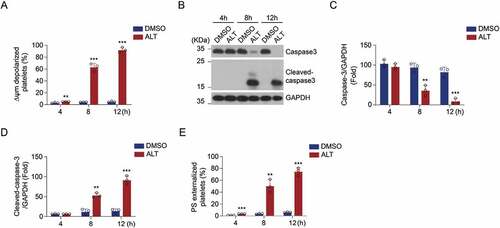
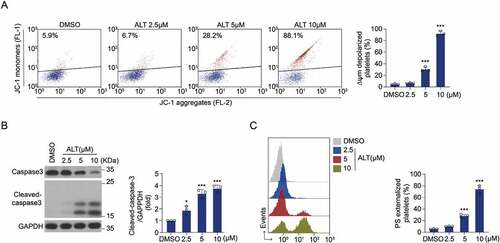
The effect of ALT on platelet was also dependent on incubation concentration. The ALT-induced ΔΨm depolarization in human platelets increased in a dose-dependent manner (~90% at 10 μM; ). Moreover, ALT activated caspase-3 in human platelets in a dose-dependent manner (), thereby driving PS exposure (). Results from murine platelets further confirmed this (Supplemental Figure S2). Collectively, these data indicate that ALT induces platelet apoptosis and the efficacy depends on incubation time and concentration.
Figure 2. ALT dose-dependently induces human platelet apoptosis. Washed human platelets were incubated with the indicated concentrations of ALT (2.5 μM, 5 μM and 10 μM) or vehicle control (DMSO) at 37°C for 8 h, and platelet apoptosis was measured. (a) ΔΨm depolarization was detected and quantitatively analyzed by flow cytometry. (b) Caspase-3 activity was evaluated using western blot analysis, and the densitometric analysis of the immunoblot for cleaved caspase-3 was performed. (c) PS exposure was detected and calculated by flow cytometry. Data presented are from three independent experiments and expressed as the mean ± SD; * p < .05, ** p < .01, and *** p < .001 versus the control with one-way ANOVA.
ALT initiates Akt activation in platelets and ALT-induced apoptosis is diminished in Akt-deficient platelets
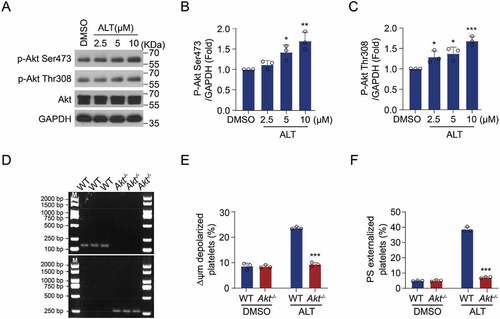
The PI3K/Akt signaling pathway has been reported to play an essential role in platelet apoptosis [Citation26]. In addition, ALT was proved to modulate Akt activity during the anti-inflammatory and anti-tumor processes [Citation2,Citation33]. Therefore, we hypothesized that ALT induces platelet apoptosis by activating the Akt pathway. To test this, Akt activity was measured in platelets treated by ALT. We found that Akt activity improved following ALT treatment, indicated by elevated phosphorylation of Akt at Ser473 and Thr308 (). We further confirmed the role of Akt in ALT-induced platelet apoptosis using genetic ablation platelets. Akt deletion (Akt-/-) mice were generated and the deficiency of Akt was examined (). The results showed that ALT-induced ΔΨm depolarization and PS externalization were markedly reduced in Akt-/- murine platelets compared with those in WT platelets (). These data suggest that ALT-induced platelet apoptosis is associated with elevated Akt activity.
Figure 3. Akt activity is improved by ALT and ALT-induced apoptosis is diminished in Akt-deficient platelets. (a–c) Washed human platelets were incubated with the indicated concentrations of ALT (2.5 μM, 5 μM, and 10 μM) or vehicle control (DMSO) at 37°C for 8 h. The activity of Akt was measured by the western blot analysis (a), and a histogram showing the relative normalized expression of p-Akt Ser473 (b) and Thr308 (c) was constructed. (d-f) Washed WT and Akt-/- murine platelets were incubated with ALT (5 μM) or vehicle control (DMSO) at 37°C for 8 h. Akt deletion of mice was confirmed by PCR (d), while ΔΨm depolarization (e) and PS exposure (f) were assessed by flow cytometry. Data presented are from three independent experiments and expressed as the mean ± SD; * p < .05, ** p < .01 and *** p < .001 versus the control with one-way ANOVA for (a–c) and versus the control with a two-tailed Student’s t-test for (d–f).
ALT induces apoptosis by activating Akt signaling pathways independent of platelet activation
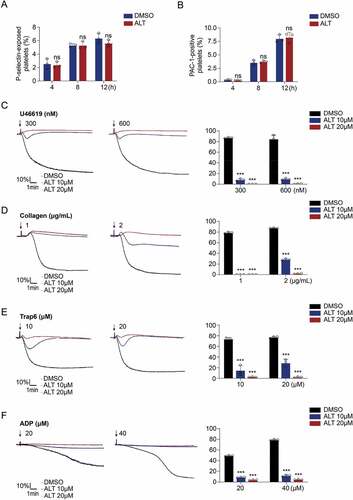
On the other hand, the PI3K/Akt signaling pathway is also involved in the regulation of platelet activation [Citation34,Citation35]. It was consequently hypothesized that ALT may affect platelet activation. To test this, the surface expression of P-selectin and αIIbβ3 activation were examined to assess platelet activation. However, the results showed that ALT did not increase the levels of P-selectin and activated αIIbβ3 in human () or murine resting platelets (Supplemental Figure S3a,b). Conversely, ALT significantly inhibited platelets activation and aggregation stimulated by U46619, collagen, Trap 6 or ADP (). Taken together, these data suggest that ALT induces platelet apoptosis through downstream molecules of Akt, different from those which regulate platelet activation.
Figure 4. ALT does not induce human platelet activation. (a,b) Washed human platelets were incubated with ALT (5 μM) or vehicle control (DMSO) at 37°C for 4 h, 8 h or 12 h, and platelet activation was measured at different time points. Platelet activation was assessed using CD62P (a) and PAC-1 antibodies (b) by flow cytometry. Data presented are from three independent experiments and expressed as the mean ± SD. (c–f) Washed human platelets were incubated with ALT (5 μM, 10 μM) or vehicle (DMSO) at 37°C for 4 h, and then were stimulated with indicated concentrations of U46619 (c), collagen (d), Trap6 (e), and ADP (f) at 37°C under constant stirring. Platelets aggregation was monitored using a CHRONO-LOG aggregometer. Histograms of maximal platelets aggregation under the indicated conditions are shown as mean ± SD of three independent experiments. * p < .05, ** p < .01, *** p < .001 and ns (not significant) versus the control with a two-tailed Student’s t-test for (a,b) and versus control group with two-way ANOVA prior to Bonferroni post hoc test for (c–f).
PKA inhibition plays an important role in ALT-induced platelet apoptosis
Next, we explored the downstream executors involved in ALT-induced platelet apoptosis. Our previous work demonstrated that PKA activity reduction is responsible for apoptotic events in aged platelets or platelets from patients ITP (Immune thrombocytopenia), sepsis or diabetes [Citation36]. Therefore, we hypothesized that PKA was involved in ALT-induced platelet apoptosis. To test this, the PKA activity of platelets was detected after ALT treatment. We found that PKA activity declined with increasing concentrations of ALT, as indicated by the dephosphorylation of PKA substrate, vasodilator-associated stimulated phosphoprotein (VASP) at Ser157 and Bcl-xL/Bcl-2-associated death promoter (Bad) at Ser155 (). On the other hand, the expression of Bak/Bax and Bcl-xL didn’t change in ALT-treated platelets (). Moreover, evidence suggests that the phosphorylation of p53 inactivates Bcl-xL, leading to platelet apoptosis [Citation17]. However, the phosphorylation of p53 was not altered by ALT treatment (). These data indicate that PKA activity is reduced in platelets following ALT application.
Figure 5. ALT reduces platelet PKA activity and PKA activation rescues platelet from ALT-induced apoptosis. (a–d) Washed human platelets were incubated with the indicated concentrations of ALT (2.5 μM, 5 μM and 10 μM) or vehicle control (DMSO) at 37°C for 8 h. The activity of PKA was evaluated using western blot analysis (a), with phosphorylated VASP Ser157 (b) and Bad Ser155 (c). The expression of classical apoptosis-related proteins, p-P53, Bcl-xL, Bak and Bax, were detected with various primary antibodies via western blot analysis (d). (e–j) Washed human platelets were pre-treated with different concentrations of PKA activator forskolin (FSK) (2.5 μM, 5 μM and 10 μM) or vehicle control (DMSO) at 37°C for 10 min followed by incubation with 5 μM ALT at 37°C for 8 h. PKA activity was measured using western blot analysis (e), with the phosphorylation of VASP at Ser157 (f) and Bad at Ser155 (g); caspase-3 was also evaluated with western blot analysis (h). Platelet ΔΨm depolarization (i) and PS exposure (j) were detected by flow cytometry. (k–m) Washed platelets from the WT or Bad−/− mice were incubated with 5 μM ALT or vehicle control (DMSO) at 37°C for 8 h. The deficiency of Bad in platelets was examined using western blot analysis (k); Platelet ΔΨm depolarization (l) and PS exposure (m) were detected by flow cytometry. Data presented are from three independent experiments and expressed as the mean ± SD; * p < .05, ** p < .01, and *** p < .001 versus the control with one-way ANOVA for (a–j) and versus the control with a two-tailed Student’s t-test for (k–m).
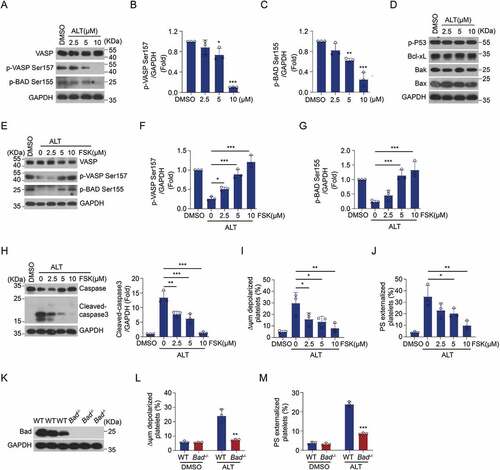
The connection between PKA inhibition and ALT-mediated platelet apoptosis was then elucidated. Forskolin, a well-known PKA activator, was utilized to selectively elevate PKA activity. We then examined whether the application of forskolin affects ALT-induced platelet apoptosis. As expected, forskolin dose-dependently reversed ALT-induced PKA inhibition, as indicated by the improved phosphorylation of VASP Ser157 and Bad Ser155 (). Simultaneously, forskolin markedly decreased ALT-induced platelet apoptotic events: caspase-3 activation, ΔΨm depolarization, and PS exposure (). We further verified the critical role of PKA in ALT-induced platelet apoptosis with Bad-deficient platelets (Bad-/-), which lacked the PKA substrate (). The results showed that ALT-induced platelet apoptosis was markedly reduced in Bad-/- murine platelets when compared to that in WT platelets (). Taken together, PKA inhibition plays a vital role in ALT-induced platelet apoptosis.
ALT-activated P13K/Akt signaling pathways induce platelet apoptosis by suppressing PKA activity
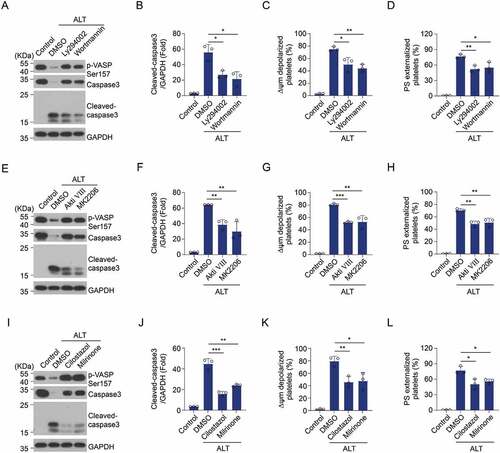
The molecular mechanism of Akt activation and PKA inhibition contributing to ALT-incurred platelet apoptosis was explored. We have reported that Akt reduces PKA activity and regulates PKA-mediated platelet apoptosis through the activation of PDE3A in carbamazepine and anti-GPIbα antibody-induced platelet apoptosis [Citation25,Citation26]. Therefore, it is possible that the PI3K/Akt signaling pathway regulates PKA activity through PDE3A in ALT-treated platelets. To test this, platelets were incubated with PI3K or Akt inhibitors prior to ALT treatment. Inhibitors of PI3K (Ly294002 and Wortmannin) and Akt (Akti VIII and MK2206) were found to markedly elevate PKA activity in ALT-treated platelets (). Correspondingly, the ALT-induced apoptotic events were markedly reduced by PI3K or Akt inhibitors (). Moreover, PDE3A inhibitors (Milrinone and Cilostazol) increased PKA activity () and reduced apoptotic events in ALT-treated platelets (). These data indicate that ALT activates the PI3K/Akt/PDE3A signaling pathway, which results in platelet apoptosis through PKA inhibition.
Figure 6. Selective inhibition of the PI3K/Akt/PDE3A signaling pathway reverses ALT-induced PKA suppression and platelet apoptosis. Washed human platelets were pre-treated with PI3K inhibitors (20 μM Ly294002 and 100 nM Wortmannin) (a–d), Akt inhibitors (2 μM Akti VIII and 6 μM MK2206) (e–h), PDE3A inhibitors (10 μM Milrinone and 10 μM Cilostazol) (i–l) or vehicle control (DMSO) at 22°C for 10 min and then incubated with 5 μM ALT at 37°C for 8 h. The phosphorylation of VASP at Ser157 and caspase-3 activity were evaluated using western blot analysis; ΔΨm depolarization and PS exposure of platelets were detected by flow cytometry. Data presented are from three independent experiments and expressed as the mean ± SD; * p < .05, ** p < .01, and *** p < .001 versus the control with a two-tailed Student’s t-test.
ALT accelerates apoptosis-dependent platelet clearance and reduces the count of platelets in circulation
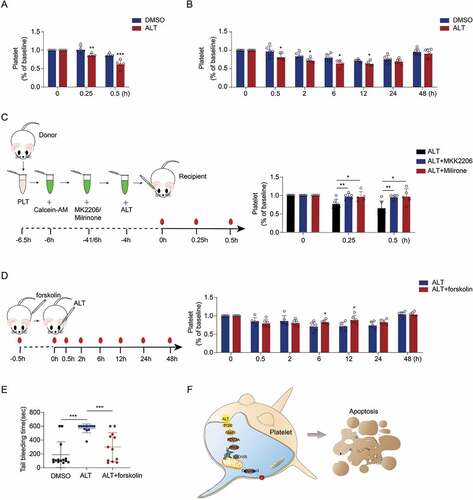
The lifespan of platelets and their counts in circulation are strictly controlled within physiological ranges. Physical and chemical factors triggered PS exposure as an “eat-me” signal leading to the phagocytosis of apoptotic cells in vivo [Citation37]. Therefore, we investigated whether ALT affects platelet clearance in vivo. As expected, calcein-labeled platelets pre-incubated with ALT were removed from circulation more rapidly compared with those in control group (). We further verified ALT-initiated platelet clearance and investigated its effect on platelet count. As shown in , after intravenous injection of ALT, platelet count in mice showed a decreasing trend from 0.5 h and declined to an absolute minimum at 12 h. Platelet count began to recover at 24 h and became comparable to that in the control group around 48 h after injection. These data demonstrate that ALT accelerates platelet clearance in vivo, leading to a drop in platelet count.
Figure 7. ALT accelerates apoptosis-dependent platelet clearance and reduces the count of platelets in circulation. (a) Calcein-labeled murine platelets were incubated with 5 μM ALT or vehicle control (DMSO) at 37°C for 4 h and then transfused into WT mice; n = 6 for each group. The baseline is the percentage of calcein-positive platelets in circulation tested immediately after platelet infusion. (b) C57BL/6J mice were i.v. injected with 2 mg/kg ALT or vehicle control (DMSO) and platelet counts were determined at the indicated time points; n = 6 for each group. The baseline is defined as the platelet count before ALT injection. (c) Calcein-labeled murine platelets were pre-treated with 10 μM milrinone, 10 μM cilostazol or vehicle control (DMSO) at 37°C for 10 min and then incubated with 5 μM ALT at 37°C for 4 h. These platelets were then transfused to C57BL/6J mice and the percentage of calcein-positive platelets remaining in circulation was evaluated at the indicated time points by flow cytometry; n = 6 for each group. The baseline is defined as the percentage of calcein-positive platelets in circulation tested immediately after transfusion. (d) C57BL/6J mice were i.v. injected with 5 mg/kg PKA activator forskolin or vehicle control (DMSO) for 30 min and were then i.v. injected with 2 mg/kg ALT. Platelet counts were then detected at the indicated time points; n = 6 for each group. The baseline is defined as the platelet count before the ALT injection. (e) C57BL/6J mice were i.v. injected with 5 mg/kg PKA activator forskolin or vehicle control (DMSO) for 30 min and were then i.v. injected with 2 mg/kg ALT. Tail bleeding time was recorded to cessation of bleeding in response to tail injury. (f) Schematic illustration of ALT-induced platelets apoptosis: the ALT initiates Akt activation, the downstream effector of PI3K, and then Akt activates PDE3A, leading to PKA inhibition and Bad release from the combination with Bcl-xL. Released Bad induces caspase-3 mediated apoptosis leading to PS externalization. Data presented are the mean ± SD; * p < .05, ** p < .01, and *** p < .001 versus the control with two-way ANOVA prior to the Bonferroni post-hoc test for (a–d) and versus the control with a two-tailed Student’s t-test for (e).
Inhibition of the P13K/Akt/PDE3A signaling pathway or activation of PKA reverses ALT-incurred platelet clearance and platelet count reductions
As described above, the PI3K/Akt/PDE3A signaling pathway activated by ALT results in platelet apoptosis through PKA inhibition in vitro. To test the role of the PI3K/Akt/PDE3A signaling pathway in ALT-induced platelet clearance, calcein-labeled platelets were pre-treated with Akt or PDE3A inhibitors before transfusion. We found that Akt inhibitor MK2206 or PAE3A inhibitor Milirone markedly reduced ALT-induced platelet clearance in vivo (). Moreover, PKA activator forskolin diminished the side effects of ALT on platelet count (). Furthermore, ALT significantly impaired the hemostatic ability of platelets indicated by prolonged tail bleeding time after ALT injection, which could be partly rescued by forskolin (). Taken together, these data demonstrate that ALT accelerates platelet clearance by activating the PI3K/Akt/PDE3A signaling pathway and PKA inhibition, which contributes to platelet count decline in vivo.
Discussion
ALT is an essential active component in many traditional Chinese medicine formulas [Citation1]. Despite the various pharmacological activities of ALT, most research focus on its therapeutic potential in inflammatory conditions and tumors [Citation2–4]. The anti-tumor activity of ALT has been confirmed in various cancers, but its side effects are still not fully understood [Citation12]. In this study, we report for the first time that ALT induces platelet apoptosis through activating Akt pathway. Activated Akt facilitates platelet apoptosis through activating PDE3A and PDE3A-mediated PKA inhibition (). The Apoptotic platelets are removed more rapidly from circulation, accounting for lower platelet counts. In addition, the efficacy of ALT depends on incubation time and working concentration, and the concentration and incubation time used in this study are referenced from those in cancer treatment experiments. Therefore, our study reveals possible side effects of ALT treatment and suggests relevant mechanisms as well, providing the reference for ALT application in clinical practice.
Malignant tumors are characterized by high incidence and fatality rates and impose a significant economic burden globally. Tumor progression can be retarded by early surgery and radiotherapy treatments, while chemotherapy is usually the only option available for advanced tumors [Citation38,Citation39]. In the past several decades, natural products, including traditional Chinese medicine, have been recognized as a vast reservoir from which to develop lead compounds with therapeutic potential for malignant diseases. However, side effects are common in chemotherapy and can severely reduce the patients’ quality of life. To avoid adverse outcomes, a reduction in the drug dosage or even a cessation in administration is often required, which ultimately diminishes the effectiveness of the treatment. Different from conventional systemic cytotoxic drugs, ALT was reported to exhibit anti-tumor activities through modulating the abnormal signaling pathways in cancer cells. With no obvious side effects, ALT was also recommended to enhance the cytotoxic effects with other anti-cancer agents such as gemcitabine and oxaliplatin in the past [Citation32,Citation40]. Akt, one of the ALT-targeted molecules, expresses in platelet and plays an essential role both in platelet apoptosis and activation [Citation26]. Therefore, we found that Akt was activated after ALT treatment and genetic ablation of Akt disrupted ALT-induced platelet apoptosis. We have revealed Akt activation induced by anti-GPIbα antibodies resulted in platelet apoptosis and activation in ITP patients [Citation26]. However, there are different opinions about the effects of Akt activation on platelet. Tian et al. found that Akt pathway activation reduces platelet apoptosis and contributes to the increase of platelet counts in solid tumor patients [Citation41]. What’s more, Zhang et al. demonstrated that P2Y12 protects platelets from apoptosis via PI3k-dependent Bak/Bax inactivation [Citation42]. Akt is a pivotal node involved in many signaling pathways, and PI3K/Akt pathway has been implicated in the signals transmission of many membrane receptors of platelet. It is possible that the effect of Akt activation on platelet apoptosis under physiological and pathological conditions depends on which membrane receptors and associated signal pathways are activated.
On the other side, the PI3K/Akt signaling pathway is thought to be involved in the regulation of platelet activation during hemostasis and thrombosis, which involves platelet-matrix interactions and platelet aggregation [Citation34,Citation35]. Besides platelet apoptosis, PS exposure is also one of the later events in platelet activation, and we demonstrated that ALT-induced PS exposure in platelets. However, ALT treatment didn’t alter platelet activation, and conversely, ALT significantly impaired platelet aggregation. These data suggest that ALT induces platelet apoptosis through the downstream of Akt, which is different from those that regulate platelet activation. We previously demonstrated that PKA determines platelet life span and survival by regulating apoptosis [Citation36]. We also elucidated the relationship between Akt and PKA in terms of platelet apoptosis regulation: Akt is located upstream of PKA and suppresses PKA activity via the activation of PDE3A [Citation25,Citation26]. Consistent with these reports, we found that PKA activity was markedly reduced after ALT application. Activation of PKA by the forskolin rescued platelets from ALT-induced apoptosis, and ALT-induced platelet apoptosis was markedly reduced in Bad-/- platelets. Moreover, PKA activator forskolin diminished the side effects of ALT on platelet count and partly rescued impaired hemostatic ability of platelets, as indicated by shortening tail bleeding time. These data suggest that PKA is a downstream apoptotic executor involved in ALT-induced platelet apoptosis. Supporting data show that the inhibition of the PI3K/Akt signaling pathway and PDE3A significantly reversed ALT-induced PKA suppression and reduced ALT-induced platelet apoptosis simultaneously. Moreover, ALT was found to accelerate the clearance of platelets in circulation, and intravenous injections of ALT led to a decline in the number of platelets in circulation. Consistent with in vitro results, selective inhibition of the PI3K/Akt/PDE3A signaling pathway and PKA activation could rescue ALT-induced platelet clearance and platelet count decline. The current study provides additional evidence to the essential role of the PI3K/Akt-PDE3A axle and PKA in the regulation of platelet apoptosis, thereby providing guidance to understanding the mechanism of drug-induced platelet disorder.
It is remarkable that the severity of ALT-induced platelet count decline is discordant with the capacity of ALT to facilitate platelet apoptosis in vitro. It can be partly explained by the fact that the steady-state number of mature platelets in circulation results from the balance between platelet production and destruction as roughly 100 billion new platelets are produced daily from bone marrow megakaryocytes [Citation15,Citation43]. In addition, the effect of ALT in vivo particularly depends on the utilization of the drug, suggesting further research is required on the pharmacokinetics of ALT. What’s more, the effect of ALT on Akt is paradoxical among different cell types, based on the available data. ALT was shown to promote Akt activation in the skeletal muscle [Citation2], while ALT was reported to exhibit anti-tumor efficacy through the suppression of Akt activity in many cancer cells [Citation6,Citation8,Citation9,Citation32]. Therefore, our work provides experimental evidence supporting the hypothesis that the effects of ALT on Akt may vary depending on cell types. Akt is a pivotal node involved in many signaling pathways participating in the regulation of various cell functions. Therefore, more research should be conducted to explore the side effects of ALT on other cells before clinical application.
Author contributions
Yueyue Sun, Mengnan Yang and Rong Yan designed the research, collected and analyzed data, and wrote the paper. Yueyue Sun, Mengnan Yang, Shujun Li, Ying Hu, Biao Yang and Xu Li performed the experiments. Kesheng Dai initiated and supervised the project, designed research, analyzed and interpreted results, and reviewed the paper. All the authors reviewed and approved the final version.
Supplemental Material
Download PDF (442.2 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/09537104.2023.2173505
Additional information
Funding
References
- Liu X, Bian L, Duan X, Zhuang X, Sui Y, Yang L. Alantolactone: a sesquiterpene lactone with diverse pharmacological effects. Chem Biol Drug Des. 2021;98(6):1131–11. Epub 2021/10/09. doi:10.1111/cbdd.13972.
- Kim M, Song K, Kim YS. Alantolactone improves prolonged exposure of Interleukin-6-Induced skeletal muscle inflammation associated glucose intolerance and insulin resistance. Front Pharmacol. 2017;8:405. Epub 2017/07/15. doi:10.3389/fphar.2017.00405.
- Dang X, He B, Ning Q, Liu Y, Guo J, Niu G, Chen M. Alantolactone suppresses inflammation, apoptosis and oxidative stress in cigarette smoke-induced human bronchial epithelial cells through activation of Nrf2/HO-1 and inhibition of the NF-κB pathways. Respir Res. 2020;21(1):95. Epub 2020/04/24. doi:10.1186/s12931-020-01358-4.
- Cai Y, Gao K, Peng B, Xu Z, Peng J, Li J, Chen X, Zeng S, Hu K, Yan Y. Alantolactone: a natural plant extract as a potential therapeutic agent for cancer. Front Pharmacol. 2021;12:781033. Epub 2021/12/14. doi:10.3389/fphar.2021.781033.
- Fu Z, Li S, Liu J, Zhang C, Jian C, Wang L, Zhang Y, Shi C. Natural product alantolactone targeting AKR1C1 suppresses cell proliferation and metastasis in non-small-cell lung cancer. Front Pharmacol. 2022;13:847906. Epub 2022/04/05. doi:10.3389/fphar.2022.847906.
- Kang X, Wang H, Li Y, Xiao Y, Zhao L, Zhang T, Zhou S, Zhou X, Li Y, Shou Z, et al. Alantolactone induces apoptosis through ROS-mediated AKT pathway and inhibition of PINK1-mediated mitophagy in human HepG2 cells. Artif Cells Nanomed Biotechnol. 2019;47(1):1961–1970. Epub 2019/05/23. doi:10.1080/21691401.2019.1593854.
- Shi C, Lan W, Wang Z, Yang D, Wei J, Liu Z, Teng Y, Gu M, Yuan T, Cao F, et al. Alantolactone inhibits cell autophagy and promotes apoptosis via AP2M1 in acute lymphoblastic leukemia. Cancer Cell Int. 2020;20(1):442. Epub 2020/09/19. doi:10.1186/s12935-020-01537-9.
- Zhang Y, Weng Q, Han J, Chen J. Alantolactone suppresses human osteosarcoma through the PI3K/AKT signaling pathway. Mol Med Rep. 2020;21:675–684. Epub 2020/01/25. doi:10.3892/mmr.2019.10882.
- Zhang X, Zhang HM. Alantolactone induces gastric cancer BGC-823 cell apoptosis by regulating reactive oxygen species generation and the AKT signaling pathway. Oncol Lett. 2019;17:4795–4802. Epub 2019/06/13. doi:10.3892/ol.2019.10172.
- He W, Cao P, Xia Y, Hong L, Zhang T, Shen X, Zheng P, Shen H, Zhao Y, Zou P. Potent inhibition of gastric cancer cells by a natural compound via inhibiting TrxR1 activity and activating ROS-mediated p38 MAPK pathway. Free Radic Res. 2019;53(1):104–114. Epub 2019/01/23. doi:10.1080/10715762.2018.1558448.
- Yao Y, Xia D, Bian Y, Sun Y, Zhu F, Pan B, Niu M, Zhao K, Wu Q, Qiao J, et al. Alantolactone induces G1 phase arrest and apoptosis of multiple myeloma cells and overcomes bortezomib resistance. Apoptosis. 2015;20(8):1122–1133. Epub 2015/06/03. doi:10.1007/s10495-015-1140-2.
- Babaei G, Aziz SGG, Bazl MR, Ansari MK. A comprehensive review of anticancer mechanisms of action of Alantolactone. Biomed Pharmacother. 2021;136:111231. Epub 2021/01/18. doi:10.1016/j.biopha.2021.111231.
- Furie B, Furie BC. Thrombus formation in vivo. J Clin Invest. 2005;115(12):3355–3362. Epub 2005/12/03. doi:10.1172/JCI26987.
- Rivera J, Lozano ML, Navarro-Nunez L, Vicente V. Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica. 2009;94(5):700–711. Epub 2009/03/17. doi:10.3324/haematol.2008.003178.
- Lefrancais E, Ortiz-Munoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, Thornton EE, Headley MB, David T, Coughlin SR, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. 2017;544(7648):105–109. Epub 2017/03/23. doi:10.1038/nature21706.
- Grozovsky R, Giannini S, Falet H, Hoffmeister KM. Regulating billions of blood platelets: glycans and beyond. Blood. 2015;126(16):1877–1884. Epub 2015/09/04. doi:10.1182/blood-2015-01-569129.
- McArthur K, Chappaz S, Kile BT. Apoptosis in megakaryocytes and platelets: the life and death of a lineage. Blood. 2018;131(6):605–610. Epub 2017/12/21. doi:10.1182/blood-2017-11-742684.
- Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128(6):1173–1186. Epub 2007/03/27. doi:10.1016/j.cell.2007.01.037.
- Lebois M, Josefsson EC. Regulation of platelet lifespan by apoptosis. Platelets. 2016;27(6):497–504. Epub 2016/04/22. doi:10.3109/09537104.2016.1161739.
- Thushara RM, Hemshekhar M, Kemparaju K, Rangappa KS, Devaraja S, Girish KS. Therapeutic drug-induced platelet apoptosis: an overlooked issue in pharmacotoxicology. Arch Toxicol. 2014;88(2):185–198. Epub 2013/12/24. doi:10.1007/s00204-013-1185-3.
- De Silva E, Kim H. Drug-induced thrombocytopenia: focus on platelet apoptosis. Chem Biol Interact. 2018;284:1–11. Epub 2018/02/08. doi:10.1016/j.cbi.2018.01.015.
- Liu L, Chen M, Zhao L, Zhao Q, Hu R, Zhu J, Yan R, Dai K. Ethanol induces platelet apoptosis. Alcohol Clin Exp Res. 2017;41(2):291–298. Epub 2017/01/13. doi:10.1111/acer.13295.
- Zhang J, Chen M, Zhang Y, Zhao L, Yan R, Dai K. Carmustine induces platelet apoptosis. Platelets. 2015;26(5):437–442. Epub 2014/06/24. doi:10.3109/09537104.2014.928676.
- Zhao L, Zhang W, Chen M, Zhang J, Zhang M, Dai K. Aspirin Induces platelet apoptosis. Platelets. 2013;24(8):637–642. Epub 2013/01/11. doi:10.3109/09537104.2012.754417.
- Xiao W, Zhou K, Yang M, Sun C, Dai L, Gu J, Yan R, Dai K. Carbamazepine induces platelet apoptosis and thrombocytopenia through protein Kinase A. Front Pharmacol. 2021;12:749930. Epub 2021/10/19. doi:10.3389/fphar.2021.749930.
- Chen M, Yan R, Zhou K, Li X, Zhang Y, Liu C, Jiang M, Ye H, Meng X, Pang N, et al. Akt-mediated platelet apoptosis and its therapeutic implications in immune thrombocytopenia. Proc Natl Acad Sci USA. 2018;115(45):E10682–E10691. Epub 2018/10/20. doi:10.1073/pnas.1808217115.
- Manikanta K, Kumar SN, Hemshekhar M, Kemparaju K, Girish KS. ASK1 inhibition triggers platelet apoptosis via p38-MAPK-mediated mitochondrial dysfunction. Haematologica. 2020;105(8):e419–e423. Epub 2019/11/30. doi:10.3324/haematol.2019.233908.
- Rukoyatkina N, Mindukshev I, Walter U, Gambaryan S. Dual role of the p38 MAPK/cPLA2 pathway in the regulation of platelet apoptosis induced by ABT-737 and strong platelet agonists. Cell Death Dis. 2013;4(11):e931. Epub 2013/11/23. doi:10.1038/cddis.2013.459.
- Paul M, Manikanta K, Hemshekhar M, Sundaram MS, Naveen S, Ramesh TN, Kemparaju K, Girish KS. Bisdemethoxycurcumin promotes apoptosis in human platelets via activation of ERK signaling pathway. Toxicol In Vitro. 2020;63:104743. Epub 2019/12/07. doi:10.1016/j.tiv.2019.104743.
- Paul M, Hemshekhar M, Thushara RM, Sundaram MS, NaveenKumar SK, Naveen S, Devaraja S, Somyajit K, West R, Basappa, et al. Methotrexate promotes platelet apoptosis via JNK-mediated mitochondrial damage: alleviation by N-Acetylcysteine and N-Acetylcysteine amide. PLoS One. 2015;10(6):e0127558. Epub 2015/06/18. doi:10.1371/journal.pone.0127558.
- Wu Y, Dai J, Zhang W, Yan R, Zhang Y, Ruan C, Dai K. Arsenic trioxide induces apoptosis in human platelets via C-Jun NH2-terminal kinase activation. PLoS One. 2014;9(1):e86445. Epub 2014/01/28. doi:10.1371/journal.pone.0086445.
- Wang J, Zhang Y, Liu X, Wang J, Li B, Liu Y, Wang J. Alantolactone enhances gemcitabine sensitivity of lung cancer cells through the reactive oxygen species-mediated endoplasmic reticulum stress and Akt/GSK3beta pathway. Int J Mol Med. 2019;44:1026–1038. Epub 2019/09/17. doi:10.3892/ijmm.2019.4268.
- Liu YR, Cai QY, Gao YG, Luan X, Guan YY, Lu Q, Sun P, Zhao M, Fang C. Alantolactone, a sesquiterpene lactone, inhibits breast cancer growth by antiangiogenic activity via blocking VEGFR2 signaling. Phytother Res. 2018;32(4):643–650. Epub 2017/12/07. doi:10.1002/ptr.6004.
- Lee SH, Meng XW, Flatten KS, Loegering DA, Kaufmann SH. Phosphatidylserine exposure during apoptosis reflects bidirectional trafficking between plasma membrane and cytoplasm. Cell Death Differ. 2013;20(1):64–76. Epub 2012/08/04. doi:10.1038/cdd.2012.93.
- Chen Z, Li T, Kareem K, Tran D, Griffith BP, Wu ZJ. The role of PI3K/Akt signaling pathway in non-physiological shear stress-induced platelet activation. Artif Organs. 2019;43(9):897–908. Epub 2019/04/12. doi:10.1111/aor.13465.
- Zhao L, Liu J, He C, Yan R, Zhou K, Cui Q, Meng X, Li X, Zhang Y, Nie Y, et al. Protein kinase a determines platelet life span and survival by regulating apoptosis. J Clin Invest. 2017;127(12):4338–4351. Epub 2017/10/31. doi:10.1172/JCI95109.
- Savill J, Gregory C. Apoptotic PS to phagocyte TIM-4: eat me. Immunity. 2007;27(6):830–832. Epub 2007/12/21. doi:10.1016/j.immuni.2007.12.002.
- Lin L, Li Z, Yan L, Liu Y, Yang H, Li H. Global, regional, and national cancer incidence and death for 29 cancer groups in 2019 and trends analysis of the global cancer burden, 1990–2019. J Hematol Oncol. 2021;14(1):197. Epub 2021/11/24. doi:10.1186/s13045-021-01213-z.
- Ferlay J, Colombet M, Soerjomataram I, Dyba T, Randi G, Bettio M, Gavin A, Visser O, Bray F. Cancer incidence and mortality patterns in Europe: estimates for 40 countries and 25 major cancers in 2018. Eur J Cancer. 2018;103:356–387. Epub 2018/08/14. doi:10.1016/j.ejca.2018.07.005.
- Cao P, Xia Y, He W, Zhang T, Hong L, Zheng P, Shen X, Liang G, Cui R, Zou P. Enhancement of oxaliplatin-induced colon cancer cell apoptosis by alantolactone, a natural product inducer of ROS. Int J Biol Sci. 2019;15(8):1676–1684. Epub 2019/07/31. doi:10.7150/ijbs.35265.
- Tian H, Huang S, Luo Q, Lin Z, Liu H, Zhang Z, Fong W, Zhao J, Yu F. Akt pathway activation reduces platelet apoptosis and contributes to the increase of platelet counts in solid tumor patients. Platelets. 2022;33:1009–1017. Epub 2022/01/25.
- Zhang S, Ye J, Zhang Y, Xu X, Liu J, Zhang SH, Kunapuli SP, Ding Z. P2Y12 protects platelets from apoptosis via PI3k-dependent Bak/Bax inactivation. J Thromb Haemost. 2013;11(1):149–160. Epub 2012/11/13. doi:10.1111/jth.12063.
- Thon JN, Italiano JE. Platelets: production, morphology and ultrastructure. Handb Exp Pharmacol . 2012;210:3–22. Epub 2012/08/25.