
Abstract
Essential thrombocythemia (ET) with double driver mutations is a rare disease. ET patients with both MPL and Type 1 CALR mutations have been reported. Here, we report the first case of an ET patient with both MPL S204P and Type 2 CALR mutations and a summary of our literature review findings. In the patient whose case is reported here, the disease progressed to an accelerated phase 3.5 months after diagnosis. CALR mutation disappeared and new mutations emerged as the disease progressed, such as ASXL1, CBL, ETV6, and PTPN11 mutations. This case highlights that screening for additional mutations using NGS should be considered in patients with ET to assess the prognosis, especially as the disease progresses.
Introduction
Polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) are classified as “classic” myeloproliferative neoplasms (MPNs) according to the 2016 World Health Organization, and these MPNs share several essential clinical features, such as abnormal clonal hematopoiesis, bone marrow hypercellularity, and a tendency for hemorrhage and thrombosis [Citation1]. The genetic mechanisms of the onset and progression of MPN have been investigated extensively in recent decades. Recurrent mutations in JAK2, CALR and MPL have been identified as phenotypic drivers of MPNs [Citation2]. Mutations in JAK2, CALR and MPL are collectively detectable in 83% of patients with ET [Citation3]. However, JAK2, CALR and MPL are found to be mutated in a mutually exclusive fashion, suggesting that they drive transformation through the same pathway. Two concomitant driver mutations, for example, JAK2 V617F and an MPL mutation [Citation4] or JAK2 V617F with a concomitant CALR mutation [Citation5], have been reported in MPN, albeit rarely. Concurrent MPL mutations were reported in 0.3% of MPN patients with CALR mutations [Citation6]. The clonal architecture of such cases has not been entirely clarified. Hence, we report the first case of ET with combined MPL S204P and Type 2 CALR mutations in China, which offers the opportunity to elucidate the contributions of genetic factors to disease pathogenesis.
Case report
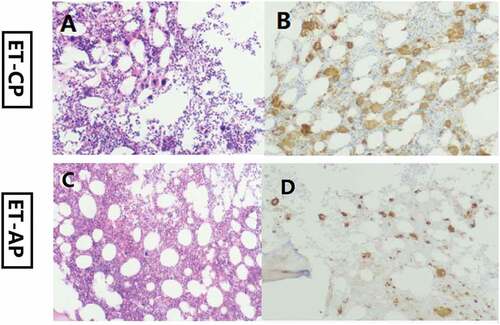
A 67-year-old woman was admitted to the First People’s Hospital of Chuzhou in November 2019 due to new-onset thrombocytosis (platelets 1097 × 109/L). She had no hepatosplenomegaly. No history of reactive thrombocytosis was found. No conventional cardiovascular risk factors (hypertension, cigarette smoking, diabetes, obesity, and dyslipidaemia) were observered. Bone marrow evaluation showed increased numbers of enlarged, mature megakaryocytes without dysplasia and fibrosis (). Flow cytometry was used to identify bone marrow cells by surface markers, CD16, CD11b, CD13, CD33, CD34, CD19, CD45, CD10, HLA-DR, CD117, CD56, CD14, CD38, CD20. No evidence of any active hematological malignancy was observered. The karyotype was noted to be normal. The patient was negative for the BCR/ABL fusion gene. Next-generation sequencing (NGS) for MDS/MPN was performed as previously described [Citation7], and the results revealed the presence of CALR (K385Nfs × 47, variant frequency [VF] = 27.8%), MPL (S204P, VF = 71.5%) and SETBP1 (W222S, VF = 50.2%) mutations. The diagnosis of ET was retained. She was rated as “low‒risk” by the International Prognostic Score for Thrombosis in ET (IPSET-thrombosis) and classified as “intermediate-risk” according to mutation-enhanced international prognostic systems in ET (MIPSS-ET). After confirmation of diagnosis, the patient was treated with hydroxyurea 500 mg twice-daily and asprin 100 mg once-daily. Peripheral blood tests demonstrated a leukocyte count of 12.0 × 109/L, a hemoglobin level of 97 g/L, and a platelet count of 36 × 109/L after treatment with hydroxyurea for 3.5 months. A bone marrow biopsy at this time showed transformation to an accelerated phase with 10% megakaryocytic dysplasia and 18% myeloblasts, and no fibrosis was observed (). There was a slight numerical increase in megakaryocytes (). NGS was repeated, and the results revealed the persistence of the MPL (S204P, VF = 94.7%) and SETBP1 (W222S, VF = 47.4%) mutations, the disappearance of the CALR (K385Nfs × 47) mutation and the acquisition of mutations in ASXL1 (E635Rfs × 15, VF = 27.9%), RUNX1 (D198 G, VF = 37.5%), CBL (c.1227 + 2T>C, VF = 53.2%), ETV6 (P214 L, VF = 7.5%), and PTPN11 (S502P, VF = 7.2%). Then, the patient was treated with decitabine in combination with cytarabine, aclarubicin hydrochloride and granulocyte colony-stimulating factor (DCAG) in March 2020. Platelet count was observed to be 33 × 109/L with Hb of 10.4 g/dL and leukocyte of 11.9 × 109/L in May 2020. After that time, the patient refused further treatment for financial reasons. The patient developed blood transfusion dependence in 2021. A peripheral blood manual differential count performed in July 2021 revealed 11% myeloblasts. The patient refused bone marrow examination. The patient received the DCAG regimen again in July and September. Peripheral blood tests demonstrated a leukocyte count of 25.3 × 109/L, a hemoglobin level of 68 g/L, and a platelet count of 3 × 109/L in October 2021. Taking into account the risks and costs, her family decided to abandon treatment. Finally, the patient died in December 2021.
Figure 1. Hematoxylin and eosin (H&E) stain of bone marrow biopsies (a, c). Immunohistochemical analysis of bone marrow demonstrating CD61+ (b, d). CP, chronic phase; AP, acceleration phase.
Discussion
The coexistence of CALR and MPL mutations in ET is a rare clonal entity. Jeromin et al. studied 685 CALR-mutated MPN patients and found that 0.3% of patients had concurrent MPL mutations [Citation6]. Our previous study on the occurrence of driver mutations in Chinese Han patients with ET did not find patients with coexisting CALR and MPL mutations [Citation8]. We summarized previously published ET cases with co-occurring CALR and MPL mutations at diagnosis in . To the best of our knowledge, this is the first report of an ET patient with MPL S204P and Type 2 CALR mutations from China. Additionally, CALR mutant disappeared during follow-up.
Table I. Clinical characteristics of essential thrombocythemia with coexisting CALR and MPL mutations at diagnosis.
The MPL S204P mutant has a milder effect on the extracellular ligand binding domain of the thrombopoietin receptor than the common W515K mutant [Citation9]. A functional study on MPL S204P revealed that it is a weak gain-of-function mutant that increases MPL signaling and confers TPO hypersensitivity but with low efficiency [Citation10]. MPL S204P also has been reported in a PMF patient [Citation9]. In particular, MPL-mutated patients had lower platelet counts [Citation11]. It is tempting to assume that the constitutive activation of MPL signaling due to MPL S204P is enough to lead to an ET phenotype but may not be sufficient to result in a platelet count up to 1097 × 10^9/l in this case. At the phenotypic level, markedly elevated platelet counts are the main hematological features of CALR-mutant ET patients [Citation12]. It is believed that the mutation allele burden dictates phenotype. A platelet count up to 1097 × 10^9/l in this case might be caused by CALR mutation.
CALR mutations in JAK2/CALR-mutant ET patients were found at a lower allelic burden than in matched CALR-mutated patients [Citation13]. The biological relevance of coexisting driver mutations in these types of cases is still unclear, and the possibility that one of the mutations is a “bystander” mutation outside the scope of the true disease cannot be excluded [Citation14]. Thompson et al. described two unusual cases of MPN patients, each harboring two driver mutations (JAK2/CALR and JAK2/MPL), and the results of single-cell DNA sequencing analysis showed that the two driver mutations occurred in independent clones [Citation14]. According to NGS data, the MPL S204P mutation (VF = 71.5%) showed clonal dominance, implying that the MPL mutant might give a stronger clonal advantage than the CALR mutant in this case. The disappearance of the CALR mutant and an increase in the MPL mutant burden were observed in this ET patient during follow-up, suggesting the biclonality of the CALR and MPL mutations. The disappearance of the CALR mutation is highly likely to be due to the clonal competition and the expansion of the MPL mutations. Cells with a MPL mutation would achieve clonal dominance much faster than those with a CALR mutation.
Leukemic transformation of MPNs is associated with dismal outcomes. Feenstra et al. did not observe disease progression or leukemic transformation in a PMF patient with MPL S204P at a 12-year follow-up [Citation9]. The acquisition of new mutations in this patient resulted in rapidly progressive disease and death. Repeat NGS revealed the persistence of a SETBP1 mutation after disease progression in the patient in our study. SETBP1 is now commonly recognized as a driver oncogene almost exclusively in myeloid neoplasms [Citation15]. SETBP1 mutation leads to SETBP1 overexpression, protecting SET from protease cleavage, ultimately leading to cell proliferation and the expansion of leukemic cells [Citation16] and playing a main role in secondary leukemogenesis [Citation17]. SETBP1 mutations were found to be strongly associated with ASXL1, CBL and RUNX1 mutations in myeloid neoplasms [Citation15,Citation16] and were mutually exclusive to JAK2 and TET2 mutations [Citation16]. We suspected that SETBP1 mutation could induce genetic instability at the stem cell level and consequently be able to favor mutations at ASXL1, CBL and RUNX1 loci, which were correlated with leukemogenesis [Citation18–20]. The mutation (p.W222S) does not locate in the hot spot region of SETBP1 and this variant is described in dbSNP with a very low MAF making it unknown significance. The VarSite database shows that the mutation (p.W222S) is likely deleterious [Citation21]. Subclonal evolution is the process by which the founding malignant clone generates subclones through the acquisition of additional driver mutations.
Different routes to leukemic transformation were reported in JAK2 mutation-positive MPNs [Citation22]. Tefferi et al. did not find an independent effect of JAK2 V617F on disease transformation [Citation23]. JAK2 V617F can disappear during leukemic transformation, suggesting that a JAK2-negative leukemic clone was derived from MPN progenitor cells without a JAK2 mutation [Citation22]. TET2 mutation can precede the JAK2 mutation and aberrant TET2 plays roles in the development of leukemia [Citation24]. TP53 deletion also cooperates with JAK2V617F expression in mice to induce AML development [Citation25]. The molecular mechanism causing the molecular complexity and their clinical impacts are still unclear. The results of the present study suggest that beyond the driver mutations in JAK2, CALR, and MPL, the additional mutations detected are compatible with disease progression.
Limitation
Our molecular approach has obvious limitations because it cannot detect mutations in noncoding sequences and gene fusions or even mutations at low VF. This analysis is inferred from the NGS quantitative data, which is not strictly equivalent to clonal genetic structure analysis based on the genotyping of progenitor colonies in vitro. Further testing should be done to detect these genes for the presence of germline mutations. In addition, it is unclear whether acquired mutations in other genes play a role in disease progression because we performed targeted sequencing of only 29 genes.
Conclusion
This report may be the first on the clonal evolution of an ET patient with co-occurring MPL S204P and Type 2 CALR mutations, and the findings will contribute to a better understanding of the genetic mechanisms by which ET develops and progresses.
Authors’ contributions
Conceptualization, JW and QZ; methodology, WF, WB and WG; formal analysis, SX and CL; investigation, QJ; data curation, SX and CL; writing—original draft preparation, JW and WF; writing—review and editing, JW and QZ; supervision, QZ.
Ethics approval statement
This case report received exemption for ethical approval by First People’s Hospital of Chuzhou because all data were collected retrospectively and anonymously. Written informed consent to publish medical information and images was obtained from the patient’s direct relatives.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–4. doi:10.1182/blood-2016-03-643544.
- Cazzola M, Kralovics R. From Janus kinase 2 to calreticulin: the clinically relevant genomic landscape of myeloproliferative neoplasms. Blood. 2014;123(24):3714–3719. doi:10.1182/blood-2014-03-530865.
- Tefferi A, Pardanani A. Myeloproliferative neoplasms: a contemporary review. JAMA Oncol. 2015;1(1):97–105. doi:10.1001/jamaoncol.2015.89.
- Jang MA, Seo MY, Choi KJ, Hong DS. A rare case of essential thrombocythemia with coexisting JAK2 and MPL driver mutations. J Korean Med Sci. 2020;35(23):e168. doi:10.3346/jkms.2020.35.e168.
- Kang MG, Choi HW, Lee JH, Choi YJ, Choi HJ, Shin JH, Suh SP, Szardenings M, Kim HR, Shin MG. Coexistence of JAK2 and CALR mutations and their clinical implications in patients with essential thrombocythemia. Oncotarget. 2016;7(35):57036–57049. doi:10.18632/oncotarget.10958.
- Jeromin S, Kohlmann A, Meggendorfer M, Schindela S, Perglerová K, Nadarajah N, Kern W, Haferlach C, Haferlach T, Schnittger S. Next-generation deep-sequencing detects multiple clones of CALR mutations in patients with BCR-ABL1 negative MPN. Leukemia. 2016;30(4):973–976. doi:10.1038/leu.2015.207.
- Zhang QG, Wang J, Gong WY, Jing QC. Clonal evolution in a chronic neutrophilic leukemia patient. Hematology. 2019;24(1):455–458. doi:10.1080/16078454.2019.1613291.
- Wang J, Zhang B, Chen B, Rf Z, Qg Z, Li J, Yang YG, Zhou M, Shao XY, Xu Y, et al. JAK2, MPL, and CALR mutations in Chinese Han patients with essential thrombocythemia. Hematology. 2017;22(3):145–148. doi:10.1080/10245332.2016.1252003.
- Milosevic Feenstra JD, Nivarthi H, Gisslinger H, Leroy E, Rumi E, Chachoua I, Bagienski K, Kubesova B, Pietra D, Gisslinger B, et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood. 2016;127(3):325–332. doi:10.1182/blood-2015-07-661835.
- Cabagnols X, Favale F, Pasquier F, Messaoudi K, Defour JP, Ianotto JC, Marzac C, Couédic Jp L, Droin N, Chachoua I, et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood. 2016;127(3):333–342. doi:10.1182/blood-2015-07-661983.
- Rumi E, Pietra D, Guglielmelli P, Bordoni R, Casetti I, Milanesi C, Sant’antonio E, Ferretti V, Pancrazzi A, Rotunno G, et al. Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative. Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood. 2013;121(21):4388–4395. doi:10.1182/blood-2013-02-486050.
- Rumi E, Pietra D, Ferretti V, Klampfl T, Harutyunyan AS, Milosevic JD, Them NC, Berg T, Elena C, Casetti IC, et al. Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative Investigators. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood. 2014;123(10):1544–1551. doi:10.1182/blood-2013-11-539098.
- Mansier O, Luque Paz D, Ianotto JC, Le Bris Y, Chauveau A, Boyer F, Conejero C, Fitoussi O, Riou J, Adiko D, et al. Clinical and biological characterization of MPN patients harboring two driver mutations, a French intergroup of myeloproliferative neoplasms (FIM) study. Am J Hematol. 2018;93(4):E84–86. doi:10.1002/ajh.25014.
- Thompson ER, Nguyen T, Kankanige Y, Yeh P, Ingbritsen M, McBean M, Semple T, Mir Arnau G, Burbury K, Lee N, et al. Clonal independence of JAK2 and CALR or MPL mutations in comutated myeloproliferative neoplasms demonstrated by single cell DNA sequencing. Haematologica. 2021;106(1):313–315. doi:10.3324/haematol.2020.260448.
- Makishima H. Somatic SETBP1 mutations in myeloid neoplasms. Int J Hematol. 2017;105(6):732–742. doi:10.1007/s12185-017-2241-1.
- Meggendorfer M, Bacher U, Alpermann T, Haferlach C, Kern W, Gambacorti-Passerini C, Haferlach T, Schnittger S. SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations. Leukemia. 2013;27(9):1852–1860. doi:10.1038/leu.2013.133.
- Hou HA, Kuo YY, Tang JL, Chou WC, Yao M, Lai YJ, Lin CC, Chen CY, Liu CY, Tseng MH, et al. Clinical implications of the SETBP1 mutation in patients with primary myelodysplastic syndrome and its stability during disease progression. Am J Hematol. 2014;89(2):181–186. doi:10.1002/ajh.23611.
- Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, Finke C, Score J, Gangat N, Mannarelli C, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861–1869. doi:10.1038/leu.2013.119.
- Guglielmelli P, Bartalucci N, Contini E, Rotunno G, Pacilli A, Romagnoli S, Mannelli L, Mannelli F, Coltro G, Pancrazzi A, et al. Involvement of RUNX1 pathway is a common event in the leukemic transformation of chronic myeloproliferative neoplasms (MPNs). Blood. 2019;134(Supplement_1):2968. doi:10.1182/blood-2019-129094.
- Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S, Tamura A, Honda H, Sakata-Yanagimoto M, Kumano K, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature. 2009;460(7257):904–908. doi:10.1038/nature08240.
- Laskowski RA, Stephenson JD, Sillitoe I, Orengo CA, Jm T. VarSite: disease variants and protein structure. Protein Sci. 2020;29(1):111–119. doi:10.1002/pro.3746.
- Beer PA, Delhommeau F, LeCouédic JP, Dawson MA, Chen E, Bareford D, Kusec R, McMullin MF, Harrison CN, Vannucchi AM, et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood. 2010;115(14):2891–2900. doi:10.1182/blood-2009-08-236596.
- Tefferi A, Guglielmelli P, Lasho TL, Coltro G, Finke CM, Loscocco GG, Sordi B, Szuber N, Rotunno G, Pacilli A, et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br J Haematol. 2020;189(2):291–302. doi:10.1111/bjh.16380.
- Lundberg P, Karow A, Nienhold R, Looser R, Hao-Shen H, Nissen I, Girsberger S, Lehmann T, Passweg J, Stern M, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. 2014;123(14):2220–2228. doi:10.1182/blood-2013-11-537167.
- Rampal R, Ahn J, Abdel-Wahab O, Nahas M, Wang K, Lipson D, Otto GA, Yelensky R, Hricik T, McKenney AS, et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc Natl Acad Sci USA. 2014;111(50):E5401–10. doi:10.1073/pnas.1407792111.
- Partouche N, Conejero C, Barathon Q, Moroch J, Tulliez M, Cordonnier C, Giraudier S. Emergence of MPLW515 mutation in a patient with CALR deletion: Evidence of secondary acquisition of MPL mutation in the CALR clone. Hematol Oncol. 2018;36(1):336–339. doi:10.1002/hon.2431.
- Bernal M, Jiménez P, Puerta J, Ruíz-Cabello F, Jurado M. Co-mutated CALR and MPL driver genes in a patient with myeloproliferative neoplasm. Ann Hematol. 2017;96(8):1339–1401. doi:10.1007/s00277-017-3023-9.