Abstract
Clinical studies have demonstrated an impairment of glucocorticoid receptor (GR)-mediated negative feedback on the hypothalamus–pituitary–adrenal (HPA) axis in patients with major depression (GR resistance), and its resolution by antidepressant treatment. Accordingly, reduced GR function has also been demonstrated in vitro, in peripheral tissues of depressed patients, as shown by reduced sensitivity to the effects of glucocorticoids on immune and metabolic functions. We and others have shown that antidepressants in vitro are able to modulate GR mRNA expression, GR protein level and GR function. This paper reviews the in vitro studies that have examined the effect of antidepressants on GR expression, number and function in human and animal cell lines, and the possible molecular mechanisms underlying these effects. Antidepressants are shown to both increase and decrease GR function in vitro, based on different experimental conditions. Specifically, increased GR function is likely to be mediated by an increased intracellular concentration of glucocorticoids, while decreased GR function seems to be the consequence of GR downregulation. We suggest that the study of the effects of antidepressants on glucocorticoid function might help clarify the therapeutic action of these drugs.
The HPA axis in major depression
The hypothalamus–pituitary–adrenal (HPA) axis is the main hormonal system involved in major depression. The HPA axis is coordinated by neuropeptides produced by neurones in the paraventricular nucleus of the hypothalamus, including the corticotropin releasing hormone (CRH) and vasopressin (Antoni Citation1993). CRH then stimulates the production of adrenocorticotropin (ACTH) from the anterior pituitary corticotrophs, which in turn stimulates the production of glucocorticoids by the adrenal gland. Glucocorticoids—cortisol in humans and corticosterone in rodents—then interact with their receptors in multiple target tissues. These include the hypothalamus and anterior pituitary, where they are responsible for the negative feedback regulation of the HPA axis through actions on paraventricular nucleus CRH/vasopressin neurones and corticotrophs, respectively. Glucocorticoids also exert their negative feedback actions at other levels in the brain, including the hippocampus, which is an upstream regulator of the HPA axis (Tasker et al. Citation2006; de Kloet et al. Citation2007). Glucocorticoids have notable actions on the neural pathways projecting to the CRH neurons from distant limbic-midbrain and cortical brain regions, like the amygdala and prefrontal anterior cingulate cortex (de Kloet et al. Citation2007). Although glucocorticoids regulate the function of almost every tissue in the body, major physiological effects of these hormones are in the regulation of energy metabolism, through increased gluconeogenesis, increased lipolysis, and increased protein degradation, and in the modulation of immune cell function (Pariante Citation2004).
The effects of glucocorticoids are mediated by nuclear intracellular receptors, namely the glucocorticoid receptor (GR) and the mineralocorticoid receptor (MR) (de Kloet et al. Citation1998). These intracellular receptors initiate transcriptional activation or repression by translocating, after ligand-binding, to the nucleus, and exert their effect either by interacting to a glucocorticoid response element (GRE) sequence in the promoter region of different glucocorticoid-regulated genes, or by interacting with other transcription factors (Mangelsdorf et al. Citation1995; de Kloet et al. Citation1998; Falkenstein et al. Citation2000). Changes in receptor number or function can be expected to alter the homeostatic function of the HPA axis.
The GR is expressed in almost all tissues in the body, although tissue and cell cycle-specific regulation of GR levels have been reported (Cidlowski et al. Citation1990; Oakley et al. Citation1996; Lu and Cidlowski Citation2005). In the brain, the GR is widely expressed, and is abundant in hypothalamic CRH neurons; it is also expressed in the anterior pituitary (de Kloet et al. Citation1998). Apart from binding glucocorticoids, the MR also binds aldosterone and is involved in the regulation of salt appetite and autonomic outflow (beyond the scope of this review). The MR is found in some hypothalamic sites, although the highest MR expression is found outside the hypothalamus, in the hippocampus, where it binds glucocorticoids at low, basal levels (de Kloet et al. Citation1998).
The MR and GR mediate different actions of glucocorticoids. The MR has a high affinity for endogenous glucocorticoid and for aldosterone, and is considered to be proactive and to regulate basal HPA activity (de Kloet et al. Citation1998; Juruena et al. Citation2004). In contrast to the MR, the GR has a high affinity for dexamethasone but a low affinity for endogenous corticosteroids. The GR is therefore considered to be more important in the regulation of the response to stress when endogenous levels of glucocorticoids are high (McEwen et al. Citation1997; de Kloet et al. Citation1998).
Classically, the MR and the GR mediate the slow, genomic actions of glucocorticoids; rapid action responses have also been described, and these could possibly include the classical intracellular receptor acting at the membrane through non-genomic signaling mechanisms (Tasker et al. Citation2006). Indeed, Karst et al. (Citation2005) reported a fast glucocorticoid action critically dependent on the MR, and suggested that a membrane location for the MR could mediate faster glucocorticoid actions when the levels of this hormone are high.
The HPA axis has been found to be hyperactive in many depressed patients and this disturbance is analogous to a sustained stress response in the absence of a stressor. The hyperactivity of the HPA axis in major depression is one of the most consistent findings in psychiatry, occurring in up to 80% of the patients when severely depressed (reviewed in Heuser et al. Citation1994; Nemeroff Citation1996; Holsboer Citation2000; McQuade and Young Citation2000; Pariante and Miller Citation2001a, Citation2003). In brief, the hyperactivity of the HPA axis is characterized by high cortisol levels in the plasma, urine and cerebrospinal fluid (Gold et al. Citation1988), impairment in the negative feedback regulation of the HPA axis (Carroll et al. Citation1981; Nemeroff Citation1996), and hyperplasia of the adrenal and pituitary glands (Axelson et al. Citation1992; Rubin et al. Citation2001). Consistent with these findings, increased volume of the pituitary in patients with affective and non-affective psychosis was shown by our group using brain magnetic resonance imaging (Pariante et al. Citation2004c, Citation2005). Other HPA axis abnormalities have been found in victims of suicide—many of whom were presumably depressed—including a downregulation of CRH receptors in the frontal cortex (Merali et al. Citation2004) and increased weight of the adrenal glands (Dumser et al. Citation1998).
Such disturbance of the HPA axis has been also verified indirectly, using tests designed to measure the integrity of the GR-mediated negative feedback mechanism, and these studies support the notion that GR-mediated negative feedback is impaired in major depression. There is a multitude of studies demonstrating non-suppression of cortisol secretion following administration of the synthetic glucocorticoid, dexamethasone (dexamethasone suppression test, DST), and more recent studies have shown a lack of inhibition of ACTH responses to CRH following dexamethasone pre-treatment (dexamethasone/CRH test) (Nemeroff Citation1996; Holsboer Citation2000). While non-suppression by dexamethasone in the DST and the dexamethasone/CRH test likely represent impaired feedback inhibition at the level of the pituitary (de Kloet et al. Citation1998), impaired responsiveness to hydrocortisone challenge in depressed patients suggests these feedback alterations occur in the brain (Young et al. Citation1991), although this latter finding has not been always replicated (Cooney and Dinan Citation1996).
The relevance of DST and the dexamethasone/CRH test comes from the finding that the responses to these tests are not only a biomarker of depression, but also a biomarker of treatment success. Indeed, efficacious antidepressant treatment is associated with resolution of the disturbance in the negative feedback in patients who are non-suppressors before treatment, with up to 75% of non-suppressor patients switching to suppressor status coincident with a treatment response (Linkowski et al. Citation1987; Heuser et al. Citation1996).
Resolution of the HPA disturbance is also seen after non-pharmacological antidepressant treatments like electroconvulsive therapy (Yuuki et al. Citation2005; Kunugi et al. Citation2006) and transcranial magnetic stimulation (Pridmore Citation1999; Reid and Pridmore Citation1999; Zwanzger et al. Citation2003). Moreover, persistence of non-suppression after antidepressant treatment is associated with high risk of early relapse and a poor outcome after discharge; specifically, in one study 90% of patients who were still non-suppressors after initial resolution of depressive symptoms had an early relapse (Zobel et al. Citation1999, Citation2001). Although normalization of the hyperactivity of the HPA axis occurs after successful antidepressant therapy, data in humans and animals (discussed below) suggest that the mechanism by which antidepressants normalize the HPA axis is at least partially independent of changes in monoaminergic neurotransmitter systems—the accepted mechanism of action of antidepressants.
In agreement with the studies mentioned above is the finding that first-degree relatives of depressed patients also show the same impaired GR-mediated negative feedback, which may therefore represent a genetic (trait) vulnerability to depressive disorders (Lauer et al. Citation1998; Modell et al. Citation1998). Moreover, a preliminary prospective study conducted in these first-degree relatives has shown that 26% of the subjects develop depression within 4 years (Ising et al. Citation2005). In addition, the importance of the impaired GR-mediated negative feedback in depression is underlined by genetic investigations which found that polymorphisms of the GR gene and of genes encoding for chaperones of the GR possibly contribute to the development of depressive symptoms and play a role in the variability of antidepressant response (Binder et al. Citation2004; van Rossum et al. Citation2006).
It is also of note that clinical studies show that manipulation of GR function has antidepressant effects. Thus, treatment with GR and MR agonists, like dexamethasone, prednisolone and cortisol, has shown their antidepressant action (Dinan et al. Citation1997; Bouwer et al. Citation2000; DeBattista et al. Citation2000). In contrast, GR antagonists, like RU486, have therapeutic effects in the treatment of psychotic depression (Belanoff et al. Citation2001) and bipolar disorder (Young et al. Citation2004). These findings could appear to contradict each other. However, the therapeutic effects of GR antagonists could also be explained by the action of these drugs in blocking negative feedback, thus causing a persistent elevation of cortisol levels. The increased cortisol levels, in turn, would increase the cortisol signal in the brain, similar to a treatment with an agonist. Consequently, during treatment with a GR antagonist, the increased cortisol levels could increase MR stimulation since this receptor is not blocked by RU486.
Interestingly, although typical major depression is associated with a hyperactive HPA system and hypersecretion of CRH, the same system may be underactive in atypical depression. It has been speculated that an underactive HPA axis might contribute to the profound fatigue and reversed neurovegetative symptoms that characterize this specific subtype of major depression (Gold and Chrousos Citation2002; Levitan et al. Citation2002; Stewart et al. Citation2005).
Why depressed patients have a hyperactive HPA axis is still a matter of debate. Some authors suggest that it is related to the abnormal GR-mediated feedback found in these patients, and thus to glucocorticoid resistance (Pariante and Miller Citation2001). This may be due to a reduced GR sensitivity, a reduced GR number, or to both, a view that is supported by reduced GR function in peripheral blood cells (Yehuda et al. Citation1993) and in the skin (Fitzgerald et al. Citation2006), and by the lack of cushingoid stigmata (Murphy Citation1991), in hypercortisolaemic depressed patients. This suggests that GR dysfunction generalizes to tissues outside the HPA axis, but the extent of this dysfunction is not known. Because of this uncertainty, in this review the GR dysfunction will be called partial glucocorticoid resistance.
Evaluation of glucocorticoid receptor function in major depression
Given the limited access to the brain GR in clinical populations, in vitro studies have been used to understand the molecular mechanisms underlying GR abnormalities in patients with major depression. Indeed, data have demonstrated similar regulation of GR in the brain and in the immune system of laboratory animals. For example, Lowy (Citation1990) has shown that treatment of rats with reserpine, the amine depleting drug that is known to induce depressive symptoms in humans and to produce dexamethasone non-suppression in rats, decreases GR levels in the hippocampus, frontal cortex and pituitary as well as in lymphocytes and spleen. Similarly, Spencer et al. (Citation1991) have found that, both in the brain and in the immune system, GR is up-regulated following adrenalectomy and down-regulated following chronic treatment with corticosterone. Therefore, many studies have relied on the measurement of peripheral GR number and function in blood cells, but it is still unknown whether these results reflect the actual values in key areas of the brain associated with clinical depression.
A detailed analysis of these studies has been presented (Pariante et al. 2001b; Pariante Citation2004, Citation2006). Briefly, depressed patients have shown some alterations of lymphocyte GR number (Gormley et al. Citation1985; Whalley et al. Citation1986). Although most studies have found no evidence of reduced GR number when measured in the whole cell, some studies found reduced GR levels in the cytoplasm, which is likely due to increased sequestration in the nucleus, as discussed previously (Pariante et al. 2001b; Pariante Citation2004). More consistently, depressed patients have shown reduced GR function when investigated in vitro in immune cells from the peripheral blood. The studies have focused on the well-known capacity of glucocorticoids to inhibit the ability of peripheral blood mononuclear cells to proliferate in response to polyclonal mitogens. Interestingly, in these studies immune cells from depressed patients showed partial glucocorticoid resistance, seen as a reduced dexamethasone-induced inhibition of this proliferative response in vitro (Lowy et al. Citation1984, Citation1988; Rupprecht et al. Citation1991; Wodarz et al. Citation1991, Citation1992). The partial glucocorticoid resistance found in lymphocytes correlates with the partial glucocorticoid resistance found with the DST mentioned above. When patients are non-suppressors in the DST, indicating in vivo partial glucocorticoid resistance, they also show reduced dexamethasone-induced inhibition of the lymphocyte proliferative response. In addition, when patients respond positively to treatment and overcome the suppression found on the DST, the sensitivity of lymphocytes to dexamethasone also returns to normal levels (Wodarz et al. Citation1992).
HPA abnormalities in depressed patients have been shown also by other techniques. A remarkable lack of translocation response of GR was found when measured by cytosolic binding following incubation with dexamethasone (Wassef et al. Citation1990; Yehuda et al. Citation2002); probing peripheral cutaneous GR function by the steroid vasoconstriction assay also revealed partial GR resistance (Fitzgerald et al. Citation2006). Consistently, in these experiments the lack of response is particularly evident in those who are non-suppressors in the DST (Calfa et al. Citation2003).
Although the glucocorticoid-mediated negative feedback action is mediated by both GR and MR, most of the studies have indeed focused on the GR instead of the MR. In order to evaluate the contributions of both receptors to the HPA disturbance in depression in vivo, we have proposed a suppressive test using prednisolone (PST), a synthetic glucocorticoid that, in contrast to dexamethasone, is more similar to the endogenous glucocorticoid cortisol in its pharmacodynamic and pharmacokinetic features (Pariante et al. Citation2002b, Citation2004a) and therefore should probe both receptors. Using the PST, we have shown that depressed patients who present partial glucocorticoid resistance of the GR, still have intact MR function (Juruena et al. Citation2006). In accordance, the only previous study that specifically examined MR function in depression using an MR antagonist—spironolactone—also found that this pathway is intact, and possibly oversensitive (Young et al. Citation2003).
The effect of antidepressants on the glucocorticoid receptor
The demonstrated efficacy of antidepressants in modifying the HPA axis when inducing the therapeutic effects suggests that the HPA axis could be a target of antidepressant action. Can antidepressants directly interact with the HPA axis? Can these drugs change the number or the function of the GR or of the MR, in order to overcome this partial glucocorticoid resistance? And if so, what are the molecular mechanisms that could be operative?
Following the initial study by Peiffer et al. (Citation1991), considerable evidence from humans, animals and in vitro work has accumulated demonstrating that antidepressants do interact with the GR and increase GR-mediated negative feedback. Specifically, studies in depressed patients, animals and cellular models, have demonstrated that, depending on the tissue, antidepressants are able to increase GR expression, enhance GR function and promote GR nuclear translocation (Pariante et al. 2001b). This review is focused on the in vitro models as these allow examination of the effects of functional GR responsiveness without the confounding pharmacodynamic or pharmacokinetic effects that are present with either acute or long-term in vivo treatment studies. A summary of the studies discussed here on the effect of antidepressants on GR is shown in Tables I–III, respectively, in human, rat and mouse cells. Like the studies of primary HPA abnormalities in depression, most of the studies on the effects of antidepressants have investigated GR only. The only study that has measured MR in vitro has found no effect of antidepressants (Lai et al. Citation2003).
Table I. In vitro effects of antidepressants on the GR and the MR in human cells.
Table II. In vitro effects of antidepressants on the GR and the MR in rat cells.
Table III. In vitro effects of antidepressants on the GR and the MR in mouse cells.
In animals, tricyclic and SSRI antidepressants increase GR mRNA expression in many cells including neuronal cell cultures (Pepin et al. Citation1989; Okugawa et al. Citation1999; Hery et al. Citation2000; Lai et al. Citation2003) and fibroblasts (Pepin et al. Citation1992a). Interestingly, increased GR expression has also been confirmed in human peripheral blood mononuclear cells (Vedder et al. Citation1999; Heiske et al. Citation2003; Funato et al. Citation2006). It is of note that the effect of antidepressants on GR mRNA levels can actually be time-dependent as reported by Heiske et al. (Citation2003). These authors found increased GR mRNA levels in human leukocytes after 2.5 h treatment with imipramine, which decreased after longer periods of incubation.
GR protein has also been quantified, either indirectly using cytosolic binding or directly using Western blot techniques. Interestingly, some of the studies that have analyzed GR mRNA expression and quantified GR protein in the same cells have detected a positive correlation of these two measurements (Pepin et al. Citation1992b; Barden Citation1996; Lai et al. Citation2003). Nevertheless, the results using GR protein have been more difficult to interpret, with studies finding both increased (Pepin et al. Citation1992a; Hery et al. Citation2000; Lai et al. Citation2003; Cai et al. Citation2005), decreased (Pariante et al. Citation1997; Hery et al. Citation2000; Pariante 2003a) or unaltered level (Pepin et al. Citation1992a; Hery et al. Citation2000). Most studies that have looked at the immediate in vitro effects, that is, within the first 24 h, have found that antidepressants reduce GR protein level (Pariante et al. Citation1997, Citation2003a; Heiske et al. Citation2003; Okuyama-Tamura et al. Citation2003). This is particularly important in the light of the studies (described below) that have evaluated GR function.
GR function has been investigated using basically two different approaches: translocation of the receptor to the nucleus, or GR-mediated gene transcription. GR translocation has been measured by immunocytochemistry and Western blot; whereas GR-mediated gene transcription has been measured using a mouse fibroblast cell line transiently transfected with the MMTV–chloramphenicol acetyltransferase (MMTV–CAT) reporter gene (LMCAT cells). Expression of the CAT reporter gene by these cells is under glucocorticoid control by virtue of several GRE residing within the MMTV promoter, which lies upstream of the CAT reporter gene (Sanchez et al. Citation1994).
Using the reporter gene technique above specified, Pepin in their pivotal paper showed that in rat fibroblasts, desipramine enhances GR function, measured as GR-mediated gene transcription after 24 h (while up-regulating GR protein after 72 h), suggesting that antidepressants directly increase GR function before (and in the absence) of GR upregulation (Pepin et al. Citation1992a). Subsequently, our group showed that 24 h treatment with desipramine in mouse fibroblasts also increases GR function: specifically, it increases GR translocation in the absence of any glucocorticoids (Pariante et al. Citation1997). Following our work, other groups confirmed that antidepressants increase GR translocation using this and other animal species, including human (Heiske et al. Citation2003; Okuyama-Tamura et al. Citation2003; Funato et al. Citation2006).
Moreover, we found that 24 h coincubation of antidepressants and dexamethasone or cortisol enhances GR-mediated gene transcription compared to cells treated with the glucocorticoids alone (Pariante et al. Citation1997, Citation2001a, Citation2003b). The increased GR-mediated gene transcription is evident in the absence of any GR upregulation, suggesting that the GR upregulation described by other authors after antidepressants is a consequence, rather than a cause, of increased receptor function (Pariante et al. Citation1997). This is consistent with the study discussed above (Pepin et al. Citation1992a), where the increased GR function was already present after treatment with desipramine for 1 day, while the increased GR number was only seen after 3 days of desipramine treatment.
It is important to mention that some studies, including from our own group, also found GR function to be reduced after antidepressant treatment (Pariante et al. Citation1997, 2001a). Mouse fibroblasts cells (LMCAT cells) show reduced GR-mediated gene transcription after antidepressant treatment when followed by (rather than coincubated with) dexamethasone (Pariante et al. Citation1997), or in the presence of another glucocorticoid hormone, corticosterone (both when coincubated with and when following the antidepressants). This finding has been replicated by other authors using various antidepressants (Augustyn et al. Citation2005; Budziszewska et al. Citation2000,2005). Also confirming our results, Miller et al. (Citation2002) have found that incubation with desipramine alone induces a small decrease in unstimulated GR-mediated gene transcription. Therefore, under some experimental conditions, antidepressants decrease rather than increase GR function.
At first, the differential effect of antidepressants on GR function can seem confusing. However, it is of note that the results agree with each other when studies are separated according to the experimental condition. When cells are pre-treated with antidepressants, before incubation with any glucocorticoids, GR function is decreased (Barden Citation1996; Pariante et al. Citation1997; Budziszewska et al. Citation2000, Citation2005; Augustyn et al. Citation2005). When cells are coincubated with antidepressants and cortisol or dexamethasone, GR function is increased, while if cells are coincubated with antidepressants and corticosterone, GR function is decreased (Pariante et al. Citation1997; Miller et al. Citation2002). The only result that reports the opposite has used a different cell culture model—LTK fibroblasts and neuroblastoma cells (Pepin et al. Citation1992a).
Interesting, and possibly relevant for the differential effect of antidepressants on glucocorticoid function, is the finding that antidepressants directly inhibit membrane steroid transporters that expel glucocorticoids out of cells. By blocking these transporters, antidepressants increase the intracellular levels of glucocorticoids: an effect that is independent of any neurotransmitter action, and which we have described in both mice fibroblasts and rat cortical neurons (Pariante et al. Citation2003a,Citationb, Citation2004b). The inhibiting effects of antidepressants on membrane steroid transporters has been shown, by us and others, for tricyclic antidepressants (Merry et al. Citation1991; Varga et al. Citation1996; Szabo et al. Citation1999; Pariante et al. Citation2003a) and newer antidepressants including fluoxetine, sertraline, paroxetine, fluvoxamine, reboxetine, citalopram and venlafaxine (Pariante et al. Citation2003b; Weiss et al. Citation2003). In support of a role for steroid transporters in the effects of antidepressants on GR function are the findings described above that coincubation of antidepressants and glucocorticoids leads to increased GR function if the glucocorticoids are substrate for the transporters (dexamethasone and cortisol), while it leads to decreased GR function if the glucocorticoid is not a substrate for the transporters (corticosterone) (Pariante et al. 2001a); moreover, we have also shown that if cells are treated with verapamil, an inhibitor of these steroid transporters, then antidepressants decrease GR function even when coincubated in the presence of dexamethasone or cortisol (Pariante et al. 2001a); and finally, antidepressants directly increase the intracellular concentrations of (radioactive) cortisol, but not of (radioactive) corticosterone (Pariante et al. Citation2003a,Citationb). Moreover, Herr et al. (Citation2003) have also shown that treatment of mouse hippocampal cells with verapamil reduces the effects of antidepressants on GR-mediated gene transcription.
Mechanisms for the effect of antidepressants on the GR: A possible model
Our group has hypothesized a possible explanation for the effect of antidepressants on GR function, as depicted in . As discussed above, this model supports a role for the membrane steroid transporters, like the multidrug resistance P-glycoprotein (MDR PGP). Overall, treatment with antidepressants (for 24 h or less) inhibits steroid transporters, induces GR translocation, and reduces GR expression (Pariante et al. Citation1997, Citation2001b, Citation2003a,Citationb). These three effects could be mediated by the same mechanism. For example, blocking of steroid transport can increase the intracellular levels of steroids taken up from the medium, thus leading to translocation of the GR. Data showing that inhibitors of the steroid transporter P-glycoprotein induce partial GR translocation in human ovarian cancer cells support this possibility (Prima et al. Citation2000); although a recent study found no effect of verapamil on GR translocation in human lymphocytes (Okuyama-Tamura et al. Citation2003). Translocation of GR, in turn, leads to the reduction in GR expression (Schaaf and Cidlowski Citation2002). Indeed, GR translocation by both GR agonists and GR antagonists has been associated with GR downregulation (Burnstein et al. Citation1994). This downregulation takes place over a few hours and is due to a reduction in the protein half-life and an inhibition of GR mRNA synthesis; it is temporary and can be followed by a subsequent upregulation (Schmidt and Meyer Citation1994). Therefore, it is possible that the inhibition of the transporters precedes (and causes, by inducing GR translocation) the GR downregulation.
Figure 1 Theoretical model of antidepressant differential effects on GR function. We hypothesise that the final in vitro effect of antidepressants on glucocorticoid receptor (GR) function depends on the experimental conditions, and especially on the ability of antidepressants to block membrane steroid transporters like P-glycoprotein (PGP) that expel glucocorticoids from the cells. In conditions that do elicit effect on the PGP (A), antidepressants lead to increased GR function by blocking steroid transporters and therefore increasing steroid level inside the cells; these effects compensate and overcome the effects of the GR downregulation. By contrast, in conditions that do not elicit effects on PGP, antidepressants decrease GR function because the effects on GR downregulation predominate (B).
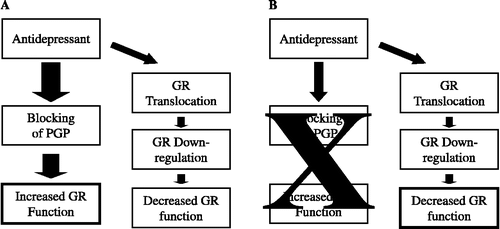
If cells are treated in experimental conditions that do elicit the effects on the transporter (), as when cells are coincubated for 24 h with antidepressants and a glucocorticoid that is expelled by the transporter, such as dexamethasone or cortisol, enhanced GR-mediated gene transcription is evident (Pariante et al. Citation1997, Citation2003a,Citationb, 2001b). This is because the increase in the intracellular levels of the glucocorticoid overcomes, and possibly precedes, the GR downregulation.
However, if cells are treated in experimental conditions that do not elicit the effects on the transporter (), the GR downregulation leads to a reduced GR-mediated gene transcription. This could explain why antidepressants give a reduction in GR-mediated gene transcription when cells are coincubated with corticosterone (Pariante et al. Citation2001a, Citation2003b), which is not a substrate of P-glycoprotein, or in the presence of the transporter inhibitor, verapamil (Pariante et al. Citation2001a, Citation2003b), or when cells are treated with antidepressants alone (Miller et al., Citation2002). Finally, pre-incubation of cells with antidepressants inhibits GR-mediated gene transcription induced by a subsequent short treatment (1.5–2 h) with dexamethasone or corticosterone (Pariante et al. Citation1997). In this case, even if the inhibition of the transporter increases the intracellular levels of the glucocorticoid, as confirmed by our in vitro experiments with radioactive cortisol (Pariante et al. Citation2003a), this is unable to compensate for the GR downregulation, possibly because of the short incubation, or possibly because the GR downregulation is present before the glucocorticoid is added (in contrast with the coincubation experiments).
Interestingly, this model is supported by our most recent study in mice showing that treatment with desipramine induces GR downregulation in P-glycoprotein knockout mice—and therefore in a condition that is similar to the in vitro experiments that do not elicit the effect on the transporter; on the other hand, desipramine induces GR upregulation in control mice—and therefore in a condition that is similar to the in vitro experiments which do elicit effects on the transporter (Yau et al. Citation2007). Of course, these animal data, taken together with the clinical data reviewed above, support the notion that the GR downregulation is a transient effect, possibly due to the initial GR activation, and remains evident only under artificial conditions such as in the P-glycoprotein knockout; while the long-term effect of antidepressants under normal conditions is that of increased GR expression and function.
Mechanisms for the effect of antidepressants on the GR: Other possible models
Undoubtedly, other molecular pathways are involved in the molecular mechanisms of the effects of antidepressants on GR, besides the effects on the membrane steroid transporters. There is now considerable evidence that phosphorylation of the GR by cAMP-dependent protein kinase has a relevant role in the regulation of GR function. For example, adenylate cyclase activators, PKA activators and phosphodiesterase type 4 inhibitors—all compounds that increase PKA activity—increase GR function in vitro (Rangarajan et al. Citation1992; Miller et al. Citation2002), and β-agonists have been shown to translocate the GR from cytoplasm to nucleus via the cAMP/protein kinase A pathway (Eickelberg et al. Citation1999). These findings are particularly intriguing in view of the finding that cultured fibroblasts from depressed patients exhibit reduced cAMP-dependent protein kinase activity (Manier et al. Citation2000). Moreover, recent work on the mechanism of action of antidepressants suggests that cAMP and protein kinase A play an important role as mediators of the psychotropic effects of these agents, possibly leading to increased neurogenesis (Rasenick et al. Citation1996; Duman et al. Citation2001). Indeed, the phosphodiesterase type 4 inhibitor rolipram has been shown not only to increase GR function alone but also to potentiate the antidepressant-induced increase of GR function (Miller et al. Citation2002). Therefore, it is possible that disruption in the cAMP/protein kinase A pathway described in major depression is linked to GR resistance in this disorder, and that antidepressants may overcome these receptor alterations via a direct effect on this pathway.
Antidepressants are also known to influence the activity of other intracellular protein kinases such as protein kinase C and the Ca2+ –calmodulin-dependent protein kinase (Silver et al. Citation1986; Nalepa and Vetulani Citation1991; Bouron and Chatton Citation1999; Budziszewska et al. Citation2000; Augustyn et al. Citation2005). These kinases seem to be involved in the antidepressant-induced decrease of GR-mediated gene transcription (and therefore possibly in the antidepressant-induced GR downregulation, although this has not been investigated yet). Some authors have shown that the effect of imipramine on the corticosterone mediated gene transcription depends partly on the PLC/PKC pathway (Budziszewska et al. Citation2000; Augustyn et al. Citation2005).
Finally, another possible pathway by which GR function is abnormal in depression could be the activation of mediators of the immune response. Pro-inflammatory cytokines, like interleukins 1 and 6, can induce partial glucocorticoid resistance and result in hyperactivity of the HPA axis (Miller et al. Citation1999; Pariante et al. Citation1999; Maddock and Pariante Citation2001). A number of studies have demonstrated that treatment with pro-inflammatory cytokines induces a decrease in GR function as evidenced by decreased sensitivity to the effects of glucocorticoids on functional endpoints and decreased GR affinity for ligand. Moreover, studies performed on peripheral cells and tissues of patients with inflammatory diseases such as asthma, ulcerative colitis, acquired immunodeficiency syndrome and rheumatoid arthritis, especially those showing resistance to the therapeutic effects of glucocorticoids, have also demonstrated reductions in GR function and affinity that are similar to those induced by cytokines (Miller et al. Citation1999; Pariante et al. Citation1999; Maddock and Pariante 2001). Indeed, major depression has also been associated with evidence of immune inflammation and increased levels of pro-inflammatory cytokines (Raison and Miller Citation2003). In turn, treatment with pro-inflammatory cytokines, or treatments that increase the production of pro-inflammatory cytokines like interferon-α for chronic viral hepatitis, can induce depressive symptoms (Pariante et al. Citation1999, Citation2002a; Raison and Miller 2003, Maddock et al. Citation2004, Citation2005). We have shown that the pro-inflammatory cytokine interleukin-1 directly blocks GR translocation and function in vitro (Pariante et al. Citation1999), an effect that is virtually opposite to that of antidepressants in the same experimental system (Pariante et al. Citation1997). Wang et al. (Citation2004) have shown that these effects of interleukin-1 are mediated by stimulating the p38 mitogen-activated protein kinase signal transduction pathway.
It is of note that a few studies have examined the effect of antidepressants on GR function in populations of psychiatric patients. For example, Yehuda et al. (Citation2004, Citation2006) have found increased GR function in leukocytes of patients with post-traumatic stress disorder, which is reduced by 3 days of in vitro treatment with sertraline. These findings are consistent with an extensive literature supporting the notion that the GR function in these patients is virtually opposite to that in depressed patients, that is, it is hypersensitive (Yehuda Citation2001; Yehuda et al. Citation2004, Citation2006). This study supports once again the effect of antidepressants on the GR and the fundamental ability of these in vitro tests to reproduce the function of GR in the HPA axis tissues.
Conclusions
In conclusion, in vitro use of antidepressants is able to extensively modulate GRs irrespective of their antidepressant structure and putative target molecule. Hence, the study of the in vitro effect of antidepressants and GR are of particular relevance to understand the molecular mechanisms underlying GR abnormalities in depressed patients and its regulation by antidepressant treatment. Moreover, this approach might prove to be successful in finding strategies to maximize therapeutic antidepressants effects.
Acknowledgements
Livia A. Carvalho is currently funded by a grant from the UK Medical Research Council (MRC). Carmine M. Pariante has been funded by the UK MRC since 1999, first as a Clinical Training Fellow, and currently as an MRC Clinician Scientist Fellow. His research is also funded by the NIHR South London and Maudsley NHS Foundation Trust & Institute of Psychiatry Specialist Biomedical Research Centre for Mental Health, the NARSAD, the APIRE, and the British Academy. The authors have no relevant financial interest to disclose.
References
- Antoni FA. Vasopressinergic control of pituitary adrenocorticotropin secretion comes of age. Front Neuroendocrinol 1993; 14: 76–122
- Augustyn M, Otczyk M, Budziszewska B, Jagla G, Nowak W, Basta-Kaim A, et al. Effects of some new antidepressant drugs on the glucocorticoid receptor-mediated gene transcription in fibroblast cells. Pharmacol Rep 2005; 57: 766–773
- Axelson DA, Doraiswamy PM, Boyko OB, Rodrigo EP, McDonald WM, Ritchie JC, et al. In vivo assessment of pituitary volume with magnetic resonance imaging and systematic stereology: Relationship to dexamethasone suppression test results in patients. Psychiatr Res 1992; 44: 63–70
- Barden N. Modulation of glucocorticoid receptor gene expression by antidepressant drugs. Pharmacopsychiatry 1996; 29: 12–22
- Basta-Kaim A, Budziszewska B, Jaworska-Feil L, Tetich M, Kubera M, Leskiewicz M, et al. Antipsychotic drugs inhibit the human corticotropin-releasing-hormone gene promoter activity in neuro-2A cells-an involvement of protein kinases. Neuropsychopharmacology 2006; 31: 853–865
- Belanoff JK, Flores BH, Kalezhan M, Sund B, Schatzberg AF. Rapid reversal of psychotic depression using mifepristone. J Clin Psychopharmcol 2001; 21: 516–521
- Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, Putz B, et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet 2004; 36: 1319–1325
- Bouron A, Chatton JY. Acute application of the tricyclic antidepressant desipramine presynaptically stimulates the exocytosis of glutamate in the hippocampus. Neuroscience 1999; 90: 729–736
- Bouwer C, Claassen J, Dinan TG, Nemeroff CB. Prednisone augmentation in treatment-resistant depression with fatigue and hypocortisolaemia: A case series. Depress Anxiety 2000; 12: 44–50
- Budziszewska B, Jaworska-Feil L, Kajta M, Lason W. Antidepressant drugs inhibit glucocorticoid receptor-mediated gene transcription—a possible mechanism. Br J Pharmacol 2000; 130: 1385–1393
- Budziszewska B, Basta-Kaim A, Kubera M, Jaworska L, Leskiewicz M, Tetich M, et al. Effect of lipopolysaccharide and antidepressant drugs on glucocorticoid receptor-mediated gene transcription. Pharmacol Rep 2005; 57: 540–544
- Burnstein KL, Jewell CM, Sar M, Cidlowski JA. Intragenic sequences of the human glucocorticoid receptor complementary DNA mediate hormone-inducible receptor messenger RNA down-regulation through multiple mechanisms. Mol Endocrinol 1994; 8: 1764–1773
- Cai W, Khaoustov VI, Xie Q, Pan T, Le W, Yoffe B. Interferon-alpha-induced modulation of glucocorticoid and serotonin receptors as a mechanism of depression. J Hepatol 2005; 42: 880–887
- Calfa G, Kademian S, Ceschin D, Vega G, Rabinovich GA, Volosin M. Characterization and functional significance of glucocorticoid receptors in patients with major depression: Modulation by antidepressant treatment. Psychoneuroendocrinology 2003; 28: 687–701
- Carroll BJ, Feinberg M, Greden JF, Tarika J, Albala AA, Haskett RF, et al. A specific laboratory test for the diagnosis of melancholia. Standardization, validation, and clinical utility. Arch Gen Psychiatry 1981; 38: 15–22
- Cidlowski JA, Bellingham DL, Powell-Oliver FE, Lubahn DB, Sar M. Novel antipeptide antibodies to the human glucocorticoid receptor: Recognition of multiple receptor forms in vitro and distinct localization of cytoplasmic and nuclear receptors. Mol Endocrinol 1990; 4: 1427–1437
- Cooney JM, Dinan TG. Preservation of hypothalamic–pituitary–adrenal axis fast-feedback responses in depression. Acta Psychiatr Scand 1996; 94: 449–453
- DeBattista C, Posener JA, Kalehzan BM, Schatzberg AF. Acute antidepressant effects of intravenous hydrocortisone and CRH in depressed patients: A double-blind, placebo-controlled study. Am J Psychiatry 2000; 157: 1334–1337
- Dinan TG, Lavelle E, Cooney J, Burnett F, Scott L, Dash A, et al. Dexamethasone augmentation in treatment-resistant depression. Acta Psychiatr Scand 1997; 95: 58–61
- Duman RS, Nakagawa S, Malberg J. Regulation of adult neurogenesis by antidepressant treatment. Neuropsychopharmacology 2001; 25: 836–844
- Dumser T, Barocka A, Schubert E. Weight of adrenal glands may be increased in persons who commit suicide. Am J Forensic Med Pathol 1998; 19: 72–76
- Eickelberg O, Roth M, Lorx R, Bruce V, Rudiger J, Johnson M, et al. Ligand-independent activation of the glucocorticoid receptor by beta2-adrenergic receptor agonists in primary human lung fibroblasts and vascular smooth muscle cells. J Biol Chem 1999; 274: 1005–1010
- Falkenstein E, Tillmann HC, Christ M, Feuring M, Wehling M. Multiple actions of steroid hormones—a focus on rapid, nongenomic effects. Pharmacol Rev 2000; 52: 513–556
- Fitzgerald P, O'Brien SM, Scully P, Rijkers K, Scott LV, Dinan TG. Cutaneous glucocorticoid receptor sensitivity and pro-inflammatory cytokine levels in antidepressant-resistant depression. Psychol Med 2006; 36: 37–43
- Funato H, Kobayashi A, Watanabe Y. Differential effects of antidepressants on dexamethasone-induced nuclear translocation and expression of glucocorticoid receptor. Brain Res 2006; 1117: 125–134
- Gold PW, Chrousos GP. Organization of the stress system and its dysregulation in melancholic and atypical depression: High vs low CRH/NE states. Mol Psychiatry 2002; 7: 254–275
- Gold PW, Goodwin FK, Chrousos GP. Clinical and biochemical manifestations of depression. Relation to the neurobiology of stress (1). N Engl J Med 1988; 319: 348–353
- Gormley GJ, Lowy MT, Reder AT, Hospelhorn VD, Antel JP, Meltzer HY. Glucocorticoid receptors in depression: Relationship to the dexamethasone suppression test. Am J Psychiatry 1985; 142: 1278–1284
- Heiske A, Jesberg J, Krieg JC, Vedder H. Differential effects of antidepressants on glucocorticoid receptors in human primary blood cells and human monocytic u-937 cells. Neuropsychopharmacology 2003; 28: 807–817
- Herr AS, Tsolakidou AF, Yassouridis A, Holsboer F, Rein T. Antidepressants differentially influence the transcriptional activity of the glucocorticoid receptor in vitro. Neuroendocrinology 2003; 78: 12–22
- Hery M, Semont A, Fache MP, Faudon M, Hery F. The effects of serotonin on glucocorticoid receptor binding in rat raphe nuclei and hippocampal cells in culture. J Neurochem 2000; 74: 406–413
- Heuser I, Yassouridis A, Holsboer F. The combined dexamethasone/CRH test: A refined laboratory test for psychiatric disorders. J Psychiatr Res 1994; 28: 341–356
- Heuser IJ, Schweiger U, Gotthardt U, Schmider J, Lammers CH, Dettling M, et al. Pituitary–adrenal system regulation and psychopathology during amitriptyline treatment in elderly depressed patients and normal comparison subjects. Am J Psychiatry 1996; 153: 93–99
- Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 2000; 23: 477–501
- Ising M, Lauer CJ, Holsboer F, Modell S. The Munich vulnerability study on affective disorders: Premorbid neuroendocrine profile of affected high-risk probands. J Psychiatr Res 2005; 39: 21–28
- Juruena MF, Cleare AJ, Pariante CM. The hypothalamic-pituitary-adrenal axis, glucocorticoid receptor function and relevance to depression. Rev Bras Psiquiatr 2004; 26: 189–201
- Juruena MF, Cleare AJ, Papadopoulos AS, Poon L, Lightman S, Pariante CM. Different responses to dexamethasone and prednisolone in the same depressed patients. Psychopharmacology (Berl) 2006; 189: 225–235
- Karst H, Berger S, Turiault M, Tronche F, Schutz G, Joels M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci USA 2005; 102: 19204–19207
- de Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev 1998; 19: 269–301
- de Kloet ER, Derijk RH, Meijer OC. Therapy insight: Is there an imbalanced response of mineralocorticoid and glucocorticoid receptors in depression?. Nat Clin Pract Endocrinol Metab 2007; 3: 168–179
- Kunugi H, Ida I, Owashi T, Kimura M, Inoue Y, Nakagawa S, et al. Assessment of the dexamethasone/CRH test as a state-dependent marker for hypothalamic–pituitary–adrenal (HPA) axis abnormalities in major depressive episode: A multicenter study. Neuropsychopharmacology 2006; 31: 212–220
- Lai M, McCormick JA, Chapman KE, Kelly PAT, Seckl JR, Yau JLW. Differential regulation of corticosteroid receptors by monoamine neurotransmitters and antidepressant drugs in primary hippocampal culture. Neuroscience 2003; 118: 975–984
- Lauer CJ, Schreiber W, Modell S, Holsboer F, Krieg JC. The Munich vulnerability study on affective disorders: Overview of the cross-sectional observations at index investigation. J Psychiatr Res 1998; 32: 393–401
- Levitan RD, Vaccarino FJ, Brown GM, Kennedy SH. Low-dose dexamethasone challenge in women with atypical major depression: Pilot study. J Psychiatry Neurosci 2002; 27: 47–51
- Linkowski P, Mendlewicz J, Kerkhofs M, Leclercq R, Golstein J, Brasseur M, et al. 24-hour profiles of adrenocorticotropin, cortisol, and growth hormone in major depressive illness: Effect of antidepressant treatment. J Clin Endocrinol Metab 1987; 65: 141–152
- Lowy MT. Reserpine-induced decrease in type I and II corticosteroid receptors in neuronal and lymphoid tissues of adrenalectomized rats. Neuroendocrinology 1990; 51: 190–196
- Lowy MT, Reder AT, Antel JP, Meltzer HY. Glucocorticoid resistance in depression: The dexamethasone suppression test and lymphocyte sensitivity to dexamethasone. Am J Psychiatry 1984; 141: 1365–1370
- Lowy MT, Reder AT, Gormley GJ, Meltzer HY. Comparison of in vivo and in vitro glucocorticoid sensitivity in depression: relationship to the dexamethasone suppression test. Biol Psychiatry 1988; 24: 619–630
- Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell 2005; 18: 331–342
- Maddock C, Pariante CM. How does stress affect you? An overview of stress, immunity, depression and disease. Epidemiol Psichiatr Soc 2001; 10: 153–162
- Maddock C, Baita A, Orru MG, Sitzia R, Costa A, Muntoni E, et al. Psychopharmacological treatment of depression, anxiety, irritability and insomnia in patients receiving interferon-alpha: A prospective case series and a discussion of biological mechanisms. J Psychopharmacol 2004; 18: 41–46
- Maddock C, Landau S, Barry K, Maulayah P, Hotopf M, Cleare AJ, et al. Psychopathological symptoms during interferon-alpha and ribavirin treatment: effects on virologic response. Mol Psychiatry 2005; 10: 332–333
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, et al. The nuclear receptor superfamily: The second decade. Cell 1995; 83: 835–839
- Manier DH, Shelton RC, Ellis TC, Peterson CS, Eiring A, Sulser F. Human fibroblasts as a relevant model to study signal transduction in affective disorders. J Affect Disord 2000; 61: 51–58
- McEwen BS, Biron CA, Brunson KW, Bulloch K, Chambers WH, Dhabhar FS, et al. The role of adrenocorticoids as modulators of immune function in health and disease: Neural, endocrine and immune interactions. Brain Res Brain Res Rev 1997; 23: 79–133
- McQuade R, Young AH. Future therapeutic targets in mood disorders: The glucocorticoid receptor. Br J Psychiatry 2000; 177: 390–395
- Merali Z, Du L, Hrdina P, Palkovits M, Faludi G, Poulter MO, et al. Dysregulation in the suicide brain: mRNA expression of corticotropin-releasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. J Neurosci 2004; 24: 1478–1485
- Merry S, Hamilton TG, Flanigan P, Freshney RI, Kaye SB. Circumvention of pleiotropic drug resistance in subcutaneous tumours in vivo with verapamil and clomipramine. Eur J Cancer 1991; 27: 31–34
- Miller AH, Pariante CM, Pearce BD. Effects of cytokines on glucocorticoid receptor expression and function. Glucocorticoid resistance and relevance to depression. Adv Exp Med Biol 1999; 461: 107–116
- Miller AH, Vogt GJ, Pearce BD. The phosphodiesterase type 4 inhibitor, rolipram, enhances glucocorticoid receptor function. Neuropsychopharmacology 2002; 27: 939–948
- Modell S, Lauer CJ, Schreiber W, Huber J, Krieg JC, Holsboer F. Hormonal response pattern in the combined DEX-CRH test is stable over time in subjects at high familial risk for affective disorders. Neuropsychopharmacology 1998; 18: 253–262
- Murphy BE. Steroids and depression. J Steroid Biochem Mol Biol 1991; 38: 537–559
- Nalepa I, Vetulani J. Involvement of protein kinase C in the mechanism of in vitro effects of imipramine on generation of second messengers by noradrenaline in cerebral cortical slices of the rat. Neuroscience 1991; 44: 585–590
- Nemeroff CB. The corticotropin-releasing factor (CRF) hypothesis of depression: New findings and new directions. Mol Psychiatry 1996; 1: 336–342
- Oakley RH, Sar M, Cidlowski JA. The human glucocorticoid receptor beta isoform expression, biochemical properties, and putative function. J Biol Chem 1996; 271: 9550–9559
- Okugawa G, Omori K, Suzukawa J, Fujiseki Y, Kinoshita T, Inagaki C. Long-term treatment with antidepressants increases glucocorticoid receptor binding and gene expression in cultured rat hippocampal neurones. J Neuroendocrinol 1999; 11: 887–895
- Okuyama-Tamura M, Mikuni M, Kojima I. Modulation of the human glucocorticoid receptor function by antidepressive compounds. Neurosci Lett 2003; 342: 206–210
- Pariante CM. Depression, stress and the adrenal axis. J Neuroendocrinol 2003; 15: 811–812
- Pariante CM. Glucocorticoid receptor function in vitro in patients with major depression. Stress 2004; 7: 209–219
- Pariante CM. The glucocorticoid receptor: Part of the solution or part of the problem?. J Psychopharmacol 2006; 20: 79–84
- Pariante CM, Miller AH. Glucocorticoid receptors in major depression: Relevance to pathophysiology and treatment. Biol Psychiatry 2001; 49: 391–404
- Pariante CM, Landau S, Carpiniello B. Interferon alpha-induced adverse effects in patients with a psychiatric diagnosis. N Engl J Med 2002a; 347: 148–149
- Pariante CM, Kim RB, Makoff A, Kerwin RW. Antidepressant fluoxetine enhances glucocorticoid receptor function in vitro by modulating membrane steroid transporters. Br J Pharmacol 2003b; 139: 1111–1118
- Pariante CM, Dazzan P, Danese A, Morgan KD, Brudaglio F, Morgan C, et al. Increased pituitary volume in antipsychotic-free and antipsychotic-treated patients of the AEsop first-onset psychosis study. Neuropsychopharmacology 2005; 30: 1923–1931
- Pariante CM, Hye A, Williamson R, Makoff A, Lovestone S, Kerwin RW. The antidepressant clomipramine regulates cortisol intracellular concentrations and glucocorticoid receptor expression in fibroblasts and rat primary neurones. Neuropsychopharmacology 2003a; 28: 1553–1561
- Pariante CM, Makoff A, Lovestone S, Feroli S, Heyden A, Miller AH, et al. Antidepressants enhance glucocorticoid receptor function in vitro by modulating the membrane steroid transporters. Br J Pharmacol 2001a; 134: 1335–1343
- Pariante CM, Papadopoulos AS, Poon L, Checkley SA, English J, Kerwin RW, et al. A novel prednisolone suppression test for the hypothalamic–pituitary–adrenal axis. Biol Psychiatry 2002b; 51: 922–930
- Pariante CM, Papadopoulos AS, Poon L, Cleare AJ, Checkley SA, English J, et al. Four days of citalopram increase suppression of cortisol secretion by prednisolone in healthy volunteers. Psychopharmacology (Berl) 2004a; 177: 200–206
- Pariante CM, Pearce BD, Pisell TL, Owens MJ, Miller AH. Steroid-independent translocation of the glucocorticoid receptor by the antidepressant desipramine. Mol Pharmacol 1997; 52: 571–581
- Pariante CM, Pearce BD, Pisell TL, Sanchez CI, Po C, Su C, et al. The proinflammatory cytokine, interleukin-1alpha, reduces glucocorticoid receptor translocation and function. Endocrinology 1999; 140: 4359–4366
- Pariante CM, Pearce BD, Pisell TL, Su C, Miller AH. The steroid receptor antagonists RU40555 and RU486 activate glucocorticoid receptor translocation and are not excreted by the steroid hormones transporter in L929 cells. J Endocrinol 2001b; 169: 309–320
- Pariante CM, Thomas SA, Lovestone S, Makoff A, Kerwin RW. Do antidepressants regulate how cortisol affects the brain?. Psychoneuroendocrinology 2004b; 29: 423–447, 2003 Curt Richter Award Paper
- Pariante CM, Vassilopoulou K, Velakoulis D, Phillips L, Soulsby B, Wood SJ, et al. Pituitary volume in psychosis. Br J Psychiatry 2004c; 185: 5–10
- Peiffer A, Veilleux S, Barden N. Antidepressant and other centrally acting drugs regulate glucocorticoid receptor messenger RNA levels in rat brain. Psychoneuroendocrinology 1991; 16: 505–515
- Pepin MC, Beaulieu S, Barden N. Antidepressants regulate glucocorticoid receptor messenger RNA concentrations in primary neuronal cultures. Brain Res Mol Brain Res 1989; 6: 77–83
- Pepin MC, Govindan MV, Barden N. Increased glucocorticoid receptor gene promoter activity after antidepressant treatment. Mol Pharmacol 1992a; 41: 1016–1022
- Pepin MC, Pothier F, Barden N. Antidepressant drug action in a transgenic mouse model of the endocrine changes seen in depression. Mol Pharmacol 1992b; 42: 991–995
- Pridmore S. Rapid transcranial magnetic stimulation and normalization of the dexamethasone suppression test. Psychiatry Clin Neurosci 1999; 53: 33–37
- Prima V, Depoix C, Masselot B, Formstecher P, Lefebvre P. Alteration of the glucocorticoid receptor subcellular localization by non steroidal compounds. J Steroid Biochem Mol Biol 2000; 72: 1–12
- Raison CL, Miller AH. When not enough is too much: The role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry 2003; 160: 1554–1565
- Rangarajan PN, Umesono K, Evans RM. Modulation of glucocorticoid receptor function by protein kinase A. Mol Endocrinol 1992; 6: 1451–1457
- Rasenick MM, Chaney KA, Chen J. G protein-mediated signal transduction as a target of antidepressant and antibipolar drug action: Evidence from model systems. J Clin Psychiatry 1996; 57(Suppl 13)49–55
- Reid PD, Pridmore S. Dexamethasone suppression test reversal in rapid transcranial magnetic stimulation-treated depression. Aust N Z J Psychiatry 1999; 33: 274–277
- van Rossum EF, Binder EB, Majer M, Koper JW, Ising M, Modell S, et al. Polymorphisms of the glucocorticoid receptor gene and major depression. Biol Psychiatry 2006; 59: 681–688
- Rubin R, Dinan TJ, Scott LV. The neuroendocrinology of affective disorders. Hormones, brain and behaviour, D Pfaff, AP Arnold, AM Etgen, SE Fahrbach, PL Moss, RT Rubin. Academic Press, New York, NY 2001; 467–514
- Rupprecht R, Kornhuber J, Wodarz N, Lugauer J, Gobel C, Riederer P, et al. Lymphocyte glucocorticoid receptor binding during depression and after clinical recovery. J Affect Disord 1991; 22: 31–35
- Sanchez ER, Hu JL, Zhong S, Shen P, Greene MJ, Housley PR. Potentiation of glucocorticoid receptor-mediated gene expression by heat and chemical shock. Mol Endocrinol 1994; 8: 408–421
- Schaaf MJM, Cidlowski JA. Molecular mechanisms of glucocorticoid action and resistance. J Steroid Biochem Mol Biol 2002; 83: 37–48
- Schmidt TJ, Meyer AS. Autoregulation of corticosteroid receptors. How, when, where, and why?. Receptor 1994; 4: 229–257
- Silver PJ, Sigg EB, Moyer JA. Antidepressants and protein kinases: Inhibition of Ca2+-regulated myosin phosphorylation by fluoxetine and iprindole. Eur J Pharmacol 1986; 121: 65–71
- Spencer RL, Miller AH, Stein M, McEwen BS. Corticosterone regulation of type I and type II adrenal steroid receptors in brain, pituitary, and immune tissue. Brain Res 1991; 549: 236–246
- Stewart JW, Quitkin FM, McGrath PJ, Klein DF. Defining the boundaries of atypical depression: Evidence from the HPA axis supports course of illness distinctions. J Affect Disord 2005; 86: 161–167
- Szabo D, Szabo G, Jr., Ocsovszki I, Aszalos A, Molnar J. Anti-psychotic drugs reverse multidrug resistance of tumor cell lines and human AML cells ex-vivo. Cancer Lett 1999; 139: 115–119
- Tasker JG, Di S, Malcher-Lopes R. Minireview: Rapid glucocorticoid signaling via membrane-associated receptors. Endocrinology 2006; 147: 5549–5556
- Varga A, Nugel H, Baehr R, Marx U, Hever A, Nacsa J, et al. Reversal of multidrug resistance by amitriptyline in vitro. Anticancer Res 1996; 16: 209–211
- Vedder H, Bening-Abu-Shach U, Lanquillon S, Krieg JC. Regulation of glucocorticoid receptor-mRNA in human blood cells by amitriptyline and dexamethasone. J Psychiatr Res 1999; 33: 303–308
- Wang X, Wu H, Miller AH. Interleukin 1alpha (IL-1alpha) induced activation of p38 mitogen-activated protein kinase inhibits glucocorticoid receptor function. Mol Psychiatry 2004; 9: 65–75
- Wassef A, Smith EM, Rose RM, Gardner R, Nguyen H, Meyer WJ. Mononuclear leukocyte glucocorticoid receptor binding characteristics and down-regulation in major depression. Psychoneuroendocrinology 1990; 15: 59–68
- Weiss J, Dormann SM, Martin-Facklam M, Kerpen CJ, Ketabi-Kiyanvash N, Haefeli WE. Inhibition of P-glycoprotein by newer antidepressants. J Pharmacol Exp Ther 2003; 305: 197–204
- Whalley LJ, Borthwick N, Copolov D, Dick H, Christie JE, Fink G. Glucocorticoid receptors and depression. Br Med J (Clin Res Ed) 1986; 292: 859–861
- Wodarz N, Rupprecht R, Kornhuber J, Schmitz B, Wild K, Braner HU, et al. Normal lymphocyte responsiveness to lectins but impaired sensitivity to in vitro glucocorticoids in major depression. J Affect Disord 1991; 22: 241–248
- Wodarz N, Rupprecht R, Kornhuber J, Schmitz B, Wild K, Riederer P. Cell-mediated immunity and its glucocorticoid-sensitivity after clinical recovery from severe major depressive disorder. J Affect Disord 1992; 25: 31–38
- Yau JL, Noble J, Thomas S, Kerwin R, Morgan PE, Lightman S, et al. The antidepressant desipramine requires the ABCB1 (Mdr1)-type P-glycoprotein to upregulate the glucocorticoid receptor in mice. Neuropsychopharmacology 2007
- Yehuda R. Biology of posttraumatic stress disorder. J Clin Psychiatry 2001; 64(Suppl 17)41–46
- Yehuda R, Boisoneau D, Mason JW, Giller EL. Glucocorticoid receptor number and cortisol excretion in mood, anxiety, and psychotic disorders. Biol Psychiatry 1993; 34: 18–25
- Yehuda R, Golier JA, Yang RK, Tischler L. Enhanced sensitivity to glucocorticoids in peripheral mononuclear leukocytes in posttraumatic stress disorder. Biol Psychiatry 2004; 55: 1110–1116
- Yehuda R, Halligan SL, Grossman R, Golier JA, Wong C. The cortisol and glucocorticoid receptor response to low dose dexamethasone administration in aging combat veterans and holocaust survivors with and without posttraumatic stress disorder. Biol Psychiatry 2002; 52: 393–403
- Yehuda R, Yang RK, Golier JA, Grossman RA, Bierer LM, Tischler L. Effect of sertraline on glucocorticoid sensitivity of mononuclear leukocytes in post-traumatic stress disorder. Neuropsychopharmacology 2006; 31: 189–196
- Young EA, Haskett RF, Murphy-Weinberg V, Watson SJ, Akil H. Loss of glucocorticoid fast feedback in depression. Arch Gen Psychiatry 1991; 48: 693–699
- Young EA, Lopez JF, Murphy-Weinberg V, Watson SJ, Akil H. Mineralocorticoid receptor function in major depression. Arch Gen Psychiatry 2003; 60: 24–28
- Young AH, Gallagher P, Watson S, Del Estal D, Owen BM, Nicol FI. Improvements in neurocognitive function and mood following adjunctive treatment with mifepristone (RU-486) in bipolar disorder. Neuropsychopharmacology 2004; 29: 1538–1545
- Yuuki N, Ida I, Oshima A, Kumano H, Takahashi K, Fukuda M, et al. HPA axis normalization, estimated by DEX/CRH test, but less alteration on cerebral glucose metabolism in depressed patients receiving ECT after medication treatment failures. Acta Psychiatr Scand 2005; 112: 257–265
- Zobel AW, Yassouridis A, Frieboes RM, Holsboer F. Prediction of medium-term outcome by cortisol response to the combined dexamethasone-CRH test in patients with remitted depression. Am J Psychiatry 1999; 156: 949–951
- Zobel AW, Nickel T, Sonntag A, Uhr M, Holsboer F, Ising M. Cortisol response in the combined dexamethasone/CRH test as predictor of relapse in patients with remitted depression. A prospective study. J Psychiatr Res 2001; 35: 83–94
- Zwanzger P, Baghai TC, Padberg F, Ella R, Minov C, Mikhaiel P, et al. The combined dexamethasone-CRH test before and after repetitive transcranial magnetic stimulation (rTMS) in major depression. Psychoneuroendocrinology 2003; 28: 376–385