
Abstract
High-throughput DNA sequencing-based approaches continue to revolutionise our understanding of microbial ecosystems, including those associated with fermented foods. Metagenomic and metatranscriptomic approaches are state-of-the-art biological profiling methods and are employed to investigate a wide variety of characteristics of microbial communities, such as taxonomic membership, gene content and the range and level at which these genes are expressed. Individual groups and consortia of researchers are utilising these approaches to produce increasingly large and complex datasets, representing vast populations of microorganisms. There is a corresponding requirement for the development and application of appropriate bioinformatic tools and pipelines to interpret this data. This review critically analyses the tools and pipelines that have been used or that could be applied to the analysis of metagenomic and metatranscriptomic data from fermented foods. In addition, we critically analyse a number of studies of fermented foods in which these tools have previously been applied, to highlight the insights that these approaches can provide.
1. Introduction: microbiome research and its relevance to food
The growth and activities of microbial communities associated with food influence its biological state, for example by promoting preservation through fermentation or degradation by spoilage. Since ancient times, fermentation-associated microorganisms and human nutrition have been linked, with the earliest records dating back to 7000 BC. Fermentation of food is still a routinely practiced form of food production across the globe, producing culturally significant foods that are consumed daily by millions of people (Selhub et al. Citation2014). Fermented foods and beverages are classified by the International Scientific Association for Probiotics and Prebiotics (ISAPP) as “foods made through desired microbial growth and enzymatic conversions of food components” (Marco et al. Citation2021). These enzymatic processes cause significant changes in the properties of foods, including dairy, meat, fish, vegetable, fruit and cereal substrates (Kabak and Dobson Citation2011), providing a stabilising effect, while adding flavour, aroma and distinctive features to the foods (Marco et al. Citation2017).
Fermented foods have recently attracted renewed interest among Western consumers, particularly because of an enhanced appreciation of their associated health benefits. These health benefits are attributed to the food’s nutritional content, the fermenting microorganisms themselves and the by-products (postbiotics) of their metabolic activities. From a nutritional perspective, fermentation may remove some anti-nutrients such as allergens, and typically improves the micronutrient content of most foods by increasing the bioavailability of minerals and vitamins. Additionally, macronutrients such as carbohydrates and proteins may be more digestible following fermentation (Nout Citation2014; Şanlier et al. Citation2019). Some studies support the hypothesis that fermented foods host microbial species/strains with health-promoting functionality, using food as a transport matrix and thus improving health outcomes (Bove et al. Citation2013; Walsh et al. Citation2016; Marco et al. Citation2017).
By-products of fermentation have a significant effect on the sensory and nutritional properties of food (Chaves-López et al. Citation2014; Şanlier et al. Citation2019). Some of these by-products, such as bioactive peptides and exopolysaccharides, have been associated with reduced occurrence of conditions such as diabetes, obesity, cardiovascular disease and hypertension (Martinez-Villaluenga et al. Citation2017; Nampoothiri et al. Citation2017). Despite evidence of the health benefits provided by some fermented foods, there is a lack of appropriately designed human trials to determine the effects of specific fermented foods on human health (Gille et al. Citation2018). Additionally, most health claims lack a sufficient mechanistic understanding, highlighting the need for further research to understand the methods by which fermented food microorganisms and/or their by-products might contribute to health, as well as the consumption level of the fermented food required for these health benefits to be conferred (Şanlier et al. Citation2019). Despite the need for more clinical studies, our understanding of the microbial components of fermented foods has progressed significantly with recent advances in culture-independent high-throughput sequencing approaches. The expanded use of shotgun metagenomics and metatranscriptomics to study a considerable variety of microbial environments has provided a deep insight into the microbial composition, functional potential and gene expression in foods (Cao et al. Citation2017).
2. Shotgun metagenomics
Whole metagenome shotgun sequencing (WMGS) provides an untargeted sequencing-based approach to assess metagenomic DNA from biological samples. Historically, culture-dependent approaches were used to characterise microbial communities associated with fermented foods. However, these methods are limited to culturable microorganisms, failing to provide insights for the yet to be cultured or difficult-to-culture microbes (Zepeda Mendoza et al. Citation2015). Such microbes can play significant roles in fermentation and prior to the introduction of high-throughput sequencing (HTS), few experimental applications existed to examine their physiology (Solden et al. Citation2016). For example, in Pu-erh tea, a fermented beverage typically containing an array of species corresponding to multiple genera, only a select number of microorganisms have been successfully cultured, namely Aspergillus sp. and Blastobotrys sp., due to difficulties in replicating the fermentation environment in laboratory settings (Abe et al. Citation2008; Tian et al. Citation2013). Most studies analysing the compositional structure of microbial communities to date have used targeted sequencing approaches such as 16S rDNA gene sequencing, as opposed to WMGS, with the routine utilisation of WMGS initially being impeded by the associated costs and computational challenges (Cao et al. Citation2017; De Filippis et al. Citation2017). Such issues continue to be a major consideration with respect to the application of WMGS for large-scale longitudinal studies (Hillmann et al. Citation2018). However, this is offset by the fact that WMGS sequencing offers several advantages.
Whole metagenome shotgun sequencing can provide both a functional and species level taxonomic profile of bacterial, viral, archaeal and eukaryotic taxa, and recover fragmented draft genomes and genes present in the microbial ecosystem. In comparison, amplicon sequencing typically achieves a genus-level compositional insight and provides little functional insight for bacterial, archaeal or eukaryotic taxa (depending on the target marker selected), and fails to detect viral members due to an absence of appropriate phylogenetic single marker sequences (Quince et al. Citation2017; Walsh et al. Citation2018; Beier et al. Citation2017). The strength of WMGS is highlighted when considering strain level phylogenetic reconstructions at the population level. Previously this was only possible through time-consuming sequencing of isolates, but several papers have reported that the adoption of WMGS can produce comparable results (Truong et al. Citation2017). Other key factors influencing the uptake of WMGS include further reductions in the cost of sequencing and the development of shallow shotgun metagenome sequencing (SSMS). Shallow shotgun metagenome sequencing provides an economic approach, by sequencing at a limited depth and has been demonstrated to produce comparable species and functional outputs to shotgun sequencing for a number of well-characterised and simulated human metagenomes (Hillmann et al. Citation2018).
2.1. Overview of bioinformatic approaches for metagenomic data
An ever-increasing array of bioinformatic tools, pipelines and databases are available for the interpretation of metagenomic datasets ( and and ). The selection of these tools prior to experimentation is typically driven by the scientific question, sample type, properties of the tools and availability of reference databases (). Before reviewing the bioinformatic tools available, a preliminary PubMed search was carried out using the query string “"Algorithms*[MESH Terms] OR Software* [MESH Terms] AND High-Throughput Nucleotide Sequencing* [MESH Terms] OR Metagenomics*[MESH Terms] OR Metagenomics/methods* [MESH Terms]" to acquire peer-reviewed research papers introducing novel and improved tools designed for the interpretation of metagenomic data. Records were assessed initially using title/abstract analysis, followed by full-text screening. Publications deemed appropriate were selected for citation analysis using the relative citation ratio (RCR) metric (Hutchins et al. Citation2016, Citation2019) as calculated by iCite (https://icite.od.nih.gov/). Through this analysis, a measure of the influence that established tools (2 years and older) had on the research community was obtained. 1–3 highly cited tools were selected per bioinformatic category, to demonstrate how underlying principles of bioinformatic tools (see below) are applied ( and ).
Figure 1. Relative citation ratio scores for selected peer-reviewed research papers, introducing established metagenomic tools (2 years and older) with the highest influence on the research community. Peer-reviewed research papers were selected through a literature mining methodology and assessed using a citation analysis as described above. Peer-reviewed research papers discussed Bioinformatic tools with applications in ![]()
Figure 2. Annual growth and availability in reference data available for the interpretation of metagenomic data including reference genomes in the NCBI database (https://www.ncbi.nlm.nih.gov/). (A) Annual growth (from 2005 to 2021) in reviewed research papers introducing novel and improved tools designed for the interpretation of metagenomic data, metagenomes available in the NCBI GenBank database and reference genomes in the NCBI GenBank and NCBI RefSeq (Tatusova et al. Citation2014; Clark et al. Citation2016; (B) Available Metagenome, NCBI Genbank and NCBI Refseq reference data separated by assembly level. (C) Available Metagenome, NCBI Genbank and NCBI RefSeq reference data separated by domain. (D) Cladogram of bacterial and fungal species found in milk kefir, species information obtained from (Bourrie et al. Citation2016). Tip labels represent the species detected, e.g. Lentilactobacillus parabuchneri, tip colour represents the phylum, and shape represents the kingdom of the species. Presence/absence heatmap displays the availability of reference genomes for each of the listed species in both NCBI RefSeq and NCBI GenBank databases. Metagenome, Refseq and Genbank derived data was generated and analysed in R using the Biomartr package (Drost and Paszkowski Citation2017). Visualisation was performed using the ggplot2 package (Wickham Citation2016) and ggtree package (Yu Citation2020).
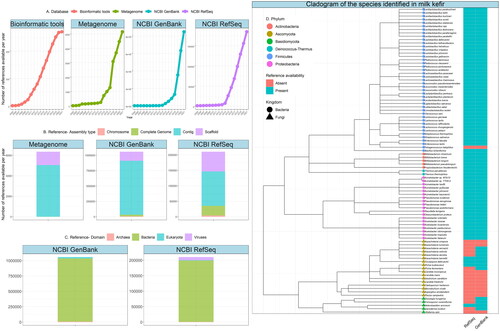
Table 1. Source codes, synopsis, tutorial and basic characteristics of the reviewed bioinformatic tools. Basic characteristics include RCR citation (a measure of the influence that publications introducing tools have on the research community), advantages and disadvantages of the tool e.g. extent of documentation.
These bioinformatic tools facilitate the analysis of millions of sequences in parallel from a diverse range of metagenomes or can be designed specifically for a niche application, such as the pathogen profiling of metagenomes obtained from archaeological sites (Hübler et al. Citation2019). Most tools employ heuristic approaches, due to the general complexity of metagenomic datasets. This data complexity arises due to the considerable volume of information that can be generated and the frequent need to integrate results obtained from multiple tools, which are often used in a non-standardised fashion (Tamames and Puente-Sánchez 2019). Most bioinformatic tools available for the interpretation of metagenomic data are tailored for short-read sequencing data, reflecting the widespread use of Illumina platforms within the research community ( and ). Whole metagenome shotgun sequencing tools can be broadly assigned to two distinct approaches, referred to as “read-based” and “assembly-based” approaches, which can be used separately or in parallel, depending on the study design. Many of the “read-based” and “assembly-based” tools ( and ), while tailored to the complexity of metagenomics data can serve dual purposes and be applied to Metatranscriptional data. Caution is advised when considering the dual application of such tools, as many have not been sufficiently tested for their efficacy in classifying Metatranscriptional data (Shakya et al. Citation2019). For a detailed review of bioinformatic tools applied to Metatranscriptional data please see (Shakya et al. Citation2019).
2.2. Short-read approaches
Short-read tools profile microbial communities at the compositional and functional level (). Short-read taxonomic classification tools function by aligning or mapping sequences to informative subsections of meta(genomic) data, e.g. taxon specific genes and k-mer features (short strings of DNA sequences). The short read taxonomic tool MetaPhlAn3 (metagenomic phylogenetic analysis 3) (Beghini et al. Citation2021) functions by aligning reads to an internal database of clade-specific and quasi marker genes through BLASTn (Altschul et al. Citation1990). Quasi markers expand the scope of analysis enabling the profiling of viral and eukaryotic reads. The distinct nucleotide composition of marker genes reduces the occurrence of false positives and negatives, allowing a simplified workflow without needing to modify or error check the initial metagenomic dataset (Beghini et al. Citation2021). Kraken2 (Wood et al. Citation2019) is another popular taxonomic classification tool that uses k-mers to profile metagenomic reads or their translated protein sequences. The minimiser content of both k-mers contained in query sequences and Kraken’s internal database are compared. Minimisers are nucleotide/protein sequences that are shared by multiple k-mers (Wood and Salzberg Citation2014; Wood et al. Citation2019).
Functional short read approaches often incorporate taxonomic identifications to construct a customised database of gene or protein features, known to be attributed to the identified taxa. Reads or k-mers are aligned or matched respectively to this customised database (Silva et al. Citation2016) and unmapped reads are further examined against larger protein databases containing annotated sequences to identify homologs (Zepeda Mendoza et al. Citation2015). These tools provide a summary-level characterisation of a metagenome, estimating feature abundance profiles (Gloor et al. Citation2017; Calle Citation2019) and assigning reads to the most likely microbial lineage (Segata et al. Citation2012; Silva et al. Citation2016). The selective methodology reduces the time and computational requirements compared to traditional mapping approaches that attempt to label every read sequence from a metagenome (Truong et al. Citation2015). HUMAnN3 (HMP Unified Metabolic Analysis Network) (Beghini et al. Citation2021) is a short read functional profiling tool, querying metagenomic and metatranscriptomic datasets using a multi-search phase methodology. Initially, reads are aligned against a customised pangenome database of functionally annotated genes, built from the taxonomic identifications of MetaPhlAn3. By incorporating MetaPhlAn3, functional units can be linked to their taxonomic source. Unmapped reads are translated and queried against a selectable protein database through DIAMOND by default. SuperFocus (Silva et al. Citation2016) also applies a similar methodology and can build a reduced seed database informed by the taxonomic identifications of FOCUS (Silva et al. Citation2014) for the initial alignment phase (Silva et al. 2016).
2.3. Metagenome assembly-based approaches
2.3.1. Metagenomic assembly
Assembly methods merge the consecutive k-mer content of metagenomic reads, based on their overlapping sequence similarity, into single contiguous sequences (contigs) of the shortest possible length. Assembly is performed to simplify data analysis processes, such as homology-based searches, as a single read contains limited information. A commonly used metagenomic assembly technique is global assembly, which attempts to construct all the genomes present within a sample (Ayling et al. Citation2020). Most popular metagenomic assemblers construct De Bruijn graphs (Pevzner et al. Citation2001) and are tailored to assemble short-reads ( and ), failing to reproduce the same results with longer reads, e.g. all IDBA (iterative de Bruijn graph assembler) methods available incorporate the stepwise use of increasing k-mer values to build a de Bruijn graph (Peng et al. Citation2012).
Metagenomic assembly tools employ different algorithmic approaches to address the caveats of assembling microbial communities. Such caveats include uneven sequencing depth and coverage, sequencing errors, repeat structures and strain mixtures, all of which contribute to the development of error prone and fragmented contigs (Nurk et al. Citation2017; Ayling et al. Citation2020). The extension of the IDBA algorithms IDBA-UD (Peng et al. Citation2012) is used to assemble de novo paired-end reads of uneven coverage, and accounts for errors in metagenomic data through multiple approaches. Approaches include a flexible cut-off value for removing erroneous contigs that is determined by the sequence depth of adjacent contigs. Errors are further removed via contig alignment. Low depth repeat patterns are resolved using the dual ends of paired reads. Paired-end reads that are unaligned on one end but are uniquely aligned to contigs of high confidence are identified and grouped. Local assembly is then performed on the unaligned end to resolve repeat structures (Peng et al. Citation2012). MetaSPAdes (Nurk et al. Citation2017) attempts to reconstruct species level consensus sequences using coverage information, extending the sequence based on the highest coverage values of approaching edges (k-mers). Furthermore, coverage ratios of all adjacent edges are examined. Those of a low coverage ratio are detached from their source vertex (k-1-mers), but the information is retained to enable the construction of strain contigs. MetaSPAdes then uses these strain contigs to influence its prediction of hypothetical reads, using them to resolve repeat structures (Nurk et al. Citation2017).
2.3.2. Metagenomic binning
The assembly methods discussed above typically achieve contig-level resolution only, because of multiple limiting factors. These factors include repetitive genomic sequences, strain level variation, sequencing errors and low coverage of sequences due to technological limitations, all of which contribute to fragmentation after the assembly process. Fragmented contigs that fall short of chromosome level resolution are often insufficient representations of microbial populations, and can hamper insights into the physiology of microbial communities (Chen et al. Citation2020). Thus, additional bioinformatic tools such as binners ( and ) are required to further profile the informational content of reads (Alneberg et al. Citation2014). Metagenomic binning involves classifying reads and/or contigs of interest into separate groups referred to as OTU’s (operational taxonomic units), e.g. genera or species-level clusters. Binning methods are typically used to recover Metagenomic-Assembled Genomes (MAGs), which are fragmented draft genomes that can be used for a diverse range of analyses, including taxonomic assessment that extends to unknown species, and functional comparisons/associations. Binning algorithms are separated into two stages, i.e. read extraction and assignment. Reads/contigs are typically extracted based on similarity in sequence composition/features, e.g. oligonucleotide frequency and abundance profiles, all of which display taxon specific patterns (Sedlar et al. Citation2017). A typical method to assess oligonucleotide frequency is to determine the tetra-nucleotide frequency (TNF) content of reads, which is employed in metagenomics assemblers such as MetaBAT (Metagenome Binning with Abundance and Tetra-nucleotide frequencies) (Kang et al. Citation2015) and MetaBAT2 (Kang et al. Citation2019). Tetra-nucleotide frequency is the frequency at which a set of unique k-mers, four base pairs in length, e.g. AGTC, appear in the reads to be binned. Tetra-nucleotide frequencies provide an insight into phylogeny as closely related genomes can often contain a similar TNF content (Pride et al. Citation2003).
CONCOCT (Alneberg et al. Citation2014) is a binning tool that groups contigs based on their abundances and k-mer frequency. Fragmented contigs form two vectors, referred to as the coverage vector and the composition vector. The coverage vector is the amount of reads that are available for a given base in the genomic sequence, while the composition vector is a concatenation of k-mer frequencies of any selected length and their respective complement (Alneberg et al. Citation2014). As described previously, the extraction strategies used by binning tools rely on multiple parameters. While most tools share similar extraction strategies, the algorithms employed at the assignment stage distinguish the binning tools (Sedlar et al. Citation2017). Supervised and unsupervised binning methods are examples of widely used assignment techniques in metagenomics. Supervised methods compare reads or contigs to reference databases and their accuracy is influenced by the completeness of the respective database. Unsupervised methods use machine learning algorithms to bin read or contigs based on abundance profiles or sequence composition (Wu YW et al. Citation2016; Wang et al. Citation2019). MaxBin 2 (Wu YW et al. Citation2016) performs binning using an expectation-maximization algorithm, which assigns scaffolds to their respective bins based on median values of single-copy marker genes and probabilities scores (Wu YW et al. Citation2016). Genome binning can be performed before or after assembly and, regardless of the stage, it improves downstream analysis by providing more information from sequencing reads, which can be summed to calculate a taxonomic profile.
2.3.3. Downstream analysis of MAGs
The application of genome binning methods often results in the construction of MAGs with varying levels of completion and contamination. These metrics are often not provided by the binning tool and other downstream analysis tools such as CheckM (Parks et al. Citation2015) must be applied. CheckM examines the single-copy genes including duplicates of MAGs to determine their number. Completion is reported based on the number of single-copy genes compared to the expected number and contamination is determined by the percentage of duplicates (Parks et al. Citation2015). Good quality MAGs represent composite genomes containing the majority of the total gene content of a representative species (Imelfort et al. Citation2014; Kang et al. Citation2015). GOLD (Genomes Online Database) contains 18,945 MAGs (April 2022), but this is only a fraction of the total of >200,000 MAGs that are publicly available (Asnicar et al. Citation2020). High quality MAGs are close approximations of individual genomes and provide a comprehensive data source for applications such as comparative genomics (Mukherjee et al. Citation2019) and reference data (Olm et al. Citation2021). Analysis of MAGs is the final step in assembly-based workflows and the beginning of biology-based analysis. Phylogenetic placement involves surveying the gene content of MAGs to detect taxonomically informative marker genes (Asnicar et al. Citation2020). GTDB-Tk (Chaumeil et al. Citation2019) initially classifies the domain of MAGs by comparing the gene content of MAGs predicted using Prodigal (Prokaryotic Dynamic Programming Genefinding Algorithm) (Hyatt et al. Citation2010) to a reference set of bacterial and 122 archaeal marker genes using HMMER (Eddy Citation2011). The domain with the highest number of marker genes is chosen and the reference marker gene sets are aligned again using HMMER. The output of this pipeline is a concatenated MSA used for phylogenetic placement by pplacer (Matsen et al. Citation2010; Chaumeil et al. Citation2019).
Annotation of MAGs typically involves identifying all coding genes of a MAG and deducing their possible biological function through alignment with functionally annotated reference sequences and or translated homology searches. Multiple webservers and standalone tools exist specifically for the analysis of MAGs (Dong and Strous Citation2019). Prokka (Seemann Citation2014) relies on established prediction tools and databases to perform genome annotation. Prediction-based tools such as Prodigal compare genome queries against reference genomes at the nucleotide level (Hyatt et al. Citation2010). Through this search, Prokka can acquire positional information about contig bound features, i.e. coding sequences (CDs), and acquire their protein output. Gene annotation is performed by a translated search against multiple databases in a hierarchical fashion. The following databases are queried sequentially, a user selectable database of annotated proteins (optional), UniProt (Apweiler et al. Citation2004), RefSeq, Pfam (Punta et al. Citation2012) and other hidden Markov model databases. Genomic features in question are assigned an identity by the first accurate match between the query sequence and one of the mentioned databases, if a match occurs further searches are not performed (Seemann Citation2014).
2.4. Advantages and disadvantages of the short read and assembly-based approaches
Current bioinformatic approaches fail to achieve 100% accuracy and have several limitations and flaws that are difficult to address. Given the general short-read nature of WMGS, short read-based approaches commonly utilise sequence similarity and alignment length criteria separately or in parallel, which can determine if reads are evenly distributed across a reference genome/marker gene or are recruited to uninformative subsections of the genome, and represent a false positive result. Such requirements ensure accuracy and limit the quantity of false positives but restrict the sensitivity of the tool and as such only features of high confidence are identified. Given this, short read -based approaches excel at identifying well documented and highly conserved features such as ARGs but are limited when profiling novel features sharing little resemblance to those contained in reference databases (Menzel et al. Citation2015; Sunagawa et al. Citation2015). For example, it was noted that the short-read marker gene-based classification tool MetaPhlAn2 failed to accurately profile metagenomic reads belonging to the Brettanomyces genus due to a complete absence of Brettanomyces reference marker genes in MetaPhlAn2. Reference availability remains an issue in the updated versions of the tool (April 2022) (Verce et al. Citation2019). Furthermore, marker genes make up only a small proportion of a microbial genome and as such have limited profiling capabilities if a species is present at low abundance compared to host and environmental DNA (Hübler et al. Citation2019). Similar issues are also reported for short-read k-mer-based approaches, which can only compare query sequences to known k-mers of a reference database. K-mer-based approaches such as Kraken2 are prone to false positive results generated by closely related genomes with similar k-mer content to true identifications (Wood et al. Citation2019)
Assembly-based approaches achieve more satisfactory results when profiling novel environments, allowing for the construction of unclassified composite genomes. Assembly-based approaches are useful for the analysis of viral metagenomes (viromes), in particular viral prophage, which are dominated by poorly annotated sequences with few reference genomes available (54,352 in the Genomes – NCBI Datasets, April 2022 and ) (Aggarwala et al. Citation2017). Nonetheless, assembly-based approaches produce incomplete draft genomes containing contaminated sequences that hinder downstream binning applications. Constructed assemblies are further limited by sample type, strain-level diversity, population heterogeneity, sequence repeats and a requirement for an average 100-fold coverage difference, compared to short-read approaches to detect low abundance species (Prosser Citation2015; Boyd et al. Citation2018). These limitations cause inconsistencies in results, which vary according to the tool used and restrict comparisons between studies. Machine learning allows for moderate constraints in cut off values, increasing the sensitivity of such tools. However, the implementation of machine learning classifiers is hampered by the availability of suitable features, e.g. most reference AR data is derived from clinical bacterial isolates and represent a poor database for training machine learning methods (Wallace et al. Citation2017).
Assembly-based approaches can often only produce complete genomes for the most dominant species, producing low resolution fragmented assemblies for lower abundant microbial taxa and fail to capture all the functional diversity of a microbiota; which would require the assembly and functional annotation of hundreds or thousands of microorganisms in complex communities such as the ocean. While capturing a complete functional and compositional profile of a complex environment can be a computationally challenging process, certain studies may wish to focus on certain aspects of an environment, such as the reconstruction of cholesterol-lowering genes, in such cases a targeted assembly-based approach may be employed guided by a reference database (Guo et al. Citation2019) ().
Table 2. Description of short read-based and metagenome assembly-based approaches available for the interpretation of metagenomic data.
2.5. Specialised bioinformatics tools
Most bioinformatic tools available report on a broad range of taxonomic and or functional features detected in a metagenome ( and ), which often meet the needs of the researcher. However, in some cases, the research question is too specific and specialised bioinformatic tools are required to interpret the data. The proliferation of genomic and molecular information () enables the profiling of more specific compositional and functional features, which have been restricted in the past due to a lack of appropriate databases that can unify curated data, sequencing coverage requirements and appropriate algorithms. For example, the profiling of functional features such as carbohydrate-active enzymes, requires databases that reflect the bio-curation efforts of researchers (Zhang H et al. Citation2018). Such specialised bioinformatic tools are becoming available and a number of assembly-based and short read approaches can be used to predict specific compositional elements and/or functional traits such as predominate strains, viral and or pathogenic taxa and antibiotic resistance genes (ARGs). Other specialised tools exist to profile functional features, such as bacteriocin and CRISPR-Cas systems, which were not included in this review, but are often included in pipelines designed for the downstream analysis of MAGs (Almeida and De Martinis Citation2019).
2.5.1. Strain level analysis
Strain profiling represents the pinnacle of metagenomics analysis. A comprehensive understanding of strain variants is needed to address ambiguities arising from substantial genetic variation that strains of the same species can possess (Segata Citation2018). For example, different strains of Escherichia coli have vastly different impacts on host health, with some strains displaying beneficial probiotic properties and others being classified as pathogenic (Leimbach et al. Citation2013). Ambiguities with respect to species-level resolution impact insights at the functional level and can prevent association with different host/microbial phenotypes, limiting the application of metagenomics in clinical and environmental settings. Several tools are available for profiling microbial communities at higher genomic resolution and represent a mixture of short read and assembly-based approaches.
Available tools attempt to introduce methods that can overcome the challenges imposed by genetic variation within a population, e.g. data complexities arising from the presence of multiple polymorphic sites at varying frequencies in genes/genomes. Tools often distinguish between strains using compositional features for whole genomes or marker genes, such as single-nucleotide variants (SNVs), overall similarity to strain-reference genomes or shared gene composition (Van Rossum et al. Citation2020). StrainPhlAn (Beghini et al. Citation2021) examines the nucleotide differences of consensus marker genes obtained using MetaPhlAn3 (Truong et al. Citation2015), while PanPhlAn3 (pangenome-based phylogenomic analysis) (Beghini et al. Citation2021) utilises gene family coverage information to compute species unique gene presence/absence profiles. Abundant gene families of similar coverage are grouped together and are assumed to be from the same genome. Comparisons of gene presence/absence between reference genomes and reference strains allow for the identification of novel or existing strains, and their genetic repertoire, which is used to infer functionality (Scholz et al. Citation2016; Beghini et al. Citation2021).
2.5.2. Viral detection
Viruses that use microorganisms as hosts for viral replication are universally present in biomes, including fermented foods (Hendrix Citation2002; Zahn and Halter Citation2018). In such environments, viruses can interact with residential microbes, contributing to lytic infections and horizontal gene transfer, influencing their composition and genetic makeup, respectively (Weinbauer and Rassoulzadegan Citation2004). Changes driven by viruses can pose a distinct threat to the production of certain fermented foods, which rely on the viability and functionality of the microorganisms involved. Viruses can influence the overall functionality of the fermenting ecosystem, affecting the sensory and safety properties of the final product (Garneau and Moineau Citation2011; Fernández et al. Citation2017). Despite the recognised importance of bacteriophages (phage, i.e. viruses that target bacteria) in microbial ecosystems, further research into phage biology, diversity, and interactions is needed to mitigate the negative impact that phage may have in these environments. Viral discovery has increased rapidly in recent years due to technological advancements in genomics and metagenomics (). Metagenomic datasets capture viral sequences contained in actively infected cells and their genomes, as well as those located outside of the cellular environment, and as such represent a unique opportunity to investigate phage biology, diversity, and interactions with host microorganisms (Ren et al. Citation2017; Roux Citation2019). However, despite the widespread acceptance of the utility of viral metagenomics, robust bioinformatic tools are still needed to address the complexity associated with viral profiling that arises due to the intra-species genetic diversity, short size, and absence of marker sequences in viral features (Roux et al. Citation2017). Virfinder (Ren et al. Citation2017) employs a k-mer-based machine learning approach to identify viral sequences in metagenomic data. Viruses often contain similarities in their k-mer profile to other viral sequences and its host, compared to non-host species. By employing a k-mer-based approach, the tool benefits from improved prediction accuracy, compared to alignment-based techniques and is more likely to characterise shorter sequences and those lacking reference genomes. VirSorter (Guo et al. Citation2021) is designed to detect viral elements in high-throughput sequencing datasets using an iterative process. VirSorter examines input sequences using a sliding window plot providing a quantitative measure for each sequence of one of the following features; presence of viral hallmark genes, enrichment of viral-like or non-Caudovirales genes, uncategorised and short-reads, as well as depletions in PFAM-associated genes and strand switching. These preselected features allow for the identification of both viral-specific, viral-like and non-viral features in metagenomics data that are leveraged in downstream probability calculations (Roux et al. Citation2015).
2.5.3. Antimicrobial resistance
Antimicrobial resistance (AMR) refers to genetic adaptations in microorganisms that confer resilience to an antimicrobial agent. Adaptations commonly occur by horizontal gene transfer or chromosomal mutation, and their incidence rate is heavily influenced by antibiotic exposure (Gillings and Stokes Citation2012; Blair et al. Citation2015). The widespread distribution of ARGs, existing AMR mechanisms to all known treatments and limited availability of novel antibiotics poses a threat to public health and medical practices, for example by impacting the safety of surgical procedures (Blair et al. Citation2015; Macesic et al. Citation2017). Many factors have contributed to the spread of AMR, such as excessive antibiotic usage, limited antibiotic discovery pipelines and ineffective detection methods which prevent appropriate treatments (Ventola Citation2015; Brown and Wright Citation2016). Current detection methods such as antimicrobial susceptibility testing (AST) and PCR, are laborious, time-consuming and fail to capture the complete resistome (the entire AMR gene collection of a metagenome) (Satlin et al. Citation2016; Guitor et al. Citation2019). Metagenomics represents a potential culture-free surveillance and monitoring application, allowing for the in silico detection and profiling of AMR mechanisms present in a resistome. Additionally, metagenomics enables comparisons of resistomes within and between different target populations, revealing patterns of transmission (Berglund et al. Citation2019). However, it should be noted that the presence of a putative AMR gene does not always correspond with resistance and the corollary is also true as resistance may be conferred by a gene not already assigned as a resistance determinant.
Most bioinformatic approaches examining AMR elements in environmental samples use alignment-based search tools such as BLAST and Bowtie2 to compare raw reads or predicted open reading frames (ORFs) from assemblies against existing ARG databases, which are subsequently assigned an annotation from the “best hits” obtained. Several reference databases such as CARD (Comprehensive Antibiotic Resistance Database) (Alcock et al. Citation2019), ARDB (Liu B and Pop 2008) and ResFinder (Zankari et al. Citation2012) are commonly used for such alignments. ARG-ANNOT (Antibiotic Resistance Gene-ANNOTation) (Gupta et al. Citation2014) is an example of a bioinformatic tool that employs a local alignment-methodology to predict and identify AR features in bacteria (Gupta et al. Citation2014). Machine-learning algorithms provide an alternative approach to detect AR elements by training a prediction model on a complex dataset. The dataset provides a representative sample of similarity distributions found in nature of features in ARG reference databases. DeepARGdeepARG-LS and deepARG-SS (Arango-Argoty et al. Citation2018) are deep learning prediction models used to detect AR features in raw reads and predicted genes, respectively, from metagenomic data. Both models were created using a machine learning methodology. Models were trained and evaluated using a distance distribution. The distribution represents sequence similarity values between hypothetical ARGs provided by UNIPROT and known ARGs taken from the ARGminer, CARD, ARDB, UniProt and SARG databases (Arango-Argoty et al. Citation2018).
2.5.4. Genome scale metabolic models
Functional profiling tools have been discussed, which predict metabolic genes/pathways present in a MAG ( and ). Such tools provide a description of metabolic features, but in isolation cannot provide a complete understanding of the phenotypic properties of the studied organism. Such genome assessments can however inform the creation of genome-scale metabolic models (GEMs). These GEMs are mathematical constructs that predict, for the organism studied, the relationship between metabolic genes, enzymes, metabolites and metabolic reactions, storing the metabolic features, set of reactions between them and the association biomass equations in separate metrics (Zhang C and Hua 2016). GEMs provide a representation of the metabolic capabilities that the organism may be capable of based on their repertoire of metabolic genes and/or experimentally obtained information. Such knowledge can advance culturing approaches by providing insights into the supplemented nutrients required by an organism to sustain growth in different substrates/mediums. GEMs can further highlight knowledge gaps in the metabolism of the species in question (Lieven et al. Citation2020). Organism-specific GEMs can be combined with other GEMs recovered from the same environment and expanded to community level models to provide insights into cross feeding and/or nutrient competition between members of the same community (Zomorrodi and Maranas Citation2012; Zelezniak et al. Citation2015). The construction of high quality GEMs can be a laborious process requiring extensive manual curation, including reviewing each metabolic gene, metabolite and pathway association, which is further complicated by the diverse functional roles of enzymes that can take part in multiple reactions, or alternatively the requirement of multiple enzymes in certain reactions and missing reactions (Cuevas et al. Citation2016). Missing reactions are often addressed by gap filling, which involves inferring missing reactions based on the metabolites present in the metabolic network (Karlsen et al. Citation2018). The manual curation step has in the past been reported to take up to two years to complete (Thiele and Palsson Citation2010). A number of steps are available that can help to automate the construction of GEMs, such as literature mining and comparisons to reference templates housed in repositories such as BioModels (Li C et al. Citation2010) and BiGG (Norsigian et al. Citation2019). A number of bioinformatic tools are available for the automated construction of GEMs that can be scaled to metagenomic applications (Mendoza et al. Citation2019), such as CarveMe (Machado et al. Citation2018) and Metage2Metabo (Belcour et al. Citation2020). CarveMe introduces an automated top-down approach to genome scale metabolic modelling. A high quality reference model is used as a template and tailored towards the organism of interest through genome annotations, which are used to inform the removal of features from the template models (Machado et al. Citation2018).
2.6. Limitations of current comparative performance methods for bioinformatic tools
As described above, a range of tools can be used and combined to profile a metagenome, all of which employ different reference sources and classification algorithms, contributing to performance differences. Walsh et al. (Citation2018) reported that short read bioinformatic approaches can have a statistically significant impact on the results of taxonomic and functional profiling, highlighting the importance of questioning and validating when possible the choice of bioinformatic tools (Walsh et al. Citation2018).
While bioinformatic tools and algorithms are rapidly improving, it is often difficult to identify which tools are optimal for specific applications due to the fast pace development of these tools, the absence of a uniform evaluation strategy and an appropriate comparison model, such as a real metagenomic dataset derived from multiple sample types (Dong and Strous Citation2019; Wang et al. Citation2019; Meyer et al. Citation2021). Most published studies introducing novel or improved bioinformatic tools implement comparative performance methods for the assessment of the proposed tool against other established bioinformatic tools. Such studies employ a diverse range of evaluation techniques, performance metrics and sample datasets preventing the generation of uniform results that can be compared across studies (Sczyrba et al. Citation2017).
In 2014, the critical assessment of metagenome interpretation (CAMI) was proposed that, through collaboration with the metagenomic community, was intended to develop uniform and reproducible datasets, standards, and strategies for the evaluation of bioinformatic applications (McHardy et al. Citation2014). A number of bioinformatic tools have been developed through the CAMI initiative to evaluate the performance of short-read (Meyer et al. Citation2019), assembly (Mikheenko et al. Citation2016) and binning (Meyer et al. Citation2018) tools applied to metagenomic data. These tools rely on simulated shotgun metagenomic datasets generated using the metagenomic simulator CAMISIM (Fritz et al. Citation2019). Through CAMISIM, users can create custom made synthetic microbial communities built from input genomes or taxonomic profiles or recreate existing benchmark datasets for the evaluation of different bioinformatic tools. However, the lack of a detailed user manual hinders the applicability of the former approach. Users can apply these stimulated datasets to the tools, reviewed extensively in Meyer et al. (Citation2021) to identify tools with optimal performance for the benchmarked dataset. An alternative approach is to sequence DNA mixtures of known origin. Such an approach allows for technical variation and biases introduced during data generation and is particularly suitable for the simpler microbial communities found in fermented food (Meyer et al. Citation2021).
Recent publications (Sczyrba et al. Citation2017; Meyer et al. Citation2018; Fritz et al. Citation2019; Meyer et al. Citation2019; Ye et al. Citation2019) utilised the CAMI benchmarking platform to evaluate the performance of bioinformatic tools used in metagenomic analysis. Unfortunately, these publications failed to account for either parameter settings or variability in sample type, and assessed only a select number of established tools (Murovec et al. 2019).
Whole metagenome shotgun sequencing-based profiling is a relatively novel field with many improvements still needed, such as the need for advancements in tools supporting long read and hybrid sequencing. Currently, the key problem faced by bioinformatic analysis is the availability of reference data (), the selection and integration of multiple tools and parameter settings for optimal classification accuracy and recall. The utilisation of multiple tools of the same category, or the development of tools that use integrated results from different bioinformatic applications as input data. e.g. BinSanity (Graham et al. Citation2017) MetaMeta (Piro et al. Citation2017), MetaWRAP (Uritskiy et al. Citation2018) and bioBakery workflows (McIver et al. Citation2017) may help improve the accuracy of analysis (Walsh et al. Citation2018).
3. Applications of bioinformatic approaches to fermented food microbiomes
3.1. Research into the fermentation process
Many fermentation processes are uncharacterised or not fully understood and, as a result, the primary application of bioinformatics tools is often to enhance knowledge about these systems. Studies employing these tools will typically report on population composition at the genus or species level including putative new species and, occasionally, will reach strain level resolution (). To date, compositional profiles of many fermented foods have been acquired through bioinformatic analysis of kefir, kimchi, wine, beer, meat, and cocoa metagenomic datasets (Jung et al. Citation2011; Bellon et al. Citation2015; Flores et al. Citation2015; Meersman et al. Citation2015; Walsh et al. Citation2016; Verce et al. Citation2019). Through functional analysis, it is possible to detect and profile potential functional and metabolic features present in the fermentation process and assign them to a contributing species (Beier et al. Citation2017). Many studies are applying metagenomics tools beyond their feature profiles to better understand diverse aspects of fermented food microbiomes such as uncovering the functional features driving co-occurrence and succession patterns. Here we refer to a subset of studies as examples of the novel insights that bioinformatic tools and pipelines can provide into fermented food microbiomes ().
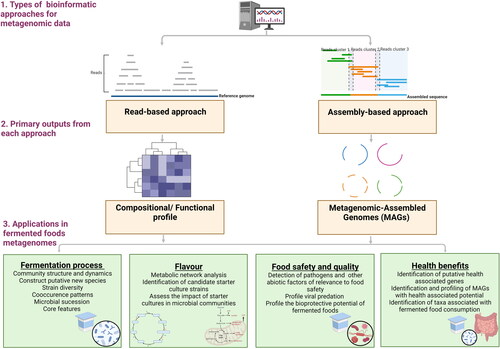
Figure 3. Conceptual overview of the primary steps, outputs and applications associated with the application of read-based and assembly-based approaches to fermented food metagenomic data. Figure was produced using BioRender (BioRender, 2021) https://biorender.com/
3.2. Community structure and dynamics
Firstly, we discuss a representative case study that highlights the results that can be expected from applying both short-read and assembly-based approaches to examine a fermentation ecosystem. Through a combination of short-read alignment and assembly-based approaches Verce et al. Citation2019, examined the microbiota of water kefir at 24 and 72 h of fermentation. Multiple short-read compositional tools and databases were used in combination to produce a taxonomic profile. The tools Kraken, BLASTn, BLASTx, Kaiju (Menzel et al. Citation2016) and metagenomic recruitment plotting were used to perform compositional classification using a k-mer or alignment strategy, while MetaPhlAn3, rRNA selector (Lee et al. Citation2011) and ITSx (Bengtsson-Palme et al. Citation2013) facilitated classification using a marker genes strategy. Each tool varied slightly in taxa assignment, providing differing species level results but agreeing at the genus level detecting Lactobacillus, Oenococcus, Bifidobacterium, Saccharomyces and Brettanomyces as the dominant bacteria and yeast genera. Metagenomic recruitment plotting was the preferred compositional approach to further profile the microbial community to the species level, resulting in the identification of a novel Oenococcus species, Candidatus Oenococcus aquikefiri. In addition, assembly-based methods were employed to acquire water kefir-based MAGs, including the novel species Candidatus O. aquikefiri. The workflow involved contig assembly using MEGAHIT (Li D et al. Citation2016), followed by annotation using Prokka, which informed further annotation by HMMER using various databases specific for carbohydrate-active enzymes. Binning was carried out by CONCOCT, and the resulting contigs were mapped using BWA-MEM (Li H Citation2013) to reference genomes, selected by the results of short-read taxonomic profiling. The genes identified were linked to roles in amino acid, vitamin, and cofactor biosynthesis and were further assigned to their taxonomic source. For example, genes for glycerol biosynthesis were located on assembled contigs of the species Saccharomyces cerevisiae. Identified genes were also linked to metabolites with roles in carbohydrate, pyruvate, citrate, and malate metabolism. For example, the protein-coding genes known to encode for glycerol kinase and glycerol-3-phosphate dehydrogenase were located on contigs of the species Lactobacillus, indicating their potential to use glycerol as part of their metabolism and suggesting a possible interaction with S. cerevisiae (Verce et al. Citation2019). Overall, the interpretations made from the bioinformatic methods applied in this study advance knowledge into the microbial taxa that can be found in water kefir, their functional contributions to the fermentation ecosystem and suggested the potential of interspecies interactions during the fermentation process.
3.2.1. Microbial succession
It is difficult to characterise the microbial ecosystems associated with a fermentation process using a single fermented sample due to the occurrence of temporal changes. Shifts in the abundance, diversity and functionality of microbial communities often occur along the fermentation process. Bioinformatic tools in combination with a longitudinal study design involving the profiling of samples representing different time points of a fermentation, can provide insights into how microbial communities change and adapt over the fermentation period. Such shifts in community structure are referred to as microbial succession (Wolfe et al. Citation2014). Bioinformatic tools have been used in this manner in a number of publications focussing on the fermented foods kefir, soy sauce and cheese rinds to name a few (Sulaiman et al. Citation2014; Wolfe et al. Citation2014; Walsh et al. Citation2016). Walsh et al. Citation2016; Verce et al. Citation2019 identified a clear pattern of microbial succession in kefir throughout the 24-h fermentation period using the taxonomic profiling tools, Kraken and MetaPhlAn2. At 8 h, Lactobacillus kefiranofaciens was the dominant microbial resident in kefir, but as the fermentation progressed its proportions decreased and the relative abundance of Leuconostoc mesenteroides increased considerably becoming the dominant bacterial resident at 24 h. Further investigation using the functional profiling tool HUMAnN2, revealed the presence of aromatic amino acid biosynthesis genes in Leuc. mesenteroides and their absence in L. kefiranofaciens, suggesting a potential mechanism driving microbial succession (Walsh et al. Citation2016). Insights into microbial succession are particularly important to assess the distribution of inoculated species and strains throughout fermentation. Bertuzzi et al. Citation2018 profiled microbial communities associated with surface ripened cheese and their corresponding smear cultures using the taxonomic profiling tool, Kaiju and strain profiling tool PanPhlAn and performed functional analysis via Super-focus. Through this bioinformatic workflow they identified patterns of microbial succession in two distinct surface ripened cheeses at the species level, which were further profiled to provide insights into the strain level distributions. Such analysis revealed the present/absence of inoculated smear cultures strains during the ripening process (Bertuzzi et al. Citation2018).
3.2.2. Core feature detection
Bioinformatic analysis of metagenomics datasets derived from a fermented sample at different time points as opposed to a single representative sample can reveal core functions present throughout the fermentation process. Sulaiman et al. (Citation2014) used metagenome assembly-based approaches, specifically CLC Genomic Workbench to assemble contigs from metagenomic reads, derived from different stages of fermentation from zero to 6 months. Prodigal and AUGUSTUS (Stanke and Morgenstern Citation2005) were used to predict ORFS from prokaryotic and eukaryotic contigs respectively. Contigs were used as references to map unassembled reads, to determine gene abundance values. Predicted genes were then functionally classified using the KEGG classification workflow provided by MEGAN (Beier et al. Citation2017). Through this bioinformatic approach, they observed core functional features, in soy sauce fermentation that were consistently recovered after 6 months of fermentation (Sulaiman et al. Citation2014).
3.2.3. Evaluate the effectiveness of laboratory-based techniques specific for fermented foods
Bioinformatic tools can be used to assess and compare certain laboratory-based techniques. Dugat-Bony et al. (Citation2020) evaluated the effectiveness of four viral extraction methods designed specifically for cheese environments. Viral DNA extracted from treated samples of Epoisses cheese, and vesicle DNA were selected for WMGS-based analysis. Sequence reads were processed by a global assembly-based approach, using SPAdes (Bankevich et al. Citation2012) to produce contigs from paired-end reads. VirSorter compared quality assessed contigs against the Viromes database and a catalogue of non-redundant genes predicted from viral metagenomes, identifying contigs likely to be of viral origin. A subset of the contigs were selected based on sequence coverage, viral origin, and circular characteristics, with contigs matching one or more of the selection criteria included in the finished contig dataset. The final contig dataset represents the first Epoisses cheese surface virome acquired for a cheese environment. Contigs were characterised using PHASTER (Arndt et al. Citation2016) and an alignment-based approach involving Blast searches which queried contig sequences against all nucleotide and viral specific information contained in the NCBI database. The characterisation tools separated the contigs into four distinct categories, namely putative phage encoding contigs, unclassified contigs, putative and plasmid-derived contigs. An abundance table was acquired by mapping sequencing reads from each of the extraction methods to the putatively viral contigs through Bowtie2. The abundance table then informed statistical testing using Spearman correlations, to evaluate the impact of the different extraction protocols on the composition of the virome. Reads from each extraction method were further examined by SortMeRNa v2.0 (Kopylova, Noé and Touzet Citation2012) to detect ribosomal DNAs, which served as indicators of microbial contamination. The computational workflow that examined the viral profile and microbial contamination levels identified the best performing extraction method, but further wet lab techniques are required to validate its effectiveness (Dugat-Bony et al. Citation2020).
3.3. Flavour
The metabolism of microbial communities in fermented foods and beverages plays an essential role in the development of flavour, a combination of aroma and taste sensations. An in-depth knowledge of the metabolic activity and network of microbial communities during fermentation is necessary to understand, at a systems level, how microbes contribute to the organoleptic properties of food.
Bioinformatic approaches can provide a systems level understanding and be employed to provide a theoretical basis for the improvement of fermentation processes without the use of detailed genome scale metabolic models (Melkonian et al. Citation2019), which is of particular importance for industrial applications with increasing quality requirements. Potential avenues of improvement include the selection of starter cultures, appropriate nutrients, environmental conditions and cross‐contamination events with production environments, all of which can contribute to the sensory characteristics of fermented foods. The identification of such improvements depends on the ability to characterise the microbial community in question and is thus applicable to bioinformatic analysis where careful consideration into the functional characteristics of microbial strains is possible.
3.3.1. Metabolic network analysis
Short-read and assembly profiling tools can be used to perform metabolic network analysis as demonstrated by Wu L-H et al. (Citation2017). The microbial metabolic network of vinegar pei, a starter culture mixture used to produce Zhenjiang vinegar, was reconstructed using multiple bioinformatic tools, detailed below. The short-read tool MetaCV (Liu J et al. Citation2013) profiled the vinegar samples at the taxonomic and functional level, using the KEGG database for gene assignment. Metavelvet assembled contigs (Afiahayati et al. 2015) were analysed using FragGeneScan (Rho et al. Citation2010) to detect contig bound ORFs, which were subsequently annotated by BLASTx using the Genbank, NR and KEGG databases. All genes assigned a KO term were mapped to their respective KEGG pathways by the tool KEGGMapper and assigned an EC number (Kanehisa et al. 2013). This assembly-based analysis revealed the metabolic pathways and enzymes that are potentially involved in flavour production in cereal vinegar, which were traced to contributing microbial members at the genus level by MetaCV (Wu L-H et al. Citation2017).
3.3.2. Identification of candidate starter culture strains
Variability during fermentation prevents the production of uniform products with consistent flavour. As such, the production of many fermented foods would benefit from the identification of an appropriate starter culture to minimise variability. Starter cultures are inoculums of selected microbial strains, which are deliberately introduced into a food matrix to ensure a more controlled fermentation process, ideally producing an homogeneous product with enhanced nutritional and sensory qualities (Liang et al. Citation2018; Laranjo et al. Citation2019). Starter cultures routinely used to produce fermented foods, such as cheese, and can influence cheese-based microbiomes and, in turn, flavour and appearance (Bertuzzi et al. Citation2018). Liang et al. (Citation2018) identified potential sources of starter cultures from an assembly-based approach that examined industrially produced paocai. Paocai is a dish composed of an assortment of vegetables including cabbage, radish, long beans and peppers, all of which are fermented together in a jar with a paocai brine solution (Chen and Narbad Citation2018). Metagenomic reads were assembled using IDBA-UD, and ORFs were predicted and translated from the assemblies using Prodigal, followed by clustering based on sequence identity and coverage values using CD-HIT (Huang et al. Citation2010). Clustering was used to construct a non-redundant gene catalog from input sequences obtained in the clustering phase. The gene catalog subsequently informed searches against the NCBI non-redundant (NR) protein sequences and KEGG databases using diamond and GhostKOALA (Kanehisa et al. Citation2016), respectively. From this analysis the authors were able to identify the species Lactobacillus paralimentarius and Lactobacillus alimentarius in Paocai that, on the basis of their abundance and functional roles, were predicted to substantially contribute to a successful fermentation process and could potentially serve as reservoirs for selecting candidate starter culture strains (Liang et al. Citation2018).
Bioinformatic tools can compare functional subsystems found in metagenomes or metagenomic contigs to isolated starter cultures or potential candidates to investigate the potential of a particular isolate to be used as a starter. Illeghems et al. (Citation2015) examined the cocoa bean fermentation process through WMGS of a single representative sample after a 30-h fermentation. Using the data generated from this analysis, they performed a sequence-based comparison of candidate starter culture strains and Genovo-assembled contigs, using EDGAR (Edgar Citation2004). This analysis revealed the proportion of CDs shared between the candidate starter cultures and the metagenomic contigs, as well as a description of their functional categories. Based on shared gene content, the potential contribution of candidate starter cultures to the overall fermentation process was revealed, e.g. Limosilactobacillus fermentum 222 was 82.7% identical in gene content to the collective gene content of the metagenomic contigs, highlighting its usefulness as a starter culture (Illeghems et al. Citation2015). Further research into the metabolic activities of computationally identified or inspected strains, and their association with flavour compounds are needed to validate their potential as starter cultures (Liang et al. Citation2018).
3.3.3. Assess the impact of starter cultures and fermentation parameters in microbial communities
Taxonomic and functional profiling tools can be used to study changes in taxonomic membership and gene content due to a trait/response of interest (Calle Citation2019). Zepeda-Mendoza et al. (Citation2018) employed a short read based, taxonomic and functional pipeline to profile multiple microbial communities in control and strain inoculated samples of Cabernet Sauvignon wine. Profiling was performed to examine how inoculated strains of Oenococcus oeni and Brettanomyces bruxellensis interact with microbial communities and thus effect the flavour of Cabernet Sauvignon wine. Taxonomic profiling was performed using MG mapper (Petersen et al. Citation2017). The functional annotation workflow included contig assembly using IDBA-UD, gene prediction by Prodigal, and KO (KEGG Orthology) assignment through a translated homology search against the KEGG database using BLASTx. Univariate differential abundance testing using pairwise fisher exact tests, highlighted taxa, KO terms and their associated KEGG pathways inferred to be differentially abundant in the strain inoculated samples compared to spontaneously fermented control samples (Zepeda-Mendoza et al. Citation2018).
Bioinformatic approaches can also be employed to detect how additional factors such as raw ingredients, e.g. herbs and spices and manufacturing conditions, influence the microbial communities and its metabolic activities during fermentation and in turn, the organoleptic properties of the fermented product. Bertuzzi et al. (Citation2018) profiled microbial communities associated with surface ripened cheese and their corresponding smear cultures using a previously described pipeline. Through this bioinformatic analysis they observed a correlation between salting the surface of ripening cheese and the increased relative abundance of osmotic stress resistance and the osmoprotectant-related gene families (Bertuzzi et al. Citation2018). Link the volatilome profile to contributing taxa and metabolic pathways
Despite the capacity to determine differentially abundant metabolic pathways in fermented foods, it is often unclear which metabolic pathways generate the appropriate volatile organic compounds in the necessary quantity for a successful fermentation process. Bioinformatic profiling coupled with metabolomics can be used to reveal links between meta pathways and fermentation-based volatilomes to detect formation pathways that may be preferentially used and their microbial contributors (Illeghems et al. Citation2015). Li Z et al. (Citation2018) employed a combination of metagenomic and metabolomic-based approaches to examine interactions between microbial communities and flavour production in Pu-erh tea. The metabolomic workflow involved volatile organic compounds (VOCs) identification by headspace (HS) solid phase microextraction (SPME) and subsequent analysis by gas chromatography mass spectrometry (GC-MS). VOCs represent a group of evaporated carbon based chemicals emitted from products or processes. Examples of VOCs include aldehydes, ketones, acids, benzene derivatives and hydrocarbons. The metabolomic analysis detected five dominant flavours in the fermentation process of Pu-erh tea, namely methoxy phenolic compounds, theabrownin, alcohol and caravone. The Pu-erh tea microbiome was further examined using metagenomics. An assembly-based approach was taken to characterise the microbial community. Sequence reads were assembled using Soapdenovo (Luo et al. Citation2012) and the resulting assemblies were subjected to gene prediction and annotation using MetaGeneMark and DIAMOND, respectively. DIAMOND performed multiple translated searches against the NR, KEGG, eggNOG and CAZy databases. This multi-omic approach allowed for a detailed understanding of the factors associated with the biosynthesis of methoxy-phenolic compounds; these included genes, enzymes (methyltransferase) and their taxonomic affiliations inferred from detected protein families. In addition to identification, this workflow also monitored the contribution of the identified microorganisms at the genus level to community production of methyltransferase during fermentation by recalculating their relative abundance using the total sum abundance of the enzyme at two distinct timepoints. The biological interactions involved in the production of the 4 other dominant compounds were also examined in this manner, which revealed similar results, extending the knowledge into metabolic pathways that contribute to the dominant flavours of Pu-erh tea (Li Z et al. Citation2018).
This method can be further applied to strain level analysis. Ferrocino et al. (Citation2018) computationally profiled the microbial communities in both spontaneously fermented and strain inoculated samples of Salame Felino, a type of Italian cured pork sausage. Computational profiling was achieved through taxonomic classification by MetaPhlAn2 and a multistage assembly approach. Reads were assembled using Velvet, followed by gene prediction via MetaGeneMark. Subsequently genes were clustered through Usearch (Edgar Citation2010) based on sequence identity and alignment length criteria, creating a gene catalogue. The gene catalogue was annotated by querying its content against the NCBI-NR database using mBLASTX. Lastly, meta pathway analysis was performed using MEGAN, which assigned the identified genes to KEGG pathways. VOCs generated during the fermentation process were examined using metabolomics, which involved VOC extraction using HS SPME and analysis by GC/MS. Similarly to Zepeda-Mendoza et al. (Citation2018), the bioinformatic pipeline used by Ferrocino et al. (Citation2018), coupled with statistical analysis (Spearman’s rank-order correlation), revealed differences in gene content and metapathways between the Salame samples examined, which, in combination with metabolomics data, could be further linked to expressed volatile compounds. For example, bioinformatic profiling reported a higher abundance of KEGG genes encoding acetate kinase and butanediol dehydrogenase in the strain inoculated samples that correlated with elevated levels of acetic acid and reduced product acceptance. A higher concentration of acetic acid may have contributed to their inferior sensory properties compared to control samples reported during a liking test. Similarly, other sources have reported less fragrant products when employing starter cultures in the fermentation process (Sulaiman et al. Citation2014). Such results highlight the application of this bioinformatic approach in assessing the potential impact of strains on fermentation mechanisms, which contribute to flavour production (Ferrocino et al. Citation2018).
3.4. Food safety and quality
Foodborne illness can result in sickness and death, the full impact of which cannot be quantified due to limitations in surveillance methods (World Health Organisation 2017). There is an associated need for swift and accurate techniques for the detection of foodborne pathogens and spoilage microorganisms. Metagenomic tools can identify pathogenic or spoilage-causing microorganisms, which can be further examined computationally to reveal insights of relevance to food safety. For example, such tools can monitor changes in the diversity or proportion of undesirable microbes in food to anticipate food borne diseases and or microbial spoilage (Ercolini et al. Citation2011; Kable et al. Citation2016). In terms of exploratory potential, several short read and assembly based bioinformatic tools can detect and trace foodborne pathogens to the strain level (Stasiewicz et al. Citation2015; Scholz et al. Citation2016; Truong et al. Citation2017).
3.4.1. Detection of pathogens other abiotic factors of relevance to food safety
Several short read bioinformatic tools were employed in a proof-of-concept study to demonstrate that metagenomic shotgun sequencing can detect pathogenic strains in the dairy product nunu. Initially, metagenomic data was taxonomically profiled using MethPhlAn2, revealing that the majority of samples were dominated by the potentially pathogenic species Streptococcus infantarius. It was with some concern that the presence of the species Escherichia coli and Klebsiella pneumoniae was also detected at varying abundances across the samples. Three short-read bioinformatic tools, MetaMLST (Zolfo et al. Citation2017), PanPhlAn, and StrainPhlAn were used to detect strains of E. coli and K. pneumoniae present in 10 nunu samples. StrainPhlAn detected several E. coli and K. pneumoniae marker genes while MetaMLST found an E. coli strain in one sample and K. pneumoniae strains in seven samples. PanPhlAn identified E. coli strains in two nunu samples and several K. pneumoniae strains (Walsh et al. Citation2017). E. coli and K. pneumoniae specific marker genes detected by StrainPhlAn were aligned against E. coli and K. pneumoniae reference genomes, respectively, and the outputs were visualised as phylogenetic trees created using GraPhlAn (Asnicar et al. Citation2015). Phylogenetic analysis reveal that an E. coli strain detected in one of the nunu samples was closely related to the outbreak associated strain E. coli O139:H28 E24377A and shared the same ShET2 enterotoxin-encoding gene. Two K. pneumoniae strains showed antibiotic resistance properties and were closely related to K. pneumoniae KpQ3, another outbreak associated strain (Walsh et al. Citation2017).
Other abiotic factors of relevance to food safety include food production practices and food processing environments, which potentially introduce opportunistic microorganisms to food and can contribute to the development of AR (Oniciuc et al. Citation2019). Alexa et al. Citation2020 employed functional metagenomics to inspect a cheese production chain using a representative recombinant library to identify potential sources of pathogens, ARGs and virulence factors. The library consisted of cloned DNA fragments obtained from cheese production facilities, as well as raw cheese and milk samples. Recombinant clones (9216) were randomly selected from each sample category and sequenced using a Nextseq 500. Filtered paired-end reads were taxonomically profiled by Kraken2 revealing phylum and species level profiles, interestingly the production environment displayed the highest microbial diversity of all tested samples, containing multiple species of low abundance. Functional annotation involved two alignment methods used specifically to examine the AR potential of the sample. Bowtie2 was used to align reads against the MEGARes database (Lakin et al. 2016) to detect ARGs. BLASTx was employ to detect any gene with quorum quenching activity by performing a translated search against a custom built protein database, detailing all enzymes recorded by LaSarre and Federle (Citation2013) to inhibit quorum sensing. Detected ARGs were characterised into nine categories, and separated by sample type, which upon comparison displayed a similar resistome profile between samples. A limited number of ARGs could be assigned to their species of origin (11%), but highlighted the potential of E. coli and Lactococcus lactis as reservoirs for multi-drug resistant genes. Notably, raw cheese samples seem to contain a high fraction of Lc. lactis with AR potential compared to other samples. The BLASTx based annotation identified homologues to genes with predicted quorum quenching activity for example QSDH was reported to have the highest content of homologous genes (Alexa et al. Citation2020). Taxonomic profiling assigned most QQ genes to their host species or strains (72.7%) identifying members such as Lc. lactis, and Stenotrophomonas sp. LM091, which based on their gene content may help regulate the spread of AR in the dairy food chain (Alexa et al. Citation2020).
3.4.2. Profile viral predation
As noted above, viruses are known contributors to fermentation failure despite this, for many fermented foods interactions between viral and host communities are poorly described. Many well-studied fermented foods such as cheese, milk –kefir and water kefir ( and Figure S1) lack detailed insights into viral based interactions despite extensive research efforts over the last decade (Dugat-Bony et al. Citation2020). Colombo et al. (Citation2018) performed a shotgun metagenomics analysis on airborne virus-like particles (VLP) in two cheese production plants to assess their aptitude for microbial contamination, which is of relevance to cheese production given the prolonged exposure of certain cheese surfaces to air during ripening (Salazar et al. Citation2018). An assembly-based approach was taken involving contig assembly via MIRA and gene prediction of the assemblies using Prodigal. Predicted ORFs were subjected to a BLAST + search against a custom-built viral database. Further searches, using BLASTn, BLASTp and BLASTx were employed to compare viral and microbial data, using the NCBI NR nucleotide and protein databases, as well as CARD. Through this analysis detected ORFs were linked to taxonomic members of viral and bacterial origin, for example ORFs mapped to Legionella pneumophila and the human Papillomavirus, raising some safety concerns. ORFs were further assigned to AR categories based on gene function and antibiotic drug targets. The frequency of ARGs genes was calculated by dividing the representative ARG-related ORFs by the total ORFs content detected. Viral taxonomy was assessed to determine potential interactions with bacteria at the genus level, highlighting that the virome can target both fermenting and non-fermenting bacteria. Interactions were quantified using the same ORF based method employed to determine the frequency of antibiotic resistant genes (Colombo et al. Citation2018).
3.4.3. Profile the bioprotective potential of fermented foods
Bioinformatic tools can reveal functionalities in fermented foods, some of which have a protective effect against spoilage and disease. Features include the occurrence of CRISPRs elements, the production of bacteriocins and other antimicrobials, highlighting the bioprotective potential of some foods. (Illeghems et al. Citation2015) detected such functions in a spontaneous cocoa bean fermentation process using the following computational workflow. Metagenomic data representing the fermentation process was assembled using Newbler, CABOG (Miller et al. Citation2008), Genovo (Laserson et al. Citation2011) and CAMERA. Notably Genova had the best performance statistics in terms of contig length, abundance and predicted functional genes as determined through the gene finding tools Glimmer (Delcher et al. Citation1999), GeneMarks and Augustus. Genovo-assembled contigs were selected for further annotation through GenDB. GenDB detected five bacteriocin encoding genes, six CRISPR elements and reported minimal evidence for the presence of antibiotic resistant genes (Illeghems et al. Citation2015).
3.5. Health benefits
Foods with added benefits beyond their nutritional value that possibly contribute to overall health and wellbeing are referred to as functional foods, with many fermented foods falling under this category. The market value of fermented functional foods continues to increase annually, achieving its most rapid growth in recent years (Behera et al. Citation2018; Shahbandeh Citation2019). Despite this growth, the further development and marketing of fermented functional foods may be limited by a lack of sufficient research to provide the evidence base required for robust health or functional claims.
To date, numerous studies have examined the health promoting properties of fermented food, with yogurt being examined most extensively (Taylor et al. Citation2020). Such studies offer interesting insights into the effects of fermented foods, but often fail to elucidate the mechanisms driving such effects. Metagenomics can be used to profile the gene and metabolic content of microbial communities to provide further evidence of the features driving any observed changes attributed to fermented foods.
3.5.1. Identification of putative health associated genes
Bioinformatic applications can be used to provide insights into the effects of fermented foods on human health. For example, fermenting microorganisms can be computationally assessed for the presence or absence of putative health promoting genes (Leech et al. Citation2020). Leech et al. Citation2020 investigated the microbial component of 58 diverse fermented foods and non-fermented milk samples using shotgun metagenomics. BAGEL (de Jong et al. Citation2006) compared Prodigal predicted genes to the BAGEL4 bacteriocin database to identify potential bacteriocin producing genes. Only four fermented food samples completely lacked gene clusters associated with bacteriocin production, with 55 putative bacteriocin-encoding gene clusters detected. Additional putative health-promoting genes were detected from UniRef gene clusters obtained through HUMAnN2 using a list of search terms. Health associated genes were detected and grouped into three categories; host colonisation, intestinal survival and host modulation with a notable difference in the number of detected colonisation genes in the fermented samples as opposed to the unfermented samples (Leech et al. Citation2020). Such insights can help to predict if fermenting microorganisms contribute to the health promoting properties of fermented foods.
3.5.2. Identify and profile MAGs with health associated potential
Pasolli et al. (Citation2020) compared MAGS derived from fermented milk products (n = 303) to those of human (n = 9445) and reference genomes, to examine the colonisation potential of lactic acid bacteria (LAB) found in dairy based fermented foods. Metagenomes were taxonomically profiled using MetaPhlAn2, and their compositions were compared to identify LAB species found in both sets of samples. Streptococcus thermophilus and Lc. lactis were the most prevalent species in the gut, and an additional 37-foodborne species were also detected at varying prevalence. Most overlapping LAB species shared a relative abundance of <2% with a small number of exceptions. Metadata associated with the human samples were used to identify correlations between lifestyle, age and geography and the prevalence of food associated LAB. Strong correlations were detected between the three categories of metadata and the LAB species. MAGs were then reconstructed by reassembling sequencing reads using a combination of IDBA-UD and metaSPAdes and binning by MetaBat2. Reconstructed MAGs and GenBank reference genomes were integrated together and clustered into species level bins based on sequence similarity values determined using MASH. Species-level bins showed that 52.4% of fermented food MAGs clustered with species detected in the human gut. Species-level bins were further visualised via phylogenetic trees and multidimensional scaling plots. PhyloPhlAn3 and GraPhlAn built phylogenies, while FastANI was employed for the multidimensional scaling plots. The phylogenies enabled a genomic comparison of the species detected in different samples. Notably, the phylogenetic analysis of S. thermophilus strains revealed no unique subclades between the sample types, providing evidence that gut ecosystems may acquire S. thermophilus through the diet. Other strains such as Lactobacillus delbrueckii subsp. bulgaricus and Lactobacillus delbrueckii subsp. lactis also displayed a similar pattern when phylogenetically assessed (Pasolli et al. Citation2020).
3.5.3. Identify taxa associated with fermented food consumption
Metagenomic analysis can assess the impact of fermented food consumption in shaping the composition of host microbiomes. A recent intervention study examined the impact of fermented food consumption on the composition of the gut microbiome (Taylor et al. Citation2020). Participants of the longitudinal study also took part in the American Gut Project (https://microsetta.ucsd.edu/) and were selected based on their answer to a fermented plant question from the American gut survey. Participants were separated into consumers of fermented foods and non-consumers based on their answer to the aforementioned survey (McDonald et al. Citation2018). A further fermented food questionnaire was given to the participants, which captured the types of food consumed over the 4-week intervention study. Foods include fermented dairy products such as kefir and yogurt, fermented grains such as miso and fermented vegetables such as sauerkraut and kimchi. Stool samples were collected from 115 participants each week for a period of four weeks. All stool samples collected were subjected to 16S rDNA sequencing and metabolomic analysis with a subset of samples derived from the same single time point chosen for shotgun sequencing. SHOGUN (Hillmann et al. Citation2020) was used in combination with bowtie2 for taxonomic annotations and quality filtering, NCBI RefSeq (O'Leary et al. Citation2016) was used as a reference database. Species level taxonomic identifications with an abundance >0.01 in at least one sample were reported in a biological Observation Matrix (BIOM) format taxa table. Firstly, a literature research was conducted to identify taxa that were commonly found in fermentation environments and could also be found in the metagenomic samples. The species Lactobacillus acidophilus, Levilactobacillus brevis, Limosilactobacillus fermentum, Lc. lactis, Leuc. mesenteroides, Lacticaseibacillus paracasei, Lactiplantibacillus plantarum and Lacticaseibacillus rhamnosus were identified as a result of the literature search and were classified as set 1. The taxa of set 1 were compared to 40 taxa that were found across all samples, referred to as set 2. The log ratio of both sets of taxa was computed for the consumer and non-consumer group using churro (Fedarko et al. Citation2020). Notably the consumer group had a higher log ratio of both sets of taxa compared to the non-consumer group, suggesting that the consumption of fermented foods is associated with the taxa contained in set one. To assess overall differences between the groups, differential abundance testing was performed using the statistical tool songbird (Morton et al. Citation2019) which ranks taxa that have changed the most between groups. The log ratio of 40 of the highest (set 5) and lowest ranked species (set 6) associated with fermented food consumption were calculated using churro, with a higher log ratio of the set 5 to set 6 displayed by the consumer group. Such analysis identified compositional differences between consumers and non-consumers of fermented foods and identifies taxa that could be potential contributing to such differences. Furthermore the metagenomics data was integrated with metabolomics data to identify occurrence patterns between the taxa associated with the consumption of fermented foods and a isomer of the putative health promoting fatty acid conjugated linoleic acid (CLA) (Taylor et al. Citation2020).
4. Conclusion
Distinct bioinformatic tools applied in combination can be used to profile fermentation ecosystems, allowing for a detailed understanding of complex fermentation processes and the potential roles of individual species in the fermentation environment (Illeghems et al. Citation2015; Beier et al. Citation2017). The assessment of microbial communities in this manner has significantly advanced our understanding of mixed populations, and the microorganism’s individual contributions to ecosystems. Metagenomic-associated methods, when applied to fermented foods, have resulted in the documentation of fermentation processes and production methods associated with many traditional foods. Such studies have shared and preserved knowledge concerning production practices, and in some cases have assessed/applied such insights to novel applications. Metabolic network analysis (see Section 3) may in the future provide insights into mechanisms needed to customise pathways, ensuring the generation of amino acids that are associated with a desirable taste while avoiding those that lead to off flavours, allowing for increased product acceptance. For example, the production of glutamic acid in a sufficient quantity to promote or enhance the umami taste of a food. Despite, the insights provided by WMGS and metabolomics into flavour production, experimental validations such as, sensory analysis will be key to confirming any hypotheses generated by WMGS (Wu L-H et al. Citation2017).
Organisations such as the Food Safety and Inspection Service (FSIS), an agency of the United States Department of Agriculture, have recognised that sequencing-based approaches have the potential to become future microbial pathogen and spoilage detection tools (Bauer et al. Citation2014). Metagenomics is gathering considerable interest in such applications, and It is technologically feasible that in the future real time in situ metagenomics analysis will be possible for field research (Albrecht et al. Citation2020). Current limitations of metagenomics in assessing the safety of food and food processing environments include its inability to provide sufficient gene expression level data and to definitively validate all taxonomic and functional outputs as associated with pathogenic microorganisms. Metagenomics is restricted to the detection of features with homology to those previously described and included in the available databases. Such homology-based inferences are hampered by the limited number of reference pathogens in current databases, which cannot yet account for all taxa of interest (Alexa et al. Citation2020).
Despite this, given appropriate sequencing depth and coverage, shotgun metagenomics can provide a valuable overview of potential pathogens, virulence factors, and antimicrobial resistance genes and their source (). Shotgun metagenomics provides a mechanistic understanding of the microbes in question furthering knowledge relating to associated pathogenic and/or spoilage processes and any additional factors that contribute to their occurrence, which can for now be used to complement culture dependent techniques (Hazards et al. Citation2019), until validated and certified shotgun approaches for industry are established.
Few studies exist that have applied WMGS to assess the health-promoting aspects of fermented foods, and to our knowledge no metagenomic specific tool currently available, is designed to link taxonomic and functional outputs with biomarkers of health. The absence of health-related tools and publications is reflective of the limited mechanistic understanding of how fermented foods can promote health. A recent milk kefir review reported the findings of various animal models and in vitro studies, specifically investigating the effects of kefir consumption on cholesterol metabolism, cancer, wound healing, the immune system and the gut microbiome (Bourrie et al. Citation2016). While the publications discussed in the review showed promising results, few were able to provide mechanistic insights, which could be integrated with metagenomics tools and studies (Slattery et al. Citation2019).
Metagenomics analysis will continue to benefit from the development and improvement of tools and sequencing platforms, as the field continues to embrace the ideas and principles of computer science. One significant improvement is database expansion, with an increasing number of genomes and metagenomes sequenced each year (), allowing for improved annotations that can scale to bigger datasets. Tools such as InStrain (Olm et al. Citation2021) allow for the flexible construction of customised reference databases, enabling the incorporation of genomes, with publically available and or environmental specific MAGs. Such tools would be particularly suitable for fermented foods such as milk and water kefir, where there is a notable absence in the availability of Eukaryotic reference genomes ( and Figure S1).
Despite this positive trend, the forecast advances in computational tools will largely be accessible through command line interfaces at institutes with sufficient computational infrastructures. Future tool developers should incorporate user-friendly functionalities or preferably a graphical user interface (GUI) in their tools to facilitate the participation of laboratory oriented microbiologists (Liu Y-X et al. Citation2021). Concerning tool development, no single tool is available to date that encompasses all the steps required to interpret a metagenome, and gold standard methods capable of full-fledged metagenomics analyses have not yet been developed. Therefore performing bioinformatic analysis to meet a publishable scientific standard requires the researcher to identify, install and configure bioinformatic tools that will fulfil the research requirements and construct personal scripts to ensure the outputs of the various tools align (Uritskiy et al. Citation2018).
Given the interest in this field as well as the developments in sequencing and bioinformatics, it is reasonable to assume that gold standard methods are on the horizon and will ensure accurate comparability between studies. In fact, the first steps have been taken towards this, with the development of MAGO (metagenome-assembled genomes orchestra) (Murovec et al. 2019), MetaWRAP (Uritskiy et al. Citation2018) and bioBakery workflows (Beghini et al. Citation2021), all of which provides a standardised automated approach for metagenomic analysis, incorporating many separate softwares into an integrated workflow. Other tools have also been developed such as Squeezemeta that, similar to MAGO, provides an integrated standardised metagenomic and metatranscriptomic workflow, which is amenable to a standard local computer (Tamames and Puente-Sánchez 2019).
In conclusion, further research is required to identify and develop gold standard methods for metagenomics analysis that will allow for accurate standardised workflows for foods, fermented or otherwise and other environments. Such workflows should incorporate all steps required in a short-read and or assembly-based approach; ensuring comparability between studies by controlling for intrinsic differences in the selection of bioinformatics tools (Sczyrba et al. Citation2017; Angers-Loustau et al. Citation2018; Couto et al. Citation2018; Gruening et al. Citation2018). Further research should begin by benchmarking the performance of tools across different biological ecosystems to assess their aptitude on the selected environment and develop new parameters to optimise performance based on use case scenarios. Such research would allow for the cataloguing of bioinformatic tools, enabling researchers to make informed decisions on their application.
Supplemental Material
Download JPEG Image (2.2 MB){kind=link}
Acknowledgements
We thank members of the Vision 1 laboratory, for helpful discussions and a critical review of the manuscript.
Disclosure statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Funding
References
- Abe M, Takaoka N, Idemoto Y, Takagi C, Imai T, Nakasaki K. 2008. Characteristic fungi observed in the fermentation process for Puer tea. Int J Food Microbiol. 124(2):199–203.
- Aggarwala V, Liang G, Bushman FD. 2017. Viral communities of the human gut: metagenomic analysis of composition and dynamics. Mob DNA. 8(1):12.
- Albrecht B, Bağcı C, Huson DH. 2020. MAIRA- real-time taxonomic and functional analysis of long reads on a laptop. BMC Bioinf. 21(Suppl 13):390.
- Alcock BP, Raphenya AR, Lau TTY, Tsang KK, Bouchard M, Edalatmand A, Huynh W, Nguyen A-LV, Cheng AA, Liu S, et al. 2019. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48(D1):D517–D525.
- Alexa EA, Walsh CJ, Coughlan LM, Awad A, Simon CA, Ruiz L, Crispie F, Cotter PD, Alvarez-Ordóñez A. 2020. Dairy products and dairy-processing environments as a reservoir of antibiotic resistance and quorum-quenching determinants as revealed through functional metagenomics. mSystems. 5(1):e00723-19.
- Almeida OGG, De Martinis ECP. 2019. Bioinformatics tools to assess metagenomic data for applied microbiology. Appl Microbiol Biotechnol. 103(1):69–82.
- Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, Lahti L, Loman NJ, Andersson AF, Quince C. 2014. Binning metagenomic contigs by coverage and composition. Nat Methods. 11(11):1144–1146.
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215(3):403–410.
- Angers-Loustau A, Petrillo M, Bengtsson-Palme J, Berendonk T, Blais B, Chan KG, Coque TM, Hammer P, Heß S, Kagkli DM, et al. 2018. The challenges of designing a benchmark strategy for bioinformatics pipelines in the identification of antimicrobial resistance determinants using next generation sequencing technologies. F1000Res. 7:ISCB Comm J-459.
- Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, et al. 2004. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 32(Database issue):D115–D119.
- Arango-Argoty G, Garner E, Pruden A, Heath LS, Vikesland P, Zhang L. 2018. DeepARG: a deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome. 6(1):23.
- Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS. 2016. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44(W1):W16–21.
- Asnicar F, Thomas AM, Beghini F, Mengoni C, Manara S, Manghi P, Zhu Q, Bolzan M, Cumbo F, May U, et al. 2020. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat Commun. 11(1):2500.
- Asnicar F, Weingart G, Tickle TL, Huttenhower C, Segata N. 2015. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ. 3:e1029.
- Ayling M, Clark MD, Leggett RM. 2020. New approaches for metagenome assembly with short reads. Brief Bioinform. 21(2):584–594.
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Bauer N, Evans P, Leopold B, Levine J, White P. 2014. White paper: current and future development and use of molecular subtyping by USDA-FSIS. Washington (DC): Food Safety and Inspection Service, U.S. Department of Agriculture. Available from: https://www.fsis.usda.gov/wps/wcm/connect/6c7f71fd-2c0c-4ff0-b2bc-4977c7947516/Molecular-Subtyping-White-Paper.pdf?MOD=AJPERES.
- Beghini F, McIver LJ, Blanco-Míguez A, Dubois L, Asnicar F, Maharjan S, Mailyan A, Manghi P, Scholz M, Thomas AM, et al. 2021. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife. 10:e65088.
- Behera S, Bal P, Das S, Panda S, Mohanty N. 2018. Advances in microbial fermentation and fermented food for health. In: Panda S, Shetty P, editors. Innovations in technologies for fermented food and beverage industries. Food microbiology and food safety. Cham: Springer; p. 53–69.
- Beier S, Tappu R, Huson DH. 2017. Functional analysis in metagenomics using MEGAN 6. In: Charles TC, Liles MR, Sessitsch A, editors. Functional metagenomics: tools and applications. Cham: Springer International Publishing; p. 65–74.
- Belcour A, Frioux C, Aite M, Bretaudeau A, Hildebrand F, Siegel A. 2020. Metage2Metabo, microbiota-scale metabolic complementarity for the identification of key species. eLife. 9:e61968.
- Bellon JR, Yang F, Day MP, Inglis DL, Chambers PJ. 2015. Designing and creating Saccharomyces interspecific hybrids for improved, industry relevant, phenotypes. Appl Microbiol Biotechnol. 99(20):8597–8609.
- Bengtsson-Palme J, Ryberg M, Hartmann M, Branco S, Wang Z, Godhe A, De Wit P, Sánchez-García M, Ebersberger I, de Sousa F, et al. 2013. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol Evol. 4(10):914–919.
- Berglund F, Österlund T, Boulund F, Marathe NP, Larsson DGJ, Kristiansson E. 2019. Identification and reconstruction of novel antibiotic resistance genes from metagenomes. Microbiome. 7(1):52.
- Bertuzzi AS, Walsh AM, Sheehan JJ, Cotter PD, Crispie F, McSweeney PLH, Kilcawley KN, Rea MC. 2018. Omics-based insights into flavor development and microbial succession within surface-ripened cheese. mSystems. 3(1):e00211-17.
- Blair JMA, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJV. 2015. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol. 13(1):42–51.
- Bourrie BCT, Willing BP, Cotter PD. 2016. The microbiota and health promoting characteristics of the fermented beverage kefir. Front Microbiol. 7:647.
- Bove P, Russo P, Capozzi V, Gallone A, Spano G, Fiocco D. 2013. Lactobacillus plantarum passage through an oro-gastro-intestinal tract simulator: carrier matrix effect and transcriptional analysis of genes associated to stress and probiosis. Microbiol Res. 168(6):351–359.
- Boyd JA, Woodcroft BJ, Tyson GW. 2018. GraftM: a tool for scalable, phylogenetically informed classification of genes within metagenomes. Nucleic Acids Res. 46(10):e59–e59.
- Brown ED, Wright GD. 2016. Antibacterial drug discovery in the resistance era. Nature. 529(7586):336–343.
- Calle ML. 2019. Statistical analysis of metagenomics data. Genom Inform. 17(1):e6.
- Cao Y, Fanning S, Proos S, Jordan K, Srikumar S. 2017. A review on the applications of next generation sequencing technologies as applied to food-related microbiome studies. Front Microbiol. 8:1829.
- Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. 2019. GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics. 36(6):1925–1927.
- Chaves-López C, Serio A, Grande-Tovar CD, Cuervo-Mulet R, Delgado-Ospina J, Paparella A. 2014. Traditional fermented foods and beverages from a microbiological and nutritional perspective: the Colombian heritage. Compr Rev Food Sci Food Saf. 13(5):1031–1048.
- Chen LX, Anantharaman K, Shaiber A, Eren AM, Banfield JF. 2020. Accurate and complete genomes from metagenomes. Genome Res. 30(3):315–333.
- Chen W, Narbad A. 2018. Lactic acid bacteria in foodborne hazards reduction. Cham: Springer.
- Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. 2016. GenBank. Nucleic Acids Res. 44(D1):D67–D72.
- Colombo S, Arioli S, Gargari G, Neri E, Della Scala G, Mora D. 2018. Characterization of airborne viromes in cheese production plants. J Appl Microbiol. 125(5):1444–1454.
- Couto N, Schuele L, Raangs EC, Machado MP, Mendes CI, Jesus TF, Chlebowicz M, Rosema S, Ramirez M, Carriço JA, et al. 2018. Critical steps in clinical shotgun metagenomics for the concomitant detection and typing of microbial pathogens. Sci Rep. 8(1):13767. PubMed. (Accessed 2018/09//).
- Cuevas DA, Edirisinghe J, Henry CS, Overbeek R, O'Connell TG, Edwards RA. 2016. From DNA to FBA: how to build your own genome-scale metabolic model. Front Microbiol. 7:907.
- De Filippis F, Parente E, Ercolini D. 2017. Metagenomics insights into food fermentations. Microb Biotechnol. 10(1):91–102.
- de Jong A, van Hijum SA, Bijlsma JJ, Kok J, Kuipers OP. 2006. BAGEL: a web-based bacteriocin genome mining tool. Nucleic Acids Res. 34(Web Server issue):W273–9.
- Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27(23):4636–4641.
- Dong X, Strous M. 2019. An integrated pipeline for annotation and visualization of metagenomic contigs. Front Genet. 10:999–999.
- Drost H-G, Paszkowski J. 2017. Biomartr: genomic data retrieval with R. Bioinformatics. 33(8):1216–1217.
- Dugat-Bony E, Lossouarn J, De Paepe M, Sarthou AS, Fedala Y, Petit MA, Chaillou S. 2020. Viral metagenomic analysis of the cheese surface: a comparative study of rapid procedures for extracting viral particles. Food Microbiol. 85:103278.
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797.
- Eddy SR. 2011. Accelerated Profile HMM Searches. PLOS Computational Biology. 7(10):e1002195.
- Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 26(19):2460–2461.
- Ercolini D, Ferrocino I, Nasi A, Ndagijimana M, Vernocchi P, La Storia A, Laghi L, Mauriello G, Guerzoni ME, Villani F. 2011. Monitoring of microbial metabolites and bacterial diversity in beef stored under different packaging conditions. Appl Environ Microbiol. 77(20):7372–7381.
- Fedarko MW, Martino C, Morton JT, González A, Rahman G, Marotz CA, Minich JJ, Allen EE, Knight R. 2020. Visualizing ’omic feature rankings and log-ratios using Qurro. NAR Genom Bioinform. 2(2):lqaa023.
- Fernández L, Escobedo S, Gutiérrez D, Portilla S, Martínez B, García P, Rodríguez A. 2017. Bacteriophages in the dairy environment: from enemies to allies. Antibiotics. 6(4):27.
- Ferrocino I, Bellio A, Giordano M, Macori G, Romano A, Rantsiou K, Decastelli L, Cocolin L. 2018. Shotgun metagenomics and volatilome profile of the microbiota of fermented sausages. Appl Environ Microbiol. 84(3):e02120-17.
- Flores M, Corral S, Cano-García L, Salvador A, Belloch C. 2015. Yeast strains as potential aroma enhancers in dry fermented sausages. Int J Food Microbiol. 212:16–24.
- Fritz A, Hofmann P, Majda S, Dahms E, Dröge J, Fiedler J, Lesker TR, Belmann P, DeMaere MZ, Darling AE, et al. 2019. CAMISIM: simulating metagenomes and microbial communities. Microbiome. 7(1):17.
- Garneau JE, Moineau S. 2011. Bacteriophages of lactic acid bacteria and their impact on milk fermentations. Microb Cell Fact. 10(Suppl 1):S20.
- Gille D, Schmid A, Walther B, Vergères G. 2018. Fermented food and non-communicable chronic diseases: a review. Nutrients. 10(4):448.
- Gillings MR, Stokes HW. 2012. Are humans increasing bacterial evolvability? Trends Ecol Evol. 27(6):346–352.
- Gloor GB, Macklaim JM, Pawlowsky-Glahn V, Egozcue JJ. 2017. Microbiome datasets are compositional: and this is not optional. Front Microbiol. 8:2224.
- Graham ED, Heidelberg JF, Tully BJ. 2017. BinSanity: unsupervised clustering of environmental microbial assemblies using coverage and affinity propagation. PeerJ. 5:e3035.
- Gruening B, Sallou O, Moreno P, da Veiga Leprevost F, Ménager H, Søndergaard D, Röst H, Sachsenberg T, O'Connor B, Madeira F, et al. 2018. Recommendations for the packaging and containerizing of bioinformatics software. F1000Res. 7:ISCB Comm J-742.
- Guitor AK, Raphenya AR, Klunk J, Kuch M, Alcock B, Surette MG, McArthur AG, Poinar HN, Wright GD. 2019. Capturing the resistome: a targeted capture method to reveal antibiotic resistance determinants in metagenomes. Antimicrob Agents Chemother. 64(1):e01324-19.
- Guo J, Bolduc B, Zayed AA, Varsani A, Dominguez-Huerta G, Delmont TO, Pratama AA, Gazitúa MC, Vik D, Sullivan MB, et al. 2021. VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome. 9(1):37.
- Guo J, Quensen JF, Sun Y, Wang Q, Brown CT, Cole JR, Tiedje JM. 2019. Review, evaluation, and directions for gene-targeted assembly for ecological analyses of metagenomes. Front Genet. 10:957.
- Gupta SK, Padmanabhan BR, Diene SM, Lopez-Rojas R, Kempf M, Landraud L, Rolain JM. 2014. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother. 58(1):212–220.
- Hazards E, Panel B, Koutsoumanis K, Allende A, Alvarez-Ordóñez A, Bolton D, Bover-Cid S, Chemaly M, Davies R, De Cesare A, et al. 2019. Whole genome sequencing and metagenomics for outbreak investigation, source attribution and risk assessment of food-borne microorganisms. EFSA J. 17(12):e05898.
- Hendrix RW. 2002. Bacteriophages: evolution of the majority. Theor Popul Biol. 61(4):471–480.
- Hillmann B, Al-Ghalith GA, Shields-Cutler RR, Zhu Q, Gohl DM, Beckman KB, Knight R, Knights D, Rawls JF. 2018. Evaluating the information content of shallow shotgun metagenomics. mSystems. 3(6):e00069-18.
- Hillmann B, Al-Ghalith GA, Shields-Cutler RR, Zhu Q, Knight R, Knights D. 2020. SHOGUN: a modular, accurate and scalable framework for microbiome quantification. Bioinformatics. 36(13):4088–4090.
- Huang Y, Niu B, Gao Y, Fu L, Li W. 2010. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 26(5):680–682.
- Hübler R, Key FM, Warinner C, Bos KI, Krause J, Herbig A. 2019. HOPS: automated detection and authentication of pathogen DNA in archaeological remains. Genome Biol. 20(1):280.
- Hutchins BI, Baker KL, Davis MT, Diwersy MA, Haque E, Harriman RM, Hoppe TA, Leicht SA, Meyer P, Santangelo GM. 2019. The NIH open citation collection: a public access, broad coverage resource. PLoS Biol. 17(10):e3000385.
- Hutchins BI, Yuan X, Anderson JM, Santangelo GM. 2016. Relative citation ratio (RCR): a new metric that uses citation rates to measure influence at the article level. PLoS Biol. 14(9):e1002541.
- Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 11:119–119.
- Illeghems K, Weckx S, De Vuyst L. 2015. Applying meta-pathway analyses through metagenomics to identify the functional properties of the major bacterial communities of a single spontaneous cocoa bean fermentation process sample. Food Microbiol. 50:54–63.
- Imelfort M, Parks D, Woodcroft BJ, Dennis P, Hugenholtz P, Tyson GW. 2014. GroopM: an automated tool for the recovery of population genomes from related metagenomes. PeerJ. 2:e603.
- Jung JY, Lee SH, Kim JM, Park MS, Bae J-W, Hahn Y, Madsen EL, Jeon CO. 2011. Metagenomic analysis of kimchi, a traditional Korean fermented food. Appl Environ Microbiol. 77(7):2264–2274.
- Kabak B, Dobson AD. 2011. An introduction to the traditional fermented foods and beverages of Turkey. Crit Rev Food Sci Nutr. 51(3):248–260.
- Kable ME, Srisengfa Y, Laird M, Zaragoza J, McLeod J, Heidenreich J, Marco ML. 2016. The core and seasonal microbiota of raw bovine milk in tanker trucks and the impact of transfer to a milk processing facility. MBio. 7(4):e00836–16.
- Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. 2014. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42(Database issue):D199–D205.
- Kanehisa M, Sato Y, Morishima K. 2016. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 428(4):726–731.
- Kang DD, Froula J, Egan R, Wang Z. 2015. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 3:e1165.
- Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, Wang Z. 2019. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 7:e7359.
- Karp PD, Latendresse M, Paley SM, Krummenacker M, Ong QD, Billington R, Kothari A, Weaver D, Lee T, Subhraveti P, et al. 2016. Pathway Tools version 19.0 update: software for pathway/genome informatics and systems biology. Brief Bioinform. 17(5):877-90. PMID: 26454094
- Kopylova E, Noé L, Touzet H. 2012. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 28(24):3211-3217.
- Karlsen E, Schulz C, Almaas E. 2018. Automated generation of genome-scale metabolic draft reconstructions based on KEGG. BMC Bioinf. 19(1):467.
- Lakin SM, Dean C, Noyes NR, Dettenwanger A, Ross AS, Doster E, Rovira P, Abdo Z, Jones KL, Ruiz J, et al. 2017. MEGARes: an antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. 45(D1):D574–D580.
- Laranjo M, Potes ME, Elias M. 2019. Role of starter cultures on the safety of fermented meat products. Front Microbiol. 10:853.
- Laserson J, Jojic V, Koller D. 2011. Genovo: de novo assembly for metagenomes. J Comput Biol. 18(3):429–443.
- LaSarre B, Federle MJ. 2013. Exploiting quorum sensing to confuse bacterial pathogens. Microbiology and Molecular Biology Reviews. 77(1):73–111.
- Lee J-H, Yi H, Chun J. 2011. rRNASelector: a computer program for selecting ribosomal RNA encoding sequences from metagenomic and metatranscriptomic shotgun libraries. J Microbiol. 49(4):689–691.
- Leech J, Cabrera-Rubio R, Walsh AM, Macori G, Walsh CJ, Barton W, Finnegan L, Crispie F, O’Sullivan O, Claesson MJ, et al. 2020. Fermented-food metagenomics reveals substrate-associated differences in taxonomy and health-associated and antibiotic resistance determinants. mSystems. 5(6):e00522-20.
- Leimbach A, Hacker J, Dobrindt U. 2013. E. coli as an all-rounder: the thin line between commensalism and pathogenicity. Curr Top Microbiol Immunol. 358:3–32.
- Li C, Donizelli M, Rodriguez N, Dharuri H, Endler L, Chelliah V, Li L, He E, Henry A, Stefan MI, et al. 2010. BioModels database: an enhanced, curated and annotated resource for published quantitative kinetic models. BMC Syst Biol. 4(1):92.
- Li D, Luo R, Liu CM, Leung CM, Ting HF, Sadakane K, Yamashita H, Lam TW. 2016. MEGAHIT v1.0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods. 102:3–11.
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. [Preprint].
- Li Z, Feng C, Luo X, Yao H, Zhang D, Zhang T. 2018. Revealing the influence of microbiota on the quality of Pu-erh tea during fermentation process by shotgun metagenomic and metabolomic analysis. Food Microbiol. 76:405–415.
- Liang H, Chen H, Ji C, Lin X, Zhang W, Li L. 2018. Dynamic and functional characteristics of predominant species in industrial paocai as revealed by combined DGGE and metagenomic sequencing. Front Microbiol. 9:2416–2416.
- Lieven C, Beber ME, Olivier BG, Bergmann FT, Ataman M, Babaei P, Bartell JA, Blank LM, Chauhan S, Correia K, et al. 2020. MEMOTE for standardized genome-scale metabolic model testing. Nat Biotechnol. 38(3):272–276.
- Liu B, Pop M. 2009. ARDB—antibiotic resistance genes database. Nucleic Acids Res. 37(Database issue):D443–D447.
- Liu J, Wang H, Yang H, Zhang Y, Wang J, Zhao F, Qi J. 2013. Composition-based classification of short metagenomic sequences elucidates the landscapes of taxonomic and functional enrichment of microorganisms. Nucleic Acids Res. 41(1):e3.
- Liu Y-X, Qin Y, Chen T, Lu M, Qian X, Guo X, Bai Y. 2021. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell. 12(5):315–330.
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience,. 1(1):18.
- Macesic N, Polubriaginof F, Tatonetti NP. 2017. Machine learning: novel bioinformatics approaches for combating antimicrobial resistance. Curr Opin Infect Dis. 30(6):511–517.
- Machado D, Andrejev S, Tramontano M, Patil KR. 2018. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 46(15):7542–7553.
- Marco ML, Heeney D, Binda S, Cifelli CJ, Cotter PD, Foligné B, Gänzle M, Kort R, Pasin G, Pihlanto A, et al. 2017. Health benefits of fermented foods: microbiota and beyond. Curr Opin Biotechnol. 44:94–102.
- Marco ML, Sanders ME, Gänzle M, Arrieta MC, Cotter PD, De Vuyst L, Hill C, Holzapfel W, Lebeer S, Merenstein D, et al. 2021. The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on fermented foods. Nat Rev Gastroenterol Hepatol. 18(3):196–208.
- Martinez-Villaluenga C, Peñas E, Frias J. 2017. Chapter 2 - bioactive peptides in fermented foods: production and evidence for health effects. In: Frias J, Martinez-Villaluenga C, Peñas E, editors. Fermented foods in health and disease prevention. Boston: Academic Press; p. 23–47.
- Matsen FA, Kodner RB, Armbrust EV. 2010. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinf. 11(1):538.
- McDonald D, Hyde E, Debelius JW, Morton JT, Gonzalez A, Ackermann G, Aksenov AA, Behsaz B, Brennan C, Chen Y, et al. 2018. American gut: an open platform for citizen science microbiome research. mSystems. 3(3):e00031–18.
- McHardy A, Sczyrba A, Rattei T. 2014. The critical assessment of metagenome interpretation (CAMI) competition. Nat Methods. 19:429–440.
- McIver LJ, Abu-Ali G, Franzosa EA, Schwager R, Morgan XC, Waldron L, Segata N, Huttenhower C. 2017. bioBakery: a meta’omic analysis environment. Bioinformatics. 34(7):1235–1237.
- Meersman E, Steensels J, Paulus T, Struyf N, Saels V, Mathawan M, Koffi J, Vrancken G, Verstrepen KJ. 2015. Breeding strategy to generate robust yeast starter cultures for cocoa pulp fermentations. Appl Environ Microbiol. 81(18):6166–6176.
- Melkonian C, Gottstein W, Blasche S, Kim Y, Abel-Kistrup M, Swiegers H, Saerens S, Edwards N, Patil KR, Teusink B, et al. 2019. Finding functional differences between species in a microbial community: case studies in wine fermentation and kefir culture. Front Microbiol. 10:1347.
- Mendoza SN, Olivier BG, Molenaar D, Teusink B. 2019. A systematic assessment of current genome-scale metabolic reconstruction tools. Genome Biol. 20(1):158.
- Menzel P, Gudbergsdottir SR, Rike AG, Lin L, Zhang Q, Contursi P, Moracci M, Kristjansson JK, Bolduc B, Gavrilov S, et al. 2015. Comparative metagenomics of eight geographically remote terrestrial hot springs. Microb Ecol. 70(2):411–424.
- Menzel P, Ng KL, Krogh A. 2016. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat Commun. 7(1):11257.
- Meyer F, Bremges A, Belmann P, Janssen S, McHardy AC, Koslicki D. 2019. Assessing taxonomic metagenome profilers with OPAL. Genome Biol. 20(1):51.
- Meyer F, Hofmann P, Belmann P, Garrido-Oter R, Fritz A, Sczyrba A, McHardy AC. 2018. AMBER: assessment of metagenome BinnERs. GigaScience. 7(6):giy069.
- Meyer F, Lesker T-R, Koslicki D, Fritz A, Gurevich A, Darling AE, Sczyrba A, Bremges A, McHardy AC. 2021. Tutorial: assessing metagenomics software with the CAMI benchmarking toolkit. Nat Protoc. 16:1785–1801.
- Mikheenko A, Saveliev V, Gurevich A. 2016. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics. 32(7):1088–1090.
- Miller JR, Delcher AL, Koren S, Venter E, Walenz BP, Brownley A, Johnson J, Li K, Mobarry C, Sutton G. 2008. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 24(24):2818–2824.
- Morton JT, Marotz C, Washburne A, Silverman J, Zaramela LS, Edlund A, Zengler K, Knight R. 2019. Establishing microbial composition measurement standards with reference frames. Nat Commun. 10(1):2719.
- Mukherjee S, Stamatis D, Bertsch J, Ovchinnikova G, Katta HY, Mojica A, Chen IMA, Kyrpides NC, Reddy T. 2019. Genomes OnLine database (GOLD) v.7: updates and new features. Nucleic Acids Res. 47(D1):D649–D659.
- Murovec B, Deutsch L, Stres B. 2020. Computational framework for high-quality production and large-scale evolutionary analysis of metagenome assembled genomes. Mol Biol Evol. 37(2):593–598.
- Nampoothiri KM, Beena DJ, Vasanthakumari DS, Ismail B. 2017. Chapter 3 – health benefits of exopolysaccharides in fermented foods. In: Frias J, Martinez-Villaluenga C, Peñas E, editors. Fermented foods in health and disease prevention. Boston: Academic Press; p. 49–62.
- Norsigian CJ, Pusarla N, McConn JL, Yurkovich JT, Dräger A, Palsson BO, King Z. 2019. BiGG Models 2020: multi-strain genome-scale models and expansion across the phylogenetic tree. Nucleic Acids Res. 48(D1):D402–D406.
- Nout MJR. 2014. Food technologies: fermentation. In: Motarjemi Y, editor. Encyclopedia of food safety. Waltham: Academic Press; p. 168–177.
- Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. 2017. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27(5):824–834.
- O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. 2016. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44(D1):D733–45.
- Olm MR, Crits-Christoph A, Bouma-Gregson K, Firek BA, Morowitz MJ, Banfield JF. 2021. inStrain profiles population microdiversity from metagenomic data and sensitively detects shared microbial strains. Nat Biotechnol. 39(6):727–736.
- Oniciuc E-A, Likotrafiti E, Alvarez-Molina A, Prieto M, López M, Alvarez-Ordóñez A. 2019. Food processing as a risk factor for antimicrobial resistance spread along the food chain. Curr Opin Food Sci. 30:21–26.
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25(7):1043–1055.
- Pasolli E, De Filippis F, Mauriello IE, Cumbo F, Walsh AM, Leech J, Cotter PD, Segata N, Ercolini D. 2020. Large-scale genome-wide analysis links lactic acid bacteria from food with the gut microbiome. Nat Commun. 11(1):2610.
- Peng Y, Leung HC, Yiu SM, Chin FY. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 28(11):1420–1428.
- Petersen TN, Lukjancenko O, Thomsen MCFM, Sperotto M, Lund O, Møller Aarestrup F, Sicheritz-Pontén T. 2017. MGmapper: reference based mapping and taxonomy annotation of metagenomics sequence reads. PLoS One. 12(5):e0176469.
- Pevzner PA, Tang H, Waterman MS. 2001. An Eulerian path approach to DNA fragment assembly. Proc Natl Acad Sci U S A. 98(17):9748–9753.
- Piro VC, Matschkowski M, Renard BY. 2017. MetaMeta: integrating metagenome analysis tools to improve taxonomic profiling. Microbiome. 5(1):101.
- Pride DT, Meinersmann RJ, Wassenaar TM, Blaser MJ. 2003. Evolutionary implications of microbial genome tetranucleotide frequency biases. Genome Res. 13(2):145–158.
- Prosser JI. 2015. Dispersing misconceptions and identifying opportunities for the use of 'omics’ in soil microbial ecology. Nat Rev Microbiol. 13(7):439–446.
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. 2012. The Pfam protein families database. Nucleic Acids Res. 40(Database issue):D290–D301.
- Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. 2017. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol. 35(9):833–844.
- Ren J, Ahlgren NA, Lu YY, Fuhrman JA, Sun F. 2017. VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome. 5(1):69.
- Rho M, Tang H, Ye Y. 2010. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res. 38(20):e191.
- Roux S, Emerson JB, Eloe-Fadrosh EA, Sullivan MB. 2017. Benchmarking viromics: an in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ. 5:e3817.
- Roux S, Enault F, Hurwitz BL, Sullivan MB. 2015. VirSorter: mining viral signal from microbial genomic data. PeerJ. 3:e985.
- Roux S. 2019. A viral ecogenomics framework to uncover the secrets of nature’s “microbe whisperers”. mSystems. 4(3):e00111-19.
- Salazar JK, Carstens CK, Ramachandran P, Shazer AG, Narula SS, Reed E, Ottesen A, Schill KM. 2018. Metagenomics of pasteurized and unpasteurized gouda cheese using targeted 16S rDNA sequencing. BMC Microbiol. 18(1):189.
- Şanlier N, Gökcen BB, Sezgin AC. 2019. Health benefits of fermented foods. Crit Rev Food Sci Nutr. 59(3):506–527.
- Satlin MJ, Cohen N, Ma KC, Gedrimaite Z, Soave R, Askin G, Chen L, Kreiswirth BN, Walsh TJ, Seo SK. 2016. Bacteremia due to carbapenem-resistant Enterobacteriaceae in neutropenic patients with hematologic malignancies. J Infect. 73(4):336–345.
- Sato K, Sakakibara Y. 2015. MetaVelvet-SL: an extension of the Velvet assembler to a de novo metagenomic assembler utilizing supervised learning. DNA Res. 22(1):69–77.
- Scholz M, Ward DV, Pasolli E, Tolio T, Zolfo M, Asnicar F, Truong DT, Tett A, Morrow AL, Segata N. 2016. Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat Methods. 13(5):435–438.
- Sczyrba A, Hofmann P, Belmann P, Koslicki D, Janssen S, Dröge J, Gregor I, Majda S, Fiedler J, Dahms E, et al. 2017. Critical assessment of metagenome interpretation—a benchmark of metagenomics software. Nat Methods. 14(11):1063–1071.
- Sedlar K, Kupkova K, Provaznik I. 2017. Bioinformatics strategies for taxonomy independent binning and visualization of sequences in shotgun metagenomics. Comput Struct Biotechnol J. 15:48–55.
- Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 30(14):2068–2069.
- Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C. 2012. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods. 9(8):811–814.
- Segata N. 2018. On the road to strain-resolved comparative metagenomics. mSystems. 3(2):e00190-17.
- Selhub EM, Logan AC, Bested AC. 2014. Fermented foods, microbiota, and mental health: ancient practice meets nutritional psychiatry. J Physiol Anthropol. 33(1):2.
- Shahbandeh M. 2019. Global functional food market revenue 2019–2025.
- Shakya M, Lo C-C, Chain PSG. 2019. Advances and challenges in metatranscriptomic analysis. Front Genet. 10:904.
- Silva GG, Green KT, Dutilh BE, Edwards RA. 2016. SUPER-FOCUS: a tool for agile functional analysis of shotgun metagenomic data. Bioinformatics. 32(3):354–361.
- Silva GGZ, Cuevas DA, Dutilh BE, Edwards RA. 2014. FOCUS: an alignment-free model to identify organisms in metagenomes using non-negative least squares. PeerJ. 2:e425.
- Slattery C, Cotter PD, O'Toole PW. 2019. Analysis of health benefits conferred by Lactobacillus species from kefir. Nutrients. 11(6):1252.
- Solden L, Lloyd K, Wrighton K. 2016. The bright side of microbial dark matter: lessons learned from the uncultivated majority. Curr Opin Microbiol. 31:217–226.
- Stanke M, Morgenstern B. 2005. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33(Web Server issue):W465–W467.
- Stasiewicz MJ, den Bakker HC, Wiedmann M. 2015. Genomics tools in microbial food safety. Curr Opin Food Sci. 4:105–110.
- Sulaiman J, Gan HM, Yin WF, Chan KG. 2014. Microbial succession and the functional potential during the fermentation of Chinese soy sauce brine. Front Microbiol. 5:556.
- Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, Djahanschiri B, Zeller G, Mende DR, Alberti A, et al. 2015. Structure and function of the global ocean microbiome. Science. 348(6237):1261359.
- Tamames J, Puente-Sánchez F. 2018. SqueezeMeta, a highly portable, fully automatic metagenomic analysis pipeline. Front Microbiol. 9:3349.
- Tatusova T, Ciufo S, Fedorov B, O'Neill K, Tolstoy I. 2014. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 42(Database issue):D553–D559.
- Taylor BC, Lejzerowicz F, Poirel M, Shaffer JP, Jiang L, Aksenov A, Litwin N, Humphrey G, Martino C, Miller-Montgomery S, et al. 2020. Consumption of fermented foods is associated with systematic differences in the gut microbiome and metabolome. mSystems. 5(2):e00901-19.
- Thiele I, Palsson B. 2010. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 5(1):93–121.
- Tian J, Zhu Z, Wu B, Wang L, Liu X. 2013. Bacterial and fungal communities in Pu’er tea samples of different ages. J Food Sci. 78(8):M1249–M1256.
- Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, Segata N. 2015. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 12(10):902–903.
- Truong DT, Tett A, Pasolli E, Huttenhower C, Segata N. 2017. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Research. 27(4):626–638.
- Uritskiy GV, DiRuggiero J, Taylor J. 2018. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 6(1):158.
- Van Rossum T, Ferretti P, Maistrenko OM, Bork P. 2020. Diversity within species: interpreting strains in microbiomes. Nat Rev Microbiol. 18(9):491–506.
- Ventola CL. 2015. The antibiotic resistance crisis: part 1: causes and threats. P T. 40(4):277–283.
- Verce M, De Vuyst L, Weckx S. 2019. Shotgun metagenomics of a water kefir fermentation ecosystem reveals a novel Oenococcus species. Front Microbiol. 10(479):479.
- Wallace JC, Port JA, Smith MN, Faustman EM. 2017. FARME DB: a functional antibiotic resistance element database. Database. 2017:baw165.
- Walsh AM, Crispie F, Daari K, O'Sullivan O, Martin JC, Arthur CT, Claesson MJ, Scott KP, Cotter PD. 2017. Strain-level metagenomic analysis of the fermented dairy beverage nunu highlights potential food safety risks. Appl Environ Microbiol. 83(16):1–13.
- Walsh AM, Crispie F, Kilcawley K, O'Sullivan O, O'Sullivan MG, Claesson MJ, Cotter PD. 2016. Microbial succession and flavor production in the fermented dairy beverage kefir. mSystems. 1(5):e00052-16.
- Walsh AM, Crispie F, O'Sullivan O, Finnegan L, Claesson MJ, Cotter PD. 2018. Species classifier choice is a key consideration when analysing low-complexity food microbiome data. Microbiome. 6(1):50–50.
- Wang Z, Wang Y, Fuhrman JA, Sun F, Zhu S. 2019. Assessment of metagenomic assemblers based on hybrid reads of real and simulated metagenomic sequences. Briefings Bioinf. 21(3):777–790.
- Weinbauer MG, Rassoulzadegan F. 2004. Are viruses driving microbial diversification and diversity? Environ Microbiol. 6(1):1–11.
- Wickham H. 2016. ggplot2: elegant graphics for data analysis. Cham: Springer.
- Wolfe BE, Button JE, Santarelli M, Dutton RJ. 2014. Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell. 158(2):422–433.
- Wood DE, Lu J, Langmead B. 2019. Improved metagenomic analysis with Kraken 2. Genome Biol. 20(1):257.
- Wood DE, Salzberg SL. 2014. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15(3):R46.
- World Health Organization. 2017. The burden of foodborne diseases in the WHO European Region. https://www.euro.who.int/__data/assets/pdf_file/0005/402989/50607-WHO-Food-Safety-publicationV4_Web.pdf
- Wu L-H, Lu Z-M, Zhang X-J, Wang Z-M, Yu Y-J, Shi J-S, Xu Z-H. 2017. Metagenomics reveals flavour metabolic network of cereal vinegar microbiota. Food Microbiol. 62:23–31.
- Wu YW, Simmons BA, Singer SW. 2016. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 32(4):605–607.
- Ye SH, Siddle KJ, Park DJ, Sabeti PC. 2019. Benchmarking metagenomics tools for taxonomic classification. Cell. 178(4):779–794.
- Yu G. 2020. Using ggtree to visualize data on tree-like structures. Curr Protoc Bioinformatics. 69(1):e96.
- Zahn JA, Halter MC. 2018. Surveillance and elimination of bacteriophage contamination in an industrial fermentation process. In: Savva R, editor. Bacteriophages. London: IntechOpen.
- Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. 2012. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 67(11):2640–2644.
- Zelezniak A, Andrejev S, Ponomarova O, Mende DR, Bork P, Patil KR. 2015. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc Natl Acad Sci U S A. 112(20):6449–6454.
- Zepeda Mendoza ML, Sicheritz-Pontén T, Gilbert MT. 2015. Environmental genes and genomes: understanding the differences and challenges in the approaches and software for their analyses. Brief Bioinform. 16(5):745–758.
- Zepeda-Mendoza ML, Edwards NK, Madsen MG, Abel-Kistrup M, Puetz L, Sicheritz-Ponten T, Swiegers JH. 2018. Influence of Oenococcus oeni and Brettanomyces bruxellensis on wine microbial taxonomic and functional potential profiles. American Journal of Enology and Viticulture. 69(4):321–333.
- Zhang C, Hua Q. 2015. Applications of genome-scale metabolic models in biotechnology and systems medicine. Front Physiol. 6:413.
- Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, Yin Y. 2018. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46(W1):W95–W101.
- Zolfo M, Tett A, Jousson O, Donati C, Segata N. 2017. MetaMLST: multi-locus strain-level bacterial typing from metagenomic samples. Nucleic Acids Res. 45(2):e7.
- Zomorrodi AR, Maranas CD. 2012. OptCom: a multi-level optimization framework for the metabolic modeling and analysis of microbial communities. PLoS Comput Biol. 8(2):e1002363.