
Abstract
Associations between per- and polyfluoroalkyl substances (PFASs) and increased blood lipids have been repeatedly observed in humans, but a causal relation has been debated. Rodent studies show reverse effects, i.e. decreased blood cholesterol and triglycerides, occurring however at PFAS serum levels at least 100-fold higher than those in humans. This paper aims to present the main issues regarding the modulation of lipid homeostasis by the two most common PFASs, PFOS and PFOA, with emphasis on the underlying mechanisms relevant for humans. Overall, the apparent contrast between human and animal data may be an artifact of dose, with different molecular pathways coming into play upon exposure to PFASs at very low versus high levels. Altogether, the interpretation of existing rodent data on PFOS/PFOA-induced lipid perturbations with respect to the human situation is complex. From a mechanistic perspective, research on human liver cells shows that PFOS/PFOA activate the PPARα pathway, whereas studies on the involvement of other nuclear receptors, like PXR, are less conclusive. Other data indicate that suppression of the nuclear receptor HNF4α signaling pathway, as well as perturbations of bile acid metabolism and transport might be important cellular events that require further investigation. Future studies with human-relevant test systems would help to obtain more insight into the mechanistic pathways pertinent for humans. These studies shall be designed with a careful consideration of appropriate dosing and toxicokinetics, so as to enable biologically plausible quantitative extrapolations. Such research will increase the understanding of possible perturbed lipid homeostasis related to PFOS/ PFOA exposure and the potential implications for human health.
1. Introduction
Per- and polyfluoroalkyl substances (PFASs) are man-made substances with unique physicochemical properties, such as oil and water repellence, high temperature and chemical resistance, and emulsifying/surfactant properties. Because of these properties, PFASs have been in use since the 1950s for a wide range of industrial and consumer applications, including food contact materials, water-repellent fabrics, waxes, fire-fighting foams, shampoos and cosmetics, as well as insecticides. Several long-chain PFASs, including the well-known perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS), are extremely persistent in the environment and tend to bioaccumulate (OECD Citation2015). Measurable blood concentrations of PFOA and PFOS, and to a lesser degree other PFASs, have been found in populations worldwide (US EPA Citation2016a, Citation2016b; Ballesteros et al. Citation2017; ATSDR Citation2018; EFSA CONTAM Panel Citation2018a, Citation2020). Moreover, this class of substances has been associated with various adverse health effects in humans, including serum lipid perturbations, immunotoxicity, and developmental toxicity (US EPA Citation2016a, Citation2016b; ATSDR Citation2018; EFSA CONTAM Panel Citation2018a, Citation2020).
Despite agreements to phase out the production of certain PFASs by industry, part of the European population is still exposed to levels of PFASsFootnote1 exceeding the tolerable weekly intake (TWI) recently proposed by the EFSA CONTAM Panel (Citation2020), based on effects in humans. Furthermore, alternative PFASs are increasingly being used without sufficient knowledge on their potential hazards and sources of emissions. Thus, PFASs are a public health concern deserving attention from health authorities and policy makers.
One of the human health concerns associated with PFAS exposure is potential perturbation of triglyceride (TG) and cholesterol homeostasis. PFASs, have been repeatedly found to be positively associated with increased blood cholesterol concentrations, and in some cases TGs, in numerous human epidemiological studies. Increased serum cholesterol (total cholesterol of >5.2 mmol/L, i.e. >200 mg/dL) (Leritz et al. Citation2016; Piepoli et al. Citation2016; Ference et al. Citation2017), and in particular its low density lipoprotein (LDL) fraction, is a well-established risk factor for cardiovascular disease (CVD), including ischemic heart disease and ischemic stroke (Piepoli et al. Citation2016; Ference et al. Citation2017; Borén et al. Citation2020). The use of cholesterol-lowering drugs such as statins has been shown to decrease the risk of CVD (Piepoli et al. Citation2016; Ference et al. Citation2017). Moderate hypertriglyceridemia (>1.5 mmol/L) is also considered a CVD risk factor, albeit with a smaller correlation when compared to the correlation between hypercholesterolemia and CVD (Nordestgaard and Varbo Citation2014; Piepoli et al. Citation2016; Sandesara et al. Citation2019). Consequently, even a small increase in serum lipids caused by PFASs can be considered a potential human health hazard.
In contrast to the evidence from human data, rodent studies with PFASs, commonly performed with high doses, have demonstrated decreased serum cholesterol and TG levels, accompanied by increased intrahepatic lipid (mainly TG) concentrations (Seacat et al. Citation2003; Loveless et al. Citation2006; Curran et al. Citation2008; DeWitt et al. Citation2009; NTP Citation2019a, Citation2019b). Next to this, liver toxicity is one of the most frequently reported effects manifested as hypertrophy, steatosis, and in some cases, even necrosis (RIVM Citation2018; NTP Citation2019a, Citation2019b). The divergent results regarding blood lipids between rodents and humans raise debate about the human relevance of rodent data on lipid perturbation, but also about the causality of the human findings on PFAS-associated elevated serum lipids (EFSA CONTAM Panel Citation2018b).
Despite the fact that perturbed lipid homeostasis associated with PFAS exposure has received substantial attention, clear understanding of the mechanisms involved in both animals and humans, is still lacking. This is partly due to distinct species differences, pertaining to the combination of toxicokinetics and toxicodynamics, which have obscured the evaluation of causal pathways and their interpretation in the context of human health. Additionally, many studies focused on peroxisome proliferator-activated receptor alpha (PPARα)-mediated mechanisms, and less attention has been given to other possible mechanisms explaining the observed effects, such as interactions with other transcription factors.
The goal of the present paper is to present the state of the art knowledge on the disturbance of cholesterol and TG homeostasis by PFASs, and to bring forward the most important issues pertaining to this topic. Possible explanations for the findings and discrepancies observed between different lines of evidence are identified, with an emphasis on the underlying mechanisms, especially those that could be relevant for humans. Elucidating the mechanism through which PFASs might induce lipid perturbations would assist in explaining the epidemiological findings, as well as establishing the human relevance of experimental data. For this purpose, this review presents i) a summary of the main findings on PFAS-mediated lipid dysregulation, as recorded in epidemiological and animal studies, ii) an overview of the most important related mechanistic knowledge, as derived from mechanistic rodent studies and in vitro human-relevant test systems, and iii) the importance of PFAS species-specific toxicokinetics. The aim of the work is neither to perform a systematic review nor to evaluate the quality and reliability of all available data, since this has been previously performed (EFSA CONTAM Panel Citation2018a, Citation2020), and hence, information used is mainly derived from studies that are highlighted in existing reviews and reports published by various agencies (RIVM Citation2018; EFSA CONTAM Panel Citation2018a, Citation2020; Pizzurro et al. Citation2019), complemented with some recent scientific publications. The focus is on the two main congeners of the PFAS group, PFOS (, left) and PFOA (, right). Furthermore, this paper provides some recommendations on how to address the identified issues and fill the knowledge gaps, and lays down important factors that require careful consideration when designing new studies. Altogether, this paper aims to contribute to a better understanding of PFAS-mediated lipid perturbations and the issues involved in their interpretation for human health risk assessment.
Figure 1. Chemical structure of PFOS (left) and PFOA (right).
2. PFOS and PFOA: lipid homeostasis perturbations
2.1. Effects observed in human studies
Both, PFOS and PFOA (further referred to as “PFOS/PFOA” and/or PFASs), have been repeatedly found to be positively associated with increased blood cholesterol concentrations in multiple human epidemiological studies (EFSA CONTAM Panel, Citation2018a). A few examples, which are representative for these findings, are shown in . The epidemiological evidence mainly comprises cross-sectional associations between serum PFOS/PFOA and increased levels of cholesterol in blood, with a few examples of longitudinal studies (EFSA CONTAM Panel Citation2018a). Most studies have used general population samples with the “normal” range of PFOS/PFOA concentrations for that country at that time (Nelson et al. Citation2010; Eriksen et al. Citation2013; Geiger et al. Citation2014; Starling et al. Citation2014) and some have used specific populations with occupational exposure (Olsen et al. Citation2003; Sakr et al. Citation2007a; Sakr et al. Citation2007b) or contaminated community drinking water supplies (Steenland et al. Citation2009; Frisbee et al. Citation2010; Canova et al. Citation2020; Li et al. Citation2020). Exposure to the chemicals was in general for several decades. In the majority of these studies, the general pattern observed was a significant increase in the total serum cholesterol or low density lipoprotein cholesterol (LDL-C) associated with increased blood levels of PFOS and/or PFOA, while the results reported for high density lipoprotein cholesterol (HDL-C) were inconsistent. For the general population studies, the magnitude of the increase in total serum cholesterol, based on highest versus lowest quantiles, was around 5% (Steenland et al. Citation2009: PFOS +6.4%, PFOA +5.5%; Nelson et al. Citation2010: PFOS +6.8%; Eriksen et al. Citation2013: PFOS +4.9%, PFOA +5.6%; Li et al. Citation2020: PFOA, PFOS +7–9%), which may correspond to a clinically relevant increase in the risk of CVD (Piepoli et al. Citation2016; Ference et al. Citation2017).
Table 1. Representative human studies reporting associations between serum levels of PFOS and/or PFOA and serum levels of lipids*.
The largest study is on 46 000 adults from the C8 cohort in the mid-Ohio valley, in which residents were exposed for many decades to various PFOA levels through contaminated drinking water and via food, and show a wide range of serum concentrations (Steenland et al. Citation2009). This study showed median blood PFOS and PFOA levels of 20 and 27 ng/mL, respectively. Notably, for PFOA very high blood levels (up to ∼18 000 ng/mL) were observed in part of the population. Much of the increase is observed at low PFOS/PFOA serum levels and seems to level off at higher levels (above about 50 ng/mL), as also shown by the modeling of the data (EFSA CONTAM Panel Citation2018a). Another large population with PFAS exposure from contaminated drinking water, predominantly PFOA, is in the Veneto region of Italy (Canova et al. Citation2020). A cross sectional analysis of PFASs and lipids was carried out in nearly 16 000 people, between 20 and 39 years. The median PFOA serum concentration was 35.8 ng/ml, and the pattern broadly consistent with the C8 study, i.e. increasing cholesterol with PFOA concentration and a steeper slope at lower concentrations. Another recent study on a community, living in a PFAS-polluted area and exhibiting raised serum levels of mainly PFOS (and other PFASs) and to a lesser degree PFOA, also reported positive associations with serum cholesterol (Li et al. Citation2020). In addition to the cross-sectional analyses associating concurrent serum measurements of PFASs and lipids, the authors included an ecological component showing higher cholesterol in the exposed community compared to subjects sampled in a nearby, non-exposed community.
In contrast to the community studies, the reported magnitude of the effect on cholesterol is lower in workers at much higher serum concentrations, e.g. a + 2–3% increase in cholesterol per increase in serum PFOA levels of 1000 ng/mL (Sakr et al. Citation2007b) with exposure for several years and higher serum concentrations of PFOS/PFOA (mean or median levels ≥1000 ng/mL, PFOA: 7–92 300 ng/mL, PFOS: 20–6240 ng/mL) (Olsen et al. Citation2003; Olsen et al. Citation2007; Sakr et al. Citation2007a; Sakr et al. Citation2007b). Olsen and Zobel (Citation2007) re-analyzed the data from 2003 (Olsen et al. Citation2003) and after some exclusions, e.g. people using cholesterol lowering drugs, no longer observed an association between PFOS/PFOA and total cholesterol and LDL-C. Positive associations between increased serum levels of TGs and PFOS and/or PFOA were also recorded in both workers and the general population, but in relatively few studies (Olsen et al. Citation2003; Olsen and Zobel Citation2007; Steenland et al. Citation2009).
Although the associations between serum levels of total cholesterol, LDL-C and TGs and serum levels of PFOS/PFOA have been recorded repeatedly, the causality of these exposure-effect relationships is still an issue requiring further scientific inquiry (EFSA CONTAM Panel Citation2020). In addition, the available evidence for an association between PFOS/PFOA exposure and an associated adverse outcome, i.e. CVD, is missing (EFSA CONTAM Panel Citation2020).
An important limitation of most of the studies, is that they were cross-sectional in design, and so the direction of causality is unknown and may be vulnerable to confounding affecting serum concentrations of both PFOS/PFOA and cholesterol. An example of potential confounding is related to the enterohepatic cycling of PFOS/PFOA and bile acids. PFOS/PFOA have been shown to be excreted to the bile and it was estimated that thereafter, most of the PFOS/PFOA must undergo extensive enterohepatic re-absorption from the gastrointestinal tract to explain the long half-lives in humans (Harada et al. Citation2007; Fujii et al. Citation2015) (see Section 4). In line with this, absorption of PFOS/PFOA was shown to be mediated by the transporters that also participate in absorption of bile acids (Zhao et al. Citation2015). Given that differences in the absorption of bile acids due to genetic factors, such as interindividual variations, food composition or medicines can result in altered levels of serum cholesterol, it is plausible that confounding related to excretion and re-absorption in the enterohepatic cycling process may play a role in the cross-sectional associations observed for PFOS/PFOA and total serum cholesterol (EFSA CONTAM Panel Citation2020).
A few studies had a longitudinal design (see example in ), and as such were subject to a smaller risk of confounding. For example, in a longitudinal study within the C8 cohort, the incidence of the diagnosis of increased serum cholesterol levels was related to the modeled serum PFOA in the population. The exposure model was based on the water concentrations and intake, not individual measurements, and thus was not vulnerable to the confounding described above. A modest, but significant, increase of serum cholesterol levels in relation to modeled PFOA intake was found (Winquist and Steenland Citation2014). The same study assessed CVD in relation to PFOA and did not find an association. A subgroup of subjects in the C8 study participated in a longitudinal follow-up study with repeated blood testing about 4 years after the first survey (Fitz-Simon et al. Citation2013), showing a general decline of serum PFOS/PFOA levels by an average of about 60% reflecting the half-life of approximately 3 years. The mean total cholesterol level did not fall, but was slightly increased, which may be explained by increasing age or change in life-style. When stratifying the group according to the extent of decrease in serum PFOS or PFOA levels, it was shown that those with the highest decrease in PFOS/PFOA showed a relative decrease in serum cholesterol levels, compared to the group with the lowest decrease in PFOS/PFOA. These results also suggest that the effect of PFOS/PFOA on cholesterol levels is reversible. The similarity in the direction of results across different study designs (cross sectional, ecologic and longitudinal) supports a causal role for PFOA in increasing cholesterol. On the other hand, one would expect an exposure-related increase in cardiovascular risk, but there is little evidence for this.
A recently published human study (Convertino et al. Citation2018) does not seem to support the findings regarding increased cholesterol, as observed in a large number of epidemiological studies. This was a clinical phase 1 dose-escalation study with 49 cancer patients, who were administered for 6 weeks very high doses of PFOA, resulting in serum levels of 150 000–230 000 ng/mL (). The authors reported a subsequent dose-dependent reduction in total cholesterol and LDL-C levels in blood. However, this study is probably of little relevance for the general and worker population, since it was conducted in a small population of late-stage cancer patients, whose metabolic activity may differ considerably from healthy individuals. In addition, high doses of PFOA were applied for a limited time period.
In parallel to cholesterol changes, an increased incidence of mildly elevated serum levels of the liver enzyme alanine transferase (ALT) associated with PFOA exposure was recorded (Lin et al. Citation2010; Gallo et al. Citation2012; Darrow et al. Citation2013; Gleason et al. Citation2015; Salihovic et al. Citation2018; Jain and Ducatman Citation2019; Nian et al. Citation2019). Some studies reported similar findings for PFOS (Lin et al. Citation2010; Gallo et al. Citation2012; Salihovic et al. Citation2018). Nevertheless, the magnitude of the associations between serum ALT and PFOA (and PFOS) levels was small (∼3%). In addition, the observed changes in ALT were not accompanied by observable adverse health effects, such as liver damage and metabolic disorders (EFSA CONTAM Panel Citation2018a, Citation2020).
2.2. Effects observed in animal toxicity studies
The interpretation of perturbations in lipid homeostasis observed in human studies becomes more challenging when considering the apparent lack of similar effects in rodent models. In fact, rodent data in general demonstrate opposite findings, i.e. a hypolipidemic effect characterized by decreased levels of serum cholesterol (∼20–40%) and TGs (∼30–80%) after exposure to PFOS/PFOA. Some representative studies are presented in . It should be noted that the purpose of this manuscript is not to perform a comprehensive review; thus, lists only examples of typical studies. In rodents, decreases in serum cholesterol and TGs have been observed after repeated exposure (starting already at exposure durations of 2–4 weeks) and at doses between 0.3 and 10 mg/kg bw/d, which resulted in serum levels of 50 000–500 000 ng/mL (Loveless et al. Citation2006; Curran et al. Citation2008; DeWitt et al. Citation2009; Minata et al. Citation2010; Bijland et al. Citation2011; Yan et al. Citation2015; NTP Citation2019a, Citation2019b; see some information in ). These PFAS serum levels are much higher than those levels associated with increased serum lipids in humans (observed at mean serum concentrations as low as 20–30 ng/mL; ). For PFOS, decreases in serum cholesterol were also reported after longer exposure durations (13–14 weeks) (Seacat et al. Citation2003; Butenhoff et al. Citation2012), whereas for PFOA these endpoints were not examined in longer-term studies (Perkins et al. Citation2004; Butenhoff et al. Citation2012). Very few investigations in animals used PFOS/PFOA doses that were low enough to have given serum concentrations like those seen in humans. At these low exposure levels, serum lipids were not affected by PFOS/PFOA treatment in rodents (Seacat et al. Citation2003; Yan et al. Citation2015; Pouwer et al. Citation2019). Nevertheless, only one of these studies illustrates a dose–response (Pouwer et al. Citation2019), discussed further in Section 3.1.2, whereas only a single dose level was applied in the other two.
Table 2. Example studies in animals reporting on lipid perturbations induced by PFOS/PFOA. Only induced effects are reported in the table.
Most investigations on the effects of PFOS/PFOA have been performed in rats and mice, with a few exceptions, in which monkeys have been used (examples in ). PFOS lowered the serum cholesterol in cynomolgus monkeys after repeated exposure, when administered at doses comparable to those in the high dose rodent studies (serum: 15 000–70 000 ng/mL) (Seacat et al. Citation2002; Chang et al. Citation2017). A 6-month oral PFOA administration in monkeys (serum: 70 000–160 000 ng/mL) produced a mild increase in circulating TGs, whereas blood cholesterol appeared unaffected (Butenhoff et al. Citation2002).
In parallel to the hypolipidemic effects in the blood, other lipid disturbances observed include enhanced intrahepatic accumulation of lipids, mainly TGs, in rodents for both PFOS (Bijland et al. Citation2011; Wan et al. Citation2012; Wang et al. Citation2014), and PFOA (Nakagawa et al. Citation2012; Tan et al. Citation2013; Wang et al. Citation2013; Das et al. Citation2017; Hui et al. Citation2017; Wu et al. Citation2018; Schlezinger et al. Citation2020) (see for examples). The liver appears to be a major target organ for both compounds in rats and mice, as indicated by increased liver weight, hypertrophy of centrilobular hepatocytes, induction of peroxisomal and mitochondrial ß-oxidation, and in some cases necrosis. Liver damage in rodents is also indicated by increased serum transaminases (Curran et al. Citation2008; Son et al. Citation2008; Yu et al. Citation2009; Elcombe et al. Citation2012; NTP Citation2019a, Citation2019b). Similarly, in primates the liver appears to be a target organ for PFOS/PFOA, with effects manifested as increased liver weights with hepatocellular hypertrophy and vacuolation (Butenhoff et al. Citation2002; Seacat et al. Citation2002; Chang et al. Citation2017). It has been speculated that the observed liver damage, like steatosis and necrosis, can be attributed to the alterations in the hepatic lipid metabolism (EFSA CONTAM Panel Citation2020).
2.3. Interpretation of human versus rodent data
Several population studies have repeatedly found correlations between increased blood levels of PFOS/PFOA and elevated blood total cholesterol and LDL-C, (and to a lesser extent TGs). Nevertheless, these findings have not been linked to a corresponding adverse health effect and are inconsistent with toxicological animal studies, where high doses of PFOS/PFOA were found to lower serum cholesterol and TGs, and increase liver lipids. These apparent divergent findings thus present the health risk assessors a conundrum.As noted above, some representative studies on these findings are described in and . For a complete picture of the epidemiological and animal data the reader is referred to the EFSA CONTAM Panel Opinions (Citation2018a, Citation2020).
Considering the large differences in exposure levels between humans and laboratory animals and in order to facilitate the discussion, serum levels of PFOS/PFOA together with externally administered doses are mentioned here, when available. Furthermore, it should be highlighted that not only PFOS/PFOA serum concentrations are of importance in such evaluations, but also the related hepatic concentrations (also reported when available). A relatively higher retention of PFOS/PFOA in the liver in one species compared to another could also play a role in the different outcomes.
Next to the exposure levels, exposure duration may also be divergent, i.e. several decades for humans versus several (2–14) weeks for animalsFootnote2. Consequently, one could argue that in humans, PFOS/PFOA chronic exposure leads to a different lipid response and balance, whereas this is not the case for rats exposed for shorter periods. It cannot be excluded that such differences may also contribute to the differential responses between the two species. It shall be noted here that irrespective of the shorter exposure duration, data indicate that a serum steady-state concentration is also reached in the rat for both compounds (Gomis et al. Citation2018).
Apart from the exposure levels and exposure duration, other reasons are known or suspected to be implicated in the observed differences, including differences in mechanisms underlying the observed effects and in PFAS species-specific toxicokinetics. An understanding of the causal pathway that may lead from chemical exposure to potential adverse outcomes could assist in a better understanding of the epidemiological data. An overview of such mechanisms, which may explain the PFOS/PFOA-mediated lipid disturbances, is presented below. Information discussed stems from mechanistic rodent studies (including genetically modified mice) and in vitro test systems performed with human relevant material, such as human hepatocytes. Next to this, PFOS/PFOA species-specific toxicokinetics issues are presented.
3. Mechanistic pathways involved in PFAS-induced lipid perturbations
3.1. Species differences in lipoprotein homeostasis
3.1.1. General information on lipoprotein circulation
The liver is the primary organ tightly controlling lipid homeostasis, in humans, as well as in other primates and rodents, to ensure a balance between influx, generation, and efflux of lipids. Main functions of the liver with respect to lipid homeostasis include the fatty acid β-oxidation for energy supply, cholesterol biosynthesis and lipogenesis. Circulation of the lipids through the body occurs via specific carrier molecules, i.e. the lipoproteins, also synthetized in the liver (Dietschy et al. Citation1993; Kwiterovich Citation2000) (). Lipoproteins contain a hydrophobic core comprising cholesteryl-esters and TGs, and a amphipathic part, which consists of apolipoproteins and phospholipids (Imes and Austin Citation2013).
Figure 2. Overview of the lipid circulation throughout the human body with their carrier molecules, the lipoproteins. LPL: lipoprotein lipase; LDL: low-density lipoprotein; VLDL: very low-density lipoprotein; TGs: triglycerides; ApoB: apolipoprotein B. Created with BioRender.com.
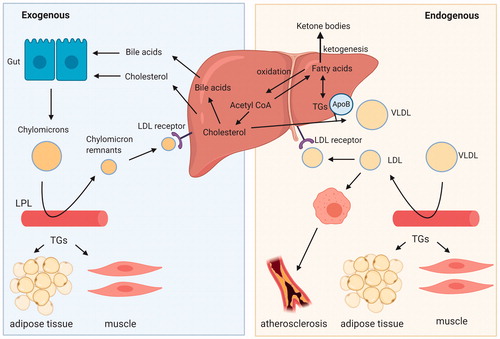
After a meal, the intestine releases chylomicrons, which are mainly composed of TGs and to a lesser extent cholesteryl-esters. Most of the TGs are cleared in the adipose tissue and muscle through the action of lipoprotein lipase. The leftover TGs and the cholesteryl-esters are taken up by the liver as part of chylomicron remnants (). The liver uses the cholesterol to synthesize bile acids, which together with cholesterol are secreted into the bile. During fasting, the liver serves as a sink for circulating adipose tissue-derived free fatty acids, which are either fully oxidized or converted into ketone bodies. In addition, incoming fatty acids are esterified into TGs and stored within lipid droplets or secreted as very low-density lipoproteins (VLDL) for delivery of primarily TGs to the peripheral tissues (Zhang et al. Citation2014). In the blood, VLDL are further metabolized through the removal of the TG portion into LDL, the latter being the main carrier of cholesterol to many tissues including the liver, and taken up via the LDL-receptor (LDLR). On the other hand, the HDL particles participate in the reverse cholesterol transport pathway, i.e. acquiring excess cholesterol effluxed from peripheral tissues and returning it to the liver. The main apolipoprotein in VLDL and LDL particles is apolipoprotein B (apoB) and in HDL apolipoprotein A-I (apoA-I) (Feingold Citation2000; Imes and Austin Citation2013; Marques et al. Citation2018). Disturbances in these metabolic pathways can promote fatty-liver disease and lead to alterations in plasma lipid levels (Adiels et al. Citation2008).
The above described processes comprise some general characteristics of lipid homeostasis that overall are well-conserved across species (Dietschy et al. Citation1993; Bergen and Mersmann Citation2005). Nevertheless, several aspects of lipid homeostasis are known to be specific for humans or rodents. These include differences pertaining to lipoprotein metabolism (Dietschy and Turley Citation2002; Princen et al. Citation2016), which ultimately results in different proportions of the circulating lipoproteins amongst species (Bergen and Mersmann Citation2005; Lee-Rueckert et al. Citation2016; Kaabia et al. Citation2018). Hence, in mice and rats, serum cholesterol is for the major part confined to HDL, while the levels of cholesterol carried by VLDL and LDL are low. In contrast, in humans and non-human primates, the majority of cholesterol is contained in the apoB-containing lipoproteins LDL and to a lesser extent VLDL, thereby resulting in a higher proportion of LDL relative to HDL in the blood (Krause and Princen Citation1998; Princen et al. Citation2016). This occurs due to a faster LDL clearance pathway in rodents compared to humans (Dietschy et al. Citation1993; Dietschy and Turley Citation2002), and the complete absence of cholesteryl ester transfer protein (CETP) in rats and mice. CETP is a central element in lipoprotein metabolism and is responsible for the transfer of cholesteryl-esters from HDL to apoB-containing lipoproteins in exchange for TGs (Chapman et al. Citation2010; Morton and Izem Citation2014; Princen et al. Citation2016). Consequently, the choice of the animal model should be carefully considered.
3.1.2. Species differences in lipoprotein homeostasis in relation to PFAS lipid-related effects
A few studies attempted to clarify the relevance of such species-specific differences for the observed PFOS/PFOA lipid-disturbing effects (Bijland et al. Citation2011; Pouwer et al. Citation2019). For this, the genetically engineered mouse model APOE*3-Leiden.CETP (Westerterp et al. Citation2006) was used, which mimics human lipoprotein metabolism and the response to clinically used hypolipidemic drugs, such as statins, fibrates, niacin and the novel PCSK9-inhibitors (Zadelaar et al. Citation2007; Ason et al. Citation2014; Kühnast et al. Citation2015; Pouwer et al. Citation2020). At the two highest doses tested, both PFOS (86 000–125 000 ng/mL, 4–6 weeks) and PFOA (90 000–150 000 ng/mL, 4–6 weeks) induced hypolipidemia in the blood, which was characterized by decreased levels of TGs (50–70%) and total cholesterol (30–60%) (mainly the non-HDL fraction) (Bijland et al. Citation2011; Pouwer et al. Citation2019). These findings are in line with other studies with PFOS/PFOA conducted in wild-type mice and rats, with similar dose levels and exposure durations. However, in the wild-type animals the decrease in cholesterol is presumed to be mainly due to the HDL fraction. Unfortunately, most of the animal studies did not discriminate between the lipoproteins and mainly measured total cholesterol. Concurrently, PFOS exposure enhanced intrahepatic TG and cholesterol concentrations in APOE*3-Leiden.CETP mice (Bijland et al. Citation2011), while such lipid changes were not seen with PFOA at a similar dose (Pouwer et al. Citation2019). Mechanistic studies revealed that the decreased serum lipid levels occurred through PFOS/PFOA-enhanced (lipoprotein lipase-mediated) VLDL-TG clearance and PFOS/PFOA-decreased hepatic VLDL-TG and apoB production. The observations were further supported by gene expression alterations and pathway analysis confirming the changes in lipoprotein metabolism measured (Bijland et al. Citation2011; Pouwer et al. Citation2019). It should be noted that these effects were only seen at doses and respective serum levels that are several orders of magnitude higher than those relevant in humans (), whereas they were absent at lower, human-relevant environmental or occupational serum levels (50–2000 ng/mL); only PFOA was tested at these low doses (Pouwer et al. Citation2019).
These findings from the studies using the APOE*3-Leiden.CETP model indicate that the known differences in lipoprotein metabolism between humans and rodents, as discussed above, cannot sufficiently explain the observed discrepancy in PFOS/PFOA-induced lipid perturbations. Although the APOE*3-Leiden.CETP mouse has a humanized lipoprotein metabolism, it does not integrate other species differences that possibly play a fundamental role in the respective lipid perturbations (see next sections). On the other hand, the findings observed could be interpreted otherwise, and one could hypothesize that substantial differences in serum PFOA concentrations (at least two or three orders of magnitude) are indeed the main determinant of the interspecies differences reported (the slight reduction in cholesterol reported for cancer patients with very high serum PFOA (Convertino et al. Citation2018) is consistent with this finding. Perhaps at such high serum levels different pathways come into play, both in humans and animals. Accordingly, exposure to PFOA at low doses may not have a significant effect on serum lipid homeostasis, as illustrated by the findings from the APOE*3-Leiden.CETP mouse. The resulting uncertainty regarding the causality of the epidemiological observations and PFOS/PFOA exposure could be reduced by further elucidation of the mechanism(s) involved.
Additionally, when evaluating the different effects of PFOS/PFOA on blood lipids between humans and rodents, it is important to realize that rodent chow contains much less fat and almost no cholesterol when compared to the high-fat Western type diet of humans. For this reason, some studies were performed with rodents fed with a more human-relevant diet (Bijland et al. Citation2011; Wang et al. Citation2014; Rebholz et al. Citation2016; Pouwer et al. Citation2019), in order to delineate whether dietary factors are responsible for the absence of the increased blood lipid effect of PFASs in rodents fed conventionally.
Wang et al. (Citation2014) treated BALB/c mice with PFOS combined with a normal or high fat diet (). Indeed, in the control animals, fed with the high-fat diet alone, a significant increase in blood cholesterol (HDL and LDL), together with an increase in hepatic fat content, was reported (Wang et al. Citation2014). Nevertheless, unlike the controls, the PFOS-treated mice exhibited reduced levels of serum lipids and lipoproteins, independent of the dietary regimen. Administration of a Western-type diet together with PFOS or PFOA was also employed with the aforementioned studies on the APOE*3-Leiden.CETP mice (Bijland et al. Citation2011; Pouwer et al. Citation2019). Similarly, blood cholesterol and TGs were decreased in PFOS or PFOA-treated animals. These results suggest that the dietary fat does not interfere with the PFOS/PFOA-induced lipid perturbations observed in rodents. On the other hand, one single study demonstrated different results, where C57BL/6 mice showed increased blood cholesterol (35% in males, 70% in females), when receiving PFOA together with a cholesterol/lipid-rich diet, in comparison to the animals treated only with the lipid rich diet (Rebholz et al. Citation2016). A less pronounced increase in blood cholesterol (20%) was seen in male BALB/c mice, whereas blood cholesterol remained unaffected in the PFOA-treated BALB/c female mice when compared to control animals being on the high fat diet alone. The increased cholesterol was contained in the (large) HDL fraction, as expected for the rodents. Unfortunately, only one dose level was applied, while a control group fed on standard chow was not included. Overall, no conclusive differences were identified that can fully justify the contrasting lipid disturbances in rodents versus humans upon PFAS exposure; still, it cannot be excluded that diet might play a role, but effects need to be further clarified.
3.2. The role of PPARα in PFOS/PFOA-induced lipid perturbations
3.2.1. General information on the PPARα
The regulation of hepatic lipid and cholesterol metabolism occurs largely at the level of gene transcription by nutrient-sensitive transcription factors, encompassing several nuclear receptors. One of the main nuclear receptors involved in the regulation of hepatic lipid metabolism is PPARα, which is primarily activated by fatty acids and various fatty acid derivatives (Göttlicher et al. Citation1992). The activation of PPARα in rodent and human hepatocytes induces the expression of numerous genes involved in various pathways of lipid metabolism, such as fatty acid storage, β-oxidation, and transport (Kersten Citation2014; Kersten and Stienstra Citation2017). For example, PPARα serves as direct molecular target of fibrate drugs, which are used in the treatment of dyslipidemia and lower blood lipid levels by inducing lipoprotein lipase-mediated VLDL-TG clearance (Schoonjans et al. Citation1996; Chapman et al. Citation2010; Fabbrini et al. Citation2010; Kim and Kim Citation2020).
3.2.2. Lipid homeostasis and activation of PPARα by PFOS/PFOA in rodents
PFOS/PFOA structurally resemble fatty acids and are well-established ligands of PPARα in the rat and mouse liver (Perkins et al. Citation2004; Wolf et al. Citation2008; Rosen et al. Citation2010; Elcombe et al. Citation2012; Wolf et al. Citation2012, Citation2014; Rosen et al. Citation2017). Consequently, activation of the PPARα signaling pathway upon exposure to PFOS or PFOA is believed to be, at least partly, responsible for the observed perturbations of lipid homeostasis in animals (DWQI Citation2017, Citation2018; EFSA CONTAM Panel Citation2018a). In fact, gene expression studies conducted on liver samples from PFOS/PFOA-exposed rodents revealed that a substantial proportion of the up- or down-regulated genes (e.g. Cyp4a1, Acox1) are under the control of the PPARα receptor (Rosen, Abbott, et al. Citation2008; Ren et al. Citation2009; Rosen et al. Citation2010, Citation2017; Pouwer et al. Citation2019). In terms of the PPARα activation potency, PFOA was shown to be more potent when compared to PFOS, both in reporter gene assays and in gene expression studies with rat hepatocytes. (Takacs and Abbott Citation2007; Wolf et al. Citation2008; Bjork and Wallace Citation2009; Wolf et al. Citation2012). Also, in the recent NTP (Citation2019a, Citation2019b) studies in male rats, PFOA appeared to be a more potent inducer of Acox1 and Cyp4a1 gene expression in livers than PFOS, despite a lower accumulation in liver.
As prototypical PPARα agonists, PFOS/PFOA induce the mitochondrial and peroxisomal ß-oxidation of fatty acids for their degradation to acyl-CoA-moieties in the rodent liver (Rosen et al. Citation2010; Bijland et al. Citation2011; Wan et al. Citation2012; Wang et al. Citation2014; Pouwer et al. Citation2019). Furthermore, they induce the fatty acid transport across the mitochondrial membrane (Rosen et al. Citation2010; Bijland et al. Citation2011; Wang et al. Citation2014; Pouwer et al. Citation2019). In parallel, they decrease the hepatic VLDL-TG and apoB production, disturbing as such the hepatic secretion of TGs (and indirectly cholesterol) into the blood. Furthermore, they promote lipoprotein lipase-mediated lipolysis of TG-rich plasma lipoproteins (Bijland et al. Citation2011; Pouwer et al. Citation2019). These processes appear to contribute to the lowered blood TG levels but also to the enhanced hepatic TG concentrations in PFOS/PFOA-treated rodents.
PPARα is also known to play a role in cholesterol homeostasis, including inhibition of cholesterol and bile acid synthesis in mice and man) (Post et al. Citation2001; Li and Chiang Citation2009), regulation of HDL metabolism and promotion of reverse cholesterol transport (Li and Glass Citation2004; Ory Citation2004; Li and Chiang Citation2009). Nevertheless, the role of PPARα in the PFAS-induced changes on blood and liver cholesterol in rodents is still elusive. This is further discussed in Section 3.4.
Although the general view remains that the PPARα plays a pivotal role in PFOS/PFOA-induced lipid disturbances in rats and mice (ATSDR Citation2018; EFSA CONTAM Panel Citation2018a), some evidence suggests its role is of less importance. Actually, the effects observed upon PFOS/PFOA exposure are not per se consistent with effects of other well-studied PPARα activators, such as fibrates and Wyeth (WY)-14643. For example, typical PPARα activators commonly do not cause liver steatosis in rodents at comparable doses and exposure durations (Larter et al. Citation2012; Pawlak et al. Citation2015), contrary to what is seen after exposure to PFOS/PFOA.
Some information from PPARα-null mice studies further support the notion for the involvement of PPARα-independent pathways in the PFOS/PFOA-exerted lipid disturbances. However, it is important to emphasize that knocking out the receptor itself in mice affects lipid metabolism, leading to steatosis in the liver of control PPARα-null mice (Howroyd et al. Citation2004; Corton et al. Citation2014; Das et al. Citation2017), which might interfere with the interpretation of the results obtained for PFOS/PFOA treated PPARα-null mice. Still, it has been shown that PPARα-null mice exhibit hepatic lipid accumulation and/or alterations in genes linked to lipid metabolism upon exposure to PFOA (Rosen, Abbott, et al. Citation2008; Rosen, Lee, et al. Citation2008; Minata et al. Citation2010; Nakagawa et al. Citation2012; Das et al. Citation2017) or PFOS (Rosen et al. Citation2010), which counterargues that these effects should be attributed to PPARα activation. Nakagawa et al. (Citation2012), for example, demonstrated that the liver steatosis in the PFOA-exposed PPARα-null mice is more prominent when compared to the WT mice (1 and 5 mg/kg bw/d, 6 weeks). In that study, control PPARα-null mice showed only a slight and not statistically significant increase in hepatic TG accumulation, contrary to what is commonly seen with such knock-out animals (Howroyd et al. Citation2004; Corton et al. Citation2014; Das et al. Citation2017). Overall, data on PFOA-exposed PPARα-null mice seem to corroborate the contribution of other PPARα-independent signaling pathways in the lipid disturbances induced by PFOS/PFOA in rodents.
3.2.3. Are PPARα-mediated effects in rodents relevant for human health?
The importance of the PPARα receptor in human liver has been questioned in the past, due to the perceived low expression of PPARα in humans and minimal responsiveness of human liver cell lines to PPARα activation (Tugwood et al. Citation1996; Auboeuf et al. Citation1997; Palmer et al. Citation1998). Accordingly, the potential human relevance of the PFOS/PFOA-induced lipid perturbations seen in rodents, and, at least partially, driven by activation of the PPARα pathway, has been subject to debate (DWQI Citation2017, Citation2018; EFSA CONTAM Panel Citation2018a). However, later research indicates that the quantitative expression of PPARα is similar in human and mouse liver (Kersten and Stienstra Citation2017) and that in human hepatocytes and liver slices, PPARα is able to effectively induce the expression of genes involved in numerous lipid metabolic pathways. Still, remains to a lesser extent compared to mouse or rat hepatocytes and mouse liver slices (Okyere et al. Citation2014; Corton et al. Citation2014; Janssen et al. Citation2015; Liss and Finck Citation2017; Heusinkveld et al. Citation2018). Indeed, studies using chimeric mice, harboring murine as well as human hepatocytes in the liver, underscore the more modest PPARα-mediated gene trans-activation in human hepatocytes compared to their murine counterparts (de la Rosa Rodriguez et al. Citation2018). Apart from these quantitative interspecies differences, qualitative differences have also been illustrated recently, after comparisons of PPARα signaling transcriptional networks in primary human hepatocytes and rats (McMullen et al. Citation2020). Such differences could in principle result in differential responses in humans and rats when exposed to PPARα-ligands.
With respect to the activation of the human PPARα (hPPARα) by PFOS/PFOA, studies with hPPARα expressing mice suggest a lower response to PFOA, when compared to their WT counterpart. This is seen by lower increase in transcripts and protein levels of PPARα target genes (Nakamura et al. Citation2009; Nakagawa et al. Citation2012). Still, in combination with these gene expression changes, PFOA-treated hPPARα mice showed increased lipid accumulation in liver (Nakagawa et al. Citation2012; Schlezinger et al. Citation2020). Actually, despite the reduced responsiveness of hPPARα to PFOA, hPPARα mice appeared to be substantially more susceptible to liver steatosis than the WT mice, as shown by larger increases in hepatic TG levels (Nakagawa et al. Citation2012). This further supports that the PFOA-induced liver steatosis, specifically the increase in TG levels, might be driven by PPARα-independent pathways. With respect to cholesterol, blood levels remained unaffected by the treatment in hPPARα mice, contrary to the WT mice that showed the typical decrease, when exposed to PFOA. Similar studies with PFOS have not been identified in the literature.
The activation of the hPPARα by PFOA, but also PFOS, was likewise seen with in vitro assays performed in human liver cells, such as human primary hepatocytes, or human liver cell lines (HepG2 and HepaRG) (Bjork et al. Citation2011; Beggs et al. Citation2016; Behr et al. Citation2020; Louisse et al. Citation2020). These studies support the activation of PPARα signaling, at concentrations commonly ranging from 10 µM to 100 µM (PFOA: ∼4000–40 000 ng/mL, PFOS: ∼5000–50 000 ng/mL). These concentrations are high when compared directly to the serum levels recorded even at the highly exposed populations or at workers in occupational settings. However, in one study, gene expression network analysis showed a simulation of the PPARα signaling already at a concentration of 1 µM for PFOA (Buhrke et al. Citation2015). As seen in vivo for PFOA, in vitro studies comparing responses upon PFOS/PFOA exposure between rodent and human primary hepatocytes support the view that induction of PPARα transcriptional responses are more pronounced in rodent than in human hepatocytes (Bjork and Wallace Citation2009; Bjork et al. Citation2011).
As already stressed for rodents, some in vitro studies have demonstrated differences with respect to the hPPARα activation potency between PFOS and PFOA. Again, PFOA seems a more potent activator of the hPPARα than PFOS in reporter gene assays (Takacs and Abbott Citation2007; Wolf et al. Citation2008; Citation2012), but also in gene expression studies with human hepatocytes (Bjork et al. Citation2011; Buhrke et al. Citation2015; Louisse et al. Citation2020). These differences can be also related to the differences in cellular uptake. For example, cellular uptake of the PFASs in HepG2 cells was shown to be low for PFOA (0.24%), but 10-fold lower for PFOS (0.04%) (at a concentration of 10 µM, 10% serum), with absolute cellular concentrations of 39 and 4 nmol/mg protein for PFOA and PFOS, respectively (Rosenmai et al. Citation2018). In that study, PFOA induced PPARα-mediated reporter gene expression at relatively high concentrations (30 and 100 µM) whereas PFOS did not induce PPARα-mediated reporter gene expression, possibly reflecting the differences in cellular uptake, but perhaps also in PPARα affinity. However, preliminary data on human HepaRG cells (own unpublished data) indicate the reverse, i.e. PFOS accumulating more in the cells than PFOA. To our knowledge, data on cellular uptake of PFASs are currently very limited. It should be emphasized here that overall the lack of information on this aspect is an important limitation of these in vitro data. Such measurements would in principle assist in more appropriate comparisons on actual exposure levels, since the nominal concentrations applied in the in vitro systems might not be a good proxy for serum levels. As such it is difficult to assess at this state whether the effective concentrations in vitro are relevant for human exposure.
3.2.4. Conclusions
The role of PPARα activation by PFOA in the observed lipid perturbations in rodents, but also its relevance for human health, has been extensively studied, including examinations in hPPARα and PPARα-null mice, and in rodent and human hepatocytes. For PFOS less data are available. Overall, PFOA and to a lesser extent PFOS activate PPARα, both its murine and human version, and the observed lipid alterations may depend to a certain extent on the PPARα-signaling pathway. Effects on PPARα-null mice indicate, however, the involvement of other pathways. With respect to the human situation, the large differences in exposure scenarios when compared to rodents, combined with the reduced hPPARα responsiveness to PFOS/PFOA, warrant the need for careful consideration when comparing rodent and human findings. It cannot be excluded that in humans higher exposure is required for the manifestation of the effects on lipid metabolism, but it should be stressed that certain PFASs accumulate to a higher extent in humans than in rats and mice (see Section 4 on toxicokinetics). As such, due to the life-long exposure of humans to such substances, along with high exposure rates, a certain critical body burden necessary to affect lipid homeostasis by this pathway might be achieved.
3.3. Other nuclear receptors potentially involved in PFOS/PFOA-mediated lipid disturbances
3.3.1. PXR, CAR and other signaling pathways
As discussed above, it is suggested that PPARα-independent signaling pathways are also involved in the lipid disturbances induced by PFOS/PFOA. In particular the transactivation of other nuclear receptors by PFOS/PFOA, such as PPARγ, constitutive androstane receptor (CAR), pregnane X receptor (PXR), liver X receptor (LXR) and farsenoid X receptor (FXR), have been studied in rats and mice. It has been suggested that the nuclear receptors PPARγ (Rosen, Lee, et al. Citation2008), CAR (Rosen, Lee, et al. Citation2008; Ren et al. Citation2009; Abe et al. Citation2017; Schlezinger et al. Citation2020) and PXR (Ren et al. Citation2009; Bjork et al. Citation2011; Pouwer et al. Citation2019), are also activated by PFOS/PFOA in the murine liver. These receptors are in general associated with cholesterol and TG homeostasis (Ory Citation2004; Yin et al. Citation2011; Yan et al. Citation2015), implying that they might also play a role in the effects induced by PFOS/PFOA. Considering that gene expression is rarely dependent on a sole transcription factor, and that cross-talk between various transcription factors is known to occur, PFOS/PFOA effects in rodents are probably a result of multiple inter-linked pathways.
In vitro studies with human relevant material reported somewhat contradicting results, which could also be the outcome of variable experimental designs, i.e. different concentrations, exposure durations, cell systems etc. In human primary hepatocytes (Bjork et al. Citation2011), in HepaRG (Abe et al. Citation2017) and in HepG2 cells (Zhang et al. Citation2017), multiple nuclear receptors (CAR, PXR, LXR) were activated by PFOS and PFOA, as illustrated by increased expression in some selected marker genes. Yet, other gene expression studies in human hepatocytes and/or reporter gene assays have shown that PFOS/PFOA may activate to a very limited (if any) extent all these receptors, including PPARγ and FXR (Vanden Heuvel et al. Citation2006; Buhrke et al. Citation2015; Behr et al. Citation2020; Louisse et al. Citation2020). Louisse et al. (Citation2020) compared the effects of PFOS/PFOA on gene expression in HepaRG cells with the effects of a known LXR-agonist and a FXR-agonist (data from Wigger et al. Citation2019), suggesting that PFOS/PFOA do not activate these receptors.
3.3.2. Disruption of HNF4a signaling pathway by PFOS/PFOA
Amongst the other nuclear receptors, of particular interest is the hepatocyte nuclear factor HNF4α, that seems to be affected by PFOS/PFOA (Yan et al. Citation2015; Beggs et al. Citation2016; Pouwer et al. Citation2019). HNF4α is considered a master regulator of liver-specific gene expression and essential for liver development and liver function, including lipid homeostasis (Hayhurst et al. Citation2001; Yin et al. Citation2011; Yeh et al. Citation2019). Dysregulation of HNF4α function has been associated with a large number of human diseases, including nonalcoholic fatty liver disease (Yeh et al. Citation2019). There is cross-talk between HNF4α and other nuclear receptors like PPARα, for which both antagonism and synergism have been reported (Chamouton and Latruffe Citation2012; Lu Citation2016). There is also evidence for inhibitory cross-talk between PXR and HNF4α as well as CAR and HNF4α in hepatic lipid metabolism. While HNF4α is a transcriptional activator of CYP7A1, the rate-limiting enzyme in bile acid biosynthesis, PPARα and PXR inhibit CYP7A1 expression, probably by competing with HNF4α for a common transcriptional coactivator (Li and Chiang Citation2005; Miao et al. Citation2006). Similarly, CAR downregulates HNF4α target genes (Miao et al. Citation2006). Repression of CYP7A1 results in decreased transformation of cholesterol into bile acids (see Section 3.4.2), leading to lipid accumulation in the liver and increased LDL-C levels in humans (Laskar et al. Citation2017).
In studies with mice, PFOS and PFOA exerted reduction in the HNF4α protein expression (10 and 3 mg/kg bw/d, respectively), after a short, i.e. 7-day exposure, while HNF4α mRNA levels were not affected (Beggs et al. Citation2016). Upon a longer exposure to PFOA (1.25 and 5 mg/kg bw/d, 28 days), HNF4α mRNA levels were slightly decreased; still, this reduction of the transcription factor was not reflected in representative target genes (Yan et al. Citation2015). In the humanized APOE*3-Leiden.CETP mouse, the expression of HNF4α mRNA was mildly increased after treatment with a similar dose, i.e. 3.2 mg/kg bw/d (serum levels ∼90 000 ng/mL at 4 weeks), whereas in silico prediction of transcription factor activity based on the expression changes of known target genes was decreased (Pouwer et al. Citation2019).
Data from in vitro assays with human cells exposed to PFOS/PFOA also point toward a downregulation of the HNF4α pathway. A proteomic study with human HepG2 cells (Scharmach et al. Citation2012) showed inhibition of HNF4α signaling upon exposure to 25 µM of PFOA (10 000 ng/mL). Such effects were also seen in primary human hepatocytes after a 96-h treatment with PFOS or PFOA (Beggs et al. Citation2016), with protein levels of HNF4α (but not mRNA levels) decreasing at the highest concentration tested (10 µM; ∼4000 ng/mL). PFOA-induced inhibition of HNF4α in primary human hepatocytes was also observed in another study, albeit at higher concentrations (25 and 100 µM; 10 000–42 000 ng/mL) (Buhrke et al. Citation2015). In human HepaRG cells, Behr et al. (Citation2020) reported a downregulation of HNF4α gene expression at concentrations of 50 µM and above after a 24- or 48-h exposure. In another HepaRG study, HNF4A was not significantly downregulated by 100 µM PFOS/PFOA, but expression of CYP7A1 was decreased (Louisse et al. Citation2020).
3.3.3. Conclusions
In conclusion, there is evidence indicating the involvement of other nuclear receptors important in lipid homeostasis, such as PXR, in the PFOS/PFOA-induced lipid dysregulation in rodents. With respect to human liver cells, such data are limited and hence, their relevance for the potential induced lipid perturbations by PFOS/PFOA in humans, is not clear. Regarding the HNF4α pathway there are some indications that it might be involved in potential effects of PFOS/PFOA on cholesterol and lipid homeostasis. However, this evidence is not so strong and more investigations are required to potentially support this mechanism. In addition, it remains unclear whether in reality PFOS/PFOA exposures result in serum levels at which suppression of the HNF4α pathway is likely to occur. More in vitro studies on primary human hepatocytes and liver cell lines would help elucidate this further.
3.4. Mechanisms linked to disturbance of cholesterol homeostasis
3.4.1. Cholesterol biosynthetic pathway and hepatic uptake
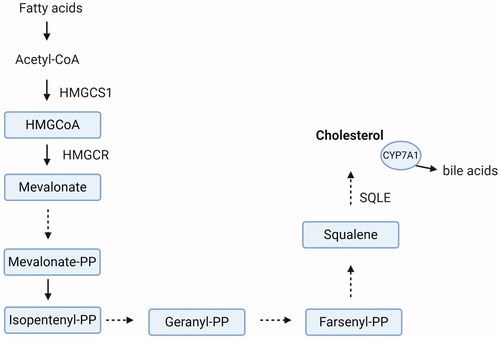
Regarding PFOS/PFOA-induced changes in cholesterol observed in humans, it is of interest to also consider a possible direct effect on the intrahepatic cholesterol biosynthetic pathway, and/or perturbation on its import to/export from the liver. In the liver, regulation of cholesterol levels is achieved through a negative feedback mechanism, in which hepatic cholesterol accumulation suppresses its de novo synthesis, and concurrently, the liver’s uptake of cholesterol from the blood (Brown and Goldstein Citation1997; Feingold Citation2000; DeBose-Boyd Citation2008). The expression of genes that are involved in de novo cholesterol synthesis, but also uptake, is under control of the hepatic transcription factor sterol regulatory element-binding proteins (SREBP) (Horton et al. Citation2002; Jeon and Osborne Citation2012; Shao and Espenshade Citation2012). Amongst these genes are the HMGCR (3-hydroxy-3-methyl-glutaryl (HMG)-coenzyme A reductase), encoding the rate-limiting enzyme of the cholesterol biosynthetic pathway (converts HMG-CoA to mevalonate; ), as well as the gene encoding the LDL receptor (LDLR). The LDLR is the main receptor involved in cholesterol uptake from the blood to the liver via endocytosis, and its activity regulates the plasma levels of cholesterol (Brown and Goldstein Citation1997). SREBP stimulates in parallel the hepatic cholesterol synthesis and clearance from the blood and thus, the balance between these two processes determines ultimately the levels of cholesterol in the liver and serum circulation (Brown and Goldstein Citation1997; Horton et al. Citation2002). Cholesterol export from the liver into the circulation occurs via the VLDL particles, which are metabolized into LDL-C in the blood.
Figure 3. The principal steps of the hepatic cholesterol biosynthetic pathway. HMGCS1: HMG-CoA synthase 1; HMGCR: HMG-CoA reductase. HMGCS1 catalyzes the condensation of acetyl-CoA with acetoacetyl-CoA to HMG-CoA. In a following step HMG-CoA is converted by HMGCR to mevalonate. Subsequently, several enzymatic reactions are required for the synthesis of cholesterol. SQLE: Squalene epoxidase. SQLE catalyzes the first oxygenation step in sterol biosynthesis. CYP7A1: cholesterol 7-alpha hydroxylase. CYP7A1 catalyzes the transformation of excess cholesterol into bile acids. Created with BioRender.com.
Exposure to high PFOS/PFOA doses has been demonstrated to decrease the hepatic VLDL-TG and apoB production in the liver of the APOE*3-Leiden.CETP mouse concomitantly with enhanced lipoprotein lipase-mediated VLDL clearance (Bijland et al. Citation2011; Pouwer et al. Citation2019), resulting in decreased cholesterol and TG levels in serum. In these studies with the APOE*3-Leiden.CETP mouse, hepatic accumulation of cholesterol and TGs was only seen upon PFOS exposure and not with PFOA (see Sections 3.1.2).
There are some reports on the effect of PFOS/PFOA on intrahepatic cholesterol synthesis, as well as on hepatic cholesterol uptake from the bloodstream. In murine liver, PFOA and PFOS were shown to enhance SREBP activity, as indicated by an increased expression on both the transcriptional and protein level. In parallel, a significant upregulation of relevant target genes, such as HMGCR and LDLR, was detected (Rosen et al. Citation2010: 10 mg/kg bw/d, 7 days; Yan et al. Citation2015: 1.25–20 mg/kg bw/d, 4 weeks). However, histopathological examinations did not reveal hepatic lipid accumulation in the case of PFOS (Rosen et al. Citation2010), while total hepatic cholesterol levels were reduced after PFOA exposure (Yan et al. Citation2015). In contrast to the two aforementioned studies, others reported reduced expression of certain SREBP target genes, accompanied by elevated intrahepatic cholesterol levels. In rats, both PFOS/PFOA lowered hepatic cholesterol synthesis, as reflected by a reduced activity of liver HMGCR enzyme (Haughom and Spydevold Citation1992) or mRNA levels (Guruge et al. Citation2006: 5 mg/kg bw/d, 3 weeks). Similarly, the expression of HMGCR and LDLR was decreased by PFOA in both WT and hPPARα mice (Schlezinger et al. Citation2020: 0.7 mg/kg b/d, 6 weeks). Despite the lowered expression of the biosynthetic genes, exposures to PFOS or PFOA led to a pronounced hepatic cholesterol accumulation. These data imply that increased intrahepatic cholesterol, as seen in rodents after exposure to PFOS/PFOA, might not be directly related to de novo cholesterol biosynthesis. Instead, the effects could be the consequence of other impaired pathways, such as the secretion as VLDL particles (Section 3.1.2) and/or cholesterol metabolism into bile acids (Section 3.4.2).
In vitro results with human cells pertaining to affected genes involved in cholesterol synthesis and PFOS/PFOA seem to be somewhat inconsistent. Using a human fetal liver cell line (L-02), Peng et al. (Citation2013) combined a gene expression and metabolomics analysis, and reported an effect of PFOA on cholesterol biosynthesis. Measurement of cholesterol suggested a concentration-dependent increase in intracellular levels (significant at the high concentration: 120 µM, 72 h). In addition, several cholesterol biosynthesis genes were upregulated at the same concentration. Opposing to these findings, Behr et al. (Citation2020) reported a downregulation of such genes (e.g. HMGCR, SQLE, and LDLR) and the transcription factor SREBP in HepaRG cells after 24 or 48 h, at concentrations ≥10 and 25 µM for PFOA and PFOS, respectively. Intracellular cholesterol levels were not affected. Similar results were obtained for PFOA and PFOS in a recent transcriptomics study with HepaRG cells (Louisse et al. Citation2020; 100 µM, 24 h), showing a downregulation of gene sets related to cholesterol biosynthesis and SREBP signaling. In primary human hepatocytes, PFOA induced a concurrent upregulation (e.g. MVK: mevalonate kinase, PMVK: phosphomevalonate kinase) and downregulation (e.g. SQLE, FDFT1: farnesyl-diphosphate farnesyltransferase 1) of few cholesterol biosynthesis genes, whereas most remained unaffected (Buhrke et al. Citation2015; 100 µM, 24 h).
3.4.2. Intrahepatic disturbances in the enterohepatic cycle and bile acid formation
Another possible explanation for the PFOS/PFOA-induced changes in blood and liver cholesterol is perturbation of bile acid synthesis from cholesterol. Excess cholesterol in the liver is stored, exported or converted into bile acids; the predominant pathway in human liver is the classic bile acid synthesis pathway, which is initiated by the rate-limiting enzyme cholesterol 7-alpha hydroxylase (CYP7A1) (Princen et al. Citation1997; Chiang Citation2017). The enzyme’s gene expression and the bile acid synthesis rate are inhibited by bile acids, which return to the liver through the enterohepatic circulation (Thompson Citation1996; Chiang Citation1998; Li and Chiang Citation2009). Hence, an elevated hepatic re-uptake of bile acids induces a negative feedback loop via the farnesoid X receptor (FXR) to lower the de novo synthesis of bile acids from cholesterol, by CYP7A1 inhibition. Alterations in serum bile acid levels suggest either a direct disruption of the bile acid flow or/and a disturbance of the intrahepatic bile acid synthesis from cholesterol (Thompson Citation1996).
In rats, PFOS/PFOA have been shown to increase the levels of serum bile acids (PFOS at 2.5 and PFOA at 5 mg/kg bw/d) after a 28-day exposure (NTP Citation2019a, Citation2019b). In APOE*3-Leiden.CETP mice, PFOS, inhibited bile acid excretion in the feces (Bijland et al. Citation2011: 3 mg/kg bw/d, 4–6 weeks). In addition, downregulation of hepatic CYP7A1 gene expression upon exposure to PFOS (Bijland et al. Citation2011; Wang et al. Citation2014: 5 mg/kg bw/d, 2 weeks) or PFOA (Pouwer et al. Citation2019; Schlezinger et al. Citation2020) was seen. These findings show impairment of the bile flow and synthesis, through which PFOS/PFOA may affect cholesterol homeostasis. It should be noted that next to the FXR the main transcription factors regulating CYP7A1 include HNF4α and PPARα (Chen et al. Citation2001; Kir et al. Citation2012), which have already been suggested as molecular target of PFOS and PFOA (see Sections Section 3.2 and Section 3.3).
Reduction of CYP7A1 expression has been also demonstrated in vitro, in human hepatocytes (Beggs et al. Citation2016: 10 µM, 96 h) and in HepaRG cells (Behr et al. Citation2020: 10 µM, 48 h; Louisse et al. Citation2020: 100 µM, 24 h).
Interference of PFOS/PFOA with the enterohepatic cycling may also play a role. PFOS/PFOA have been shown to be excreted in the bile (Harada et al. Citation2007; Fujii et al. Citation2015), and thereafter, are believed to be substantially re-absorbed from the gastrointestinal tract (see Section 4). Both substances have been reported to share the same transporters (e.g. NTCP: Na+/taurocholate co-transporting polypeptide, ASBT: apical sodium-dependent bile salt transporter, OATPs: organic anion transporting polypeptides) as bile acids for excretion via bile into the intestine and re-absorption in the ileum (Zhao et al. Citation2015, Citation2017). Therefore, PFOS/PFOA may alter the absorption of bile acids through competition for the same transporter, interfering as such with the negative feedback control of the conversion of cholesterol to bile acids and perturbing cholesterol levels. For example, losses of bile acids are compensated by enhanced bile acid synthesis from cholesterol, the mechanism behind the cholesterol lowering effect of the drug cholestyramine. This resin binds bile acids in the gastrointestinal lumen to prevent reabsorption and indirectly lowers serum cholesterol levels via enhanced conversion of cholesterol to bile acids, which in turn leads to activation of SREBP-mediated LDLR expression. Interestingly, in rats application of cholestyramine also strongly increased the excretion of PFOA via feces (Genuis et al. Citation2010, Citation2013).
With respect to the enterohepatic circulation in humans, differences in the absorption of bile acids due to genetic factors, food composition or medicines can lead into altered levels of serum cholesterol. For example, dietary fiber intake was recently reported to be associated with lower PFAS serum concentrations in humans (Dzierlenga et al. Citation2020). Consequently, it is plausible that confounding related to excretion and re-absorption in the enterohepatic cycling process may play a role in the associations for PFOS/PFOA and total serum cholesterol reported repeatedly in the cross-sectional epidemiological studies. Nevertheless, confounding due to this biological mechanism is till now only a postulation with no available supporting evidence (see Section 2.1) (EFSA CONTAM Panel Citation2020).
3.4.3. Conclusions
Collectively, there are indications that PFOS/PFOA influence different aspects of cholesterol metabolism, including biosynthesis, import/export from the liver and conversion into bile acids. Despite this, the molecular events leading to the alterations in serum cholesterol that may be caused by PFOS/PFOA exposure in animals and humans remain unclear. More insight into the mechanisms involved is needed in order to understand the molecular events that are potentially triggered by PFOS/PFOA and how these may ultimately lead to cholesterol alterations in blood and/or liver. Additionally, it should be noted once more that for the interpretation of such findings the exposure levels should be taken into consideration, which are different between animals and humans. For interpretation of data from in vitro assays difference in the free fraction between the in vitro assay and the in vivo situation has to be considered. Currently, this is hindered by lack of data on the free fraction of PFOS/PFOA in the medium of different in vitro studies and/or the related cellular concentrations in in vitro systems.
4 Species differences in toxicokinetic properties of PFOS and PFOA
Apart from the toxicodynamic differences analyzed above, toxicokinetic differences have been reported for the PFASs, with most data on PFOS and PFOA. In general, both chemicals are well absorbed from the intestinal tract and are excreted unmetabolized (US EPA Citation2016a, Citation2016b; ATSDR Citation2018; EFSA CONTAM Panel Citation2018a). Once absorbed, PFOS/PFOA bind extensively to serum albumin (>90%), as shown in several species (Ehresman et al. Citation2007; Han et al. Citation2012; Beesoon and Martin Citation2015), while also binding to the liver fatty acid binding protein (L-FABP) has also been reported for the rat and human (Luebker et al. Citation2002; Woodcroft et al. Citation2010).
With regard to organ distribution it is often mentioned that PFOS/PFOA accumulate in the liver and kidney (EFSA CONTAM Panel Citation2018a). However, as shown recently, PFOA does not deposit preferentially in the liver (NTP Citation2019b), based on the average liver:plasma partition coefficient (PC) in male rats after a 28-day exposureFootnote3 (range across doses: 0.87–1.17). This is somewhat lower than what has been previously shown in other studies in the male rat with a single PFOA exposure (showing PCs of for example 2.2 and 0.8 at a low and high dose (Kudo et al. Citation2007) and 2.3 (Kim et al. Citation2016). The NTP finding is important considering the repeated exposure, which is not commonly applied in toxicokinetic studies. For PFOS current evidence indicates a higher retention in the liver (Seacat et al. Citation2003; Curran et al. Citation2008; NTP Citation2019a). In this case, the liver: plasma PCs obtained for example from a 14-week exposure (Seacat et al. Citation2003) are in the range of 6.3–12.2 across doses, but lower after shorter (NTP Citation2019a: 4 weeks, range across doses: 2.74–3.76) and single (Kim et al. Citation2016; mean: 2.6) exposures. With regard to the kidney, neither of the two substances show accumulation in the rat, with kidney: plasma PCs of ∼0.4–1 for PFOA (Kudo et al. Citation2007; Kim et al. Citation2016; Dzierlenga et al. Citation2020) and ∼0.3–1 for PFOS (Kim et al. Citation2016; Huang et al. Citation2019).
Human data are unfortunately very limited in number (Olsen et al. Citation2003; Ericson et al. Citation2007; Perez et al. Citation2013). In order to facilitate a preliminary comparison with the rat data, human organ:plasma PCs were calculated (= 20) based on the available information. It should be mentioned though that for these calculations data of PFOS/PFOA levels in the tissues and blood plasma do not stem from the same study (see , furthermore note the high variability of organ measurements), i.e. they come from different persons; they are, however, from the same region. The results suggest a substantial higher distribution to the liver for both PFOA and PFOS in a substantial part of the human population, probably reflecting the long human exposure period. Calculated kidney:plasma PCs, in humans versus rats, are comparable for PFOA, whereas PFOS seems to accumulate more in some human kidneys compared to the rat kidney (Kudo et al. Citation2007; Kim et al. Citation2016; Huang et al. Citation2019; Dzierlenga et al. Citation2020). For the time being, and in the absence of more information, these data imply that at comparable blood concentrations a substantial part of the human population may have higher intrahepatic levels of PFOS/PFOA and higher intrarenal PFOS levels when compared to rodents.
Table 3. PFOS/PFOA organ concentrations in humans and calculated human organ:plasma partition coefficients (based on mean/median organ concentrations).
Species differences also exist regarding the elimination and excretion mechanisms. An overview of the blood terminal half-lives is presented in , designating much longer half-lives in humans as compared to rodents and monkeys. PFOS shows accumulating properties in all species, with an elimination half-life in the range of a month for the rat and mouse (20–40 days), and with a remarkable half-life of ∼ 5 years recorded in humans. In the case of PFOS, limited differences are observed between males and females of the same species. PFOA also shows high accumulation potential in many species, except for the rat (0.15–2 days). In addition, in the rat a remarkable gender difference has been observed for PFOA, which is briefly discussed below.
Table 4. Information of terminal half-lives for PFOS and PFOA in various species.
In most species, urinary clearance seems to be the primary elimination pathway (EFSA CONTAM Panel Citation2018a). For PFOA, clear differences have been reported between male and female rats pertaining to the renal elimination and they have been linked specifically to the active protein-mediated transport that governs tubular secretion and re-absorption (from the pre-urine back to the kidney and blood circulation) (Han et al. Citation2012). Sex-hormone mediated expression of organic anion transporting polypeptide (Oatp)1a1, located on the apical tubular membrane, was demonstrated to play a role in the observed renal re-absorption of PFOA in the male rat (Yang et al. Citation2009). However, more transporters, including organic anion transporters (oats) may be involved (Kudo et al. Citation2002). This information provides an explanation on the observed faster renal excretion of PFOA in female as opposed to male rats.
Elimination of PFOS/PFOA in humans is thought to be primarily via urinary excretion. However, there is a clear lack of studies on fecal excretion (EFSA CONTAM Panel Citation2018a). PFOS and PFOA are shown to be highly excreted in the bile; still, most of the quantity excreted into the gut is believed to undergo extensive enterohepatic re-absorption (>97%) (Harada et al. Citation2007; Fuji et al. Citation2015). Renal re-absorption via kidney transporters has been demonstrated for PFOS and PFOA (Nakagawa et al. Citation2009; Han et al. Citation2012). Such re-absorption processes, both renal and intestinal, are believed to contribute substantially to the observed long elimination half-lives of both PFOS and PFOA in humans.
Overall, from a kinetic perspective there are species- (and gender)-dependent differences, primarily regarding the terminal half-life, intra-hepatic and intra-renal concentrations and excretion patterns for PFOS/PFOA. These differences further complicate the extrapolation of rodent data to the human situation. Kinetic differences have to be carefully considered prior to such extrapolations, by scaling of rodent data to humans and vice versa. For risk assessment purposes it is important to consider body burdens or serum levels rather than the exposure levels. Toxicokinetic modeling, based on available data from animal and human studies, may provide a better basis for such extrapolations. In vitro kinetic studies may also provide insight into the various input parameters for such models. It is emphasized here that with regard to the in vitro toxicity assays toxicokinetics are also very important to consider, prior to extrapolations of effective doses to humans. A direct comparison of the nominally applied concentration of PFOS/PFOA in vitro with the respective human PFOS/PFOA blood levels is not necessarily a good approach. Given that in vitro and in vivo exposure situations differ fundamentally, extrapolations from these cell systems to humans are complex and shall not be performed without integration of the kinetic aspects.
Conclusions and recommendations
Many epidemiological studies have shown associations between increased blood levels of PFOS/PFOA and increased blood total cholesterol, and in some cases TGs. Exposure to the substances have occurred for several decades. Nonetheless, many of these studies are cross-sectional and consequently, the extent to which the relationships between PFOS/PFOA exposure and these altered levels of blood lipids are causal remains uncertain. Also, there are no associations with related adverse outcomes, like CVD. Even so, given the very small changes in the involved risk factors, such effects could be possibly detected only in very large studies. The recorded associations could also be the result of confounding related to excretion and re-absorption in the enterohepatic cycling process of PFOS/PFOA and bile acids, which can affect serum cholesterol levels. However, until now this remains only a postulation that requires experimental evidence.
Intriguingly, studies with shorter durations and high exposures of PFOS/PFOA in rodents and in some cases monkeys, have demonstrated opposite effects, i.e. decreased serum cholesterol and TGs. Such effects occur at much higher (at least >100-fold) serum levels and are commonly accompanied by enhanced intrahepatic lipid (mainly TG) concentrations. This complicates the interpretation of the human findings. In order to support (or not) a causal inference and to elucidate whether such findings are a real health concern for humans, a clear mechanistic understanding relevant for humans is essential.
Mechanistic evidence discussed in this manuscript stems from studies performed primarily with rodents and with human liver-derived cells. In rodents, most of the studies focus on the role of PPARα, and its activation by PFOS/PFOA appears to play, at least partially, a role in PFOS/PFOA-induced lipid perturbations, but it is not the sole mechanism. With respect to humans, studies in hPPARα mice demonstrate a reduced responsiveness of the human PPARα to PFOA when compared to rodents. The same is recorded for both PFOS/PFOA in human liver cells. This, together with the large differences in exposure levels and durations between animals and humans, indicates that comparisons between rodent and human findings shall be done with caution. Also, other pathways that do not directly involve PPARα seem to play a role in the PFOS/PFOA-induced lipid disturbances, as shown in rodents and rodent-derived hepatocytes. These relate to the activation of other nuclear receptors important for lipid homeostasis, such PXR and CAR. Nonetheless, studies with PFOS/PFOA on human hepatocytes indicate contradicting results, rendering the relevance of these receptors for humans uncertain. A possible role of these receptors remains to be clarified.
In addition, available data suggest that the effect of PFOS/PFOA on cholesterol and lipid homeostasis may also be mediated via suppression of the HNF4α pathway. Furthermore, there are indications that PFOS/PFOA may affect the cholesterol levels, by interfering with its metabolism and specifically its transformation into bile acids (including interference with CYP7A1), as well as the transport of the latter. Such observations are indeed valuable for better understanding of the mode of action, but they require further elucidation. In summary, the underlying mechanism of PFOS/PFOA-induced lipid disturbances seems to be rather complex and hitherto, not fully delineated.
Similarly, there is no simple mechanistic explanation for the differences in findings between animals and humans. The discrepancy in effects between rodents and humans may be related to profound interspecies differences in physiology regarding lipid homeostasis, and/or PFAS-species differences in toxicokinetics, as well as basic nutrition. These differences and potential interpretations are discussed throughout the manuscript (summarized in ).
Figure 4. Summarized human- and rodent-specific differences related to PFOS/PFOA exposure, as well as species-specific differences with respect to lipoprotein metabolism and nutrition. CETP: cholesteryl ester transfer protein; LDL: low-density lipoprotein; HLDL: high-density lipoprotein; TGs: triglycerides; PPARα: peroxisome proliferator-activated receptor α; PXR: pregnane X receptor; CAR: constitutive androstane receptor; PPARγ: peroxisome proliferator-activated receptor γ (Created with BioRender.com.
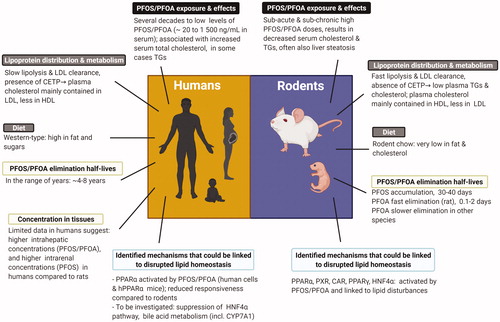
The explanation for the observed differences in health effects in rodents versus humans may also lie with the large differences in exposure levels and durations between animals and humans. Cholesterol and TG changes in humans are recorded after chronic exposure and at serum concentrations of PFOS/PFOA at least two to three orders of magnitude lower, when compared to the respective serum concentrations in rodents. This is mainly due to the higher doses commonly used for the performance of the animal studies, while animal studies using low doses, resulting in serum PFOS/PFOA levels that are comparable to the human situation, are scarce. One single 4-week study using more relevant exposure levels in APOE*3-Leiden.CETP mice showed recently that environmental (approximately 50 ng/ml) or occupational (approximately 1500 ng/ml) levels of PFOA exposure, representative for exposed community populations and fluorochemical production workers respectively, did not increase plasma cholesterol and TG levels, whereas exposure to high PFOA levels (90 000–150 000 ng/ml) did decrease TGs, total cholesterol and non-HDL-C levels and increased HDL-C level (Pouwer et al. Citation2019). This is in accordance with the slight reduction in cholesterol reported for cancer patients exposed for 6 weeks to very high PFOA levels (Convertino et al. Citation2018), although the interpretation of these data is difficult due to some methodological issues (see Section 2.1). Perhaps at such high serum levels and such exposure durations, both in humans and animals, different pathways come into play, than at the much lower concentrations and longer exposure durations observed in background populations and even in areas with increased exposure. Therefore, there are indeed few indications that the discrepancy in findings between humans and rodents might be the result of the large differences in exposure conditions. Nevertheless, it must be highlighted that PFOS/PFOA accumulate much more in humans than in rodents, as illustrated by the terminal half-lives measured in occupational workers and a highly exposed population (). In addition, human data (although limited) suggest that at comparable blood concentrations humans may have higher intrahepatic levels of PFOS/PFOA and higher intrarenal PFOS levels when compared to rodents. The life-long exposure of humans to PFOS/PFOA could possibly lead to continuously elevated body burdens, sufficient to cause effects on lipid homeostasis. Overall, it appears that the interpretation of the existing rodent data on PFOS/PFOA-induced lipid perturbations, with respect to the human situation, is complex.
In the case of the in vitro experiments with human hepatocytes, only single short exposures are generally used, attempting to mimic effects occurring in vivo after repeated chronic exposures. In addition, only nominal concentrations applied into the cell cultures are reported, whereas actual intracellular concentrations are rarely reported. These nominal concentrations in the culture medium are commonly much higher, when compared directly to serum PFOS/PFOA levels associated with increased cholesterol and TGs in humans. However, it is unclear whether these PFOS/PFOA in vitro concentrations constitute an appropriate surrogate for serum levels, especially considering the very high protein binding of these compounds. Quantitative in vitro to in vivo extrapolations (QIVIVE) would assist in translating effect levels observed in the in vitro test systems into the equivalent human PFOS/PFOA serum levels. This shall be done with the integration of kinetics, while preferably the cellular uptake of the chemicals shall be determined experimentally.
An important new asset to delineate the species differences and the inherent differences in signaling pathways between rodents and humans is to make use of mice with a humanized chimeric liver (Tateno et al. Citation2004). In these mice >80% of the mouse hepatocytes are replaced by human hepatocytes. The chimeric mice exhibit a “humanized” circulating lipoprotein cholesterol profile with an LDL-C/HDL-C ratio similar to that observed in humans, as well as bile acid regulation more characteristic of humans (Ellis et al. Citation2013). Importantly, with respect to the substantial species differences in PPARα expression and the affinity of PFOS/PFOA for the PPARα receptor, the expression levels of human PPARα are similar as in humans and their interaction with other relevant transcription factors have a human context. The same applies to other relevant biological processes. These mice have been used to elucidate the discrepancy in circulating cholesterol induced by obeticholic acid, an FXR-agonist and clinical candidate for treatment of NASH, between rodent models and humans, where obeticholic acid increased LDL-C in humans and consistently reduced total cholesterol levels in rodents (Papazyan et al. Citation2018). Studies with these mice, and importantly with different escalating exposure levels relevant to humans, may help elucidate the mechanism of action of PFOS/PFOA relevant for humans.
Together with studies on chimeric mice, further in vitro investigations with human hepatocytes may help clarify the pathway underlying the potential PFOS/PFOA-induced lipid perturbations. Specifically, more information is needed on the involvement of the HNF4α signaling pathway, as well as interference of PFOS/PFOA with cholesterol transformation into bile acids. Still, given the specific limitations of such in vitro models, the extrapolation of the effects to humans shall be done carefully by taking into consideration the dosing and integrating the kinetic aspects. The latter can be achieved with the use of physiologically based kinetic modeling, together with measurements of the actual intracellular concentrations of the compounds. If such studies are finetuned to the human situation and interpreted in the context of the intact human, they can generate valuable information that will contribute to a better understanding of PFAS-mediated lipid perturbations and the issues involved in their interpretation for human health risk assessment.
Supplemental Material
Download MS Excel (20.7 KB)Acknowledgements
This work is conducted within the HBM4EU project. HBM4EU represents a joint effort of 28 countries, the European Environment Agency and the European Commission, co-funded by Horizon 2020. The main aim of the initiative is to coordinate and advance human biomonitoring in Europe. HBM4EU provides evidence of the actual exposure of citizens to chemicals and the possible health effects to support policy making. The project involves collaboration between several Commission services, EU agencies, national representatives, stakeholders and scientists, demonstrating how research funding can build bridges between the research and policy worlds. The authors acknowledge Shalenie den Braver-Sewradj and Wieneke Bil, National Institute for Public Health and the Environment (RIVM), Bilthoven, The Netherlands, for their critical reading of the paper and valuable comments. The authors cordially thank the reviewers the Editor selected, whose identity was anonymous, for their thoughtful insight and comments towards the improvement of the manuscript.
Declaration of interest
Hans M.G. Princen does not have competing interests other than employment in contract facilities at TNO Metabolic Health Research which received previously funds from 3 M Company for contract services. The authors report no conflicts of interest. The employment affiliations of the authors are shown on the cover page. None of the authors have been involved in legal or regulatory matters related to the contents of the article.
Additional information
Funding
Notes
1 EFSA CONTAM Panel has considered four PFASs members for the calculation of a TWI: PFOS, PFOA, PFHxS (perfluorohexanesulfonic acid) and PFNA (perfluorononanoic acid) (EFSA CONTAM Panel 2020).
2 One decade in human life would correspond to approximately 12 weeks for the rat considering its two-year life span.
3 Liver levels were not analyzed in female rats.
References
- Abe T, Takahashi M, Kano M, Amaike Y, Ishii C, Maeda K, Kudoh Y, Morishita T, Hosaka T, Sasaki T, et al. 2017. Activation of nuclear receptor CAR by an environmental pollutant perfluorooctanoic acid. Arch Toxicol. 91:2365–2374.
- Adiels M, Olofsson SO, Taskinen MR, Borén J. 2008. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 28:1225–1236.
- Ason B, van der Hoorn JWA, Chan J, Lee E, Pieterman EJ, Nguyen KK, Di M, Shetterly S, Tang J, Yeh WC, et al. 2014. PCSK9 inhibition fails to alter hepatic LDLR, circulating cholesterol, and atherosclerosis in the absence of ApoE. J Lipid Res. 55:2370–2379.
- ATSDR. 2018. Toxicological profile for perfluoroalkyls. Draft for Public Comment June 2018. https://www.atsdr.cdc.gov/toxprofiles/tp200.pdf
- Auboeuf D, Rieusset J, Fajas L, Vallier P, Frering V, Riou JP, Staels B, Auwerx J, Laville M, Vidal H. 1997. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and liver X receptor-alpha in humans: no alteration in adipose tissue of obese and NIDDM patients. Diabetes. 46:1319–1327.
- Ballesteros V, Costa O, Iniguez C, Fletcher T, Ballester F, Lopez-Espinosa MJ. 2017. Exposure to perfluoroalkyl substances and thyroid function in pregnant women and children: a systematic review of epidemiologic studies. Environ Int. 99:15–28.
- Bartell SM, Calafat AM, Lyu C, Kato K, Ryan PB, Steenland K. 2010. Rate of decline in serum PFOA concentrations after granular activated carbon filtration at two public water systems in Ohio and West Virginia. Environ Health Perspect. 118:222–228.
- Beesoon S, Martin JW. 2015. Isomer-specific binding affinity of perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA) to serum proteins. Environ Sci Technol. 49:5722–5731.
- Beggs KM, McGreal SR, McCarthy A, Gunewardena S, Lampe JN, Lau C, Apte U. 2016. The role of hepatocyte nuclear factor 4-alpha in perfluorooctanoic acid- and perfluorooctanesulfonic acid-induced hepatocellular dysfunction. Toxicol Appl Pharmacol. 304:18–29.
- Behr AC, Kwiatkowski A, Ståhlman M, Schmidt FF, Luckert C, Braeuning A, Buhrke T. 2020. Impairment of bile acid metabolism by perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS) in human HepaRG hepatoma cells. Arch Toxicol. 94:1673–1686.
- Behr AC, Plinsch C, Braeuning A, Buhrke T. 2020. Activation of human nuclear receptors by perfluoroalkylated substances (PFAS). Toxicol in Vitro. 62:104700.
- Bergen WG, Mersmann HJ. 2005. Comparative aspects of lipid metabolism: impact on contemporary research and use of animal models. J Nutr. 135:2499–2502.
- Bijland S, Rensen PC, Pieterman EJ, Maas AC, van der Hoorn JW, van Erk MJ, Havekes LM, Willems van Dijk K, Chang SC, Ehresman DJ, et al. 2011. Perfluoroalkyl sulfonates cause alkyl chain length-dependent hepatic steatosis and hypolipidemia mainly by impairing lipoprotein production in APOE*3-Leiden CETP mice. Toxicol Sci. 123:290–303.
- Bjork JA, Butenhoff JL, Wallace KB. 2011. Multiplicity of nuclear receptor activation by PFOA and PFOS in primary human and rodent hepatocytes. Toxicology. 288:8–17.
- Bjork JA, Wallace KB. 2009. Structure-activity relationships and human relevance for perfluoroalkyl acid-induced transcriptional activation of peroxisome proliferation in liver cell cultures. Toxicol Sci. 111:89–99.
- Borén J, Chapman MJ, Krauss RM, Packard CJ, Bentzon JF, Binder CJ, Daemen MJ, Demer LL, Hegele RA, Nicholls SJ, et al. 2020. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 41:2313–2330.
- Brede E, Wilhelm M, Goen T, Muller J, Rauchfuss K, Kraft M, Holzer J. 2010. Two-year follow-up biomonitoring pilot study of residents’ and controls’ PFC plasma levels after PFOA reduction in public water system in Arnsberg, Germany. Int J Hyg Environ Health. 213:217–223.
- Brown MS, Goldstein JL. 1997. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 89:331–340.
- Buhrke T, Krüger E, Pevny S, Rößler M, Bitter K, Lampen A. 2015. Perfluorooctanoic acid (PFOA) affects distinct molecular signalling pathways in human primary hepatocytes. Toxicology. 333:53–62.
- Butenhoff J, Costa G, Elcombe C, Farrar D, Hansen K, Iwai H, Jung R, Kennedy G Jr, Lieder P, Olsen G, et al. 2002. Toxicity of ammonium perfluorooctanoate in male cynomolgus monkeys after oral dosing for 6 months. Toxicol Sci. 69:244–257.
- Butenhoff JL, Chang SC, Olsen GW, Thomford PJ. 2012. Chronic dietary toxicity and carcinogenicity study with potassium perfluorooctanesulfonate in Sprague Dawley rats. Toxicology. 293:1–15.
- Butenhoff JL, Kennedy GL Jr, Chang SC, Olsen GW. 2012. Chronic dietary toxicity and carcinogenicity study with ammonium perfluorooctanoate in Sprague-Dawley rats. Toxicology. 298:1–13.
- Butenhoff JL, Kennedy GL Jr, Hinderliter PM, Lieder PH, Jung R, Hansen KJ, Gorman GS, Noker PE, Thomford PJ. 2004. Pharmacokinetics of perfluorooctanoate in cynomolgus monkeys. Toxicol Sci. 82:394–406.
- Canova C, Barbieri G, Zare Jeddi M, Gion M, Fabricio A, Daprà F, Russo F, Fletcher T, Pitter G. 2020. Associations between perfluoroalkyl substances and lipid profile in a highly exposed young adult population in the Veneto Region. Environ Int. 145:106117.
- Chamouton J, Latruffe N. 2012. PPARα/HNF4α interplay on diversified responsive elements. Relevance in the regulation of liver peroxisomal fatty acid catabolism. Curr Drug Metab. 13:1436–1453.
- Chang S, Allen BC, Andres KL, Ehresman DJ, Falvo R, Provencher A, Olsen GW, Butenhoff JL. 2017. Evaluation of serum lipid, thyroid, and hepatic clinical chemistries in association with serum perfluorooctanesulfonate (PFOS) in cynomolgus monkeys after oral dosing with potassium pfos. Toxicol Sci.156:387–401.
- Chang SC, Noker PE, Gorman GS, Gibson SJ, Hart JA, Ehresman DJ, Butenhoff JL. 2012. Comparative pharmacokinetics of perfluorooctanesulfonate (PFOS) in rats, mice, and monkeys. Reprod Toxicol. 33:428–440.
- Chapman MJ, Le Goff W, Guerin M, Kontush A. 2010. Cholesteryl ester transfer protein: at the heart of the action of lipid-modulating therapy with statins, fibrates, niacin, and cholesteryl ester transfer protein inhibitors. Eur Heart J. 31:149–164.
- Chen W, Owsley E, Yang Y, Stroup D, Chiang JY. 2001. Nuclear receptor-mediated repression of human cholesterol 7alpha-hydroxylase gene transcription by bile acids. J Lipid Res. 42:1402–1412.
- Chiang JY. 1998. Regulation of bile acid synthesis. Front Biosci. 3:d176–193.
- Chiang JY. 2017. Recent advances in understanding bile acid homeostasis. F1000Res. 6:2029–2029.
- Convertino M, Church TR, Olsen GW, Liu Y, Doyle E, Elcombe CR, Barnett AL, Samuel LM, MacPherson IR, Evans TRJ. 2018. Stochastic Pharmacokinetic-pharmacodynamic modeling for assessing the systemic health risk of perfluorooctanoate (PFOA). Toxicol Sci. 163:293–306.
- Corton JC, Cunningham ML, Hummer BT, Lau C, Meek B, Peters JM, Popp JA, Rhomberg L, Seed J, Klaunig JE. 2014. Mode of action framework analysis for receptor-mediated toxicity: the peroxisome proliferator-activated receptor alpha (PPARα) as a case study. Crit Rev Toxicol. 44:1–49.
- Curran I, Hierlihy SL, Liston V, Pantazopoulos P, Nunnikhoven A, Tittlemier S, Barker M, Trick K, Bondy G. 2008. Altered fatty acid homeostasis and related toxicologic sequelae in rats exposed to dietary potassium perfluorooctanesulfonate (PFOS). J Toxicol Environ Health A. 71:1526–1541.
- Darrow LA, Stein CR, Steenland K. 2013. Serum perfluorooctanoic acid and perfluorooctane sulfonate concentrations in relation to birth outcomes in the mid-Ohio Valley, 2005–2010. Environ Health Perspect. 121:1207–1213.
- Das KP, Wood CR, Lin MT, Starkov AA, Lau C, Wallace KB, Corton JC, Abbott BD. 2017. Perfluoroalkyl acids-induced liver steatosis: effects on genes controlling lipid homeostasis. Toxicology. 378:37–52.
- de la Rosa Rodriguez MA, Sugahara G, Hooiveld G, Ishida Y, Tateno C, Kersten S. 2018. The whole transcriptome effects of the PPARα agonist fenofibrate on livers of hepatocyte humanized mice. BMC Genomics. 19:443.
- DeBose-Boyd RA. 2008. Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 18:609–621.
- DeWitt JC, Shnyra A, Badr MZ, Loveless SE, Hoban D, Frame SR, Cunard R, Anderson SE, Meade BJ, Peden-Adams MM, et al. 2009. Immunotoxicity of perfluorooctanoic acid and perfluorooctane sulfonate and the role of peroxisome proliferator-activated receptor alpha. Crit Rev Toxicol. 39:76–94.
- Dietschy JM, Turley SD, Spady DK. 1993. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J Lipid Res. 34:1637–1659.
- Dietschy JM, Turley SD. 2002. Control of cholesterol turnover in the mouse. J Biol Chem. 277:3801–3804.
- DWQI. 2017. New Jersey Drinking Water Quality Institute Health Effects Subcommittee. Health-based maximum contaminant level support document: perfluorooctanoic acid (PFOA).
- DWQI. 2018. New Jersey Drinking Water Quality Institute Health Effects Subcommittee. Health-based maximum contaminant level support document: perfluorooctane sulfonate (PFOS).
- Dzierlenga AL, Robinson VG, Waidyanatha S, DeVito MJ, Eifrid MA, Gibbs ST, Granville CA, Blystone CR. 2020. Toxicokinetics of perfluorohexanoic acid (PFHxA), perfluorooctanoic acid (PFOA) and perfluorodecanoic acid (PFDA) in male and female Hsd:Sprague dawley SD rats following intravenous or gavage administration. Xenobiotica. 50:722–732.
- Dzierlenga MW, Keast DR, Longnecker MP. 2020. The concentration of several perfluoroalkyl acids in serum appears to be reduced by dietary fiber. medRxiv. 2020.2007.2015.20154922.
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), Schrenk D, Bignami M, Bodin L, Chipman JK, del Mazo J, Grasl-Kraupp B, Hogstrand C, Hoogenboom LR, Leblanc JC, Nebbia CS, et al. 2020. Scientific Opinion on the risk to human health related to the presence of perfluoroalkyl substances in food. EFSA J. 18:6223.
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain). Knutsen HK, Alexander J, Barregard L, Bignami M, Bruschweiler B, Ceccatelli S, Cottrill B, Dinovi M, Edler L, Grasl-Kraupp B, et al. 2018a. Scientific Opinion on the risk to human health related to the presence of perfluorooctane sulfonic acid and perfluorooctanoic acid in food. EFSA J. 16:5194.
- EFSA CONTAM Panel. 2018b. Minutes of the expert meeting on perfluooroctane sulfonic acid and perfluorooctanoic acid in food assessment. https://www.efsa.europa.eu/sites/default/files/news/efsa-contam-3503.pdf
- Ehresman DJ, Froehlich JW, Olsen GW, Chang SC, Butenhoff JL. 2007. Comparison of human whole blood, plasma, and serum matrices for the determination of perfluorooctanesulfonate (PFOS), perfluorooctanoate (PFOA), and other fluorochemicals. Environ Res. 103:176–184.
- Elcombe CR, Elcombe BM, Foster JR, Chang SC, Ehresman DJ, Butenhoff JL. 2012. Hepatocellular hypertrophy and cell proliferation in Sprague-Dawley rats from dietary exposure to potassium perfluorooctanesulfonate results from increased expression of xenosensor nuclear receptors PPARα and CAR/PXR. Toxicology. 293:16–29.
- Ellis EC, Naugler WE, Parini P, Mörk LM, Jorns C, Zemack H, Sandblom AL, Björkhem I, Ericzon BG, Wilson EM, et al. 2013. Mice with chimeric livers are an improved model for human lipoprotein metabolism. PLoS One. 8:e78550.
- Ericson I, Gómez M, Nadal M, van Bavel B, Lindström G, Domingo JL. 2007. Perfluorinated chemicals in blood of residents in Catalonia (Spain) in relation to age and gender: a pilot study. Environ Int. 33:616–623.
- Eriksen KT, Raaschou-Nielsen O, McLaughlin JK, Lipworth L, Tjønneland A, Overvad K, Sørensen M. 2013. Association between plasma PFOA and PFOS levels and total cholesterol in a middle-aged Danish population. PloS One. 8:e56969.
- Fabbrini E, Mohammed BS, Korenblat KM, Magkos F, McCrea J, Patterson BW, Klein S. 2010. Effect of fenofibrate and niacin on intrahepatic triglyceride content, very low-density lipoprotein kinetics, and insulin action in obese subjects with nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 95:2727–2735.
- Fàbrega F, Kumar V, Schuhmacher M, Domingo JL, Nadal M. 2014. PBPK modeling for PFOS and PFOA: validation with human experimental data. Toxicol Lett. 230:244–251.
- Feingold KR. 2000. Introduction to lipids and lipoproteins. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, Grossman A, Hershman JM, Hofland J, Kaltsas G, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com. [Updated 2018 Feb 2]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK305896/
- Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, Hegele RA, Krauss RM, Raal FJ, Schunkert H, et al. 2017. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 38:2459–2472.
- Fitz-Simon N, Fletcher T, Luster MI, Steenland K, Calafat AM, Kato K, Armstrong B. 2013. Reductions in serum lipids with a 4-year decline in serum perfluorooctanoic acid and perfluorooctanesulfonic acid. Epidemiology (Cambridge, Mass). 24:569–576.
- Frisbee SJ, Shankar A, Knox SS, Steenland K, Savitz DA, Fletcher T, Ducatman AM. 2010. Perfluorooctanoic acid, perfluorooctanesulfonate, and serum lipids in children and adolescents: results from the C8 Health Project. Arch Pediatr Adolesc Med. 164:860–869.
- Fujii Y, Niisoe T, Harada KH, Uemoto S, Ogura Y, Takenaka K, Koizumi A. 2015. Toxicokinetics of perfluoroalkyl carboxylic acids with different carbon chain lengths in mice and humans. J Occup Health. 57:1–12.
- Gallo V, Leonardi G, Genser B, Lopez-Espinosa MJ, Frisbee SJ, Karlsson L, Ducatman AM, Fletcher T. 2012. Serum perfluorooctanoate (PFOA) and perfluorooctane sulfonate (PFOS) concentrations and liver function biomarkers in a population with elevated PFOA exposure. Environ Health Perspect. 120:655–660.
- Geiger SD, Xiao J, Ducatman A, Frisbee S, Innes K, Shankar A. 2014. The association between PFOA, PFOS and serum lipid levels in adolescents. Chemosphere. 98:78–83.
- Genuis SJ, Birkholz D, Ralitsch M, Thibault N. 2010. Human detoxification of perfluorinated compounds. Public Health. 124:367–375.
- Genuis SJ, Curtis L, Birkholz D. 2013. Gastrointestinal elimination of perfluorinated compounds using cholestyramine and Chlorella pyrenoidosa. ISRN Toxicol. 2013:657849.
- Gleason JA, Post GB, Fagliano JA. 2015. Associations of perfluorinated chemical serum concentrations and biomarkers of liver function and uric acid in the US population (NHANES), 2007-2010. Environ Res. 136:8–14.
- Gomis MI, Vestergren R, Borg D, Cousins IT. 2018. Comparing the toxic potency in vivo of long-chain perfluoroalkyl acids and fluorinated alternatives. Environ Int. 113:1–9.
- Göttlicher M, Widmark E, Li Q, Gustafsson JA. 1992. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proc Natl Acad Sci USA. 89:4653–4657.
- Guruge KS, Yeung LW, Yamanaka N, Miyazaki S, Lam PK, Giesy JP, Jones PD, Yamashita N. 2006. Gene expression profiles in rat liver treated with perfluorooctanoic acid (PFOA). Toxicol Sci. 89:93–107.
- Han X, Nabb DL, Russell MH, Kennedy GL, Rickard RW. 2012. Renal elimination of perfluorocarboxylates (PFCAs). Chem Res Toxicol. 25:35–46.
- Harada KH, Hashida S, Kaneko T, Takenaka K, Minata M, Inoue K, Saito N, Koizumi A. 2007. Biliary excretion and cerebrospinal fluid partition of perfluorooctanoate and perfluorooctane sulfonate in humans. Environ Toxicol Pharmacol. 24:134–139.
- Haughom B, Spydevold O. 1992. The mechanism underlying the hypolipemic effect of perfluorooctanoic acid (PFOA), perfluorooctane sulphonic acid (PFOSA) and clofibric acid. Biochim Biophys Acta. 1128:65–72.
- Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. 2001. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol. 21:1393–1403.
- Heusinkveld HJ, Wackers PFK, Schoonen WG, van der Ven L, Pennings JLA, Luijten M. 2018. Application of the comparison approach to open TG-GATEs: a useful toxicogenomics tool for detecting modes of action in chemical risk assessment. Food Chem Toxicol. 121:115–123.
- Horton JD, Goldstein JL, Brown MS. 2002. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 109:1125–1131.
- Howroyd P, Swanson C, Dunn C, Cattley RC, Corton JC. 2004. Decreased longevity and enhancement of age-dependent lesions in mice lacking the nuclear receptor peroxisome proliferator-activated receptor alpha (PPARalpha). Toxicol Pathol. 32:591–599.
- Huang MC, Dzierlenga AL, Robinson VG, Waidyanatha S, DeVito MJ, Eifrid MA, Granville CA, Gibbs ST, Blystone CR. 2019. Toxicokinetics of perfluorobutane sulfonate (PFBS), perfluorohexane-1-sulphonic acid (PFHxS), and perfluorooctane sulfonic acid (PFOS) in male and female Hsd:Sprague Dawley SD rats after intravenous and gavage administration. Toxicol Rep. 6:645–655.
- Hui Z, Li R, Chen L. 2017. The impact of exposure to environmental contaminant on hepatocellular lipid metabolism. Gene. 622:67–71.
- Imes CC, Austin MA. 2013. Low-density lipoprotein cholesterol, apolipoprotein B, and risk of coronary heart disease: from familial hyperlipidemia to genomics. Biol Res Nurs. 15:292–308.
- Jain RB, Ducatman A. 2019. Selective associations of recent low concentrations of perfluoroalkyl substances with liver function biomarkers: NHANES 2011 to 2014 Data on US Adults Aged ≥20 Years. J Occup Environ Med. 61:293–302.
- Janssen AW, Betzel B, Stoopen G, Berends FJ, Janssen IM, Peijnenburg AA, Kersten S. 2015. The impact of PPARα activation on whole genome gene expression in human precision cut liver slices. BMC Genomics. 16:760.
- Jeon TI, Osborne TF. 2012. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab. 23:65–72.
- Kaabia Z, Poirier J, Moughaizel M, Aguesse A, Billon-Crossouard S, Fall F, Durand M, Dagher E, Krempf M, Croyal M. 2018. Plasma lipidomic analysis reveals strong similarities between lipid fingerprints in human, hamster and mouse compared to other animal species. Sci Rep. 8:15893.
- Kersten S, Stienstra R. 2017. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie. 136:75–84.
- Kersten S. 2014. Integrated physiology and systems biology of PPARα. Mol Metab. 3:354–371.
- Kim NH, Kim SG. 2020. Fibrates revisited: potential role in cardiovascular risk reduction. Diabetes Metab J. 44:213–221.
- Kim SJ, Heo SH, Lee DS, Hwang IG, Lee YB, Cho HY. 2016. Gender differences in pharmacokinetics and tissue distribution of 3 perfluoroalkyl and polyfluoroalkyl substances in rats. Food Chem Toxicol. 97:243–255.
- Kir S, Zhang Y, Gerard RD, Kliewer SA, Mangelsdorf DJ. 2012. Nuclear receptors HNF4α and LRH-1 cooperate in regulating Cyp7a1 in vivo. J Biol Chem. 287:41334–41341.
- Krause BR, Princen HM. 1998. Lack of predictability of classical animal models for hypolipidemic activity: a good time for mice? Atherosclerosis. 140:15–24.
- Kudo N, Katakura M, Sato Y, Kawashima Y. 2002. Sex hormone-regulated renal transport of perfluorooctanoic acid. Chem Biol Interact. 139:301–316.
- Kudo N, Sakai A, Mitsumoto A, Hibino Y, Tsuda T, Kawashima Y. 2007. Tissue distribution and hepatic subcellular distribution of perfluorooctanoic acid at low dose are different from those at high dose in rats. Biol Pharm Bull. 30:1535–1540.
- Kühnast S, Fiocco M, van der Hoorn JW, Princen HM, Jukema JW. 2015. Innovative pharmaceutical interventions in cardiovascular disease: Focusing on the contribution of non-HDL-C/LDL-C-lowering versus HDL-C-raising: A systematic review and meta-analysis of relevant preclinical studies and clinical trials. Eur J Pharmacol. 763:48–63.
- Kwiterovich PO. Jr. 2000. The metabolic pathways of high-density lipoprotein, low-density lipoprotein, and triglycerides: a current review. Am J Cardiol. 86:5l–10. l.
- Larter CZ, Yeh MM, Van Rooyen DM, Brooling J, Ghatora K, Farrell GC. 2012. Peroxisome proliferator-activated receptor-α agonist, Wy 14,643, improves metabolic indices, steatosis and ballooning in diabetic mice with non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 27:341–350.
- Laskar MG, Eriksson M, Rudling M, Angelin B. 2017. Treatment with the natural FXR agonist chenodeoxycholic acid reduces clearance of plasma LDL whilst decreasing circulating PCSK9, lipoprotein(a) and apolipoprotein C–III. J. Intern. Med. 281: 575–585.
- Lee-Rueckert M, Escola-Gil JC, Kovanen PT. 2016. HDL functionality in reverse cholesterol transport – challenges in translating data emerging from mouse models to human disease. Biochim Biophys Acta. 1861:566–583.
- Leritz EC, McGlinchey RE, Salat DH, Milberg WP. 2016. Elevated levels of serum cholesterol are associated with better performance on tasks of episodic memory. Metab Brain Dis. 31:465–473.
- Li AC, Glass CK. 2004. PPAR- and LXR-dependent pathways controlling lipid metabolism and the development of atherosclerosis. J Lipid Res. 45:2161–2173.
- Li T, Chiang JY. 2005. Mechanism of rifampicin and pregnane X receptor inhibition of human cholesterol 7 alpha-hydroxylase gene transcription. Am J Physiol Gastrointest Liver Physiol. 288:G74–84.
- Li T, Chiang JY. 2009. Regulation of bile acid and cholesterol metabolism by PPARs. PPAR Res. 2009:501739.
- Li Y, Barregard L, Xu Y, Scott K, Pineda D, Lindh CH, Jakobsson K, Fletcher T. 2020. Associations between perfluoroalkyl substances and serum lipids in a Swedish adult population with contaminated drinking water. Environ Health. 19:33.
- Lin CY, Lin LY, Chiang CK, Wang WJ, Su YN, Hung KY, Chen PC. 2010. Investigation of the associations between low-dose serum perfluorinated chemicals and liver enzymes in US adults. Am J Gastroenterol. 105:1354–1363.
- Liss KH, Finck BN. 2017. PPARs and nonalcoholic fatty liver disease. Biochimie. 136:65–74.
- Lou I, Wambaugh JF, Lau C, Hanson RG Lindstrom AB, Strynar MJ, et al. (2009). Modelling single and repeated dose pharmacokinetics of PFOA in mice. Toxicol Sci. 107(2):331–341.
- Louisse J, Rijkers D, Stoopen G, Janssen A, Staats M, Hoogenboom R, Kersten S, Peijnenburg A. 2020. Perfluorooctanoic acid (PFOA), perfluorooctane sulfonic acid (PFOS), and perfluorononanoic acid (PFNA) increase triglyceride levels and decrease cholesterogenic gene expression in human HepaRG liver cells. Arch Toxicol. 94:3137–3155.
- Loveless SE, Finlay C, Everds NE, Frame SR, Gillies PJ, O'Connor JC, Powley CR, Kennedy GL. 2006. Comparative responses of rats and mice exposed to linear/branched, linear, or branched ammonium perfluorooctanoate (APFO). Toxicology. 220:203–217.
- Lu H. 2016. Crosstalk of HNF4α with extracellular and intracellular signaling pathways in the regulation of hepatic metabolism of drugs and lipids. Acta Pharm Sin B. 6:393–408.
- Luebker DJ, Hansen KJ, Bass NM, Butenhoff JL, Seacat AM. 2002. Interactions of fluorochemicals with rat liver fatty acid-binding protein. Toxicology. 176:175–185.
- Marques LR, Diniz TA, Antunes BM, Rossi FE, Caperuto EC, Lira FS, Gonçalves DC. 2018. Reverse cholesterol transport: molecular mechanisms and the non-medical approach to enhance HDL cholesterol. Front Physiol. 9:526.
- McMullen PD, Bhattacharya S, Woods CG, Pendse SN, McBride MT, Soldatow VY, Deisenroth C, LeCluyse EL, Clewell RA, Andersen ME. 2020. Identifying qualitative differences in PPARα signaling networks in human and rat hepatocytes and their significance for next generation chemical risk assessment methods. Toxicol in Vitro. 64:104463.
- Miao J, Fang S, Bae Y, Kemper JK. 2006. Functional inhibitory cross-talk between constitutive androstane receptor and hepatic nuclear factor-4 in hepatic lipid/glucose metabolism is mediated by competition for binding to the DR1 motif and to the common coactivators, GRIP-1 and PGC-1alpha. J Biol Chem. 281:14537–14546.
- Minata M, Harada KH, Kärrman A, Hitomi T, Hirosawa M, Murata M, Gonzalez FJ, Koizumi A. 2010. Role of peroxisome proliferator-activated receptor-alpha in hepatobiliary injury induced by ammonium perfluorooctanoate in mouse liver. Ind Health. 48:96–107.
- Morton RE, Izem L. 2014. Cholesteryl ester transfer proteins from different species do not have equivalent activities. J Lipid Res. 55:258–265.
- Nakagawa H, Terada T, Harada KH, Hitomi T, Inoue K, Inui K, Koizumi A. 2009. Human organic anion transporter hOAT4 is a transporter of perfluorooctanoic acid. Basic Clin Pharmacol Toxicol. 105:136–138.
- Nakagawa T, Ramdhan DH, Tanaka N, Naito H, Tamada H, Ito Y, Li Y, Hayashi Y, Yamagishi N, Yanagiba Y, et al. 2012. Modulation of ammonium perfluorooctanoate-induced hepatic damage by genetically different PPARα in mice. Arch Toxicol. 86:63–74.
- Nakamura T, Ito Y, Yanagiba Y, Ramdhan DH, Kono Y, Naito H, Hayashi Y, Li Y, Aoyama T, Gonzalez FJ, et al. 2009. Microgram-order ammonium perfluorooctanoate may activate mouse peroxisome proliferator-activated receptor alpha, but not human PPARalpha. Toxicology. 265:27–33.
- Nelson JW, Hatch EE, Webster TF. 2010. Exposure to polyfluoroalkyl chemicals and cholesterol, body weight, and insulin resistance in the general U.S. population. Environ Health Perspect. 118:197–202.
- Nian M, Li QQ, Bloom M, Qian ZM, Syberg KM, Vaughn MG, Wang SQ, Wei Q, Zeeshan M, Gurram N, et al. 2019. Liver function biomarkers disorder is associated with exposure to perfluoroalkyl acids in adults: Isomers of C8 Health Project in China. Environ Res. 172:81–88.
- Nordestgaard BG, Varbo A. 2014. Triglycerides and cardiovascular disease. Lancet. 384:626–635.
- NTP. 2019a. NTP technical report on the toxicity studies of perfluoroalkyl sulfonates (perfluorobutane sulfonic acid, perfluorohexane sulfonate potassium salt, and perfluorooctane sulfonic acid) administered by gavage to Sprague Dawley (Hsd:Sprague Dawley SD) rats. Research Triangle Park (NC): National Toxicology Program. Toxicity Report 96.
- NTP. 2019b. NTP technical report on the toxicity studies of perfluoroalkyl carboxylates (perfluorohexanoic acid, perfluorooctanoic acid, perfluorononanoic acid, and perfluorodecanoic acid) administered by gavage to Sprague Dawley (Hsd:Sprague Dawley SD) rats. Research Triangle Park (NC): National Toxicology Program. Toxicity Report 97.
- Numata J, Kowalczyk J, Adolphs J, Ehlers S, Schafft H, Fuerst P, Muller-Graf C, Lahrssen-Wiederholt M, Greiner M. 2014. Toxicokinetics of seven perfluoroalkyl sulfonic and carboxylic acids in pigs fed a contaminated diet. J Agric Food Chem. 62:6861–6870.
- OECD. 2015. Risk reduction approaches for PFASS – a crosscountry analysis. Paris (France): OECD.
- Okyere J, Oppon E, Dzidzienyo D, Sharma L, Ball G. 2014. Cross-species gene expression analysis of species specific differences in the preclinical assessment of pharmaceutical compounds. PLoS One. 9:e96853.
- Olsen GW, Burris JM, Burlew MM, Mandel JH. 2003. Epidemiologic assessment of worker serum perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA) concentrations and medical surveillance examinations. J Occup Environ Med. 45:260–270.
- Olsen GW, Burris JM, Ehresman DJ, Froehlich JW, Seacat AM, Butenhoff JL, Zobel LR. 2007. Half-life of serum elimination of perfluorooctanesulfonate,perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environ Health Perspect. 115:1298–1305.
- Olsen GW, Hansen KJ, Stevenson LA, Burris JM, Mandel JH. 2003. Human donor liver and serum concentrations of perfluorooctanesulfonate and other perfluorochemicals. Environ Sci Technol. 37:888–891.
- Olsen GW, Zobel LR. 2007. Assessment of lipid, hepatic, and thyroid parameters with serum perfluorooctanoate (PFOA) concentrations in fluorochemical production workers. Int Arch Occup Environ Health. 81:231–246.
- Ory DS. 2004. Nuclear receptor signaling in the control of cholesterol homeostasis: have the orphans found a home? Circ Res. 95:660–670.
- Palmer CN, Hsu MH, Griffin KJ, Raucy JL, Johnson EF. 1998. Peroxisome proliferator activated receptor-alpha expression in human liver. Mol Pharmacol. 53:14–22.
- Papazyan R, Liu X, Liu J, Dong B, Plummer EM, Lewis RD 2nd, Roth JD, Young MA. 2018. FXR activation by obeticholic acid or nonsteroidal agonists induces a human-like lipoprotein cholesterol change in mice with humanized chimeric liver. J Lipid Res. 59:982–993.
- Pawlak M, Lefebvre P, Staels B. 2015. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. 62:720–733.
- Peng S, Yan L, Zhang J, Wang Z, Tian M, Shen H. 2013. An integrated metabonomics and transcriptomics approach to understanding metabolic pathway disturbance induced by perfluorooctanoic acid. J Pharm Biomed Anal. 86:56–64.
- Perez F, Nadal M, Navarro-Ortega A, Fabrega F, Domingo JL, Barcelo D, Farre M. 2013. Accumulation of perfluoroalkyl substances in human tissues. Environ Int. 59:354–362.
- Perkins RG, Butenhoff JL, Kennedy GL Jr, Palazzolo MJ. 2004. 13-Week dietary toxicity study of ammonium perfluorooctanoate (APFO) in male rats. Drug Chem Toxicol. 27:361–378.
- Piepoli MF, Hoes AW, Agewall S, Albus C, Brotons C, Catapano AL, Cooney MT, Corrà U, Cosyns B, Deaton C, et al., ESC Scientific Document Group. 2016. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts) Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur Heart J. 37:2315–2381.
- Pizzurro DM, Seeley M, Kerper LE, Beck BD. 2019. Interspecies differences in perfluoroalkyl substances (PFAS) toxicokinetics and application to health-based criteria. Regul Toxicol Pharmacol. 106:239–250.
- Post SM, Duez H, Gervois PP, Staels B, Kuipers F, Princen HM. 2001. Fibrates suppress bile acid synthesis via peroxisome proliferator-activated receptor-alpha-mediated downregulation of cholesterol 7alpha-hydroxylase and sterol 27-hydroxylase expression. Arterioscler Thromb Vasc Biol. 21:1840–1845.
- Pouwer MG, Pieterman EJ, Chang SC, Olsen GW, Caspers MPM, Verschuren L, Jukema JW, Princen HMG. 2019. Dose effects of ammonium perfluorooctanoate on lipoprotein metabolism in APOE*3-Leiden.CETP Mice. Toxicol Sci. 168:519–534.
- Pouwer MG, Pieterman EJ, Worms N, Keijzer N, Jukema JW, Gromada J, Gusarova V, Princen HMG. 2020. Alirocumab, evinacumab, and atorvastatin triple therapy regresses plaque lesions and improves lesion composition in mice. J Lipid Res. 61:365–375.
- Princen H, Post S, Twisk J. 1997. Regulation of bile acid biosynthesis. Curr Pharm Des. 3:59–84.
- Princen HMG, Pouwer MG, Pieterman EJ. 2016. Comment on "Hypercholesterolemia with consumption of PFOA-laced Western diets is dependent on strain and sex of mice" by Rebholz S.L. et al. Toxicol. Rep. 2016 (3) 46–54. Toxicol Rep. 3:306–309.
- Rebholz SL, Jones T, Herrick RL, Xie C, Calafat AM, Pinney SM, Woollett LA. 2016. Hypercholesterolemia with consumption of PFOA-laced Western diets is dependent on strain and sex of mice. Toxicol Rep. 3:46–54.
- Ren H, Vallanat B, Nelson DM, Yeung LWY, Guruge KS, Lam PKS, Lehman-McKeeman LD, Corton JC. 2009. Evidence for the involvement of xenobiotic-responsive nuclear receptors in transcriptional effects upon perfluoroalkyl acid exposure in diverse species. Reprod Toxicol. 27:266–277.
- RIVM. 2018. Mixture exposure to PFAS: a relative potency factor approach. Bilthoven (The Netherlands): National Institute for Public Health and the Environment. RIVM Report 2018-0070.
- Rosen MB, Abbott BD, Wolf DC, Corton JC, Wood CR, Schmid JE, Das KP, Zehr RD, Blair ET, Lau C. 2008. Gene profiling in the livers of wild-type and PPARalpha-null mice exposed to perfluorooctanoic acid. Toxicol Pathol. 36:592–607.
- Rosen MB, Das KP, Rooney J, Abbott B, Lau C, Corton JC. 2017. PPARα-independent transcriptional targets of perfluoroalkyl acids revealed by transcript profiling. Toxicology. 387:95–107.
- Rosen MB, Lee JS, Ren H, Vallanat B, Liu J, Waalkes MP, Abbott BD, Lau C, Corton JC. 2008. Toxicogenomic dissection of the perfluorooctanoic acid transcript profile in mouse liver: evidence for the involvement of nuclear receptors PPAR alpha and CAR. Toxicol Sci. 103:46–56.
- Rosen MB, Schmid JR, Corton JC, Zehr RD, Das KP, Abbott BD, Lau C. 2010. Gene Expression Profiling in Wild-Type and PPARα-Null Mice Exposed to Perfluorooctane Sulfonate Reveals PPARα-Independent Effects. PPAR Res. 2010:1–23.
- Rosenmai AK, Ahrens L, Le Godec T, Lundqvist J, Oskarsson A. 2018. Relationship between peroxisome proliferator-activated receptor alpha activity and cellular concentration of 14 perfluoroalkyl substances in HepG2 cells. J Appl Toxicol. 38:219–226.
- Sakr CJ, Kreckmann KH, Green JW, Gillies PJ, Reynolds JL, Leonard RC. 2007a. Cross-sectional study of lipids and liver enzymes related to a serum biomarker of exposure (ammonium perfluorooctanoate or APFO) as part of a general health survey in a cohort of occupationally exposed workers. J Occup Environ Med. 49:1086–1096.
- Sakr CJ, Leonard RC, Kreckmann KH, Slade MD, Cullen MR. 2007b. Longitudinal study of serum lipids and liver enzymes in workers with occupational exposure to ammonium perfluorooctanoate. J Occup Environ Med. 49:872–879.
- Salihovic S, Stubleski J, Kärrman A, Larsson A, Fall T, Lind L, Lind PM. 2018. Changes in markers of liver function in relation to changes in perfluoroalkyl substances – a longitudinal study. Environ Int. 117:196–203.
- Sandesara PB, Virani SS, Fazio S, Shapiro MD. 2019. The forgotten lipids: triglycerides, remnant cholesterol, and atherosclerotic cardiovascular disease risk. Endocr Rev. 40:537–557.
- Scharmach E, Buhrke T, Lichtenstein D, Lampen A. 2012. Perfluorooctanoic acid affects the activity of the hepatocyte nuclear factor 4 alpha (HNF4α). Toxicol Lett. 212:106–112.
- Schlezinger J, Puckett H, Oliver J, Nielsen G, Heiger-Bernays W, Webster T. 2020. Perfluorooctanoic acid activates multiple nuclear receptor pathways and skews expression of genes regulating cholesterol homeostasis in liver of humanized PPARα mice fed an American diet. bioRxiv. 2020.2001.2030.926642
- Schoonjans K, Staels B, Auwerx J. 1996. Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. J Lipid Res. 37:907–925.
- Seacat AM, Thomford PJ, Hansen KJ, Clemen LA, Eldridge SR, Elcombe CR, Butenhoff JL. 2003. Sub-chronic dietary toxicity of potassium perfluorooctanesulfonate in rats. Toxicology. 183:117–131.
- Seacat AM, Thomford PJ, Hansen KJ, Olsen GW, Case MT, Butenhoff JL. 2002. Subchronic toxicity studies on perfluorooctanesulfonate potassium salt in cynomolgus monkeys. Toxicol Sci. 68:249–264.
- Shao W, Espenshade PJ. 2012. Expanding roles for SREBP in metabolism. Cell Metab. 16:414–419.
- Son HY, Kim SH, Shin HI, Bae HI, Yang JH. 2008. Perfluorooctanoic acid-induced hepatic toxicity following 21-day oral exposure in mice. Arch Toxicol. 82:239–246.
- Starling AP, Engel SM, Whitworth KW, Richardson DB, Stuebe AM, Daniels JL, Haug LS, Eggesbø M, Becher G, Sabaredzovic A, et al. 2014. Perfluoroalkyl substances and lipid concentrations in plasma during pregnancy among women in the Norwegian Mother and Child Cohort Study. Environ Int. 62:104–112.
- Steenland K, Tinker S, Frisbee S, Ducatman A, Vaccarino V. 2009. Association of perfluorooctanoic acid and perfluorooctane sulfonate with serum lipids among adults living near a chemical plant. Am J Epidemiol. 170:1268–1278.
- Takacs ML, Abbott BD. 2007. Activation of mouse and human peroxisome proliferator-activated receptors (alpha, beta/delta, gamma) by perfluorooctanoic acid and perfluorooctane sulfonate. Toxicol Sci. 95:108–117.
- Tan X, Xie G, Sun X, Li Q, Zhong W, Qiao P, Sun X, Jia W, Zhou Z. 2013. High fat diet feeding exaggerates perfluorooctanoic acid-induced liver injury in mice via modulating multiple metabolic pathways. PLoS One. 8:e61409.
- Tateno C, Yoshizane Y, Saito N, Kataoka M, Utoh R, Yamasaki C, Tachibana A, Soeno Y, Asahina K, Hino H, et al. 2004. Near completely humanized liver in mice shows human-type metabolic responses to drugs. Am J Pathol. 165:901–912.
- Thompson MB. 1996. Bile acids in the assessment of hepatocellular function. Toxicol Pathol. 24:62–71.
- Tugwood JD, Aldridge TC, Lambe KG, Macdonald N, Woodyatt NJ. 1996. Peroxisome proliferator-activated receptors: structures and function. Ann N Y Acad Sci. 804:252–265.
- US EPA. 2016a. Drinking Water Health Advisory for Perfluorooctanoic Acid (PFOA). Washington (DC): US EPA Office of Water (4304T) Health and Ecological Criteria Division. EPA Document Number: 822-R-16-005.
- US EPA. 2016b. Drinking Water Health Advisory for Perfluorooctane Sulfonate (PFOS). Washington (DC): US EPA Office of Water (4304T) Health and Ecological Criteria Division. EPA Document Number: 822-R-16-004.
- Vanden Heuvel JP, Thompson JT, Frame SR, Gillies PJ. 2006. Differential activation of nuclear receptors by perfluorinated fatty acid analogs and natural fatty acids: a comparison of human, mouse, and rat peroxisome proliferator-activated receptor-alpha, -beta, and -gamma, liver X receptor-beta, and retinoid X receptor-alpha. Toxicol Sci. 92:476–489.
- Wan HT, Zhao YG, Wei X, Hui KY, Giesy JP, Wong CK. 2012. PFOS-induced hepatic steatosis, the mechanistic actions on β-oxidation and lipid transport. Biochim Biophys Acta. 1820:1092–1101.
- Wang L, Wang Y, Liang Y, Li J, Liu Y, Zhang J, Zhang A, Fu J, Jiang G. 2013. Specific accumulation of lipid droplets in hepatocyte nuclei of PFOA-exposed BALB/c mice. Sci Rep. 3:2174.
- Wang L, Wang Y, Liang Y, Li J, Liu Y, Zhang J, Zhang A, Fu J, Jiang G. 2014. PFOS induced lipid metabolism disturbances in BALB/c mice through inhibition of low density lipoproteins excretion. Sci Rep. 4:4582.
- Westerterp M, van der Hoogt CC, de Haan W, Offerman EH, Dallinga-Thie GM, Jukema JW, Havekes LM, Rensen PC. 2006. Cholesteryl ester transfer protein decreases high-density lipoprotein and severely aggravates atherosclerosis in APOE*3-Leiden mice. ATVB. 26:2552–2559.
- Wigger L, Casals-Casas C, Baruchet M, Trang KB, Pradervand S, Naldi A, Desvergne B. 2019. System analysis of cross-talk between nuclear receptors reveals an opposite regulation of the cell cycle by LXR and FXR in human HepaRG liver cells. PLoS One. 14:e0220894.
- Winquist A, Steenland K. 2014. Modeled PFOA exposure and coronary artery disease, hypertension, and high cholesterol in community and worker cohorts. Environ Health Perspect. 122:1299–1305.
- Wolf CJ, Rider CV, Lau C, Abbott BD. 2014. Evaluating the additivity of perfluoroalkyl acids in binary combinations on peroxisome proliferator-activated receptor-α activation. Toxicology. 316:43–54.
- Wolf CJ, Schmid JE, Lau C, Abbott BD. 2012. Activation of mouse and human peroxisome proliferator-activated receptor-alpha (PPARα) by perfluoroalkyl acids (PFAAs): further investigation of C4-C12 compounds. Reprod Toxicol. 33:546–551.
- Wolf CJ, Takacs ML, Schmid JE, Lau C, Abbott BD. 2008. Activation of mouse and human peroxisome proliferator-activated receptor alpha by perfluoroalkyl acids of different functional groups and chain lengths. Toxicol Sci. 106:162–171.
- Woodcroft MW, Ellis DA, Rafferty SP, Burns DC, March RE, Stock NL, Trumpour KS, Yee J, Munro K. 2010. Experimental characterization of the mechanism of perfluorocarboxylic acids’ liver protein bioaccumulation: the key role of the neutral species. Environ Toxicol Chem. 29:1669–1677.
- Wu X, Xie G, Xu X, Wu W, Yang B. 2018. Adverse bioeffect of perfluorooctanoic acid on liver metabolic function in mice. Environ Sci Pollut Res Int. 25:4787–4793.
- Yan J, Chen B, Lu J, Xie W. 2015. Deciphering the roles of the constitutive androstane receptor in energy metabolism. Acta Pharmacol Sin. 36:62–70.
- Yan S, Wang J, Dai J. 2015. Activation of sterol regulatory element-binding proteins in mice exposed to perfluorooctanoic acid for 28 days. Arch Toxicol. 89:1569–1578.
- Yang CH, Glover KP, Han X. 2009. Organic anion transporting polypeptide (Oatp) 1a1-mediated perfluorooctanoate transport and evidence for a renal reabsorption mechanism of Oatp1a1 in renal elimination of perfluorocarboxylates in rats. Toxicol Lett. 190:163–171.
- Yeh MM, Bosch DE, Daoud SS. 2019. Role of hepatocyte nuclear factor 4-alpha in gastrointestinal and liver diseases. World J Gastroenterol. 25:4074–4091.
- Yin L, Ma H, Ge X, Edwards PA, Zhang Y. 2011. Hepatic hepatocyte nuclear factor 4α is essential for maintaining triglyceride and cholesterol homeostasis. Arterioscler Thromb Vasc Biol. 31:328–336.
- Yu WG, Liu W, Jin YH. 2009. Effects of perfluorooctane sulfonate on rat thyroid hormone biosynthesis and metabolism. Environ Toxicol Chem. 28:990–996.
- Zadelaar S, Kleemann R, Verschuren L, de Vries-Van der Weij J, van der Hoorn J, Princen HM, Kooistra T. 2007. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol. 27:1706–1721.
- Zhang H, Temel RE, Martel C. 2014. Cholesterol and lipoprotein metabolism: Early Career Committee contribution. Arterioscler Thromb Vasc Biol. 34:1791–1794.
- Zhang YM, Dong XY, Fan LJ, Zhang ZL, Wang Q, Jiang N, Yang XS. 2017. Poly- and perfluorinated compounds activate human pregnane X receptor. Toxicology. 380:23–29.
- Zhao W, Zitzow JD, Ehresman DJ, Chang SC, Butenhoff JL, Forster J, Hagenbuch B. 2015. Na+/Taurocholate cotransporting polypeptide and apical sodium-dependent bile acid transporter are involved in the disposition of perfluoroalkyl sulfonates in humans and rats. Toxicol Sci. 146:363–373.
- Zhao W, Zitzow JD, Weaver Y, Ehresman DJ, Chang SC, Butenhoff JL, Hagenbuch B. 2017. Organic anion transporting polypeptides contribute to the disposition of perfluoroalkyl acids in humans and rats. Toxicol Sci. 156:84–95.