
Abstract
The complete and faithful duplication of the genome is an essential prerequisite for proliferating cells to maintain genome integrity. This objective is greatly challenged by DNA damage encountered during replication, which causes fork stalling and in certain cases, fork breakage. DNA damage tolerance (DDT) pathways mitigate the effects on fork stability induced by replication fork stalling by mediating damage-bypass and replication fork restart. These DDT mechanisms, largely relying on homologous recombination (HR) and specialized polymerases, can however contribute to genome rearrangements and mutagenesis. There is a profound connection between replication and recombination: recombination proteins protect replication forks from nuclease-mediated degradation of the nascent DNA strands and facilitate replication completion in cells challenged by DNA damage. Moreover, in case of fork collapse and formation of double strand breaks (DSBs), the recombination factors present or recruited to the fork facilitate HR-mediated DSB repair, which is primarily error-free. Disruption of HR is inexorably linked to genome instability, but the premature activation of HR during replication often leads to genome rearrangements. Faithful replication necessitates the downregulation of HR and disruption of active RAD51 filaments at replication forks, but upon persistent fork stalling, building up of HR is critical for the reorganization of the replication fork and for filling-in of the gaps associated with discontinuous replication induced by DNA lesions. Here we summarize and reflect on our understanding of the mechanisms that either suppress recombination or locally enhance it during replication, and the principles that underlie this regulation.
Activities and factors preventing recombination at stalled forks
Recombination events involving donors different from the sister chromatid are potentially dangerous and can lead to genome rearrangements (Branzei & Szakal, Citation2016a,Citationb; Lambert & Carr, Citation2013; Moynahan & Jasin, Citation2010). It is therefore expected that cells would have evolved mechanisms capable to prevent such unwanted recombination events. Presumably these mechanisms are coordinated by factors present or recruited to stalled replication forks. These factors will prevent efficient assembly or initiation of homologous recombination (HR) either directly – for instance, by their ability to disrupt Rad51 filaments necessary to initiate HR –, or indirectly, by their ability to coordinate physical interactions or posttranslational modifications that would disrupt recombination initiation and maturation. Here, we begin by reviewing the mechanisms that have emerged to prevent recombination at stalled forks in yeast and mammalian cells.
In budding yeast, the most elucidated mechanism for counteracting replication-associated recombination during unperturbed replication involves the Srs2 protein and SUMOylated PCNA (Garcia-Rodriguez et al., Citation2016; Urulangodi et al., Citation2016; Watts, Citation2006). Chromatin-associated PCNA, the sliding clamp that functions as a processivity factor for DNA polymerases, is SUMOylated at a highly conserved lysine residue, K164, and to a minor extent at K127, during replication (Hoege et al., Citation2002). The SUMO modification enhances the interaction of PCNA with several factors containing SUMO Interacting Motifs (SIMs) in their structure. These SIM containing factors include the Srs2 helicase (Pfander et al., Citation2005), the alternative clamp loader replication factor C like complex, Elg1-RLC (Kubota et al., Citation2013; Parnas et al., Citation2010), and the Rad18 ubiquitin ligase (Parker & Ulrich, Citation2012) (). Srs2 can disrupt Rad51 filaments (Krejci et al., Citation2003; Papouli et al., Citation2005; Pfander et al., Citation2005; Veaute et al., Citation2003), Elg1-RLC can mediate PCNA unloading from chromatin (Kubota et al., Citation2013; Parnas et al., Citation2010), and Rad18 is critical for mono- or poly-ubiquitylation of PCNA in response to DNA damage (Hoege et al., Citation2002) (). PCNA SUMOylation also weakens the interaction between PCNA and the Eco1 acetyltransferase (Moldovan et al., Citation2006), which is required to acetylate cohesin and establish sister chromatid cohesion (Peters & Nishiyama, Citation2012). The weakened interaction between PCNA and Eco1 negatively impacts on sister chromatid cohesion (Moldovan et al., Citation2006), which favors the bias toward error-free sister chromatid recombination during replication by tethering of chromatids (Fumasoni et al., Citation2015; Tittel-Elmer et al., Citation2012) ().
Figure 1. Schematic representation of factors whose interaction with PCNA is affected by PCNA SUMOylation and the effects of these interactions on biological processes related to replication. Srs2, Elg1 and Rad18 interactions with PCNA are induced by PCNA SUMOylation, Eco1 interaction with PCNA is weakened. A color version of this figure is available online (see color version of this figure at www.tandfonline.com/ibmg).
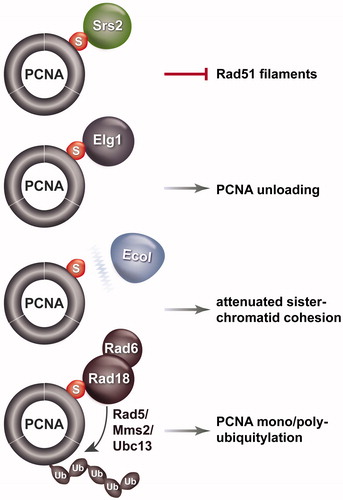
While all the above-mentioned interactions may affect recombination, so far the role of SUMOylated PCNA in inhibiting unwanted recombination is primarily understood based on its ability to attract Srs2 at stalled replication forks (Garcia-Rodriguez et al., Citation2016; Watts, Citation2006). Srs2 is a helicase similar to the bacterial UvrD helicase (Sung & Klein, Citation2006), and interacts with SUMOylated PCNA via its carboxy-terminal domain containing a PCNA-interacting peptide (PIP)-like domain followed by an SIM (Armstrong et al., Citation2012; Pfander et al., Citation2005). Srs2 recruitment to PCNA represses recombination and prevents detrimental spontaneous crossover (Papouli et al., Citation2005; Pfander et al., Citation2005; Robert et al., Citation2006), perhaps via its ability to disrupt Rad51 filaments and unwind D-loops (Krejci et al., Citation2003; Veaute et al., Citation2003) ().
Figure 2. Schematic representation of key regulators that counteract recombination at replication forks in yeast and mammalian cells. (A) The effect of Srs2 on disrupting Rad51 filaments (shown in purple) in budding yeast. (B) Roles for PARI, BLM, RecQL5 and SENP6 in counteracting recombination in mammalian cells. See text for details. A color version of this figure is available online (see color version of this figure at www.tandfonline.com/ibmg).
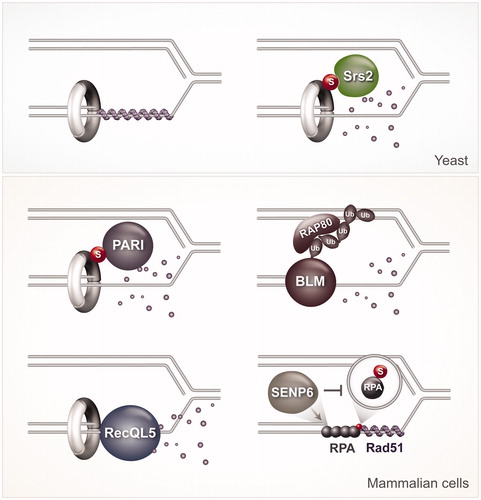
Sumoylation of PCNA at K164 was also reported in chicken DT40 cells, Xenopus laevis egg extracts, and mammalian cells (Arakawa et al., Citation2006; Gali et al., Citation2012; Leach & Michael, Citation2005; Moldovan et al., Citation2012). Functionally, SUMOylated PCNA in mammalian cells seems to act in a similar manner to the one described in yeast to prevent HR: it enhances interaction with a factor called PARI, which can also remove Rad51 filaments and lower recombination (Burkovics et al., Citation2016; Moldovan et al., Citation2012). Similar to Srs2, PARI contains a UvrD-helicase like domain, as well as PIP and SIM domains that mediate its interaction with SUMOylated PCNA (Moldovan et al., Citation2012). Recently, in a reconstituted biochemical system of DNA repair synthesis, it was shown that PARI inhibits recombination events in a manner dependent on its ability to interact with PCNA, but independently of its UvrD-like helicase domain (Burkovics et al., Citation2016). Thus, while the mechanisms by which Srs2 and PARI inhibit recombination may differ, they commonly rely on the interaction of these factors with SUMOylated PCNA. However, PCNA SUMOylation is present at much lower levels in mammalian cells compared to yeast (Gali et al., Citation2012). This raises questions on whether HR repression needs tighter regulation in yeast compared to vertebrate cells, potentially because of the high recombination frequencies of yeast (Garcia-Rodriguez et al., Citation2016). Based on the reports so far, we propose instead that mammalian cells use multiple systems, potentially specialized for diverse types of lesions, to suppress unwanted recombination during replication, as we will review below.
Two human RecQ helicases, BLM and RecQL5, orthologs of the single RecQ helicase Sgs1 in budding yeast and Rqh1 in fission yeast (Sung & Klein, Citation2006), were proposed to be critical to suppress HR by dismantling Rad51-ssDNA filaments or unwinding D-loops (Branzei & Foiani, Citation2007; Bugreev et al., Citation2007; Hu et al., Citation2007) (). Both BLM and RecQL5 interact with PCNA, although if their role in suppressing recombination involves PCNA interaction is not yet known (Sharma et al., Citation2006; Sung & Klein, Citation2006). RecQL5 interacts with PCNA and co-localizes with the replication machinery in S-phase nuclei (Kanagaraj et al., Citation2006). In response to HU treatment, which stalls replication forks by depleting dNTP pools, BLM is polyubiquitylated with K63-linked chains by the RNF8 and RNF168 ubiquitin ligases (Tikoo et al., Citation2013). This polyubiquitylation causes recruitment of BLM to stalled forks by mediating interactions with the ubiquitin interacting motifs of the adaptor protein RAP80, present at stalled forks. Constitutive association of BLM to chromatin, achieved by fusion with histone H2AX or MDC1, suppresses the elevated levels of HR associated with depletion of RNF8 or RNF18 (Tikoo et al., Citation2013). These results argue that the presence of BLM at stalled forks is critical for its ability to counteract HR.
Thus, in mammalian cells, PARI, BLM and RecQL5 can all suppress recombination during replication by disrupting RAD51 filaments (). It is currently unclear why all these helicases (and possibly more) are needed to counteract recombination seemingly at the same place. One possibility is that they preferentially respond to different replication stress cues, or different aspects of the RAD51 filament or mediators (Branzei & Foiani, Citation2007). Substantiating this view, RecQL5 knockout mouse ES cells, but not wild-type or Blm knockout cells, show a strong increase in the frequency of gross chromosomal rearrangements (GCRs), likely due to failure to regulate HR (Hu et al., Citation2007). Moreover, in vitro, Srs2, BLM and RecQL5 show differences in their mode of action. Srs2 activity is very robust and is observed even with RecA, the Escherichia coli relative of Rad51, and human RAD51 (hRAD51). On the other hand, BLM is only active toward hRAD51 and, interestingly, it can only disrupt inactive hRAD51 filaments present in an ADP-bound form. The inactivation of Rad51 is likely caused by spontaneous ATP hydrolysis (Bugreev & Mazin, Citation2004), in line with the idea that some auxiliary factors or mediators help to maintain the Rad51 filaments in an active form (Shim et al., Citation2004). Differently from BLM, however, RecQL5 is able to disrupt even tightly bound hRad51, attenuated for ATP-hydrolysis, from ssDNA (Hu et al., Citation2007). Similarly to what was reported for Srs2, the role of RecQL5 in disrupting Rad51 filaments is enhanced by RPA, which perhaps sequesters the ssDNA generated by Rad51 removal, preventing Rad51 re-nucleation on DNA (Hu et al., Citation2007).
The role of RPA itself in recombination is complex. RPA seems to be modulated by SUMOylation, at least in mammalian cells, where SUMOylated RPA was found to enhance recombination following specific types of DNA damage (Dou et al., Citation2010). During normal replication, RPA is kept in a hypoSUMOylated state, which may also function to keep recombination at bay (). The hypoSUMOylated state of RPA is achieved by stable association between RPA1 with the SUMO protease SENP6 during S phase (Dou et al., Citation2010). How this hypoSUMOylated RPA prevents recombination is not yet clear, but one possibility is that it prevents recruitment of RAD51, which contains an SIM motif necessary for hRAD51 accumulation at damaged sites (Shima et al., Citation2013). Thus, SUMOylated PCNA and hypoSUMOylated RPA act together with multiple helicases to dismantle Rad51 filaments at stressed replication forks in mammalian cells (), attesting to the importance of regulating this event to support genome stability. Interestingly, the critical players unveiled so far in suppressing unwanted recombination reflect a prominent role for SUMO modifications in orchestrating the underlying molecular events that ultimately lead to dismantling of Rad51 filaments.
DNA damage tolerance and the critical role of recombination factors for replication fork stability and restart
Prolonged stalling can lead to breakage due to the intrinsic fragility of ssDNA. Thus, stalling of replication forks can endanger replication completion and cause deleterious double strand breaks (DSBs), now recognized as an important source of genome instability (Branzei & Psakhye, Citation2016). Cells rely on the activation of pathways that promote fork reactivation and lesion bypass, which are now generally known as DNA damage tolerance (DDT) pathways. There are two main modes of DDT. One mode involves specialized trans-lesion synthesis (TLS) polymerases, which can occasionally cause mutations, and therefore this mode is known to be error-prone or mutagenic. The other mode relies on the ability to cells to use the information on the sister chromatid, using recombination (Branzei & Psakhye, Citation2016). The recombination reaction involves both recombination factors/mediators and ubiquitin ligases and conjugating activities of the Rad18/Rad6 pathway. Specifically, the Rad18 and Rad5 ubiquitin ligase activities and the Rad6, Rad5 and Mms2-Ubc13 ubiquitin conjugating activities are critical for this mode of DDT, primarily by their ability to induce PCNA poly-ubiquitylation at the conserved K164 residue (Hoege et al., Citation2002). The outcome of the recombination mode of DDT is often error-free and involves the sister chromatid (Branzei et al., Citation2008; Zhang & Lawrence, Citation2005). This recombination mode is also known as template switching, with the name deriving from the fact that a switch of templates, from the damaged strand to the undamaged sister chromatid is involved in this reaction (Branzei, Citation2011).
In replication stress mutants, there is often an increase in one or both modes of DDT. However, many replication factors actively participate in DDT. In particular, the connection between replication and recombination is very tight. Many recombination factors and mediators, such as RAD51, BRCA1 and BRCA2 are now recognized to play important roles in protecting replication fork stability (Jensen et al., Citation2010; Kolinjivadi et al., Citation2017; Lomonosov et al., Citation2003). These roles involve modulation of RAD51 to initiate recombination and to protect nascent DNA from nucleolytic activities, such as those mediated by the MRE11 nuclease (Schlacher et al., Citation2011, Citation2012; Ying et al., Citation2012) (see also below).
What DNA transitions are involved in recombination-mediated fork restart or DDT? Are they the same with the ones mediating replication fork stability? Multiple answers may apply to these questions. The reason for this complexity is that the DNA damage response (DDR) at the fork is likely to be influenced by multiple enzymatic factors, their modifications, as well as the genomic and the chromatin structural context of the locus where fork stalling occurs (Branzei & Psakhye, Citation2016; Branzei & Szakal, Citation2016a,Citationb). Here we will review the two predominant models of recombination-mediated fork stabilization and restart, namely fork reversal and postreplicative gap-filling, while summarizing critical evidence provided in support of each them.
One model of template switching proposes that a stalled replication fork can be remodeled into a reversed fork structure for stabilization or to facilitate damage bypass (Berti & Vindigni, Citation2016; Neelsen & Lopes, Citation2015). The formation of the regressed fork may involve either annealing of the ssDNA exposed at the fork with the complementary parental strand, causing exposure of the newly synthesized arm as template for DNA synthesis (, left column) or coordinated annealing of the two newly synthesized strands (, right column). The mechanistic understanding of this process is still very limited. To begin with reviewing activities proposed to take part in the latter mechanism (, right column), in vitro, several DNA translocases, such as RAD54 (Bugreev et al., Citation2011), SMARCAL1 (Betous et al., Citation2012), Mph1 (Xue et al., Citation2014) and its mammalian homolog FANCM (Gari et al., Citation2008), ZRANB3 (Ciccia et al., Citation2012; Yuan et al., Citation2012), Rad5 (Blastyak et al., Citation2007) and its mammalian homolog, HLTF (Kile et al., Citation2015), can promote fork remodeling that leads to fork reversal. Helicases such as FBH1 (Fugger et al., Citation2015), BLM and WRN (Machwe et al., Citation2006) can also promote the same in vitro reaction. However, in vivo, fork reversal functions have only been delineated so far for FBH1 (Fugger et al., Citation2015), and for SMARCAL1 (Couch et al., Citation2013) and budding yeast Rrm3 and Pif1 helicases (Rossi et al., Citation2015) when the replication checkpoint is inhibited. These latter findings on SMARCAL1, Pif1 and Rrm3 helicases promoting fork reversal when the replication checkpoint function is inhibited, together with these factors undergoing inhibitory phosphorylation dependent on the replication checkpoint, support the view that the replication checkpoint prevents fork reversal. This notion, recently reviewed in Giannattasio & Branzei (Citation2017), was initially suggested by findings in budding yeast that reported reversed forks accumulating in checkpoint mutants but being barely detectable in wild-type cells (Lopes et al., Citation2001; Sogo et al., Citation2002).
Figure 3. Schematic representation of hypothetical transitions leading to fork reversal. On the right side, remodeling of the nascent strands and the fork junction, potentially mediated by translocases or helicases, is depicted. On the left side, invasion of the RAD51-coated ssDNA exposed at the fork junction into the homologous duplex, leads to annealing of the parental strands (in black) and exposure of the nascent strand (in grey). The exposed ssDNA of the newly synthesized strand can be used as template for DNA synthesis. A color version of this figure is available online (see color version of this figure at www.tandfonline.com/ibmg).
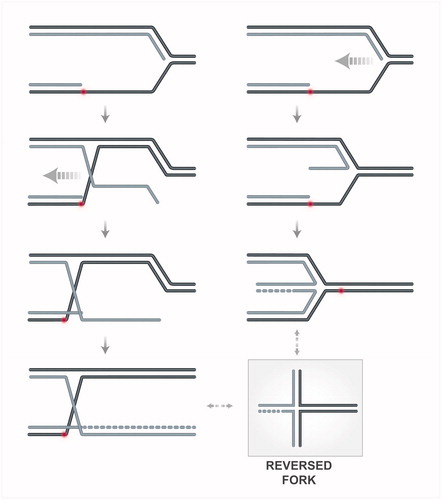
Besides the above-mentioned translocase/helicase activities that could mediate fork reversal in different circumstances, a critical role for RAD51 in fork reversal emerged, following conditions of mild genotoxic stress (Zellweger et al., Citation2015). As RAD51 can stabilize complementary ssDNA strands by paranemic pairing, and the frequency of fork reversal correlates with the amount of ssDNA at the fork junctions in both yeast and mammalian systems (Fumasoni et al., Citation2015; Rossi et al., Citation2015; Zellweger et al., Citation2015), these findings would suggest that the ssDNA at uncoupled forks is engaged by RAD51 to promote pairing with the complementary parental strand. This would then cause extrusion of the newly synthesized strand as a regressed arm, which would presumably serve as template for DNA synthesis afterwards (, left column). However, RAD51 role in fork reversal may reflect its role in stabilizing the reversed forks after they are formed – for instance, by protecting them from branch migration (Bugreev et al., Citation2006) or from nucleases such as MRE11 (Hashimoto et al., Citation2010) –, rather than an active role in their formation. Alternatively, RAD51 may cooperate with RPA in protecting ssDNA transiently exposed during replication. In this scenario, depletion of RAD51 may cause checkpoint hyperactivation, which could actively inhibit fork reversal and mediate instead other pathways of fork stabilization that do not rely on fork reversal (Couch et al., Citation2013; Rossi et al., Citation2015; Sogo et al., Citation2002).
Fork reversal events have been observed in Xenopus egg extracts, cancer cell lines and ES cells, where mild genotoxic stress and oncogene activation were shown to induce this process (Neelsen et al., Citation2013; Ray Chaudhuri et al., Citation2012; Zellweger et al., Citation2015). In budding and fission yeast, these events are rarely observed at all in wild-type cells, but are increased in several mutant backgrounds or conditions of exacerbated replication stress (Fumasoni et al., Citation2015; Rossi et al., Citation2015; Sogo et al., Citation2002). Thus, there are similarities across eukaryotic cells on the events inducing fork reversal, but also differences with regard to the kinetics or predominance of fork reversal in yeast versus mammalian cells. While different scenarios have been proposed to explain these differences (Branzei & Psakhye, Citation2016; Neelsen & Lopes, Citation2015), it is fair to say that the mechanism and frequency of fork reversal in vivo remain to date largely unknown.
Many questions remain regarding the fate of the replication machinery during formation of regressed forks and subsequently, on how the replisome is loaded for further DNA synthesis. Assuming that DNA synthesis swiftly leads to the formation of a reversed fork where the regressed arm is double stranded, the reversed fork end should be protected against unwanted fusions or ligations with other DNA ends. These issues are especially important if reversed forks persist, rather than being very transient and hardly detectable intermediates. These potential problems bring us to consider the subsequent step of reversing regressed forks to allow fork restart. The human RECQ1 helicase drives the restart of reversed replication forks, and its function is counteracted by PARP1 (Berti et al., Citation2013). Alternatively, DNA2 nuclease and WRN helicase activities were proposed to cooperate in resecting with a 5′-to-3′ polarity reversed replication forks (Thangavel et al., Citation2015). The mechanism involved in the restart may involve a DNA translocase to convert the four-way junction into functional replication forks, or alternatively, the generated 3′ ssDNA of the regressed arm could mediate invasion of the duplex ahead of the fork, in a process discussed above that is expected to be error-prone for regions containing repetitive sequences (Carr & Lambert, Citation2013).
Moreover, it is important to note that multiple activities can cleave reversed fork structures in vitro. Of these, the nuclease activity of the Mus81-Mms4 (MUS81-EME1) complex was also shown to act in vivo to process recombination intermediates (Neelsen et al., Citation2013; Szakal & Branzei, Citation2013), including reversed forks after oncogene-induced replication stress. The Mus81-Mms4 nuclease is regulated throughout the cell cycle, being activated in G2/M via Mms4 phosphorylation events mediated by the Cdc5/Plk1, Cdk1 and Cdc7-Dbf4 kinases (Gallo-Fernandez et al., Citation2012; Matos et al., Citation2011; Princz et al., Citation2017; Szakal & Branzei, Citation2013). The activities of Mus81-Mms4 and SLX4-dependent endonucleases are counteracted during replication by the replication checkpoint in yeast and mammalian cells (Froget et al., Citation2008; Giannattasio & Branzei, Citation2017; Kai et al., Citation2005; Ragland et al., Citation2013; Szakal & Branzei, Citation2013). Thus, although the model of fork reversal is attractive by its simplicity and there is evidence for situations in which fork reversal occurs in vivo, many questions remain regarding the predominance and the overall effects on genome integrity of this mode of recombination-mediated fork restart. Importantly, whether this mechanism of fork reversal is primarily activated at stalled forks in attempts to promote fork stability until an incoming fork reaches the region or if it actively participates during replication to promote lesion bypass is currently unknown.
The other main model of recombination-mediated restart by template switching envisages that replication can resume downstream of DNA damage, leaving an ssDNA gap to be filled in postreplicatively (Branzei & Szakal, Citation2016a,Citationb). This mode is intrinsic to lagging strand synthesis, due to the inherently discontinuous nature of the synthesis and maturation of Okazaki fragments. The real question here is how repriming is enabled on the leading strand. The bacterial replisome is able to re-prime and to recycle leading strand stalled replicative polymerases for downstream synthesis (Heller & Marians, Citation2006; Yeeles & Marians, Citation2011). In budding yeast, replicative helicase-coupled repriming was shown to be important for this mode of replication and DDT (Fumasoni et al., Citation2015), although how the replisome is transferred to the new primer is not yet known. It is also possible that following repriming, the DNA synthesis relies primarily on Polδ and not on the canonical leading strand replisome involving Polɛ (Vanoli et al., Citation2010). In this vein, recent work suggests a role for Polδ in replication of both leading and lagging strands (Johnson et al., Citation2015; Yeeles et al., Citation2017). In mammalian cells, a specialized protein, PrimPol, which can reprime DNA synthesis beyond DNA lesions, has been identified, in addition to the Polα-Primase activity required for bulk DNA synthesis (Bianchi et al., Citation2013; Garcia-Gomez et al., Citation2013; Mouron et al., Citation2013; Wan et al., Citation2013). PrimPol has both Primase and TLS activity, and in principle either of these activities may contribute to fork restart in vivo. Notably, however, evidence has accumulated that the primase activity of Primpol is required not only for repriming at template DNA lesions, but also at secondary structures during DNA replication (Schiavone et al., Citation2016) and when DNA synthesis is interrupted by chain terminating nucleoside analogs (Kobayashi et al., Citation2016; Sale, Citation2016). In this postreplicative mode of DDT enabled by repriming, DNA gaps are envisaged to form on both replicating strands. This was indeed confirmed by experimental observations in yeast and mammalian cells (Lehmann et al., Citation1977; Lopes et al., Citation2006). Studies in budding yeast have elucidated the subsequent steps leading to recombination products via template switching and their dissolution (reviewed in Branzei & Szakal, Citation2016a,Citationb) (), using a combination of 2D gel electrophoresis and electron microscopy, which allowed to visualize and identify the DNA structures and transitions involved in this process (Giannattasio et al., Citation2014). This study was critical for correctly assigning the DNA intermediates that mediate this process, and allows for better interpretation of previous genetic and 2D gel results that aimed at understanding template switching, as summarized in the following.
Figure 4. Schematic representation of postreplicative template switching. The process is initiated by invasion of Rad51-coated ssDNA gaps into the homologous duplex, leading to annealing of the parental strands (in black) and exposure of the nascent strand (in grey) as a template for DNA synthesis. The process leads to transient formation of double Holliday Junction-like molecules, which are swiftly processed by the Sgs1–Top3–Rmi1 complex to restore a normal replication fork structure. A color version of this figure is available online (see color version of this figure at www.tandfonline.com/ibmg).
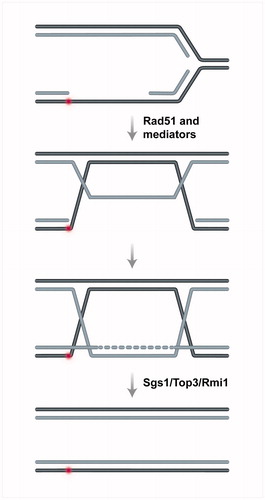
Briefly, the DNA gaps behind replication forks are further engaged by Rad51 and mediators to initiate recombination (Mankouri et al., Citation2007; Vanoli et al., Citation2010). This recombination reaction, which relies also on DNA bending activities (Gonzalez-Huici et al., Citation2014), is initiated by invasion of the Exo1 extended ssDNA gap region into the homologous duplex (Karras et al., Citation2013; Vanoli et al., Citation2010), causing exposure of the newly synthesized strand for DNA synthesis (Giannattasio et al., Citation2014). Subsequently, the D-loop structure is matured into double Holliday Junction (dHJ)-like structures, composed of annealed parental strands and plectonemically paired newly synthesized sister chromatids (Giannattasio et al., Citation2014) (). The DNA synthesis during this process relies primarily on Polδ (Vanoli et al., Citation2010). The dHJ-like intermediates are primarily resolved by the action of the Sgs1–Top3–Rmi1 (STR) complex (or BLM-TOP3α-RMI1/2, BTR complex, in humans) (Branzei et al., Citation2008; Giannattasio et al., Citation2014; Liberi et al., Citation2005), with the outcome being generally error-free (Zhang & Lawrence, Citation2005) ().
Importantly, in the postreplicative mode, the sister chromatid is generally available as donor template, and thus the errors are limited to the DNA synthesis step or to the resolution process itself (Branzei & Szakal, Citation2016a,Citationb). The action of STR/BTR is error-free, leading exclusively to non-crossover products (Branzei & Foiani, Citation2007; Sung & Klein, Citation2006), but persistent template switch recombination structures can also be resolved before anaphase by the action of the Mus81-Mms4 nuclease (Ashton et al., Citation2011; Szakal & Branzei, Citation2013), which causes the formation of both crossover and non-crossover products (Szakal & Branzei, Citation2013). In diploid cells, if the homologous chromosome is used as donor templates instead of the sister chromatid, crossover can cause loss of heterozygosity (LOH) (Moynahan & Jasin, Citation2010). Overall, considering all the above, the chances of inaccuracy in the postreplicative mode seem much lower than in the fork reversal mode. How the two pathways of recombination, fork reversal and postreplicative template switching, are regulated or how one pathway is facilitated versus the other in specific conditions remains largely unknown and an important topic of active research.
To engage or not to engage in recombination at sites of perturbed replication
If recombination is actively suppressed at replication forks (), how do cells engage in recombination following replication stress? Moreover, as TLS-mediated synthesis can promote damage-bypass as well, why do cells rely at all on recombination? Is it because recombination factors are available at the fork and intrinsic to the fork recovery (as proposed for RAD51, FANCD2, FANCA, BRCA1 and BRCA2 (Kolinjivadi et al., Citation2017)), or because TLS polymerases may need a recombination-like DNA structure to act on? These questions are subject of intensive research and the answers are likely to be complex. Although TLS is error-prone and mutagenic, it is clear that the recombination mode can also lead to errors and genome rearrangements, especially if the recombination is initiated with templates other than the sister chromatid (Branzei & Foiani, Citation2007; Carr & Lambert, Citation2013). Thus, a simple categorization of DDT on error-free and error-prone pathways, of which the former would act as default and the latter would be actively postponed as last-resort options, is likely too simplistic to be true. Looking at the experimental data for clues in this phenomenon, various findings indicate that mutagenesis is suppressed early during replication in yeast and mammalian cells (Lang & Murray, Citation2011; Stamatoyannopoulos et al., Citation2009). Ionizing radiation-induced HR is suppressed early in S phase, but observed in mid-S phase in mammalian cells, perhaps as the sister chromatids become available as templates (Cheong et al., Citation1994; Rothkamm et al., Citation2003; Saintigny et al., Citation2001; Takata et al., Citation1998). In budding yeast, using conditions of mild genotoxic stress that do not massively induce DSBs, it was observed that damage-bypass by the postreplicative mode of template switching is favored in early to mid-S phase and generally leads to an error-free outcome (Gonzalez-Huici et al., Citation2014; Karras et al., Citation2013).
These results suggest that recombination must be tightly controlled spatiotemporally to ensure availability and preferred usage of the replicated sister chromatid. Reflecting these ideas, postreplicative template switching fulfills all these conditions: it is spatially restricted in the rear of the fork at a time when the sister chromatid is available for DNA synthesis (Daigaku et al., Citation2010; Giannattasio et al., Citation2014; Karras & Jentsch, Citation2010). Moreover, establishment of cohesion favors the usage of the sister chromatid, versus other potentially available donor templates (Branzei & Szakal, Citation2016a,Citationb; Fumasoni et al., Citation2015). Additional regulations via the checkpoint also enable accurate modes of processing of the recombination intermediates during replication, while discouraging error-prone ones (Gritenaite et al., Citation2014; Szakal & Branzei, Citation2013). However, the above-mentioned findings do not exclude the idea of a “timer” that could globally activate HR in mid-S phase. This theoretical timer might be activated by signaling cascades that detect the accumulation of recombination and pro-recombination factors on chromatin and/or by the higher activity of CDK, which rises sufficiently high in mid-S to phosphorylate factors required for resection, at least for DSB end resection, which is critical in tipping the balance toward HR during DSB repair (Hustedt & Durocher, Citation2016). We should note, however, that at the moment it is not clear to what extent resection is essential for fork restart after fork stalling. Moreover, while several nucleases are required for DSB end resection and HR repair of DSBs (Hustedt & Durocher, Citation2016), a role in postreplicative template switching has only been so far established for Exo1 in budding yeast (Karras et al., Citation2013; Vanoli et al., Citation2010). A positive role for FAN1, CtIP and MRE11 nucleases in fork restart has also been proposed; however, these nuclease activities are tightly controlled by FANCD2 and HR activities, such as BRCA1 and BRCA2 (Chaudhury et al., Citation2014; Costanzo et al., Citation2001; Yeo et al., Citation2014).
The conclusion is that there is much to be learnt about how the good timing for recombination is determined and whether this would be enough to globally activate HR. However, as HR can also have deleterious consequences, it would be beneficial for the cells to combine the probabilistic timer-mediated enhancing of recombination with a deterministic local activation and restriction of HR behind the fork. Based on recent findings, we propose that this combinatorial mode of restricted activation of HR mediates recombination-mediated fork restart and DDT. In the next section, we will discuss recent findings on the mechanisms proposed to locally enhance HR at sites of perturbed replication.
SUMO- and ubiquitin-mediated interplay in activating HR at sites of perturbed replication
Recently, a SUMO-orchestrated regulatory mechanism, mediated by the conserved SUMO-like domains (SLDs)-containing S. cerevisiae protein Esc2 was shown to guide recombination specifically at sites of compromised replication (Urulangodi et al., Citation2015, Citation2016). Mechanistically, Esc2 acts by diluting out Srs2, the main activity in yeast that globally represses recombination. However, this does not cause a global enhancement in HR, only allows for local recombination events. This local effect is based on the ability of Esc2 to bind DNA structures that form at damaged replication forks, such as stalled and flapped forks (Urulangodi et al., Citation2015). In this environment, Esc2 further engages via its SLDs in regulatory interactions with SIM-containing replisome-associated proteins, including Srs2 itself. Esc2 also interacts with a conserved SUMO-targeted ubiquitin ligase (STUbL) complex, Slx5/Slx8, containing multiple SIMs (Urulangodi et al., Citation2015). STUbLs were shown to interact with SUMOylated targets and facilitate their degradation. Indeed, Esc2 and Slx5/8 mediate proteasome-dependent Srs2 turnover (), but whether this process necessarily involves Srs2 SUMOylation is not yet known (Urulangodi et al., Citation2015). Esc2 helps to dismantle the interaction between SUMOylated PCNA and Srs2, and locally reduces the concentration of Srs2 on chromatin (Urulangodi et al., Citation2015). Via its interaction with STUbL, Esc2 favors access of STUbL to Srs2. This should also allow Srs2 SUMOylation, which is induced by DNA damage (Saponaro et al., Citation2010) and inhibited by Srs2 interaction with SUMOylated PCNA (Kolesar et al., Citation2012). In this SUMOylated state, STUbL proximity could promote Srs2 degradation. Alternatively, Esc2 might bypass the requirement for SUMOylation by acting as a platform to recruit Slx5/8 to Srs2 and possibly to other substrates proximal to the replication fork. It is envisaged that by locally downregulating Srs2, Esc2 enables recruitment of Rad51 and thereby HR-mediated rescue of stalled forks ().
Figure 5. Schematic representation of local recombination enhancers. In budding yeast, Esc2 interacts with STUbL and Srs2, dismantling the interaction between SUMOylated PCNA and Srs2. This leads to Srs2 turnover and Rad51 nucleation. In mammalian cells, RPA and BLM SUMOylation induced by DNA damage and the TONSL–MMS22L complex are depicted to enhance recombination. STUbL promotes turnover of SUMOylated RPA, facilitating RAD51 filament nucleation. TONSL recognizes the H4K20me0 postreplicative chromatin mark, and facilitates, in the context of its interaction with MMS22L, RAD51-mediated recombination at regions of perturbed replication. A color version of this figure is available online (see color version of this figure at www.tandfonline.com/ibmg).
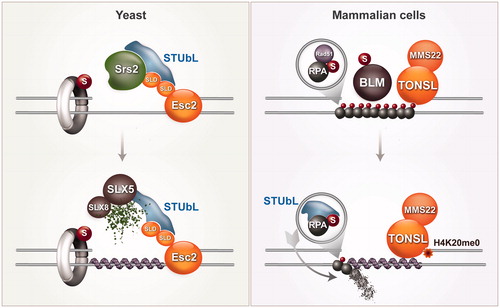
The recruitment of STUbL to regions of perturbed replication may also facilitate the transition from RPA to RAD51 on ssDNA (). RPA is kept in hypoSUMOylated state by interacting with the SUMO protease SENP6 (), but RPA SUMOylation is enhanced by DNA damage and facilitates initiation of HR (Dou et al., Citation2010). SUMOylated RPA appears to be targeted for proteosomal degradation by the mammalian STUbL RNF4 (Galanty et al., Citation2012). Consequently, in RNF4-depleted cells, RPA persists at lesions and RAD51 fails to accumulate (Galanty et al., Citation2012). Thus, SUMOylated RPA may act first to recruit RAD51 via the SIM present on RAD51 (Shima et al., Citation2013), and subsequently, RNF4-mediated turnover might facilitate the exchange between RPA and RAD51 at stalled forks (Galanty et al., Citation2012) (). RPA is SUMOylated also in yeast upon DNA damage (Cremona et al., Citation2012; Psakhye & Jentsch, Citation2012), but the functional significance of this modification and whether Esc2 and STUbL promote Rad51 recruitment by combined action on Srs2 and RPA turnover is not yet known. In addition to RPA, SUMOylation of BLM upon DNA damage also facilitates recombination (Ouyang et al., Citation2009) ().
Where is RAD51 action expected to be most critical for replication completion: at the forks or behind the forks? In yeast cells, Rad51 is not essential for viability, whereas mammalian RAD51 is required for cellular proliferation. Interestingly, however, both yeast and mammalian cells seem to critically require Rad51 function in G2, to prevent ssDNA gap accumulation in being converted to chromosome breaks (Lopes et al., Citation2006; Su et al., Citation2008). If Rad51 is particularly important in mediating postreplicative gap-filling, the question that requires attention is how the local Esc2-mediated enhancement of recombination is coupled with the readiness of the sister chromatid and recognition of a postreplicative environment. Does Esc2 preferably guide recombination at postreplicative gaps behind the fork? Esc2 may bind to factors associated to the two sides of the gap, or alternatively Esc2 may have affinity for such gapped double stranded DNA structures. In an alternative scenario, we could envisage that Esc2 binds to chromatin modifications marking newly replicated chromatin. Our understanding of post-replicative chromatin marks is relatively poor, but the recent discovery that the lack of methylation at the Lys20 residue of histone H4 (H4K20me0) represents a postreplicative mark (Saredi et al., Citation2016) has exciting implications, also because this lack of modification is specifically recognized by the TONSL subunit of the MMS22L–TONSL complex (Saredi et al., Citation2016) (). Interestingly, TONSL may be the functional ortholog of Esc2: it has ubiquitin-like domains in its C-terminus and promotes RAD51 loading in response to DNA replication stress (Duro et al., Citation2010; O'Donnell et al., Citation2010; Piwko et al., Citation2010, Citation2016).
As mammalian cells have more players with potential in repressing recombination than yeast (see ), they may also have more specialized enhancers of HR, depending on the type of lesion or replication stress cue. In this vein, another factor that deserves to be highlighted is the USP1/UAF1 complex, which promotes HR and DNA cross-link repair via its ability to deubiquitinate the Fanconi anemia protein, FANCD2 (Yang et al., Citation2011). Similarly to Esc2, UAF1 contains a tandem repeat of SLDs in its C-terminus, with the latter SLD domain interacting with an SIM on the FANCI subunit of the heterodimer FANCD2/FANCI complex. This interaction between UAF1 and FANCI is critical for FANCD2 deubiquitylation and subsequently, for FANCD2 ability to guide HR-mediated repair of DNA crosslinks (Yang et al., Citation2011). The USP1/UAF1 complex also deubiquitinates PCNA-Ub, via UAF1 binding to an SIM in hELG1. As PCNA-Ub promotes TLS-mediated damage bypass, these findings potentially suggest that the SLD-SIM interactions engaged by USP1/UAF1 may coordinate HR and TLS in response to certain types of DNA damage (Yang et al., Citation2011). This potential coordination in repair pathway choice may be similar to the roles accomplished by Esc2, which also interacts with Elg1 in yeast, although the functional consequences of this interaction remain to be elucidated (Urulangodi et al., Citation2015). What clearly Esc2 is doing, however, is to couple the formation of recombination intermediates with their processing during replication (Mankouri et al., Citation2009; Sollier et al., Citation2009) and in G2/M. The role of Esc2 in the processing of recombination intermediates may involve its stimulation of the Mus81-Mms4 nuclease (Sebesta et al., Citation2017), besides other yet to be identified factors, SLD-SIM mediated interactions and ubiquitin-mediated regulations of protein turnover. Future work will hopefully reveal how the formation and resolution of recombination intermediates are wired to each other and connected to recognition of postreplicative chromatin and cell cycle control.
Perspective
Recombination is critical to replication completion and genome integrity. SUMOylation emerged as a key regulator both in preventing recombination and in locally enhancing HR following DNA damage. The latter function seems to strongly rely on STUbL-mediated ubiquitylation events, relevant examples being Srs2 and mammalian RPA (Galanty et al., Citation2012; Urulangodi et al., Citation2015). In addition, SUMO–SIM interactions can affect the ubiquitylation pattern of certain proteins, such as FANCD2 and PCNA, affecting their activity in HR and DDT (Yang et al., Citation2011). Likely, the regulators and the relevant substrates implicated in HR modulation will be extended in the next years, and we can expect that SUMO and ubiquitin modifications will be critical in activating the change in their function. In this vein, it is important to note that several replication factors involved in HR seem to be either SUMOylated or polyubiquitylated and these modifications correlate well with their roles in suppressing or facilitating recombination. Examples include PCNA, which inhibits HR in its SUMOylated form, but facilitates recombination-mediated template switching when it is polyubiquitylated (Branzei et al., Citation2008; Papouli et al., Citation2005; Pfander et al., Citation2005), and BLM, which prevents recombination in its state of polyubiquitylation (Tikoo et al., Citation2013), but facilitates recombination when it is SUMOylated (Ouyang et al., Citation2009). The transition between these differently modified states and the modes of action of the substrate is still unclear. There is also the recurring problem in distinguishing HR regulations that pertain to stalled replication forks during unperturbed growth conditions from those induced by genotoxic stress and from more complex situations in which the genetic context and experimental conditions may cause fork collapse with formation of DSBs. The regulations pertaining on HR at collapsed replication forks may also be fundamentally different from the ones observed at DSBs induced by irradiation or other treatments. Importantly, the effects on HR or DNA dynamics deduced from observations of post-translational modifications, foci or genome-wide DNA intermediates during unperturbed replication may not reflect “global” effects on the replication process, but possibly events occurring at difficult-to-replicate sequences or specific genomic and chromatin regions. The understanding of how the above-mentioned processes and factors affect HR regulation and couple it with the sensing of postreplicative labels in the chromatin or with the recognition of structured DNA, will be a key challenge for the future and an exciting undertaking.
Disclosure statement
The authors declare no conflicts of interest.
Additional information
Funding
References
- Arakawa H, Moldovan GL, Saribasak H, et al. (2006). A role for PCNA ubiquitination in immunoglobulin hypermutation. PLoS Biol 4:e366.
- Armstrong AA, Mohideen F, Lima CD. (2012). Recognition of SUMO-modified PCNA requires tandem receptor motifs in Srs2. Nature 483:59–63.
- Ashton TM, Mankouri HW, Heidenblut A, et al. (2011). Pathways for Holliday junction processing during homologous recombination in Saccharomyces cerevisiae. Mol Cell Biol 31:1921–33.
- Berti M, Ray Chaudhuri A, Thangavel S, et al. (2013). Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 20:347–54.
- Berti M, Vindigni A. (2016). Replication stress: getting back on track. Nat Struct Mol Biol 23:103–9.
- Betous R, Mason AC, Rambo RP, et al. (2012). SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev 26:151–62.
- Bianchi J, Rudd SG, Jozwiakowski SK, et al. (2013). PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol Cell 52:566–73.
- Blastyak A, Pinter L, Unk I, et al. (2007). Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol Cell 28:167–75.
- Branzei D, Foiani M. (2007). RecQ helicases queuing with Srs2 to disrupt Rad51 filaments and suppress recombination. Genes Dev 21:3019–26.
- Branzei D, Foiani M. (2007). Template switching: from replication fork repair to genome rearrangements. Cell 131:1228–30.
- Branzei D, Psakhye I. (2016). DNA damage tolerance. Curr Opin Cell Biol 40:137–44.
- Branzei D, Szakal B. (2016a). DNA damage tolerance by recombination: molecular pathways and DNA structures. DNA Repair 44:68–75.
- Branzei D, Szakal B. (2016b). Priming for tolerance and cohesion at replication forks. Nucleus 7:8–12.
- Branzei D, Vanoli F, Foiani M. (2008). SUMOylation regulates Rad18-mediated template switch. Nature 456:915–20.
- Branzei D. (2011). Ubiquitin family modifications and template switching. FEBS Lett 585:2810–17.
- Bugreev DV, Mazin AV. (2004). Ca2+ activates human homologous recombination protein Rad51 by modulating its ATPase activity. Proc Natl Acad Sci USA 101:9988–93.
- Bugreev DV, Mazina OM, Mazin AV. (2006). Rad54 protein promotes branch migration of Holliday junctions. Nature 442:590–3.
- Bugreev DV, Rossi MJ, Mazin AV. (2011). Cooperation of RAD51 and RAD54 in regression of a model replication fork. Nucleic Acids Res 39:2153–64.
- Bugreev DV, Yu X, Egelman EH, Mazin AV. (2007). Novel pro- and anti-recombination activities of the Bloom's syndrome helicase. Genes Dev 21:3085–94.
- Burkovics P, Dome L, Juhasz S, et al. (2016). The PCNA-associated protein PARI negatively regulates homologous recombination via the inhibition of DNA repair synthesis. Nucleic Acids Res 44:3176–89.
- Carr AM, Lambert S. (2013). Replication stress-induced genome instability: the dark side of replication maintenance by homologous recombination. J Mol Biol 425:4733–44.
- Chaudhury I, Stroik DR, Sobeck A. (2014). FANCD2-controlled chromatin access of the Fanconi-associated nuclease FAN1 is crucial for the recovery of stalled replication forks. Mol Cell Biol 34:3939–54.
- Cheong N, Wang X, Wang Y, Iliakis G. (1994). Loss of S-phase-dependent radioresistance in irs-1 cells exposed to X-rays. Mutat Res 314:77–85.
- Ciccia A, Nimonkar AV, Hu Y, et al. (2012). Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol Cell 47:396–409.
- Costanzo V, Robertson K, Bibikova M, et al. (2001). Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication. Mol Cell 8:137–47.
- Couch FB, Bansbach CE, Driscoll R, et al. (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27:1610–23.
- Cremona CA, Sarangi P, Yang Y, et al. (2012). Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the mec1 checkpoint. Mol Cell 45:422–32.
- Daigaku Y, Davies AA, Ulrich HD. (2010). Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 465:951–5.
- Dou H, Huang C, Singh M, et al. (2010). Regulation of DNA repair through deSUMOylation and SUMOylation of replication protein A complex. Mol Cell 39:333–45.
- Duro E, Lundin C, Ask K, et al. (2010). Identification of the MMS22L–TONSL complex that promotes homologous recombination. Mol Cell 40:632–44.
- Froget B, Blaisonneau J, Lambert S, Baldacci G. (2008). Cleavage of stalled forks by fission yeast Mus81/Eme1 in absence of DNA replication checkpoint. Mol Biol Cell 19:445–56.
- Fugger K, Mistrik M, Neelsen KJ, et al. (2015). FBH1 catalyzes regression of stalled replication forks. Cell Rep. [Epub ahead of print]. doi: 10.1016/j.celrep.2015.02.028
- Fumasoni M, Zwicky K, Vanoli F, et al. (2015). Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polα/primase/Ctf4 complex. Mol Cell 57:812–23.
- Galanty Y, Belotserkovskaya R, Coates J, Jackson SP. (2012). RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev 26:1179–95.
- Gali H, Juhasz S, Morocz M, et al. (2012). Role of SUMO modification of human PCNA at stalled replication fork. Nucleic Acids Res 40:6049–59.
- Gallo-Fernandez M, Saugar I, Ortiz-Bazan MA, et al. (2012). Cell cycle-dependent regulation of the nuclease activity of Mus81-Eme1/Mms4. Nucleic Acids Res 40:8325–35.
- Garcia-Gomez S, Reyes A, Martinez-Jimenez MI, et al. (2013). PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell 52:541–53.
- Garcia-Rodriguez N, Wong RP, Ulrich HD. (2016). Functions of ubiquitin and SUMO in DNA replication and replication stress. Front Genet 7:87.
- Gari K, Decaillet C, Delannoy M, et al. (2008). Remodeling of DNA replication structures by the branch point translocase FANCM. Proc Natl Acad Sci USA 105:16107–12.
- Giannattasio M, Branzei D. (2017). S-phase checkpoint regulations that preserve replication and chromosome integrity upon dNTP depletion. Cell Mol Life Sci. [Epub ahead of print]. doi: 10.1007/s00018-017-2474-4
- Giannattasio M, Zwicky K, Follonier C, et al. (2014). Visualization of recombination-mediated damage bypass by template switching. Nat Struct Mol Biol 21:884–92.
- Gonzalez-Huici V, Szakal B, Urulangodi M, et al. (2014). DNA bending facilitates the error-free DNA damage tolerance pathway and upholds genome integrity. EMBO J 33:327–40.
- Gritenaite D, Princz LN, Szakal B, et al. (2014). A cell cycle-regulated Slx4–Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev 28:1604–19.
- Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V. (2010). Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17:1305–11.
- Heller RC, Marians KJ. (2006). Replication fork reactivation downstream of a blocked nascent leading strand. Nature 439:557–62.
- Hoege C, Pfander B, Moldovan GL, et al. (2002). RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419:135–41.
- Hu Y, Raynard S, Sehorn MG, et al. (2007). RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev 21:3073–84.
- Hustedt N, Durocher D. (2016). The control of DNA repair by the cell cycle. Nat Cell Biol 19:1–9.
- Jensen RB, Carreira A, Kowalczykowski SC. (2010). Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467:678–83.
- Johnson RE, Klassen R, Prakash L, Prakash S. (2015). A major role of DNA polymerase delta in replication of both the leading and lagging DNA strands. Mol Cell 59:163–75.
- Kai M, Boddy MN, Russell P, Wang TS. (2005). Replication checkpoint kinase Cds1 regulates Mus81 to preserve genome integrity during replication stress. Genes Dev 19:919–32.
- Kanagaraj R, Saydam N, Garcia PL, et al. (2006). Human RECQ5beta helicase promotes strand exchange on synthetic DNA structures resembling a stalled replication fork. Nucleic Acids Res 34:5217–31.
- Karras GI, Fumasoni M, Sienski G, et al. (2013). Noncanonical role of the 9-1-1 clamp in the error-free DNA damage tolerance pathway. Mol Cell 49:536–46.
- Karras GI, Jentsch S. (2010). The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 141:255–67.
- Kile AC, Chavez DA, Bacal J, et al. (2015). HLTF's ancient HIRAN domain binds 3′ DNA ends to drive replication fork reversal. Mol Cell 58:1090–100.
- Kobayashi K, Guilliam TA, Tsuda M, et al. (2016). Repriming by PrimPol is critical for DNA replication restart downstream of lesions and chain-terminating nucleosides. Cell Cycle 15:1997–2008.
- Kolesar P, Sarangi P, Altmannova V, et al. (2012). Dual roles of the SUMO-interacting motif in the regulation of Srs2 sumoylation. Nucleic Acids Res 40:7831–43.
- Kolinjivadi AM, Sannino V, de Antoni A, et al. (2017). Moonlighting at replication forks – a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. [Epub ahead of print]. doi: 10.1002/1873-3468.12556
- Krejci L, Van Komen S, Li Y, et al. (2003). DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423:305–9.
- Kubota T, Nishimura K, Kanemaki MT, Donaldson AD. (2013). The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol Cell 50:273–80.
- Lambert S, Carr AM. (2013). Replication stress and genome rearrangements: lessons from yeast models. Curr Opin Genet Dev 23:132–9.
- Lang GI, Murray AW. (2011). Mutation rates across budding yeast chromosome VI are correlated with replication timing. Genome Biol Evol 3:799–811.
- Leach CA, Michael WM. (2005). Ubiquitin/SUMO modification of PCNA promotes replication fork progression in Xenopus laevis egg extracts. J Cell Biol 171:947–54.
- Lehmann AR, Kirk-Bell S, Arlett CF, et al. (1977). Repair of ultraviolet light damage in a variety of human fibroblast cell strains. Cancer Res 37:904–10.
- Liberi G, Maffioletti G, Lucca C, et al. (2005). Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 19:339–50.
- Lomonosov M, Anand S, Sangrithi M, et al. (2003). Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev 17:3017–22.
- Lopes M, Cotta-Ramusino C, Pellicioli A, et al. (2001). The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412:557–61.
- Lopes M, Foiani M, Sogo JM. (2006). Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 21:15–27.
- Machwe A, Xiao L, Groden J, Orren DK. (2006). The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry 45:13939–46.
- Mankouri HW, Ngo HP, Hickson ID. (2007). Shu proteins promote the formation of homologous recombination intermediates that are processed by Sgs1-Rmi1-Top3. Mol Biol Cell 18:4062–73.
- Mankouri HW, Ngo HP, Hickson ID. (2009). Esc2 and Sgs1 act in functionally distinct branches of the homologous recombination repair pathway in Saccharomyces cerevisiae. Mol Biol Cell 20:1683–94.
- Matos J, Blanco MG, Maslen S, et al. (2011). Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 147:158–72.
- Moldovan GL, Dejsuphong D, Petalcorin MI, et al. (2012). Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol Cell 45:75–86.
- Moldovan GL, Pfander B, Jentsch S. (2006). PCNA controls establishment of sister chromatid cohesion during S phase. Mol Cell 23:723–32.
- Mouron S, Rodriguez-Acebes S, Martinez-Jimenez MI, et al. (2013). Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 20:1383–9.
- Moynahan ME, Jasin M. (2010). Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol 11:196–207.
- Neelsen KJ, Lopes M. (2015). Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 16:207–20.
- Neelsen KJ, Zanini IM, Herrador R, Lopes M. (2013). Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol 200:699–708.
- O'Donnell L, Panier S, Wildenhain J, et al. (2010). The MMS22L–TONSL complex mediates recovery from replication stress and homologous recombination. Mol Cell 40:619–31.
- Ouyang KJ, Woo LL, Zhu J, et al. (2009). SUMO modification regulates BLM and RAD51 interaction at damaged replication forks. PLoS Biol 7:e1000252.
- Papouli E, Chen S, Davies AA, et al. (2005). Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell 19:123–33.
- Parker JL, Ulrich HD. (2012). A SUMO-interacting motif activates budding yeast ubiquitin ligase Rad18 towards SUMO-modified PCNA. Nucleic Acids Res 40:11380–8.
- Parnas O, Zipin-Roitman A, Pfander B, et al. (2010). Elg1, an alternative subunit of the RFC clamp loader, preferentially interacts with SUMOylated PCNA. EMBO J 29:2611–22.
- Peters JM, Nishiyama T. (2012). Sister chromatid cohesion. Cold Spring Harbor Perspect Biol 4:a011130.
- Pfander B, Moldovan GL, Sacher M, et al. (2005). SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 436:428–33.
- Piwko W, Mlejnkova LJ, Mutreja K, et al. (2016). The MMS22L–TONSL heterodimer directly promotes RAD51-dependent recombination upon replication stress. EMBO J 35:2584–601.
- Piwko W, Olma MH, Held M, et al. (2010). RNAi-based screening identifies the Mms22L–Nfkbil2 complex as a novel regulator of DNA replication in human cells. EMBO J 29:4210–22.
- Princz LN, Wild P, Bittmann J, et al. (2017). Dbf4-dependent kinase and the Rtt107 scaffold promote Mus81-Mms4 resolvase activation during mitosis. EMBO J 36:664–78.
- Psakhye I, Jentsch S. (2012). Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 151:807–20.
- Ragland RL, Patel S, Rivard RS, et al. (2013). RNF4 and PLK1 are required for replication fork collapse in ATR-deficient cells. Genes Dev 27:2259–73.
- Ray Chaudhuri A, Hashimoto Y, Herrador R, et al. (2012). Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol 19:417–23.
- Robert T, Dervins D, Fabre F, Gangloff S. (2006). Mrc1 and Srs2 are major actors in the regulation of spontaneous crossover. EMBO J 25:2837–46.
- Rossi SE, Ajazi A, Carotenuto W, et al. (2015). Rad53-mediated regulation of Rrm3 and Pif1 DNA helicases contributes to prevention of aberrant fork transitions under replication stress. Cell Rep 13:80–92.
- Rothkamm K, Kruger I, Thompson LH, Lobrich M. (2003). Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 23:5706–15.
- Saintigny Y, Delacote F, Vares G, et al. (2001). Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J 20:3861–70.
- Sale JE. (2016). Starting over: primpol reprimes after chain termination. Cell Cycle 15:2099–100.
- Saponaro M, Callahan D, Zheng X, et al. (2010). Cdk1 targets Srs2 to complete synthesis-dependent strand annealing and to promote recombinational repair. PLoS Genet 6:e1000858.
- Saredi G, Huang H, Hammond CM, et al. (2016). H4K20me0 marks post-replicative chromatin and recruits the TONSL–MMS22L DNA repair complex. Nature 534:714–18.
- Schiavone D, Jozwiakowski SK, Romanello M, et al. (2016). PrimPol is required for replicative tolerance of G quadruplexes in vertebrate cells. Mol Cell 61:161–9.
- Schlacher K, Christ N, Siaud N, et al. (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145:529–42.
- Schlacher K, Wu H, Jasin M. (2012). A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22:106–16.
- Sebesta M, Urulangodi M, Stefanovie B, et al. (2017). Esc2 promotes Mus81 complex-activity via its SUMO-like and DNA binding domains. Nucleic Acids Res 45:215–30.
- Sharma S, Doherty KM, Brosh RM. Jr. (2006). Mechanisms of RecQ helicases in pathways of DNA metabolism and maintenance of genomic stability. Biochem J 398:319–37.
- Shim KS, Schmutte C, Tombline G, et al. (2004). hXRCC2 enhances ADP/ATP processing and strand exchange by hRAD51. J Biol Chem 279:30385–94.
- Shima H, Suzuki H, Sun J, et al. (2013). Activation of the SUMO modification system is required for the accumulation of RAD51 at sites of DNA damage. J Cell Sci 126:5284–92.
- Sogo JM, Lopes M, Foiani M. (2002). Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297:599–602.
- Sollier J, Driscoll R, Castellucci F, et al. (2009). The Saccharomyces cerevisiae Esc2 and Smc5-6 proteins promote sister chromatid junction-mediated intra-S repair. Mol Biol Cell 20:1671–82.
- Stamatoyannopoulos JA, Adzhubei I, Thurman RE, et al. (2009). Human mutation rate associated with DNA replication timing. Nat Genet 41:393–5.
- Su X, Bernal JA, Venkitaraman AR. (2008). Cell-cycle coordination between DNA replication and recombination revealed by a vertebrate N-end rule degron-Rad51. Nat Struct Mol Biol 15:1049–58.
- Sung P, Klein H. (2006). Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol 7:739–50.
- Szakal B, Branzei D. (2013). Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J 32:1155–67.
- Takata M, Sasaki MS, Sonoda E, et al. (1998). Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 17:5497–508.
- Thangavel S, Berti M, Levikova M, et al. (2015). DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 208:545–62.
- Tikoo S, Madhavan V, Hussain M, et al. (2013). Ubiquitin-dependent recruitment of the Bloom syndrome helicase upon replication stress is required to suppress homologous recombination. EMBO J 32:1778–92.
- Tittel-Elmer M, Lengronne A, Davidson MB, et al. (2012). Cohesin association to replication sites depends on rad50 and promotes fork restart. Mol Cell 48:98–108.
- Urulangodi M, Sebesta M, Menolfi D, et al. (2015). Local regulation of the Srs2 helicase by the SUMO-like domain protein Esc2 promotes recombination at sites of stalled replication. Genes Dev 29:2067–80.
- Urulangodi M, Szakal B, Branzei D. (2016). SUMO-mediated global and local control of recombination. Cell Cycle 15:160–1.
- Vanoli F, Fumasoni M, Szakal B, et al. (2010). Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genet 6:e1001205.
- Veaute X, Jeusset J, Soustelle C, et al. (2003). The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423:309–12.
- Wan L, Lou J, Xia Y, et al. (2013). hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep 14:1104–12.
- Watts FZ. (2006). Sumoylation of PCNA: wrestling with recombination at stalled replication forks. DNA Repair (Amst) 5:399–403.
- Xue X, Choi K, Bonner J, et al. (2014). Restriction of replication fork regression activities by a conserved SMC complex. Mol Cell 56:436–45.
- Yang K, Moldovan GL, Vinciguerra P, et al. (2011). Regulation of the Fanconi anemia pathway by a SUMO-like delivery network. Genes Dev 25:1847–58.
- Yeeles JT, Janska A, Early A, Diffley JF. (2017). How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol Cell 65:105–16.
- Yeeles JT, Marians KJ. (2011). The Escherichia coli replisome is inherently DNA damage tolerant. Science 334:235–8.
- Yeo JE, Lee EH, Hendrickson EA, Sobeck A. (2014). CtIP mediates replication fork recovery in a FANCD2-regulated manner. Hum Mol Genet 23:3695–705.
- Ying S, Hamdy FC, Helleday T. (2012). Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 72:2814–21.
- Yuan J, Ghosal G, Chen J. (2012). The HARP-like domain-containing protein AH2/ZRANB3 binds to PCNA and participates in cellular response to replication stress. Mol Cell 47:410–21.
- Zellweger R, Dalcher D, Mutreja K, et al. (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208:563–79.
- Zhang H, Lawrence CW. (2005). The error-free component of the RAD6/RAD18 DNA damage tolerance pathway of budding yeast employs sister-strand recombination. Proc Natl Acad Sci USA 102:15954–9.