
Abstract
A small-molecule cysteine sulfenic acid (Cys–SOH) was synthesized and isolated as stable crystals, for the first time, by utilizing a nanosized molecular cavity as a protective cradle. The cradled Cys–SOH was synthesized by direct oxidation of the corresponding cysteine thiol with H2O2 under basic conditions and its structure was established by X-ray crystallographic analysis. In the reaction of the cradled Cys–SOH with a thiol to produce the disulfide, a remarkable acceleration was observed upon the addition of an amine base. This suggests the important role of base in the reaction of Cys–SOH with thiols in biological systems. The cradled Cys–SOH was reduced to the cysteine thiol by dithiothreitol or triphenylphosphine. The high stability and sufficient reactivity of the cradled Cys–SOH indicate its usefulness as a small-molecule model compound for better understanding the chemical behavior of Cys–SOH in biological systems.
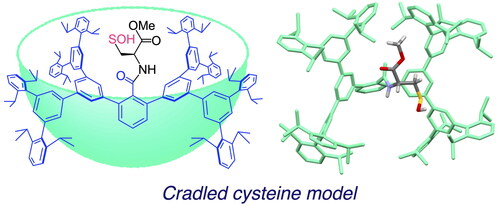
Graphical Abstract
Introduction
Sulfenic acids (R–SOH) are one of the most extensively studied reactive intermediates in the various fields of sulfur chemistry. In biological systems, cysteine sulfenic acids (Cys-SOH) are involved as crucial intermediates in a wide variety of cellular functions, including transcription regulation, signal transduction, and the regulation of oxygen metabolism and oxidative stress responses.[Citation1–6] Oxidation of cysteine thiols (Cys–SH) to Cys–SOH by reactive oxygen species (ROS) is a reversible post-translational modification, and the Cys–SOH formed can then be converted to disulfides (Cys–SSR) and cysteine sulfinic acids (Cys–SO2H) (Scheme 1). Due to the notoriously unstable nature of sulfenic acids that arises from facile dehydrative self-condensation to produce thiosulfinates, much of our knowledge of the chemistry of sulfenic acids has been obtained indirectly (Scheme 2).[Citation7]
Scheme 1. Redox processes involving cysteine sulfenic acids.
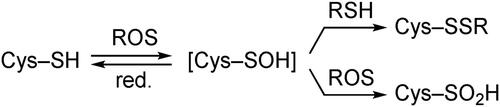
Scheme 2. Facile dehydrative self-condensation of a sulfenic acid to produce a thiosulfinate.
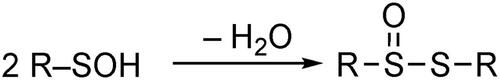
For the synthesis of stable sulfenic acids, kinetic stabilization[Citation8–16] or thermodynamic stabilization[Citation17–23] has been successfully employed. Several examples of isolable R–SOH, including cavity-shaped molecules 3[Citation13] and 4,[Citation15] which we previously reported, are shown in . In contrast to the synthesis and isolation of various non-cysteinyl R–SOH in the field of organosulfur chemistry, the chemical evidence for Cys–SOH remains elusive. Although the existence of Cys–SOH in several proteins has been supported by X-ray crystallography and mass spectrometry,[Citation24–28] there has been no report of spectroscopic observations of small-molecule Cys–SOH. In most cases, identification Cys–SOH has been based on trapping experiments using chemical probes.[Citation5] For chemical elucidation of the role of Cys–SOH in biological systems, the development of a stable model compound for Cys–SOH is highly desirable.
Figure 1. Examples of isolable non-cysteinyl R–SOH. Thermodynamically stabilized compound (1)[Citation17] and kinetically stabilized ones (2,[Citation8] 3,[Citation13] and 4[Citation15]).
![Figure 1. Examples of isolable non-cysteinyl R–SOH. Thermodynamically stabilized compound (1)[Citation17] and kinetically stabilized ones (2,[Citation8] 3,[Citation13] and 4[Citation15]).](/cms/asset/113e1222-8eb4-4935-95f0-25b1d91978b0/gpss_a_2195650_f0001_b.jpg)
The cavity-shaped substituents that we used for the synthesis of 3 and 4 were very effective for the synthesis of non-cysteinyl R–SOH, but their protective effects are limited to the directly attached reactive functionality and are likely to be insufficient to protect the exposed terminal SOH group. During the course of our studies on highly reactive species, we recently developed an expanded cavity-shaped substituent, a Bps group, with ca. 2-nm scale ().[Citation29] The expanded dendrimer-type framework of a Bps group is expected to accommodate a whole cysteine unit as shown in . In this model, a cavity-shaped benzoyl group (henceforth denoted as Bpsc) is introduced to a cysteine unit as an N-terminal protecting group. By utilizing this cradled cysteine model, we investigated the synthesis and isolation of a stable small-molecule Cys–SOH as well as the elucidation of its structure and fundamental reactivities.[Citation30]
Figure 2. Cavity-shaped substituents, Bpq and Bps groups.
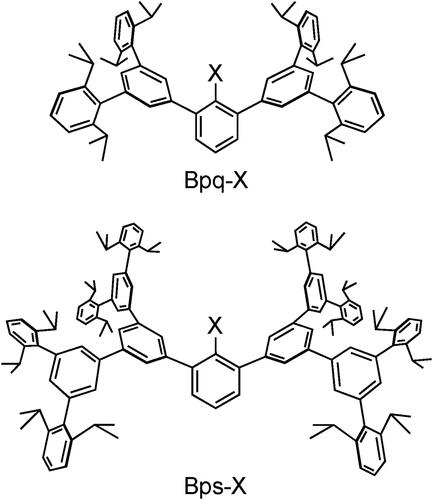
Figure 3. Cradled cysteine model for stabilization of cysteine-derived reactive intermediates such as Cys–SOH.
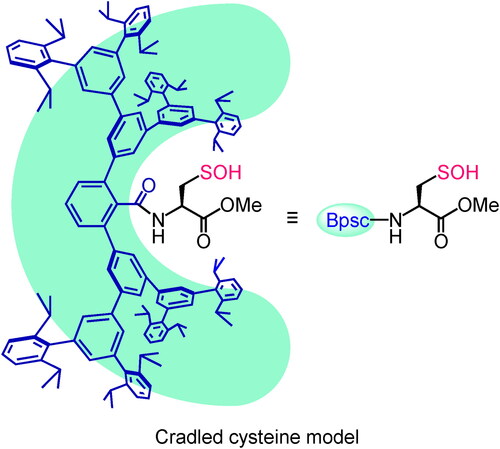
Results and discussion
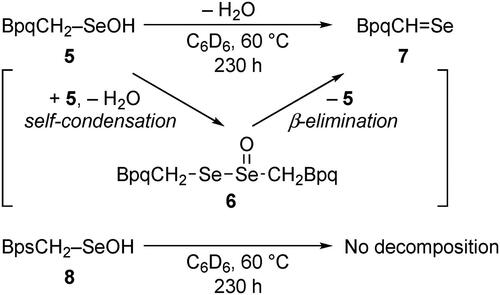
The steric protection effect of the expanded molecular cavity was evaluated by the stability of selenenic acids (R–SeOH), which are known to undergo dehydrative self-condensation even more readily than sulfenic acids.[Citation12,Citation31] We previously reported that selenenic acid 5 bearing a BpqCH2 group, a selenium derivative of sulfenic acid 4, gradually undergoes dehydrative self-condensation to selenoseleninate 6 upon heating at 60 °C in C6D6 for 230 h (Scheme 3), although 5 is isolable and almost persistent at room temperature in solution.[Citation32] The final product was found to be selenoaldehyde 7, formed by β-elimination from selenoseleninate 6. In sharp contrast, selenenic acid 8 bearing a BpsCH2 group, which has a much larger and deeper cavity than a BpqCH2 group, exhibited much higher thermal stability.[Citation29] In C6D6, almost no decomposition of 8 was observed after heating at 60 °C for 230 h, demonstrating that the large cavity-shaped framework of the Bps group in 8 effectively suppresses the bimolecular decomposition pathway of the selenenic acid. Given such a high steric protection effect of this expanded cavity-shaped framework, we expected that it will serve as a protective cradle for a cysteine unit, preventing the reactive sulfur species from undergoing bimolecular decomposition that accompanies the formation of a sulfur–sulfur linkage. Based on these results, a benzoyl group with this framework, a Bpsc group, was designed as an N-terminal protecting group for the cysteine unit bearing a reactive sulfur functionality ().
Scheme 3. Comparison of the thermal stability of BpqCH2–SeOH (5) and BpsCH2–SeOH (8).
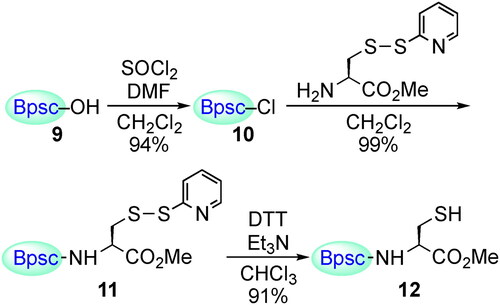
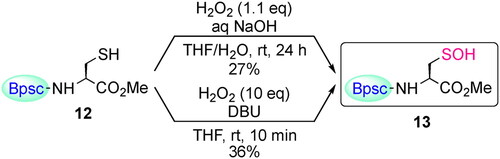
The cradled Cys–SH 12 bearing a Bpsc group was prepared from carboxylic acid 9 by the route shown in Scheme 4.[Citation30] The synthesis of Cys–SOH by direct oxidation of Cys–SH with H2O2 was then investigated. In the reaction of Cys–SH with H2O2, the initial products have been commonly assumed to be Cys–SOH, but there is no example in which this has been directly demonstrated, besides in the crystallographic monitoring of protein thiol oxidation.[Citation27,Citation28] Base or basic residues in biological systems are known to accelerate reactions of thiols with hydrogen peroxides.[Citation33–35] When Cys–SH 12 was treated with H2O2 in the presence of NaOH in THF/H2O at room temperature, the corresponding Cys–SOH 13 was formed as the major product (Scheme 5).[Citation30] Notably, Cys–SOH 13 was stable enough to be handled under standard laboratory conditions; 13 was purified by silica gel column chromatography in air and isolated as colorless crystals in 27% yield. Oxidation of 12 with H2O2 in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) also afforded 13 in the isolated yield of 36%. This is the first example of the synthesis and isolation of a small-molecule Cys–SOH. In the 1H NMR (C6D6) spectrum of 13, a singlet due to the SOH proton was observed at δ 5.36, which is readily exchangeable with D2O. The IR spectrum (CCl4) of 13 exhibited a broad absorption due to the OH stretching in the range of 3100–3400 cm−1.
Scheme 4. Preparation of Cys–SH 12.
Scheme 5. Synthesis of Cys–SOH 13 by H2O2 oxidation of Cys–SH 12.
The structure of 13 was finally established by single crystal X-ray diffraction analysis (). The cysteine moiety of 13 is anchored in a large cavity produced by the Bpsc cradle (dimensions ca. 2.3 × 1.7 nm). The S–O bond length (1.607(8) Å) is comparable to those of the primary-alkyl-substituted R–SOH 4 (S–O = 1.631(4) Å, C–S = 1.813(4) Å), which we previously reported.[Citation15] Furthermore, the bond length is significantly longer than those of sulfoxides (typical bond length: 1.50 Å),[Citation36] indicating that 13 has the sulfenyl form (R–S–O–H) rather than the sulfoxide form (R–S(O)–H). In the crystallographic analysis of Cys–SOH in proteins, the resolution has been limited to ca. 1.7 Å. The crystal structure of 13 allows detailed discussion of the structural parameters of Cys–SOH, which is expected to serve as the model for the geometric adjustment of a Cys–SOH moiety in protein crystallographic analysis.
Figure 4. Crystal structure of Cys–SOH 13: (a) stick representation (hydrogen atoms of the Bpsc group are omitted for clarity), and (b) space-filling representation.
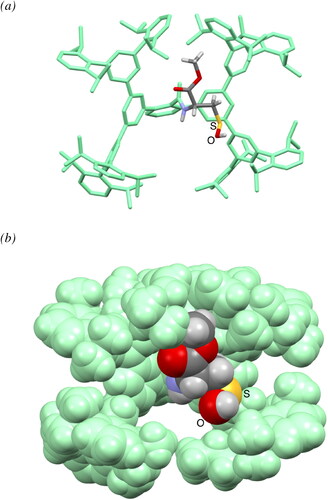
The cradled Cys–SOH 13 exhibited remarkable thermal stability: no detectable change was observed in the 1H NMR spectrum of 13 in C6D6 at 50 °C over 17 h. This result indicates that the peripheral steric protection due to the Bpsc cradle is very effective in suppressing the bimolecular decomposition process of a reactive R–SOH functionality even when it is anchored by a considerable length of linker.
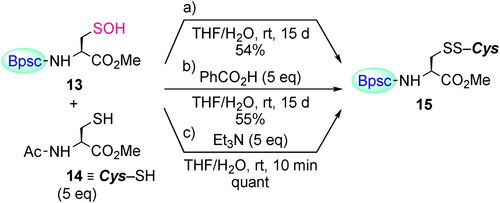
By taking advantage of the Cys–SOH with ‘shelf stability’, several fundamental reactivities of Cys–SOH were investigated. The reaction of Cys–SOH with a thiol to produce a disulfide is one of the most fundamental processes in redox regulation and protein folding.[Citation37–39] However, the chemical information for this process has been accumulated in a rather indirect fashion, that is, without direct observation of the reacting Cys–SOH. The reaction of Cys–SOH 13 with N-acetylcysteine methyl ester (14) in THF/H2O at room temperature afforded the corresponding mixed disulfide 15[Citation30] as the sole product, but the conversion of 13 to 15 was only 54% after 15 days (Scheme 6a). The addition of benzoic acid as an acid catalyst did not show any accelerating effect (Scheme 6b). In sharp contrast, the reaction in the presence of triethylamine was completed within 10 min to afford disulfide 15 quantitatively (Scheme 6c). In this reaction, triethylamine is likely to play crucial roles both in increasing the nucleophilicity of the thiol and in increasing the leaving ability of the OH group. These results suggest the importance of a basic residue in the vicinity of the –SOH functionality for facile disulfide formation in biological systems.
Scheme 6. Reaction of Cys–SOH 13 with thiol 14. The yields of 15 were estimated by no-D 1H NMR spectroscopy utilizing 1,3,5-trimethoxybenzene as an internal standard.
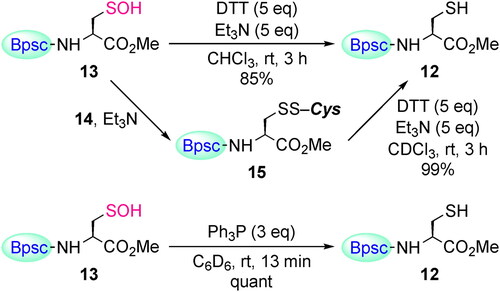
The redox process between thiols and sulfenic acids plays an important role both in biological and organic chemistry. The reduction of Cys–SOH to Cys–SH was investigated using the cradled cysteine models. When Cys–SOH 13 was treated with dithiothreitol (DTT) in the presence of triethylamine, Cys–SH 12 was obtained in 85% yield (Scheme 7), demonstrating the reversible redox processes between Cys–SH and Cys–SOH. The mixed disulfide 15 was also reduced to 12 under similar conditions. We previously reported that the non-cysteinyl RSOH 3 can be reduced to the corresponding thiol by the reaction with a phosphine.[Citation13,Citation40] Treatment of Cys–SOH 13 with triphenylphosphine in C6D6 afforded Cys–SH 12 quantitatively.
Scheme 7. Reduction of Cys–SOH 13 to Cys-SH 12.
Conclusions
By taking advantage of the cradled cysteine model, the first synthesis and isolation of a small-molecule Cys–SOH has been achieved and its structure was unambiguously established by X-ray crystallographic analysis. The reactivity of the cradled Cys–SOH with a thiol substantiated the important role of an amine base for the acceleration of the reaction of a sulfenic acid with a thiol. Recently, we also applied the Bpsc cradle to stabilize selenocysteine-derived reactive intermediates such as selenocysteine selenenic acids (Sec–SeOH) and selenocysteine selenenyl iodides (Sec–SeI) for modeling the catalytic processes of selenoenzymes.[Citation41–43] We envision that the cradled cysteine or selenocysteine models, having both high stability and sufficient reactivity, will serve as reference small-molecule models for various biologically important reactive intermediates in research efforts to improve our understanding of their chemical behavior in biological systems.
Acknowledgments
This work was partly supported by Grants-in-Aid for Scientific Research (B) (MEXT KAKENHI No. 15H03776 and 19H02698) (KG) and (C) (MEXT KAKENHI No. 22K05063) (SK). We are very grateful to Professor Adrian L. Schwan for his organization of a wonderful ISOCS-29 meeting in Guelph.
Disclosure statement
The authors report there are no competing interests to declare.
Additional information
Funding
References
- Allison, W. S. Formation and Reactions of Sulfenic Acids in Proteins. Acc. Chem. Res. 1976, 9, 293–299. DOI: 10.1021/ar50104a003.
- Poole, L. B.; Karplus, P. A.; Claiborne, A. Protein Sulfenic Acids in Redox Signaling. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 325–347. DOI: 10.1146/annurev.pharmtox.44.101802.121735.
- Paulsen, C. E.; Carroll, K. S. Orchestrating Redox Signaling Networks through Regulatory Cysteine Switches. ACS Chem. Biol. 2010, 5, 47–62. DOI: 10.1021/cb900258z.
- Paulsen, C. E.; Carroll, K. S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679. DOI: 10.1021/cr300163e.
- Gupta, V.; Carroll, K. S. Sulfenic Acid Chemistry, Detection and Cellular Lifetime. Biochim. Biophys. Acta. 2014, 1840, 847–875. DOI: 10.1016/j.bbagen.2013.05.040.
- Devarie-Baez, N. O.; Silva Lopez, E. I.; Furdui, C. M. Biological Chemistry and Functionality of Protein Sulfenic Acids and Related Thiol Modifications. Free Radic. Res. 2016, 50, 172–194. DOI: 10.3109/10715762.2015.1090571.
- Hogg, D. R. Chemistry of Sulfenic Acids and Esters. In The Chemistry of Sulfenic Acids and Their Derivatives, Patai, S. Ed.; New York: Wiley, 1990; pp 361–402. DOI: 10.1002/9780470772287.ch9.
- Nakamura, N. A Stable Sulfenic Acid, 9-Triptycenesulfenic Acid: its Isolation and Characterization. J. Am. Chem. Soc. 1983, 105, 7172–7173. DOI: 10.1021/ja00362a026.
- Yoshimura, T.; Tsukurimichi, E.; Yamazaki, S.; Soga, S.; Shimasaki, C.; Hasegawa, K. Synthesis of a Stable Sulfenic Acid, Trans-Decalin-9-Sulfenic Acid. J. Chem. Soc. Chem. Commun. 1992, 1337. DOI: 10.1039/c39920001337.
- Goto, K.; Tokitoh, N.; Okazaki, R. Synthesis of a Stable Arenesulfenic Acid Bearing a Bowl-Shaped Macrobicyclic Cyclophane Skeleton. Angew. Chem. Int. Ed. Engl. 1995, 34, 1124–1126. DOI: 10.1002/anie.199511241.
- Saiki, T.; Goto, K.; Tokitoh, N.; Okazaki, R. Synthesis and Structure of a Bridged Calix[6]Arene with a Sulfenic Acid Functionality in the Cavity. J. Org. Chem. 1996, 61, 2924–2925. DOI: 10.1021/jo960068q.
- Ishii, A.; Komiya, K.; Nakayama, J. Synthesis of a Stable Sulfenic Acid by Oxidation of a Sterically Hindered Thiol (Thiophenetriptycene-8-Thiol) and Its Characterization. J. Am. Chem. Soc. 1996, 118, 12836–12837. DOI: 10.1021/ja962995k.
- Goto, K.; Holler, M.; Okazaki, R. Synthesis, Structure, and Reactions of a Sulfenic Acid Bearing a Novel Bowl-Type Substituent: The First Synthesis of a Stable Sulfenic Acid by Direct Oxidation of a Thiol. J. Am. Chem. Soc. 1997, 119, 1460–1461. DOI: 10.1021/ja962994s.
- Goto, K.; Shimada, K.; Furukawa, S.; Miyasaka, S.; Takahashi, Y.; Kawashima, T. Formation of a Stable Sulfenic Acid by Hydrolysis of a Thionitrate and a Sulfenyl Bromide. Chem. Lett. 2006, 35, 862–863. DOI: 10.1246/cl.2006.862.
- Ishihara, M.; Abe, N.; Sase, S.; Goto, K. Synthesis, Structure, and Reactivities of a Stable Primary-Alkyl-Substituted Sulfenic Acid. Chem. Lett. 2015, 44, 615–617. DOI: 10.1246/cl.150046.
- Yukimoto, M.; Nishino, R.; Suzuki, F.; Ishihara, M.; Sugamata, K.; Minoura, M. Synthesis of a Peripherally Extended Triptycyl Group as an Aliphatic Steric Protection Group and Its Application to the Kinetic Stabilization of an Aliphatic Sulfenic Acid. Chem. Lett. 2018, 47, 425–428. DOI: 10.1246/cl.171230.
- Fries, K. alpha-Anthraquinone Sulphuric Acid [from the Chemical Institute of the University of Marburg]. Ber. Dtsch. Chem. Ges. 1912, 45, 2965–2973. DOI: 10.1002/cber.19120450323.
- Pal, B. C.; Uziel, M.; Doherty, D. G.; Cohn, W. E. Isolation and Characterization of a Pyrimidine Sulfenic Acid via Scission of the Sulfur-Sulfur Bond in the Methyl Analog of Bis(4-Thiouridine) Disulfide. J. Am. Chem. Soc. 1969, 91, 3634–3638. DOI: 10.1021/ja01041a036.
- Heckel, A.; Pfleiderer, W. Lumazinesulfenates - a New Class of Stable Sulfenic Acids. Tetrahedron Lett. 1983, 24, 5047–5050. DOI: 10.1016/S0040-4039(00)94037-1.
- Tripolt, R.; Belaj, F.; Nachbaur, E. Unexpectedly Stable Sulfenic Acid: 4,6-Dimethoxy-1,3,5-Triazine-2-Sulfenic Acid; Synthesis, Properties, Molecular and Crystal Structure. Z. Naturforsch., B: Chem. Sci. 1993, 48, 1212–1222. DOI: 10.1515/znb-1993-0909.
- Machiguchi, T.; Hasegawa, T.; Otani, H.; Yamabe, S.; Mizuno, H. Tropothione S-Oxide: The First Example of a Sulfine Charge Reversion (Umpolung). J. Am. Chem. Soc. 1994, 116, 407–408. DOI: 10.1021/ja00080a061.
- Fekner, T.; Baldwin, J. E.; Adlington, R. M.; Schofield, C. J. Unusually Stable Azetidinone Sulfenic Acids. Tetrahedron Lett. 1998, 39, 6983–6986. DOI: 10.1016/S0040-4039(98)01482-8.
- Li, X.-B.; Xu, Z.-F.; Liu, L.-J.; Liu, J.-T. Synthesis and Identification of Solution-Stable Sulfenic Acids: Perfluoroalkanesulfenic Acids. Eur. J. Org. Chem. 2014, 2014, 1182–1188. DOI: 10.1002/ejoc.201301563.
- Yeh, J. I.; Claiborne, A.; Hol,.; W. G.; J. Structure of the Native Cysteinesulfenic Acid Redox Center of Enterococcal NADH Peroxidase Refined at 2.8 Å Resolution. Biochemistry 1996, 35, 9951–9957. DOI: 10.1021/bi961037s.
- Choi, H.-J.; Kang, S. W.; Yang, C.-H.; Rhee, S. G.; Ryu, S.-E. Crystal Structure of a Novel Human Peroxidase Enzyme at 2.0 Å Resolution. Nat. Struct. Biol. 1998, 5, 400–406. DOI: 10.1038/nsb0598-400.
- Poor, C. B.; Chen, P. R.; Duguid, E.; Rice, P. A.; He, C. Crystal Structures of the Reduced, Sulfenic Acid, and Mixed Disulfide Forms of SarZ, a Redox Active Global Regulator in Staphylococcus aureus. J. Biol. Chem. 2009, 284, 23517–23524. DOI: 10.1074/jbc.m109.015826.
- van Montfort, R. L. M.; Congreve, M.; Tisi, D.; Carr, R.; Jhoti, H. Oxidation State of the Active-Site Cysteine in Protein Tyrosine Phosphatase 1B. Nature (London, U. K.) 2003, 423, 773–777. DOI: 10.1038/nature01681.
- Nakamura, T.; Yamamoto, T.; Abe, M.; Matsumura, H.; Hagihara, Y.; Goto, T.; Yamaguchi, T.; Inoue, T. Oxidation of Archaeal Peroxiredoxin Involves a Hypervalent Sulfur Intermediate. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 6238–6242. DOI: 10.1073/pnas.0709822105.
- Sase, S.; Kimura, R.; Masuda, R.; Goto, K. Model Study on Trapping of Protein Selenenic Acids by Utilizing a Stable Synthetic Congener. New J. Chem. 2019, 43, 6830–6833. DOI: 10.1039/C9NJ01072F.
- Sano, T.; Masuda, R.; Sase, S.; Goto, K. Isolable Small-Molecule Cysteine Sulfenic Acid. Chem. Commun. (Camb) 2021, 57, 2479–2482. DOI: 10.1039/d0cc08422k.
- Ishii, A.; Matsubayashi, S.; Takahashi, T.; Nakayama, J. Preparation of a Selenenic Acid and Isolation of Selenoseleninates. J. Org. Chem. 1999, 64, 1084–1085. DOI: 10.1021/jo982039g.
- Sase, S.; Kakimoto, R.; Goto, K. Synthesis of a Stable Selenoaldehyde by Self-Catalyzed Thermal Dehydration of a Primary-Alkyl-Substituted Selenenic Acid. Angew. Chem. Int. Ed. Engl. 2015, 54, 901–904. DOI: 10.1002/anie.201409485.
- Yang, K. S.; Kang, S. W.; Woo, H. A.; Hwang, S. C.; Chae, H. Z.; Kim, K.; Rhee, S. G. Inactivation of Human Peroxiredoxin I during Catalysis as the Result of the Oxidation of the Catalytic Site Cysteine to Cysteine-Sulfinic Acid. J. Biol. Chem. 2002, 277, 38029–38036. DOI: 10.1074/jbc.M206626200.
- Portillo-Ledesma, S.; Sardi, F.; Manta, B.; Tourn, M. V.; Clippe, A.; Knoops, B.; Alvarez, B.; Coitino, E. L.; Ferrer-Sueta, G. Deconstructing the Catalytic Efficiency of Peroxiredoxin-5 Peroxidatic Cysteine. Biochemistry 2014, 53, 6113–6125. DOI: 10.1021/bi500389m.
- Chauvin, J.-P. R.; Pratt, D. A. On the Reactions of Thiols, Sulfenic Acids, and Sulfinic Acids with Hydrogen Peroxide. Angew. Chem. Int. Ed. Engl. 2017, 56, 6255–6259. DOI: 10.1002/anie.201610402.
- Allen, F. H.; Kennard, O.; Watson, D. G.; Brammer, L.; Orpen, A. G.; Taylor, R. Tables of Bond Lengths Determined by x-Ray and Neutron Diffraction. Part 1. Bond Lengths in Organic Compounds. J. Chem. Soc, Perkin Trans. 2 1987, S1. DOI: 10.1039/p298700000s1.
- Rehder, D. S.; Borges, C. R. Cysteine Sulfenic Acid as an Intermediate in Disulfide Bond Formation and Nonenzymatic Protein Folding. Biochemistry 2010, 49 (35), 7748–7755. Hill, B. G.; Bhatnagar, A. Protein S-Glutathiolation: Redox-Sensitive Regulation of Protein Function. J. Mol. Cell. Cardiol. 2012, 52 (3), 559–567. DOI: 10.1016/j.yjmcc.2011.07.009.
- Zeida, A.; Trujillo, M.; Ferrer-Sueta, G.; Denicola, A.; Estrin, D. A.; Radi, R. Catalysis of Peroxide Reduction by Fast Reacting Protein Thiols. Chem. Rev. 2019, 119, 10829–10855. DOI: 10.1021/acs.chemrev.9b00371.
- Hagras, M. A.; Bellucci, M. A.; Gobbo, G.; Marek, R. A.; Trout, B. L. Computational Modeling of the Disulfide Cross-Linking Reaction. J. Phys. Chem. B 2020, 124, 9840–9851. DOI: 10.1021/acs.jpcb.0c07510.
- Goto, K.; Shimada, K.; Nagahama, M.; Okazaki, R.; Kawashima, T. Reaction of Stable Sulfenic and Selenenic Acids Containing a Bowl-Type Steric Protection Group with a Phosphine. Elucidation of the Mechanism of Reduction of Sulfenic and Selenenic Acids. Chem. Lett. 2003, 32, 1080–1081. DOI: 10.1246/cl.2003.1080.
- Masuda, R.; Kimura, R.; Karasaki, T.; Sase, S.; Goto, K. Modeling the Catalytic Cycle of Glutathione Peroxidase by Nuclear Magnetic Resonance Spectroscopic Analysis of Selenocysteine Selenenic Acids. J. Am. Chem. Soc. 2021, 143, 6345–6350. DOI: 10.1021/jacs.1c02383.
- Masuda, R.; Goto, K. Modeling of Selenocysteinederived Reactive Intermediates Utilizing a Nano-Sized Molecular Cavity as a Protective Cradle. Methods Enzymol. 2022, 662, 331–361. DOI: 10.1016/bs.mie.2021.10.018.
- Masuda, R.; Kuwano, S.; Sase, S.; Bortoli, M.; Madabeni, A.; Orian, L.; Goto, K. Model Study on the Catalytic Cycle of Glutathione Peroxidase Utilizing Selenocysteine-Containing Tripeptides: Elucidation of the Protective Bypass Mechanism Involving Selenocysteine Selenenic Acids. Bull. Chem. Soc. Jpn. 2022, 95, 1360–1379. DOI: 10.1246/bcsj.20220156.