
Pathologic activation of the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway is an oncogenic driver for many malignancies, including lymphomas [Citation1]. Although validated as a therapeutic oncologic target, the PI3K signaling pathway is also implicated in normal glucose homeostasis. Specifically, since the PI3K subunit p110α is chiefly responsible for downstream insulin receptor (INSR) signaling, PI3K signaling inhibition that includes p110α leads to severe hyperglycemia. Both compensatory endogenous insulin release as well as treatment of hyperglycemia with exogenous insulin/insulin mimetics activate INSR-associated PI3K signaling, ultimately limiting blockade of tumor-associated PI3K signaling [Citation2]. While aggressive lymphomas driven by pathologic PI3K signaling respond to combined PI3Kα and δ/γ inhibition, the antitumor effect is likely submaximal due to endogenous insulin feedback with subsequent PI3K activation, and more intense dosing of PI3K inhibitors is difficult to achieve safely due to severe hyperglycemia [Citation3].
Attempts to control PI3Kα-associated hyperglycemia have included diet, metformin, sodium-glucose cotransporter-2 (SGLT2) inhibitors, and insulin [Citation4]. Insulin is problematic as it drives PI3K signaling that supports cancer growth [Citation2]. Even endogenous insulin secretion, either occurring as feedback to PI3K induced hyperglycemia or as response to anti-hyperglycemic treatment with ‘secretagogues’, would counteract the anti-cancer effect of PI3K inhibition [Citation2]. Hypoglycemic agents that would not be predicted to activate the PI3K pathway include metformin and SGLT2 inhibitors as they do not stimulate insulin production. Disappointingly, when metformin was given to mice prior to the administration of PI3K inhibitor, it had minimal impact on PI3K induced hyperglycemia and hyperinsulinemia. SGLT2 inhibition successfully suppressed both the insulin feedback and PI3K reactivation, but SGLT2 inhibition is not without drawbacks as SGLT2 inhibitors, which increase lipolysis and contribute to dehydration, have been reported to cause euglycemic diabetic ketoacidosis (DKA) [Citation2,Citation5]. This has specifically occurred in a non-diabetic patient with metastatic breast cancer with hyperglycemia on a PI3K inhibitor [Citation6]. Finally, the ketogenic diet may be successful in curbing PI3K induced hyperglycemia and hyperinsulinemia, but this is difficult to apply in practice for most patients. Therefore, a novel strategy to maintain the blockade of tumor-associated PI3K signaling while reducing hyperglycemia is needed.
Euglycemia is normally achieved by a balance of both insulin and its opposing hormone, glucagon. Insulin is secreted during the ‘fed state’ in order to enable glucose utilization, whereas glucagon is secreted during the ‘fasting state’, promoting hepatic glycogenolysis and gluconeogenesis. In insulin deficiency, insulin resistance, or lack of insulin receptor signaling as in PI3Kα blockade, there is overall glucose underutilization similar to ‘fasting’ states, and plasma glucagon levels are elevated. In this setting of hyperglycemia, paradoxically high glucagon levels would be expected to promote hepatic glucose output, further worsening hyperglycemia [Citation7,Citation8]. This unhelpful feed-forward cycle is present in all types of diabetes, and glucagon is now recognized as a key pathogenic factor [Citation9]. Remarkably, glucagon receptor-deficient mice that have undergone beta islet cell destruction do not develop diabetes unless glucagon receptor function is restored [Citation10,Citation11]. It follows then that abrogating glucagon receptor (GCGR) signaling using an inhibitory monoclonal antibody to the glucagon receptor should ameliorate glycemic control in diabetes. This has been shown in mice, non-human primates, and in human clinical trials of patients with type 1 diabetes [Citation12–16]. Therefore, we hypothesized that inhibition of GCGR signaling, a pathway that does not activate PI3K, would be an attractive strategy to normalize PI3K inhibitor-induced hyperglycemia without disrupting the antitumor PI3K blockade.
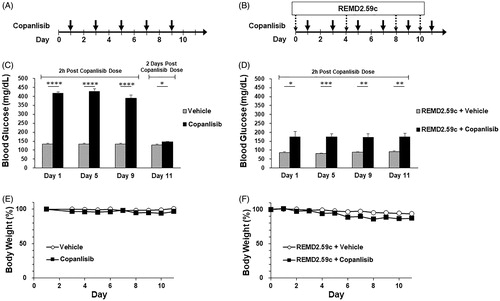
The effect of GCGR blockade on PI3K inhibitor-induced hyperglycemia was first evaluated in a non-tumor bearing mouse model. The human IgG2 anti-GCGR mAb, REMD2.59, was previously found to improve fasting blood glucose in diabetic mice [Citation17]. Here, a functionally equivalent murine IgG1 anti-GCGR mAb, REMD2.59c (REMD Biotherapeutics Inc, Camarillo CA), was administered to CB17 SCID mice receiving the PI3K inhibitor copanlisib, or vehicle only control. There were 5 mice per treatment group. Copanlisib was administered by tail vein injection every other day at the MTD of 15 mg/kg, and REMD2.59c was given by intraperitoneal injection on the day prior to the first, third, fifth, and sixth copanlisib injections (). Blood glucose levels were measured 2 h after copanlisib injection. Significant hyperglycemia was observed after copanlisib treatment alone (), and this was corrected by REMD2.59c pretreatment (), confirming the effectiveness of GCGR blockade. The corrective effect on glucose was maintained over the study period. Weight loss occurred in mice treated with copanlisib and REMD2.59c (Figure1(E,F)); less weight loss was seen in mice receiving REMD2.59c without copanlisib.
Figure 1. Blood glucose level and change of body weight in CB17 SCID mice treated with copanlisib with or without REMD 2.59c. Healthy female CB17 SCID mice, aged 6–8 weeks with body weight of normal range (20–21 g), were included in the study. Mice had free access to sterilized dry granule food and water during the entire study period. (A) Mice received vehicle or copanlisib (15 mg/kg) via tail vein injection every other day (days 1, 3, 5, 7, and 9). (B) As in A, mice received intravenous administration of copanlisib every other day (days 1, 3, 5, 7, 9 and 11). The mice also received intraperitoneal injection of vehicle or REMD2.59c mAb (7 mg/kg) on days 0, 4, 8 and 10. (C) Blood glucose levels were measured by tail vein nick using an Accu-ChekTM glucometer 2h after dosing (days 1, 5 and 9) and 2 days after final dosing (day 11) of copanlisib. Data are shown as mean ± SEM. (D) As in C, blood glucose levels were measured 2h after dosing (days 1, 3, 9 and 11). Student’s t-test was performed for the comparison of blood glucose levels. p value: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (E, F) Body weights of all animals were measured throughout the study. Body weight change, expressed in %, was calculated using the formula: BW change (%) = (BW Day x/BW Day 0) × 100%. Data are shown as mean ± SEM.
Having demonstrated that GCGR blockade controls hyperglycemia caused by PI3K inhibition with copanlisib in a mouse model, we next treated a 54-year-old non-diabetic woman with relapsed and refractory peripheral T cell lymphoma (PTCL) on an IRB-approved clinical pilot study of copanlisib in combination with the anti-human GCGR mAb REMD-477. Our patient had highly aggressive TP53 mutated PTCL NOS that rapidly progressed through multiple prior lines of therapy—CHOEP/CHOP, pralatrexate, revlimid, brentuximab vedotin, gemcitabine, duvelisib, and several week-long courses of 40 mg dexamethasone daily. As the next line of therapy, copanlisib was to be given intravenously on days 1, 8, and 15 of a 28-day cycle as per prior studies in lymphoma [Citation3,Citation18]. Following copanlisib 60 mg IV alone, glucose increased from a baseline of 100 mg/dl to a maximum of 592 mg/dl at 5 h.
In response to severe copanlisib induced hyperglycemia, REMD-477 was given prior to the second and third doses of copanlisib as a 70 mg subcutaneous injection, a dose and route well-tolerated in a previous clinical trial [Citation12]. Following pretreatment with REMD-477, copanlisib induced hyperglycemia was significantly ameliorated without significant episodes of hypoglycemia (). With improved glycemic control on REMD-477, the patient did not require any dose reductions or treatment delays.
Figure 2. Blood glucose levels in a patient treated with copanlisib without REMD-477 and then with REMD-477. A patient with relapsed/refractory peripheral T cell lymphoma (PTCL) NOS treated with 60mg IV copanlisib alone experienced hyperglycemia as shown in the left panel. The patient provided a signed informed consent to participate in an IRB-approved clinical pilot study of REMD-477 plus copanlisib. The patient received 70mg REMD-477 subcutaneously (SQ) 1 day before copanlisib in weeks one and two. Blood glucose was measured at selected time points and shown in the right panel. Solid arrows indicate the time of copanlisib administration, and dashed arrows indicate administration of REMD-477.
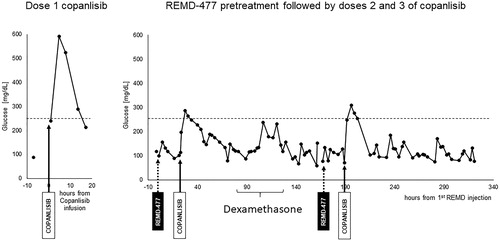
During study treatment, the patient self-administered dexamethasone 16 mg daily for two days for fatigue which contributed to transient hyperglycemia during this time. After the 3rd dose of copanlisib, she was hospitalized for one night for acute epigastric abdominal pain, a symptom she had previously experienced during her disease course prior to treatment on this clinical trial. The pain was symptomatically managed and resolved without intervention. Tumor imaging by CT of the chest abdomen and pelvis showed a treatment response with an interval decrease in all sites of lymphadenopathy without evidence of new sites of disease or splenomegaly. After one week off of treatment as per the copanlisib dosing schedule, our patient clinically progressed with painful cervical lymphadenopathy and therefore started a subsequent line of therapy.
The PI3K pathway, an important intracellular regulator in normal lymphocytes, is a clinically validated therapeutic target in the treatment of lymphoma; but as an almost ubiquitous pathway, PI3K inhibition results in various on-target toxicities that may limit patients from receiving treatment. In lymphocytes, BCR and TCR signaling pathways rely on PI3 kinases for downstream signal transduction; in lymphoma, tonic BCR and TCR signaling results in continuous PI3K activation, and subsequent neoplastic proliferation [Citation19]. While the p110α and p110β subunits are expressed in all tissues, p110δ and p110γ are restricted to B and T cells respectively [Citation20]. Selective inhibition of PI3Kδ can result in severe autoimmune effects attributed to preferential inhibition of regulatory T cell function [Citation21]. Idelalisb and duvelisib both carry boxed warnings of potentially fatal pneumonitis, hepatotoxicity, and colitis, whereas copanlisib, a pan PI3K inhibitor does not. Our patient with relapsed/refractory T cell lymphoma had previously progressed through 6 lines of therapy, including duvelisib, a selective PI3K δ/γ inhibitor. She had significant gastrointestinal toxicity on this drug, necessitating a dose reduction and ultimate discontinuation. Even though copanlisib caused grade 3 hyperglycemia in our patients, as was reported in 41% of patients in the phase III study of copanlisib, our patient had successful control of hyperglycemia with GCGR blockade with subsequent doses [Citation22]. Without delay or dose reduction, she achieved clinical and radiographic improvement in her highly refractory lymphoma.
These data support targeting the GCGR pathway as an effective way to address PI3Kα inhibitor induced hyperglycemia without counteracting tumor PI3K inhibition. Unlike SGLT2 inhibition which has been reported to cause DKA, GCGR blockade actually decreases ketogenesis and does not cause dehydration, making this toxicity unlikely [Citation14]. GCGR blockade may also enable more intensive dosing of the PI3K inhibitor, to further inhibit oncogenic PI3K signaling, and maximize the antitumor effect. Finally, by successful control of hyperglycemia, GCGR blockade would also be predicted to suppress endogenous insulin secretion, an under-recognized mechanism for tumor resistance to PI3Kα blocking agents. Our study of the GCGR mAb REMD-477 as a novel strategy to counteract PI3K inhibitor-induced hyperglycemia is an advance that may allow for more effective and safer use of potent PI3K inhibitors in the treatment of aggressive lymphomas.
Author contributions
J.W., T.O., and M.A.M. performed the research and wrote the paper. F.C., H.Y., and Z.T. performed the research. H.K.L. designed the research. All authors edited and approved the manuscript.
Disclosure statement
J.W. and T.O. have no relevant conflicts of interest. F.C. was a recent former employee of REMD Biotherapeutics and has stock options. HY and DT are current employees of REMD Biotherapeutics and have equity. HKL is a consultant for REMD Biotherapeutics.
References
- Jerkeman M, Hallek M, Dreyling M, et al. Targeting of B-cell receptor signalling in B-cell malignancies. J Intern Med. 2017;282(5):415–428.
- Hopkins BD, Pauli C, Du X, et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature. 2018;560(7719):499–503.
- Dreyling M, Morschhauser F, Bouabdallah K, et al. Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann Oncol. 2017;28(9):2169–2178.
- Esposito A, Viale G, Curigliano G. Safety, tolerability, and management of toxic effects of phosphatidylinositol 3-kinase inhibitor treatment in patients with cancer: a review. JAMA Oncol. 2019;5(9):1347–1354.
- Peters AL, Buschur EO, Buse JB, et al. Euglycemic diabetic ketoacidosis: a potential complication of treatment with sodium-glucose cotransporter 2 inhibition. Diabetes Care. 2015;38(9):1687–1693.
- Bowman C, Abramson V, Wellons M. Ketoacidosis with canagliflozin prescribed for phosphoinositide 3-kinase inhibitor-induced hyperglycemia: a case report. J Investig Med High Impact Case Rep. 2017;5(3):1–4.
- Crouthamel MC, Kahana JA, Korenchuk S, et al. Mechanism and management of AKT inhibitor-induced hyperglycemia. Clin Cancer Res. 2009;15(1):217–225.
- Brown RJ, Sinaii N, Rother KI. Too much glucagon, too little insulin: time course of pancreatic islet dysfunction in new-onset type 1 diabetes. Diabetes Care. 2008;31(7):1403–1404.
- Lee YH, Wang MY, Yu XX, et al. Glucagon is the key factor in the development of diabetes. Diabetologia. 2016;59(7):1372–1375.
- Lee Y, Wang MY, Du XQ, et al. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes. 2011;60(2):391–397.
- Lee Y, Berglund ED, Wang M-y, et al. Metabolic manifestations of insulin deficiency do not occur without glucagon action. Proc Natl Acad Sci USA. 2012;109(37):14972–14976.
- Pettus J, Reeds D, Cavaiola TS, et al. Effect of a glucagon receptor antibody (REMD-477) in type 1 diabetes: a randomized controlled trial. Diabetes Obes Metab. 2018;20(5):1302–1305.
- Okamoto H, Kim J, Aglione J, et al. Glucagon receptor blockade with a human antibody normalizes blood glucose in diabetic mice and monkeys. Endocrinology. 2015;156(8):2781–2794.
- Okamoto H, Cavino K, Na E, et al. Glucagon receptor inhibition normalizes blood glucose in severe insulin-resistant mice. Proc Natl Acad Sci USA. 2017;114(10):2753–2758.
- Yan H, Gu W, Yang J, et al. Fully human monoclonal antibodies antagonizing the glucagon receptor improve glucose homeostasis in mice and monkeys. J Pharmacol Exp Ther. 2009;329(1):102–111.
- Gu W, Yan H, Winters KA, et al. Long-term inhibition of the glucagon receptor with a monoclonal antibody in mice causes sustained improvement in glycemic control, with reversible alpha-cell hyperplasia and hyperglucagonemia. J Pharmacol Exp Ther. 2009;331(3):871–881.
- Lang S, Wei R, Wei T, et al. Glucagon receptor antagonism promotes the production of gut proglucagon-derived peptides in diabetic mice. Peptides. 2020;131(June):170349.
- Dreyling M, Santoro A, Mollica L, et al. Phosphatidylinositol 3-kinase inhibition by Copanlisib in relapsed or refractory indolent lymphoma. J Clin Oncol. 2017;35(35):3898–3905.
- Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013;12(3):229–243.
- Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. 2003;3(4):317–330.
- Chellappa S, Kushekhar K, Munthe LA, et al. The PI3K p110δ isoform inhibitor idelalisib preferentially inhibits human regulatory T cell function. J Immunol. 2019;202(5):1397–1405.
- Dreyling M, Santoro A, Mollica L, et al. Long-term safety and efficacy of the PI3K inhibitor copanlisib in patients with relapsed or refractory indolent lymphoma: 2-year follow-up of the CHRONOS-1 study. Am J Hematol. 2020;95:362–371.