
ABSTRACT
Background
Emodin is a traditional medicine that has been shown to exert anti-inflammatory and anti-oxidative effects. Previous research has indicated that emodin can alleviate myocardial remodeling and inhibit myocardial hypertrophy and fibrosis. However, the mechanism by which emodin affects myocardial fibrosis (MF) has not yet been elucidated.
Methods
Fibroblasts were treated with ANGII, and a mouse model of MF was established by ligation of the left anterior descending coronary artery. Cell proliferation was examined by a Cell Counting Kit-8 (CCK8) assay. Dihydroethidium (DHE) was used to measure reactive oxygen species (ROS) levels, and Masson and Sirius red staining were used to examine changes in collagen fiber levels. PI3K was over-expressed by lentiviral transfection to verify the effect of emodin on the PI3K/AKT/mTOR signaling axis. Changes in cardiac function in each group were examined by echocardiography.
Results
Emodin significantly inhibited fibroblast proliferation, decreased intracellular ROS levels, significantly upregulated collagen II expression, downregulated α-SMA expression, and inhibited PI3K/AKT/mTOR pathway activation in vitro. Moreover, the in vivo results were consistent with the in vitro. Emodin significantly decreased ROS levels in heart tissue and reduced collagen fibrillogenesis. Emodin could regulate the activity of PI3K to increase the expression of collagen II and downregulate α-SMA expression in part through the PI3K/AKT/mTOR pathway, and emodin significantly improved cardiac structure and function in mice.
Conclusions
This study revealed that emodin targeted the PI3K/AKT/mTOR pathway to inhibit the development of myocardial fibrosis and may be an antifibrotic agent for the treatment of cardiac fibrosis.
Introduction
Chronic heart failure caused by coronary atherosclerosis threatens human health and has attracted widespread attention. A considerable number of patients with myocardial infarction cannot achieve timely, efficient and successful reperfusion for various reasons, leading to local myocardial infarction. The myocardium in the noninfarcted area undergoes myocardial remodeling, which eventually leads to heart failure or arrhythmia, resulting in increased patient mortality rate.Citation1,Citation2 Myocardial fibrosis is an important marker of myocardial remodeling. Although therapeutic strategies have improved over the past few decades, there is no ideal treatment for myocardial fibrosis.
Oxidative stress is a common pathological feature of myocardial infarction. Several studies have shown that an increase in reactive oxygen species (ROS) after ischemia destroys cell structure, induces mitochondrial dysfunction and stimulates the apoptotic signaling cascade, leading to left ventricular (LV) dysfunction.Citation3 Excessive ROS induce cells to secrete a variety of profibrotic factors, leading to the synthesis of collagen by perivascular fibroblasts and the development of perivascular fibrosis. Then, collagen penetrates the interstitial space of myocardial cells, leading to interstitial fibrosis. Therefore, inhibiting ROS production and targeting signal transduction pathways that regulate the proliferation of cardiac fibroblasts can reduce the progression of cardiac fibrosis.
Emodin, which is an anthraquinone derivative, has attracted immense interest due to its antioxidative, antitumor, and anti-inflammatory effects. We previously showed that emodin significantly inhibited ROS production, alleviated oxidative stress damage, and inhibited malignant cell proliferation.Citation4 Previous studies have shown that emodin reduces oxidative stress damage and inflammation and alleviates the progression of various types of tissue fibrosis, including myocardial fibrosis,Citation5 liver fibrosis,Citation6 pulmonary fibrosis,Citation7 and renal fibrosis.Citation8,Citation9 However, the molecular mechanism by which emodin alleviates myocardial fibrosis has not been fully elucidated. Studies have shown that the PI3K/AKT/mTOR signaling pathway is involved in regulating cell proliferation, metabolism, differentiation and apoptosis. The PI3K/AKT/mTOR signaling pathway is significantly activated in myocardial fibrosis.Citation10 Emodin derivatives mediate changes in the PI3K/AKT/mTOR pathway to inhibit renal fibrosis.Citation9 Emodin derivatives protect against cerebral ischemia‒reperfusion injury via the PI3K/AKT/mTOR and NF-κB signaling pathways.Citation11 Therefore, we hypothesize that emodin can alleviate myocardial fibrosis by regulating the PI3K/AKT/mTOR signaling pathway.
In the present study, in vivo and in vitro models of myocardial fibrosis were used to examine the effects of emodin on myocardial fibrosis and oxidative stress, and changes in the PI3K/AKT/mTOR signaling pathway were examined. The possible mechanism associated with myocardial remodeling after muscle fibrosis was preliminarily explored.
Materials and methods
Cell culture
Cardiac fibroblasts were purchased from the Shanghai Institute of Cells Chinese Academy of Sciences, and the cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, HyClone, USA) supplemented with 10% FBS (Gibco, USA) and 1% penicillin-streptomycin solution (Hyclone, USA) at 37°C and 5% CO2. ANGII (10−7 M)(Sigma, USA) was used to establish an in vitro MF model, these methods were consistent with our previous experiments.Citation12
Cell counting Kit8 (CCK8) assay
Cardiac fibroblasts (8000 cells/well) were cultured in 96-well plates and divided into three groups. The Sham group was not treated with ANGII or emodin, the MF group was treated with ANGII (10−7 M), and the MF+Emodin group was treated with ANGII (10−7 M) plus emodin (10 μM,≥90% (HPLC)) (Sigma, USA). After 24 hours, the original medium was removed, and 110 μL of CCK8 working solution was added (10 μL CCK8 stock solution +100 μL DMEM) and incubated for 2 hours. A microplate reader (Thermo, USA) was used to measure the absorption at 450 nm and calculate the changes in cell proliferation in each group.
Dihydroethidium (DHE) staining
First, the original culture medium in each group of cells was washed away with PBS. Then, 10 μM DHE reagent (Biosharp, China) was added and incubated at 37°C for 30 min. ROS levels in each group of cells were observed by fluorescence microscopy (Olympus, IX81, Japan) at an excitation wavelength of 535 nm and an emission wavelength of 610 nm.
Western blot analysis
Total protein was extracted from cardiac fibroblasts and myocardial samples. The proteins were separated and transferred to polyvinylidene fluoride (PVDF) membranes. Then, the PVDF membranes were blocked for 2 h at room temperature and incubated overnight at 4°C with one of the following primary antibodies: anti-collagen II (Abcam, ab188570, UK), anti-SMA (Abcam, ab7817, UK), PI3K (Abcam, ab191606, UK), p-PI3K (Abcam, ab182651, UK), AKT (Abcam, ab179463, UK), p-AKT (Abcam, ab192623, UK), mTOR (Abcam, ab134903, UK), p-mTOR (CST, 5536, USA), GAPDH (Bioss, bs-0755 R, China), goat anti-mouse (Bioss, bs-0296 G, China), or goat anti-rabbit (Bioss, bs-0295 G, China). The procedures were performed as previously described, and protein expression was examined by a chemiluminescence analyzer (Azure, A300, USA).
Lentivirus transfection
The cells were cultured in a 10 cm petri dish, and when the cells were 70% confluent, medium containing a lentivirus to overexpress PI3K or the scrambled negative control (NC)(Hanbio, China) was added. The multiplicity of infection (MOI) was 20, and polybrene (6 μg/mL; Hanbio, China) was added to assist in the transfection. After 4 hours, 5 mL of medium was added. After 24 hours, medium containing the virus was removed and replaced with fresh medium, and the cells were cultured for an additional 24 hours. The cells were collected, the transfection efficiency was assessed by fluorescence microscopy, and WB was used to determine the expression of PI3K.Citation13
Animal myocardial fibrosis model
Animal experiments were approved by the Ethics Committee of the General Hospital of Western Theater Command (protocol code 2020-ky75, June 20th, 2020) and complied with the Guidelines for Animal Experiments on Laboratory Animals. Thirty-two male C57BL/6J mice (20 ± 2 g) were purchased from Chengdu Dashuo Experimental Animal Company and adaptively fed for 1 week at a temperature of 22°C with 50% humidity and a 12-hour day/night cycle. The animals were randomly divided into four groups: Sham group (n = 8), MF model group (n = 8), MF + Emodin group (n = 8), MF + Emodin + OV-PI3K group (n = 8). The MF model was established by ligating the left anterior descending coronary artery of the heart, and animals in the sham group were subjected to the same protocol, with the exception of coronary artery ligation. The MF + Emodin group was administered emodin (70 mg/kg, Sigma) by gavage once daily. The dose of emodin used was determined based on other studies.Citation8,Citation14 The MF + Emodin + OV-PI3K group was injected with the lentivirus through the tail vein 2 weeks prior to model establishment, and the remaining steps were the same as those in the MF + Emodin group.
Echocardiography
Cardiac ultrasound was used to examine the cardiac function of each group. Mice were anesthetized with 1.5% isoflurane, than chest hair was removed, and cardiac function and dimensions were measured by the Vevo 3100LT (Visual Sonics, Toronto, Canada) ultrasound system with a 40-MHz transducer. Cardiac function (left ventricular end-diastolic diameter (LVEDD), left ventricular end-systolic diameter (LVESD), left ventricular ejection fraction (EF), left ventricular fractional shortening (FS), left ventricular end-systolic pressure (LVESP), left ventricular end-diastolic pressure (LVEDP), and heart rate (HR)) was analyzed by ultrasonic software (Visual Sonics, Toronto, Canada).
Hematoxylin and eosin staining
Hearts were removed and immediately fixed in 4% paraformaldehyde for 24 h, conventionally dehydrated, paraffin embedded, sectioned at a thickness of 4 μm, and routinely deparaffinized and rehydrated. Six paraffin sections from each group were randomly selected for H&E staining (Solarbio, G1120, China). Pathological changes in the heart were observed under a light microscope (Leica, DM3000, Germany).
Masson staining
Cardiac pretreatment was performed as described in Section 2.8. Following the instructions of the Masson kit (Solarbio, G1340, China), images were taken with an optical microscope (Leica, DM3000, Germany), and Image-Pro Plus software (Media Cybernetics, Bethesda, USA) was used to calculate the degree of fibrosis in each group.
Sirius red staining
Cardiac pretreatment was the same as that described in Section 2.8. The sections were stained with iron hematoxylin staining solution (Solarbio, G1472, China) for 5 min, washed for 10 min with distilled water, stained for 15 min with Sirius red dye solution (Solarbio, G1472, China), and washed for 10 min with distilled water. Then, the sections were naturally dried, neutrally sealed with gum, photographed and processed as described in Section 2.9.
Statistical analysis
Statistical analysis was performed using IBM SPSS Statistics software V19.0 (IBM, USA). The data are expressed as the mean ± SEM. Before statistical analysis on the experimental data, the normality of the data distribution is tested using the Shapiro-Wilk test. For normally distributed data, use unpaired Student’s t-test (two tailed) to compare two groups, and use one-way ANOVA and Tukey’s post hoc test to compare the mean values of each group with the other group. For non-normally distributed data, the Mann-Whitney test is used for comparison between two groups, while the Kruskal-Wallis test and Dunnett’s multiple comparison test are used for comparison between multiple groups. p value < 0.05 was statistically significant.
Results
Emodin inhibits ANGII-induced proliferation in cardiac fibroblasts by decreasing ROS levels
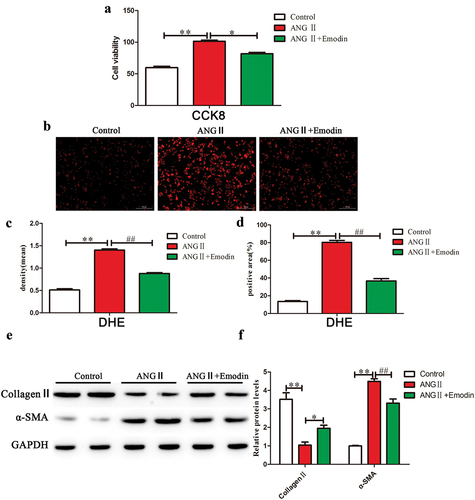
To investigate the effect of emodin on myocardial fibrosis, we used fibroblasts to evaluate the inhibitory effect of emodin on the proliferation of cardiac fibroblasts. CCK8 was used to evaluate the effect of emodin on the proliferation of cardiac fibroblasts induced by ANGII. ANGII significantly promoted the proliferation of fibroblasts compared with that in the sham group (p < .05) (). Then, we confirmed that 10 μM of emodin can significantly inhibit cell proliferation by adding different concentrations of emodin (Figure S1). Compared with that in the ANG II group, emodin significantly inhibited the proliferation of fibroblasts (p < .05) (). To evaluate the effect of emodin on ROS production, we measured the fluorescence intensity of ROS in cardiac fibroblasts using a dihydroethyl ether (DHE) probe. The results showed that ANGII induced ROS production in fibroblasts, and the effects could be reversed by emodin (). We examined the mechanism by which emodin alleviates cardiac fibrosis, the activation of fibroblasts into smooth muscle actin (α-SMA)-positive fibroblasts is a key step in interstitial fibrosis. WB analysis revealed that ANGII significantly inhibited collagen II expression (p < .05) and promoted α-SMA expression compared with those in the sham group (p < .05) (). Compared with that in the ANG II group, emodin inhibited fibroblast activation, promoted collagen II expression (p < .05), and inhibited α-SMA expression (p < .05) (). These data indicate that emodin can inhibit ANGII-induced cardiac fibroblast fibrosis.
Figure 1. Emodin inhibits fibroblast fibrosis induced by ANGII. (a) CCK8 was used to examine changes in fibroblast proliferation after emodin treatment. (b) DHE was used to examine ROS levels in cardiac fibroblasts in each group, and red indicates positive results. (c) IPP was used to measure the mean density of the cells in each group. (d) IPP was used to measure the positive area of each group of cells. (e) Western blotting was used to examine changes in the expression of collagen II and α-SMA in each group, and GAPDH was used as the internal reference protein. (f) The relative gray values in each group. For each group of six mice, the data are expressed as the mean ± standard deviation (±S). **p < .01; *p < .05; ##p < .05.
Emodin inhibits ANGII-induced cardiac fibrosis through the PI3K/AKT/mTOR signaling pathway
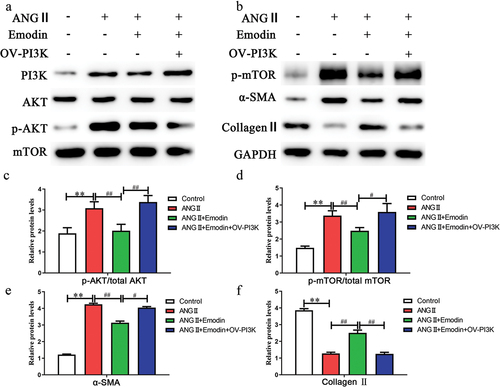
Next, we further investigated the mechanism by which emodin inhibits the proliferation of cardiac fibroblasts. The PI3K/AKT/mTOR signaling pathway is involved in a variety of tissue fibrosis processes. WB analysis revealed that ANGII activated the phosphorylation of PI3K/AKT/mTOR pathway proteins (). Compared with that in the ANG II group, emodin significantly inhibited PI3K/AKT/mTOR pathway activation. The experiment was performed by adding a lentivirus to overexpress PI3K (OV-PI3K). WB analysis revealed that emodin plus OV-PI3K reversed the inhibitory effect of emodin on the PI3K/AKT/mTOR pathway. Fibrosis-related markers were examined, and the results showed that emodin plus OV-PI3K significantly inhibited the expression of collagen II (p < .05) and promoted the expression of α-SMA (p < .05) (). Treatment with emodin plus OV-PI3K significantly reversed the effect of emodin on cardiac fibrosis. These data indicate that emodin inhibits ANGII-induced cardiac fibrosis through the PI3K/AKT/mTOR signaling pathway.
Figure 2. Emodin inhibits ANGII-induced cardiac fibrosis via the PI3K/AKT/mTOR signaling pathway (a,b) Western blotting was used to examine changes in the expression of PI3K/AKT/mTOR signaling pathway factors during cardiac fibrosis. GAPDH was used as an internal reference protein, and the gray value was calculated for p-AKT/AKT (c), p-mTOR/mTOR (d), α-SMA (e), and collagen II (F). **p < .01; ##p < .05; #p < .01.
Emodin alleviates oxidative stress damage in the heart and improves cardiac function
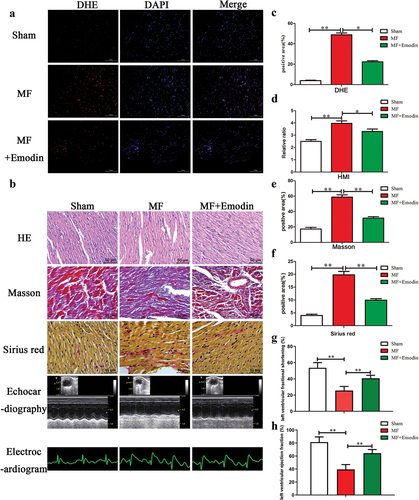
Excessive oxidative stress is an important cause of myocardial fibrosis. Firstly, we confirmed that 70 mg/kg of emodin can significantly inhibit α-SMA expression and promote collagen II expression by adding different concentrations of emodin (Figure S2A, B). ROS levels in each group were examined, and the results showed that ROS levels in the MF group (shown in red) were significantly increased compared with those in the Sham group, and emodin significantly decreased ROS production (). Electrocardiography can be used to examine the arrhythmia status of mice, and the results showed that MF induced significant arrhythmia in mice, indicating successful establishment of the model. However, emodin could improve arrhythmia, but is the effect was not statistically significant (). Cardiac ultrasound is an important method for evaluating the diagnosis and treatment of cardiac fibrosis. Emodin significantly improved left ventricular function (). HMI is a specific manifestation of myocardial fibrosis, and the results showed that after 4 weeks, HMI in the MF model group increased significantly compared with that in the sham group (2.5 ± 0.06 vs. 4.0 ± 0.08) (p < .05) (). HMI decreased significantly after emodin treatment (3.3 ± 0.08) (p < .05). Taken together, these data demonstrate that emodin alleviates oxidative stress and damage in the heart and improves heart function. Masson and Sirius red staining can be used to determine the area and degree of cardiac fibrosis, respectively. The Masson staining results showed that emodin significantly reduced myocardial fibrosis in the infarct area and reduced the degree of fibrosis in the noninfarcted area (). The Sirius red staining results were consistent with the Masson staining results, and compared with that in the MF group, the deposition of collagen fibers in the MF+Emodin group was significantly reduced (p < .05) (). These data indicate that emodin can effectively inhibit myocardial fibrosis.
Figure 3. Emodin reduces oxidative stress in the heart and inhibits collagen fiber formation. (a) DHE was used to examine ROS levels in each group. (b) HE, Masson, echocardiography, electrocardiogram and Sirius red staining were used to examine changes in collagen fiber expression and cardiac function in each group. IPP was used to calculate the positive area (c) of each group. Changes in the heart mass index after emodin treatment in the MF group (d) and changes in collagen fiber formation (e,f). Changes in cardiac function (g,h). **p < .01; *p < .05.
Emodin inhibits activation of the PI3K/AKT/mTOR signaling pathway to ameliorate myocardial fibrosis
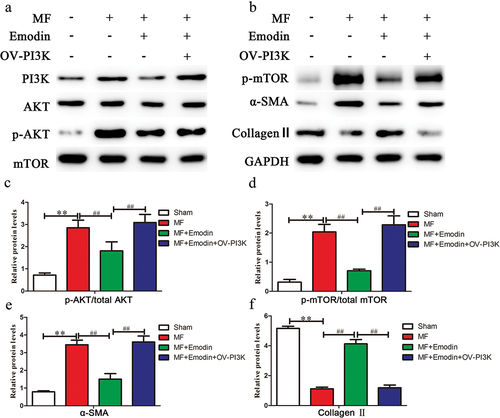
Studies have shown that the PI3K/AKT/mTOR signaling pathway plays an important role in fibrosis, by overexpressing PI3K, changes in the expression of other molecules in the signaling pathway were detected. Compared with that in the MF group, emodin significantly downregulated p-AKT and p-mTOR (p < .05) () but had no significant effect on total AKT or mTOR expression (p > .05) (). After OV-PI3K was added, emodin-mediated attenuation of fibrosis was abrogated. Fibrosis markers were examined, and the results revealed that emodin plus OV-PI3K significantly reversed the emodin-induced decrease in α-SMA (p < .05) () and reversed the emodin-induced increase in collagen II (p < .05) (). These results suggest that the PI3K/AKT/mTOR signaling pathway is one of the mechanisms by which emodin alleviates myocardial fibrosis.
Figure 4. Emodin inhibits activation of the PI3K/AKT/mTOR signaling pathway and alleviates myocardial fibrosis. (a,b) Western blotting was used to examine changes in the expression of PI3K/AKT/mTOR signaling pathway components in the heart. GAPDH was used as an internal reference protein, and the gray value of p-AKT/AKT (c), p-mTOR/mTOR (d), α-SMA (e), and collagen II (f) was determined. **p < .01; ##p < .05.
Emodin alleviates cardiac dysfunction after myocardial infarction
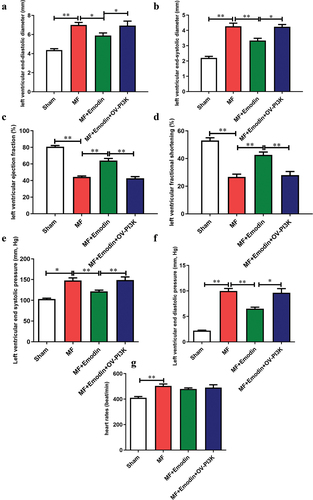
Echocardiography evaluated the effect of emodin on cardiac fibrosis. After myocardial infarction, MF group showed significant increased LVEDD, LVESD, LVESP, LVEDP (). Interestingly, emodin significantly attenuated the effects induced by MF (p < 0.05) (). Simultaneously, Compared with the Sham group, MF showed significantly inhibits EF and FS (p < 0.05) (). Emodin significantly improved EF and FS (p < 0.05) (). After OV-PI3K was added, emodin-mediated attenuation of cardiac dysfunction was abrogated (p < 0.05) (). These data indicate that emodin may significantly improve cardiac function by regulating PI3K.
Figure 5. Emodin alleviates cardiac dysfunction after myocardial infarction. Echocardiography was used to evaluate changes in cardiac function, as indicated by LVEDD (a), LVESD (b), EF (c), FS (d), LVESP (e), LVEDP (f) and HR (g). **p < .01; *p < .05.
Simplified diagram showing the protective mechanism by which emodin alleviates MF
Emodin decreases ROS levels in fibroblasts and myocardial tissue and inhibits fibroblast proliferation and collagen fibrillogenesis. Moreover, emodin can target the PI3K/Akt/mTOR signaling pathway to promote collagen II expression and inhibit α-SMA expression. These findings suggest that emodin might be useful for the treatment of MF patients.
Discussion
Myocardial fibrosis is a multifactorial process that includes inflammation, immune responses, injury, pressure overload and other factors.Citation15 The current treatment of MF is still limited by many factors. Previous studies have shown that interstitial fibrosis caused by oxidative stress is an important cause of myocardial fibrosis.Citation16 This study revealed the inhibitory effect of emodin on myocardial fibrosis and the underlying mechanism. Emodin was first shown to regulate the PI3K/AKT/mTOR pathway, which is a classic signaling pathway involved in fibrosis, and could ameliorate myocardial fibrosis. It has been confirmed that emodin significantly inhibits oxidative stress and damage in cells, inhibits the proliferation of myocardial fibroblasts induced by ANGII, downregulates PI3K/AKT/mTOR pathway activation, inhibits the expression of α-SMA and promotes the expression of collagen II, thereby alleviating myocardial fibrosis and improving cardiac function ().
Figure 6. Simplified diagram showing the mechanism by which emodin alleviates MF.
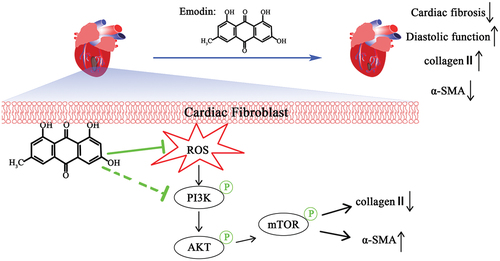
Oxidative stress, in which antioxidant enzymes cannot effectively remove excess ROS, plays a key role in MF.Citation17 ROS regulate several signaling pathways that induce MF.Citation18,Citation19 Excessive ROS directly lead to myocardial inflammation, which in turn results in myocardial hypertrophy and fibrosis.Citation20 Studies have confirmed that inhibiting ROS can effectively inhibit MF.Citation21,Citation22 Therefore, finding safe and effective antioxidants is highly important for the prevention and treatment of MF. Emodin and its derivatives have shown broad prospects for regulating various signaling pathways and tissue fibrosis. Our previous studies have shown that emodin significantly inhibits oxidative stress damage and malignant cell proliferation via the ROS/MAPK/ERK pathway.Citation4 The results of this study showed that emodin significantly inhibited the production of ROS and inhibited the proliferation of fibroblasts induced by ANGII; these results are consistent with the results of other studies.Citation23,Citation24 However, while emodin has been shown to have obvious cardioprotective effects, the specific mechanism is still unclear.
MF is results from the disproportionate accumulation of collagen secreted by fibroblasts and other extracellular matrix components, and a change in collagen II is an important component of MF.Citation25,Citation26 Studies have shown that emodin significantly promotes the expression of collagen II and inhibits the expression of α-SMA, which is similar to the effect of emodin on the alleviation of renal fibrosis.Citation8 ANGII is a key factor that induces fibrosis in various tissues. Previous studies have shown that emodin inhibits histone deacetylase activity and pathological myocardial hypertrophy.Citation27 The combination of emodin and other drugs reduces the expression of MMP-2 and TIMP-2 to inhibit pressure-induced ventricular fibrosis.Citation28 The effects of emodin and irbesartan on ventricular fibrosis in Goldblatt hypertensive rats have been studied,Citation28 but there have been few studies on MF after myocardial infarction, and the specific molecular mechanism remains unclear. MF involves the activation of multiple pathways, and studies have confirmed that continuous activation of the PI3K/AKT/mTOR signaling pathway is an important manifestation of MF.Citation10 Treatment with NVP-BEZ235 (a PI3K/mTOR inhibitor) can effectively reverse fibrosis,Citation10 suggesting that the PI3K/AKT/mTOR pathway plays an important role in MF. In addition, evidence indicates that the PI3K/AKT/mTOR pathway may be regulated by emodin.Citation29 Studies have shown that aloe-emodin, which is an emodin derivative, can alleviate renal fibrosis by inhibiting the PI3K/AKT/mTOR pathway.Citation9 Furthermore, aloe-emodin activates the TGF-β signaling pathway to improve cardiac function and inhibit the development of cardiac fibrosis and hypertrophy in rats with chronic myocardial infarction.Citation30 However, there is no direct evidence that emodin regulates MF through the PI3K/AKT/mTOR pathway. Our experimental results show for the first time that emodin inhibits MF in vivo and in vitro by negatively regulating the PI3K/AKT/mTOR pathway, thereby improving cardiac function. We directly interfered with the expression of PI3K, and the results showed that knocking down PI3K eliminated the cardioprotective effect of emodin.
Notably, the hepatotoxicity, nephrotoxicity and reproductive toxicity of emodin are still controversial. Emodin has poor oral bioavailability in mice because of its extensive glucuronidationCitation31. This topic requires additional in-depth research.
Conclusions
Our investigation demonstrated that emodin ameliorated myocardial fibrosis. Emodin decreased ROS levels in fibroblasts and myocardial tissue and inhibited fibroblast proliferation and collagen fibrillogenesis. Moreover, emodin could target the PI3K/Akt/mTOR signaling pathway to promote collagen II expression and inhibit α-SMA expression. These findings suggest that emodin inhibits MF and might be used to treat MF patients. However, several missing links remain to be examined in future studies, including the molecular mechanism by which emodin upregulates PI3K.
Author contributions
All the authors made significant contributions to the work reported, whether in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas. All the authors took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; agreed on the journal to which the article was submitted; and agreed to be accountable for all aspects of the work.
Ethics approval and consent to participate
All animal treatments were approved (approval no. 2020-ky75) by the Laboratory Animal Welfare and Ethics Committee of the General Hospital of Western Theater Command (Chengdu, China).
Supplemental Material
Download MS Word (490.5 KB)Acknowledgments
We thank Xiang-yun Wu for assistance establishing the MF model.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data presented in this study are available upon request from the corresponding author.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/10641963.2024.2326022
Additional information
Funding
References
- Van Riet EE, Hoes AW, Wagenaar KP, Limburg A, Landman MA, Rutten FH. Epidemiology of heart failure: the prevalence of heart failure and ventricular dysfunction in older adults over time. A systematic review. Eur J Heart Fail. 2016;18(3):242–10. doi:10.1002/ejhf.483.
- Rajadurai J, Tse HF, Wang CH, Yang NI, Zhou J, Sim D. Understanding the epidemiology of heart failure to improve management practices: an Asia-Pacific perspective. J Card Fail. 2017;23(4):327–39. doi:10.1016/j.cardfail.2017.01.004.
- Lee TM, Lin SZ, Chang NC. Antiarrhythmic effect of lithium in rats after myocardial infarction by activation of Nrf2/HO-1 signaling. Free Radic Biol Med. 2014;77:71–81. doi:10.1016/j.freeradbiomed.2014.08.022.
- Zhang Y, Li K, Yang J, Feng YX, Huang W, Wu XY, Zhang Y. Effect and mechanism of emodin on carotid stenosis after balloon injury in rats. J Thir Mil Med Univer. 2017;1:48–53.
- Carver W, Fix E, Fix C, Fan D, Chakrabarti M, Azhar M. Effects of emodin, a plant-derived anthraquinone, on TGF-β1-induced cardiac fibroblast activation and function. J Cell Physiol. 2021;236(11):7440–49. doi:10.1002/jcp.30416.
- Zhao XA, Chen G, Liu Y, Wu H, Chen J, Xiong Y, Tian C, Jia B, Wang G, Xia J, et al. Emodin alleviates liver fibrosis of mice by reducing infiltration of Gr1hi monocytes. Evid Based Complement Alternat Med. 2018;2018:5738101. doi:10.1155/2018/5738101.
- Tian SL, Yang Y, Liu XL, Xu QB. Emodin attenuates bleomycin-induced pulmonary fibrosis via anti-inflammatory and anti-oxidative activities in rats. Med Sci Monit. 2018;24:1–10. doi:10.12659/MSM.905496.
- Yang F, Deng L, Li J, Chen M, Liu Y, Hu Y, Zhong W. Emodin retarded renal fibrosis through regulating HGF and TGFβ-smad signaling pathway. Drug Des Devel Ther. 2020;14:3567–75. doi:10.2147/DDDT.S245847.
- Dou F, Liu Y, Liu L, Wang J, Sun T, Mu F, Guo Q, Guo C, Jia N, Liu W, et al. Aloe-emodin ameliorates renal fibrosis via inhibiting PI3K/Akt/mTOR signaling pathway in vivo and in vitro. Rejuvenation Res. 2019;3(3):218–29. doi:10.1089/rej.2018.2104.
- Yang W, Wu Z, Yang K, Han Y, Chen Y, Zhao W, Huang F, Jin Y, Jin W. BMI1 promotes cardiac fibrosis in ischemia-induced heart failure via the PTEN-PI3K/Akt-mTOR signaling pathway. Am J Physiol Heart Circ Physiol. 2019;1(1):h61–69. doi:10.1152/ajpheart.00487.2018.
- Xian M, Cai J, Zheng K, Liu Q, Liu Y, Lin H, Liang S, Wang S. Aloe-emodin prevents nerve injury and neuroinflammation caused by ischemic stroke via the PI3K/AKT/mTOR and NF-κB pathway. Food Funct. 2021;17(17):8056–67. doi:10.1039/D1FO01144H.
- Zhang Y, Zhang YL, Li W, Wang P, Gu R, Feng YX, Wei SJ, Peng K, Zhang YR, Su LN, et al. Uncoupling Protein 2 Inhibits Myointimal Hyperplasia in Preclinical Animal Models of Vascular Injury. J Am Heart Assoc. 2017;6(10):e002641. doi:10.1161/JAHA.117.006593.
- Zhang YL, Wen XD, Guo X, Huang SQ, Wang TT, Zhou PT, Li W, Zhou LF, Hu YH. Progesterone suppresses the progression of colonic carcinoma by increasing the activity of the GADD6α/JNK/c‑Jun signalling pathway. Oncol Rep. 2021;45(6):95. doi:10.3892/or.2021.8046.
- Xiao D, Zhang Y, Wang R, Fu Y, Zhou T, Diao H, Wang Z, Lin Y, Li Z, Wen L, et al. Emodin alleviates cardiac fibrosis by suppressing activation of cardiac fibroblasts via upregulating metastasis associated protein 3. Acta Pharm Sin B. 2014;9(4):724–33. doi:10.1016/j.apsb.2019.04.003.
- González A, Schelbert EB, Díez J, Butler J. Myocardial interstitial fibrosis in heart failure: biological and translational perspectives. J Am Coll Cardiol. 2018;71(15):1696–706. doi:10.1016/j.jacc.2018.02.021.
- Erasmus M, Samodien E, Lecour S, Cour M, Lorenzo O, Dludla P, Pheiffer C, Johnson R. Linking LOXL2 to cardiac interstitial fibrosis. Int J Mol Sci. 2020;21(16):5913. doi:10.3390/ijms21165913.
- Li C, Zhang J, Xue M, Li X, Han F, Liu X, Xu L, Lu Y, Cheng Y, Li T, et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc Diabetol. 2019;18(1):15. doi:10.1186/s12933-019-0816-2.
- Wu H, Li GN, Xie J, Li R, Chen QH, Chen JZ, Wei ZH, Kang LN, Xu B. Resveratrol ameliorates myocardial fibrosis by inhibiting ROS/ERK/TGF-β/periostin pathway in STZ-induced diabetic mice. BMC Cardiovasc Disord. 2016;16(1):5. doi:10.1186/s12872-015-0169-z.
- Meng Z, Li HY, Si CY, Liu YZ, Teng S. Asiatic acid inhibits cardiac fibrosis throughNrf2/HO-1 and TGF-β1/Smads signaling pathways in spontaneous hypertension rats. Int Immunopharmacol. 2019;74:105712. doi:10.1016/j.intimp.2019.105712.
- Kang LL, Zhang DM, Jiao RQ, Pan SM, Zhao XJ, Zheng YJ, Chen TY, Kong LD. Pterostilbene attenuates fructose-induced myocardial fibrosis by inhibiting ROS-Driven Pitx2c/miR-15b pathway. Oxid Med Cell Longev. 2019;2019:1243215. doi:10.1155/2019/1243215.
- Zhu W, Wu RD, Lv YG, Liu YM, Huang H, Xu JQ. BRD4 blockage alleviates pathological cardiac hypertrophy through the suppression of fibrosis and inflammation via reducing ROS generation. Biomed Pharmacother. 2020;121:109368. doi:10.1016/j.biopha.2019.109368.
- Li K, Zhai M, Jiang L, Song F, Zhang B, Li J, Li H, Li B, Xia L, Xu L, et al. Tetrahydrocurcumin ameliorates diabetic cardiomyopathy by attenuating high glucose-induced oxidative stress and fibrosis via activating the SIRT1 pathway. Oxid Med Cell Longev. 2019;2019:6746907. doi:10.1155/2019/6746907.
- Yu Y, Liu H, Yang D, He F, Yuan Y, Guo J, Hu J, Yu J, Yan X, Wang S, et al. Aloe-emodin attenuates myocardial infarction and apoptosis via up-regulating miR-133 expression. Pharmacol Res. 2019;146:104315. doi:10.1016/j.phrs.2019.104315.
- Lee EH, Baek SY, Park JY, Kim YW. Emodin in rheum undulatum inhibits oxidative stress in the liver via AMPK with Hippo/Yap signalling pathway. Pharm Biol. 2020;1(1):333–41. doi:10.1080/13880209.2020.1750658.
- Danpinid A, Luo J, Vappou J, Terdtoon P, Konofagou EE. In vivo characterization of the aortic wall stress–strain relationship. Ultrasonics. 2010;7(7):654–65. doi:10.1016/j.ultras.2010.01.003.
- Li ML, Li RN, Ma YM, Jiang B, Chen YJ, Hu WX, Qv CL, Zhang YJ, Song YY, Wang Y. MiRNA-1297 inhibits myocardial fibrosis by targeting ULK1. Eur Rev Med Pharmacol Sci. 2020;4:2070–76.
- Evans LW, Bender A, Burnett L, Godoy L, Shen Y, Staten D, Zhou T, Angermann JE, Ferguson BS. Emodin and emodin-rich rhubarb inhibits histone deacetylase (HDAC) activity and cardiac myocyte hypertrophy. J Nutr Biochem. 2020;79:108339. doi:10.1016/j.jnutbio.2019.108339.
- Chen Q, Pang L, Huang S, Lei W, Huang D. Effects of emodin and irbesartan on ventricular fibrosis in Goldblatt hypertensive rats. Pharmazie. 2014;5:374–78.
- Zheng XY, Yang SM, Zhang R, Wang SM, Li GB, Zhou SW. Emodin-induced autophagy against cell apoptosis through the PI3K/AKT/mTOR pathway in human hepatocytes. Drug Des Devel Ther 2019, 13: 3171–3180. 10.2147/DDDT.S204958.
- Yu J, Zhao X, Yan X, Li W, Liu Y, Wang J, Wang J, Yang Y, Hao Y, Liang Z, et al. Aloe-emodin ameliorated MI-induced cardiac remodeling in mice via inhibiting TGF-β/SMAD signaling via up-regulating SMAD7. Phytomedicine. 2023;114:154793. doi:10.1016/j.phymed.2023.154793.
- Dong X, Fu J, Yin X, Cao S, Li X, Lin L, Huyiligeqi NJ. Emodin: a review of its pharmacology, toxicity and pharmacokinetics. Phytother Res. 2016;8(8):1207–18. doi:10.1002/ptr.5631.