Abstract
Colon targeted drug delivery has the potential to deliver bioactive agents for the treatment of a variety of colonic diseases and to deliver proteins and peptides to the colon for their systemic absorption. Various strategies, currently available to target the release of drugs to colon, include formation of prodrug, coating of pH-sensitive polymers, use of colon-specific biodegradable polymers, timed released systems, osmotic systems, and pressure controlled drug delivery systems. Among the different approaches to achieve targeted drug release to the colon, the use of polymers especially biodegradable by colonic bacteria holds great promise. Polysaccharidases are bacterial enzymes that are available in sufficient quantity to be exploited in colon targeting of drugs. Based on this approach, various polysaccharides have been investigated for colon-specific drug release. These polysaccharides include pectin, guar gum, amylose, inulin, dextran, chitosan, and chondroitin sulphate. This family of natural polymers has an appeal to drug delivery as it is comprised of polymers with a large number of derivatizable groups, a wide range of molecular weights, varying chemical compositions, and, for the most part, low toxicity and biodegradability yet high stability. The most favorable property of these materials is their approval as pharmaceutical excipients.
High intracolonic drug concentration is required for the treatment of diseases associated within colon; i.e., ulcerative colitis, Crohn's disease, colon cancer, and amebiasis, which can be achieved by targeted delivery of drugs to colon. However, specific systemic absorption in the colonic region offers interesting possibilities for the treatment of diseases susceptible to diurnal rhythm such as asthma, arthritis, or inflammation (Yehe et al. Citation1995). The colon is also considered as the preferred absorption site for oral administration of proteins and peptide drugs due to relatively low proteolytic enzyme activities in the colon compared to upper gastrointestinal tract. The various approaches that can be exploited to target the release of drug to colon include prodrug formation, coating with pH-sensitive polymers, coating with biodegradable polymers, embedding in biodegradable matrices and hydrogels, timed release systems, osmotic systems, and bioadhesive systems ().
Strategies for colon targeted drug delivery
Utilization of the prodrug approach involves the formation of a covalent linkage between drug and carrier in such a manner that upon oral administration the moiety maintains its integrity in the hostile environment of stomach and small intestine and is converted into parent drug molecule once it reaches into the colon. Site-specific drug delivery through site-specific prodrug activation may be accomplished by some specific property at the target site, such as altered pH or high activity of certain enzymes relative to the nontarget tissues for the prodrug-drug conversion. Sulphasalazine was introduced for the treatment of rheumatoid arthritis and anti-inflammatory disease (IBD) in which sulfapyridine is linked to a salicylate radical by an azo bond (Azad Khan, Truelove, and Aronseq Citation1982). On oral administration a small amount is absorbed from stomach and small intestine and the remaining quantity reaches intact to the colon where it is split at the azo bond by colonic bacteria with liberation of sulfapyridine and 5-ASA. However, sulphapyridine seems to be responsible for most of the side effects of sulphasalazine and hence various new approaches for the treatment of IBD have emerged. The requirement for less toxic carrier moieties has led to the synthesis and testing of a number of other azo-bond prodrugs. A number of prodrugs of 5-ASA—ipsalazine, balsalazine and olsalazine—have been prepared for the treatment of colonic disorders (Chan et al. Citation1983; Willoughby et al. Citation1981, Citation1982).
Apart from azo-bond prodrugs, numerous glycosidic prodrugs have been synthesized on the basis of observations that such glycosides are cleaved by bacterial glycosidases present in the colonic mucosa (Friend Citation1992). Friend and Chang (Citation1984) prepared dexamethasone-21-β-glucoside and prednisolone-21-β-glucoside for delivery of these steroids to the colon.
The treatment of ulcerative colitis was improved by the synthesis of budenoside and dexamethasone conjugates of glucuronic acid and dextran (Mcleod et al. Citation1994a; Cui, Friend, and Fedorak Citation1994). The system showed excellent performance in ulcerative colitis and reduces the systemic toxicity of corticosteroids including adrenal suppression by immurement of activity of the drug in large intestine. Polymeric prodrugs have been formulated and examined for their efficacy in colon targeted drug delivery (Schacht, Gevaert, and Kenawy Citation1996; Schacht, Callant, and Verstraete Citation1991).
Coating of the drugs with pH-sensitive polymers provides another approach for colon-specific drug delivery. Most commonly used pH-dependent coating polymers are methacrylic acid copolymers, commonly known as Eudragits® (Rohm Pharmaceuticals, Darmstadt, Germany), more specifically Eudragit® L and Eudragit® S. These polymers contain ionizable carboxyl groups and hence dosage forms coated with these polymers remain intact in the strongly acidic pH of stomach. But as the pH increases toward alkaline side in small intestine, the coating starts to dissolve and release the drug. The critical factor that influences the performance of these polymers is the pH at which dissolution occurs. Polymers with nonesterified phthalic acid groups dissolve much faster and at a lower pH than those with acrylic or methacrylic acid groups. The presence of plasticizer (Spitael and Kinget Citation1979) and the nature of the salt (Spitael and Kinget Citation1977; Spitael, Kinget, and Naessens Citation1980) in the dissolution medium also influence the dissolution rate of Eudragit®. In addition, the permeability of the film formed may depend on the type of solvent used to dissolve Eudragit® (Spitael and Kinget Citation1980). This approach has been adopted for the delivery of mesalazine (Khan, Prebeg, and Kurjakovic Citation1999), insulin (Touitou and Rubinstein Citation1986; Morishita et al. Citation1993), and naproxen (Hardy et al. Citation1987). The disadvantage of this approach is the lack of consistency in the dissolution of the polymer at the desired site.
Depending on the intensity of the gastrointestinal motility, the dissolution of the polymer can be complete deep in the colon or at the end of the ileum. Moreover, many factors such as the presence of short chain fatty acids, residues of bile acids, carbon dioxide, or other fermentation products can reduce the colonic pH to approximately 6 and call its pH as a trigger into question. Generally, the dissolution of enteric polymers take place at terminal ileum or at ileocaecal junction when the pH exceeds 7.0 and in a short span of time drug enters into large intestine. To alleviate the problem of drug dissolution in ileum, multiparticulate systems has been reported in the literature (Rodriguez, Vila-Jato, and Torres Citation1998; Rodriguez et al. Citation2001; Markus et al. Citation2001).
Because azo reduction is a reaction that is mediated by the majority of intestinal azo bacters, CitationSaffran and co-workers (1986, 1988) were the first to exploit intestinal microflora in the design of colon targeted drug delivery systems. The authors coated insulin and lysine-vasopressin solid dosage forms (pellets, gelatin minicapsules, or simply paper strips) with copolymers of styrene and 2-hydroxyethyl methacrylate (HEMA) crosslinked with divinyl azobenzene (DVAB), postulating that this polymer would be able to protect the entrapped protein drugs in the stomach and the upper portion of the small intestine and degrade in the colon. Jain et al. (Citation2003) developed and characterized engineered commensal bacteria for site-specific targeting of galactokinase and glutamate dehydrogenase. Azo polymers were synthesized using water-soluble and water-insoluble monomers belonging to acrylate series.
In vivo screening of developed oral, colon-specific formulation, azo polymer-coated granules of engineered E. coli (56) producing galactokinase and engineered E. coli (797) producing glutamate dehydrogenase was performed on rabbits. D-galactose, which is the substrate for enzyme galactokinase and ammonium chloride, was administered intraperitoneally to all rabbits. The lowering of blood galactose level and urea level was observed with respect to variable time interval. CitationVan den Mooter, Samyn, and Kinget (1992, 1993) developed a colon targeted drug delivery system using copolymers of HEMA and methyl methacrylate (MMA), in the presence of bis(methacryloylamino)azobenzene (BMAAB). Film coatings were prepared with the azo-aromatic polymers and these films were insoluble in gastric juice and intestinal juice. On the basis of in vitro and in vivo studies, it was proved that azo polymers could be used successfully for colon targeted delivery of drugs due to the presence of azo bonds, which are cleaved by the azo reductase enzymes released by the azo bacters present in the colon.
Timed release systems are based on the strategy of suppressing the release of drug until it enters into colon. The principle in designing timed released systems is to resist the acidic environment of the stomach and to undergo a lag time of a predetermined span of time, after which release of drug take place. The lag time in this case is the time required to transit from the mouth to colon. The first formulation introduced based on this principle was Pulsincap® (MacNeil, Rashid, and Stevens Citation1990). Gazzaniga et al. (Citation1994, 1995) described a novel oral time-based drug release system for colon targeted drug delivery. The system was designed to exploit the relatively constant small intestinal transit time of dosage forms consisting of drug-containing cores coated with three polymeric layers. The outer layer dissolves at pH above 5 and then the intermediate swellable layer, made of an enteric material. The system provides the expected delayed release pattern, as also indicated by preliminary in vivo studies on rats. Several other drug delivery systems have developed that rely upon the relatively constant transit time of small intestine (Gupta, Beckert, and Price Citation2001a; Gupta et al. 2000b; Fukui, Miyamura, and Kobayashi Citation2001).
Pressure-controlled drug delivery systems have been developed to target the drug release to colon (Takaya et al. Citation1998; Shibata et al. Citation2001). Pressure-controlled colon delivery capsule (PCDC) made of ethylcellulose (EC) was prepared by coating the inner surface of gelatin capsule with water-insoluble polymer, EC. By adjusting the coating thickness of EC membrane (approximately 40 μm), colonic delivery of drug was obtained both in beagle dogs and human volunteers (Takaya et al. Citation1998). The delivery ability of a PCDC-containing caffeine as a test drug was evaluated after oral administration to healthy male human volunteers. The driving force causing PCDC disintegration in the intestinal tract is the physiological luminal pressure that results from peristalsis (Muraoka et al. Citation1998).
The OROS-CT (Alza Corporation) can be used to target the drug locally to the colon for the treatment of disease or to achieve systemic absorption that is otherwise unattainable (Theeuwes, Guittard, and Wong Citation1990). The OROS-CT system can be a single osmotic unit or may incorporate as many as 5–6 push-pull units (Swanson et al. Citation1987), each 4 mm in diameter, encapsulated within a hard gelatin capsule. Each bilayer push-pull unit contains an osmotic push layer and a drug layer, both surrounded by a semipermeable membrane. An orifice is drilled through the membrane next to the drug layer. After administration, the gelatin capsule dissolves, and due to the presence of enteric coating, the unit passes as such through the stomach. But in the small intestine enteric coating dissolves and water enters into the unit causing release of drug.
Bioadhesion has been proposed as a means of improving the performance and extending the mean residence time of colonic drug delivery systems (Chickering and Mathiowitz Citation1995; Chickering, Jacob, and Mathiowitz Citation1995). Rihova (Citation1995) used bioadhesive polymers such as HPMA copolymers that mimic bioadhesive process occurring in the guinea pig gastrointestinal tract (GIT), which are based on the presence of lectin-like structures on enterocytes and in the mucus gel layer.
INTESTINAL MICROFLORA AND METABOLIC ACTIVITY
The human GIT, the major port for the entry of drugs into humans, consists of a complex ecosystem incorporating aerobic and anaerobic microorganisms. Due to the lower pH of the stomach, its bacterial concentration, which is predominantly aerobic in nature, is low (102 CFU/ml). The number of microorganisms increases gradually on descending along the small intestine, but it rises by several orders of magnitude beyond the ileocaecal valve. The environment of human colon is anaerobic in nature consisting of mainly Bacteroids, Bifidobacteria, Eubacteria, Enterobacteria and Enterococci (Moore and Holden Citation1975). The energy requirement of colonic bacterial flora for maintaining the cellular function is derived from the fermentation of various substrates that are left indigested in the small intestine. These substrates include di and trisaccharides such as cellobiose, raffinose, stachyose, and lactulose and residues of partially digested polysaccharides such as starch and polysaccharides from endogenous sources such as mucopolysaccharides (Cummings and Englyst Citation1987; Levitt et al. Citation1987; Steggerda Citation1968). In addition to mucopolysaccharides, other substrates for fermentation are dietary fibers, which include all the non-α-glucan polymers that originate in the plant cell wall—cellulose, hemicellulose, and pectin substances (Van Soest Citation1978; Rubenstein 1990). A number of enzymes like β-D-glucosidase, β-D-galactosidase, β-xylosidase, β-arabinosidase, azo reductase, and nitro reductase are produced by colonic microflora to carry out the process of fermentation (Scheline Citation1973).
Drug delivery systems targeted to the colon that are based on the use of polysaccharides offer superiority over other systems. Polysaccharides retain their integrity and prevent the release of drug during its passage through the GIT. But when it comes in contact with colonic fluid it is confronted by the action of microorganisms and consequently entrapped drug is liberated (Potts, Clendianaings, and Ackard Citation1973; Huang, Bansleben, and Knox Citation1979; Swift Citation1992; Ratner, Gladhill, and Hobert Citation1988; Hergenrother, Wabers, and Cooper Citation1992; Park, Shalaby, and Park Citation1993).
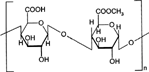
1 Chemical structure of pectin.
The rationale of this article is to explore the potential of natural polymers, polysaccharides from plant origin—amylose, pectin, guar gum; microbial origin—dextran, xanthan gum-animal origin—chitosan, chondroitin sulfate; and algal origin—alginates for colon targeted drug delivery.
PECTINS
Pectins are nonstarch linear polysaccharides that consist of α-1,4 D-galacturonic acid and 1,2 D-rhamnose with D-galactose and D-arabinose side chains having average molecular weights between 50,000 to 150,000 (). Pectin tends to produce lower viscosities than other plant gums. It is refractory to host gastric and small intestinal enzymes but is almost completely degraded by the colonic bacterial enzymes to produce a series of soluble oligalactorunates (Cummings et al. Citation1979; Englyst, Hay, and MacFarlane Citation1987). Depending on the plant source and preparation, they contain varying degrees of methyl ester substituents (Towle and Christensen Citation1973). Pectin is highly soluble in water, which put hurdles in the development of colon targeted drug delivery systems. If used alone it swells when it comes in contact with aqueous fluids of GIT and causes the release of the entrapped drug through the diffusion. This problem can be manipulated through choice of pectin type or the presence of additives (Rubinstein et al. Citation1990; Rubinstein and Radai Citation1991). Another way to alleviate this problem is by the use of hydrophobic polymers, e.g., ethylcellulose. It restricts the entry of water and consequently swelling of polymer.
Moreover, a better shielding effect can be obtained by reducing the solubility of pectin by forming its calcium salt, calcium pectinate. The degree of methylation (DM) has an essential influence on the properties of pectin, especially on its solubility and its requirements for gelation, which are directly derived from the solubility. The DM of 50% divides commercial pectins into high methoxy pectins and low methoxy pectins. These two groups of pectin are gelled by different mechanisms. High methoxy pectins require a minimum amount of soluble solids and a pH within a moderate range around 3 to form gels. Low methoxy pectins require the presence of a controlled amount of calcium ions for gelation and need neither sugar nor acid. Low methoxy pectin gelation resembles the behavior of alginate.
Rubinstein et al. (Citation1993) prepared calcium pectinate, the insoluble salt of pectin by deesterificaiton, which was then utilized for the preparation of matrix tablets incorporating indomethacin as water insoluble drug marker in the in vitro release experiments. The biodegradation of the carrier was determined by monitoring cumulative percent release of the incorporated indomethacin. Compressed tablets of pectin and indomethacin were analyzed for degradation in the presence of Pectinex 3XL, a typical pectinolytic enzyme mixture and in the presence of human colonic bacterium Bacteroids ovatus. The degradation of calcium pectinate indomethacin tablets was assessed in the presence of Pectinex 3XL and in rat caecal contents. The release of indomethacin was significantly increased in the presence of Pectinex 3XL, Bacteroids ovatus, and rat caecal contents. The weight loss of tablet mass was significantly higher in the presence of Pectinex 3XL. The exploitation of calcium pectinate carrier was based on the assumption that like pectin it can be degraded by the specific pectinolytic enzymes present in the colon but remains intact in the hostile environment of upper GIT.
Ashford et al. (Citation1993) developed a novel colon-specific drug delivery system based on polysaccharide pectin. Three sets of compression-coated tablet were prepared each comprising a core tablet that was predominantly composed of sorbitol. Sodium fluorescein as the model compound was incorporated into the core tablets for in vitro release studies. In vitro experiments demonstrated that high methoxy pectin, when applied as a compression coat, proved capable of protecting a core tablet during conditions stimulating gastrointestinal environment and was susceptible to enzymatic attack. In vivo gamma scintigraphic studies confirmed the in vitro findings. In all the volunteers, the pectin-coated tablets disintegrated in the colon indicating that site-specificity had been achieved and illustrating the potential of a colonic drug delivery system utilizing pectin. However, in the in vivo conditions, a coat of considerable thickness was required to protect the drug core. This necessitated the development of such derivatives of pectin, which were less water soluble but had the capability to be degraded by the colonic microflora.
Compression-coated core tablets of 5-ASA were prepared using pectin and HPMC (Turkoglu and Ugurlu Citation2002). Drug dissolution/system erosion/degradation studies were carried out in pH 1.2 and 6.8 buffers using a pectinolytic enzyme. The system was designed based on the gastrointestinal transit time concept, under the assumption of colon arrival times of 6 hr. They found that pectin alone was not sufficient to protect the core tablets and HPMC addition was required to control the solubility of pectin. The optimum HPMC concentration was 20% and such a system protected the cores up to 6 hr that corresponded to 25–35% erosion; after that under the influence of pectinase the system degraded faster and delivered 5-ASA to the colon. The effect of pectinolytic enzymes on the theophylline release from pellets coated with water-insoluble polymers containing high methoxy pectin or calcium pectinate has been reported by Semde et al. (Citation2000a). Theophylline pellets were coated with cellulosic (Aquacoat ECD 30, Surelease clear) or acrylic (Eudragit® NE30D, RS30D) polymer aqueous dispersions, containing 10% (related to the insoluble polymer content) of pectin HM or calcium pectinate, using a Uni-Glatt fluidized-bed coating apparatus. The results have been examined with regard to the validity of the approach based on the combination of pectin and the insoluble polymer aqueous dispersions intended for specific delivery of drugs to the colon.
Combination of pectin along with either ethyl cellulose or chitosan or hydroxypropyl methylcellulose has been successfully utilized to achieve colon targeted delivery. Combination of pectin and ethyl cellulose, when applied as film coat, has potential value as a colonic delivery system (Wakerly et al. Citation1996, Citation1997; Macleod, Fell, and Collett Citation1997). Combination of pectin and ethyl cellulose in form of aqueous dispersion was used as a coating formulation for paracetamol cores as the substrate. Drug release was controlled by the ratio of ethyl cellulose to pectin in the film coat. Increasing the proportion of ethylcellulose and coat weight reduced drug release in pH 1 and pH 7.4 media. The addition of pectinolytic enzymes to pH 6 media increased the release of drug (Wakerly et al. Citation1996). Drug release profiles were compatible with a mechanism involving the formation of channels in the film caused by pectin dissolution. Channel formation was accelerated in most of the cases by the presence of pectinolytic enzymes showing that the pectin in the mixed film was susceptible to enzymatic attack.
The mechanical and permeability properties of mixed ethylcellulose/pectin films cast from dibutyl sebacate plasticized aqueous dispersions of Aquacoat® and Pectin USP have been investigated. The films were subjected to tensile testing, elongation at break, and elastic modulus. Increasing concentrations of pectin imparted increasing brittleness and decreasing toughness to the films. Despite the inclusion of increasing quantities of the hydrophilic pectin into the films, the permeability to moisture remained essentially the same. The results imply a limit to the amount of pectin that can be included in the coating material and still produce a satisfactory film. But the protective nature of the ethyl cellulose to moisture is not compromised (Macleod et al. Citation1997).
MacLeod et al. (1999) used pectin, chitosan, and HPMC films for colon-specific drug delivery. Radiolabelled (99mTc) tablets were coated and gastrointestinal transit was assessed by γ-scintigraphy after administration to human volunteers. The results of the study revealed that tablets retain the shape during passage through stomach and small intestine while in the colon liberation of the entrapped material occurred due to degradation of the coat by colonic bacteria. Poorly soluble drug indomethacin and soluble drug paracetamol were evaluated for colon-specific delivery utilizing coating of pectin and chitosan mixtures (Fernandez-Hervas and Fell Citation1998). Considerably thick coat of pectin was required to protect the premature release of drug from core while better protection was achieved when pectin/chitosan mixtures were used at a lower coat weight. The use of pectinolytic enzymes to simulate breakdown in the colon showed that the pectin/chitosan mixture was susceptible to enzymatic breakdown and allowed drug release to occur. The pectin-HPMC envelope was found to be a promising drug delivery system for colonic delivery of 5-ASA.
Semde et al. (Citation2000b) coated theophylline pellets with Eudragit NE30D aqueous dispersions, containing various pectin HM/Eudragit RL30D ionic complexes and performed dissolution studies in presence and in absence of commercial pectinolytic enzymes. The theophylline release from the coated pellets, after an initial latency phase, occurred linearly as a function of time. The theophylline release rate depended on the high methoxy pectin content of the complexes incorporated in the coatings. The lowest theophylline release from the coated pellets was obtained when the pectin HM content of the complexes was 20% w/w (related to Eudragit RL), i.e., when the complexation between pectin HM and Eudragit RL was optimal.
Matrix tablets of the pectin as well as coated dosage form consisting of coating of pectin along with some other hydrophobic polymers has been utilized to achieve targeting to colon. Ashford et al. (Citation1994) investigated the effect of calcium on the rheological properties of pectin and the release of model compounds from pectin-based matrix tablets in conditions mimicking mouth to colon transit and in the presence and absence of pectinolytic enzymes. Solubility of low methoxy pectin was reduced by forming a calcium pectate gel in which calcium formed a crosslink between two pectin molecules in a section of the chain, which was free from the methoxy groups. As the level of calcium increased, the number of crosslinks increased until an insoluble calcium pectate gel was formed. Various parameters like pectin, the presence of calcium, and the solubility of the calcium salt influenced the release of drug. It was observed that either a high methoxy pectin formulation or low methoxy pectin with a carefully controlled amount of calcium maximized the colonic specificity by providing optimal protection of drug during its transit to the colon and a high susceptibility to enzymatic degradation.
Compression-coated and plain matrix tablet of indomethacin and insulin were prepared by Rubinstein and Radai (Citation1995). No release of indomethacin occurred from calcium pectinate and indomethacin tablets prepared as matrix as well as compression-coated tablet at pH 1.5 for 2 hr. Drug leakage was seen in matrix tablet but not in the compression-coated tablet when shaken at pH 7.4. Abrupt release of indomethacin was observed in presence of pectinolytic enzymes in both type of tablets but higher rate and percentage of release was noted in matrix tablets in comparison to compression-coated tablets after 12 hr. In vivo studies of insulin tablets in pancreatectomized dogs revealed initial drug leakage from both types of tablets; however, compression-coated tablets exhibited much better performance. The initial leakage was explained on the basis of the difficulty encountered in compression coating technique in centering the core tablet within the compression coat and thereby not giving a uniform coat thickness. Compression coating technique was efficient for coating of a water insoluble drug but for water soluble drug, an additional barrier was suggested.
Adkin et al. (Citation1997) prepared two types of enteric coated calcium pectinate matrix tablets, one using pectin as binder and the other using guar gum. Gastrointestinal transit and disintegration was seen in 10 healthy human volunteers using scintigraphic technique. It was seen that the tablets arrived intact in the colon where complete disintegration occurred for both the formulations, with tablet of guar gum exhibiting slower disintegration in comparison to the pectin tablet. The time and location of complete disintegration was more reproducible with pectin tablet as compared with guar gum tablet. Studies using rat showed colonic degradation of the polysaccharides, as significant difference was observed between degradation in antibiotic treated and nontreated rats in the two formulations. Ahrabi et al. (Citation2000a) developed pectin-based matrix tablets incorporating ropivacaine as model drug. The aim was to investigate some formulation parameters that could reduce the release of the drug in simulated gastric and intestinal fluids, increase the release in simulated caecal fluid (with pectinolytic enzymes), and improve the poor compactibility of pectins. Amidated pectin produced harder tablets than the calcium salt of pectin and was more susceptible to enzymatic degradation. Addition of ethylcellulose increased the tablet strength and the dissolution rate.
A coating of Eudragit® RL 100 reduced the release in the simulated upper GI conditions without interference with the subsequent enzymatic activity. The impact of the neutron activation procedure, i.e., incorporation of samarium oxide and neutron irradiation, on the compression properties (including the crushing strength) and in vitro dissolution of matrix tablets of amidated pectin was investigated (Ahrabi et al. Citation2000b). The neutron activation factors did not influence the compression properties of the tablets. Replacement of magnesium stearate with samarium stearate in directly compressed amidated pectin tablets to achieve both radiolabelling and lubrication resulted in a greater extent of concentration-dependent reduction of the crushing strength. Dissolution tests demonstrated that irradiation increased the release of the model drug ropivacaine from the tablets.
Semde, Amighi, and Moes (Citation1998) assessed the potential of epichlorhydrin crosslinked pectin for colon-specific drug delivery and showed that on increasing the degree of crosslinking the drug release was increased. Further, drug release was increased in presence of enzymes indicating colon targeted behavior of the formulation. An interpolymer complex of pectin with chitosan has been prepared and evaluated (Meshali and Gabar Citation1993).
In another attempt to reduce the solubility of pectin, Munjeri, Collett, and Fell (Citation1997) developed and used amidated pectins for colon targeted drug delivery. Amidated pectins are low methoxy pectins in which some of the carboxylic acid groups are amidated. They are more tolerant to pH variations and calcium levels than conventional pectins, which could make them useful in colonic delivery systems. Hydrogel beads containing indomethacin and sulphamethoxazole were prepared by allowing the droplets of solution, containing amidated pectin/indomethacin or sulphamethoxazole, to fall into agitated calcium chloride solution containing chitosan. Drug release from beads was a function of pH of media and drug loading. In simulated gastric and small intestinal conditions, drug release was greater with the more soluble sulphamethoxazole, but release of both drugs could be reduced to satisfactory levels by the formation of a chitosan polyelectrolyte complex around the beads. All the preparations released drug in simulated colonic conditions within 135 min.
Similarly, Sriamornsak and Nunthanid (Citation1998) prepared calcium pectinate gel (CPG) beads of indomethacin, by dispersing the drug in a solution of pectin and then dropping the dispersion into calcium chloride solution. The droplets instantaneously converted into gelled spheres by ionotropic gelation. The effect of several factors such as pectin type, the presence of a hardening agent, and the drug loading were investigated on the percentage of drug entrapped, size distribution, and drug release from the CPG beads. El-Gibaly (Citation2002) proposed oral delayed-release system based on zinc-pectinate gel (ZPG) microparticles as an alternative carrier to calcium pectinate beads for colonic drug delivery. The system, which consists of ketoprofen-loaded ZPG microparticles together with pectin/dextran mixtures in a tablet form, has been investigated, in vitro, using conditions chosen to simulate the pH and times likely to be encountered during transit to the colon. To find the suitable ZPG microparticles, the formulations were prepared by utilizing 2(3) factorial design and the effect of various formulation factors on the release and surface characteristics of the microparticles was studied. The results implied that the release of ketoprofen from ZPG microparticles was greatly extended with the pectinate microparticles, which were prepared with 2.5 or 3% w/v pectin, 2.75% w/v zinc acetate and 2.5% w/v drug.
Additionally, the analysis of variance results showed that the release of ketoprofen in simulated intestinal fluid (pH 7.4) was strongly affected by crosslinking agent concentration and initial drug amount, but it was not particularly affected by the amount of pectin added. The investigated drug concentration factor has significantly increased the drug entrapment efficiency. The optimum colonic drug delivery ZPG/tablet system provided the expected delayed-release sigmoidal patterns with a lag time of 4.125–4.85 hr and t50% (the time for 50% of the drug to be released) at 7.45–8.70 hr, depending on pectin/dextran ratio employed. The results also demonstrated that the untableted ZPG microparticles exhibited drug release profiles that were able to retard the release of ketoprofen in simulated intestinal fluid (pH 7.4) to be 5.28–37.82 times (depending on formulation parameters) lower than the conventional calcium pectinate beads.
AMYLOSE
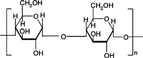
Amylose is a poly (1-4-α-D-glucopyranose) that consists of D-glucopyranose residues linked by α-(1-4) bonds (). Those substances, present naturally in the diet, have the advantages of being safe, nontoxic, and easily available. One very common dietary component that has not previously been used in this context is starch. Although, it has long been thought that starch can be broken down by pancreatic enzymes in the small intestine, in 1982 a fraction that resisted digestion in vitro was identified (Englyst, Wiggins, and Cummings Citation1982). Subsequently, various forms of starches were shown to survive passage through the human small bowel (CitationEnglyst and Cummings 1985, 1986; Englyst Citation1987). A classification of resistant starches and methods for their measurement are now available (Englyst, Kingman, and Cummings Citation1992). One form of starch, amylose, can be formed into films that are biodegradable by colonic bacteria and glucose as the test drug have shown that when mixed with EC (Ethocel®) and with additional thermal treatment, the amylose forms a coating which is resistant to gastric acid and pancreatic enzymes. But it releases the drug in simulated colonic conditions where a mixed flora of intestinal bacteria is incorporated into a batch culture fermenter (Milojevic et al. Citation1996b).
2 Chemical structure of amylose.
Milojevic et al. (Citation1996a) prepared and evaluated in vitro potential of amylose-Ethocel coating system for colon targeted delivery. Aqueous dispersions were prepared using amylose and Ethocel, which was used to coat 5-ASA pellets to make them resistant to gastric and small intestinal enzymes. Amylose and Ethocel in coating ratio of 1:4 was optimum with least drug release in stomach and intestinal fluids. In vitro release of 5-ASA from coated pellets with amylose and EC (1:4) was retarded in simulated gastric and small intestinal fluid over a period of 12 hr. It was fermented and drug was released in 4 hr in simulated colonic environment containing mixed fecal bacteria of human origin. The potential of amylose in novel colon targeted drug delivery has been investigated using glucose pellets prepared by extrusion and spheronization. Various glucose formulations were studied in which glucose and Avicel® PH101 (microcrystalline cellulose) remained the same while the binding agent, used to retard glucose release, was changed. The dissolution media (400 ml at 37 ± 0.5°C) used were water, hydrochloric acid (pH 1.2 for 3 hr), and 0.2 M phosphate buffer (pH 7.2 for 9 hr) with simulated gastric and intestinal conditions, respectively. Pellet cores containing 20% glycerol monostearate, 50% glucose, and 30% Avicel® PH101 coated with amylose-Ethocel® (1:4 w/w) exhibited gastric and small intestinal resistance but were susceptible to bacterial enzymatic attack in simulated colonic conditions. In vitro fermentation studies were carried out in a batch fermentor inoculated using fresh feces homogenized in anaerobic 0.1 ml/L sodium phosphate buffer (pH 7.0). Carbon dioxide and volatile fatty acids, end-products of colonic bacterial fermentation, were measured over a subsequent 48 hr period, which showed an early release of glucose from coated pellets.
A mixture of amylose and Ethocel (1:4) has been developed and [13C] glucose used as surrogate for drug delivery. Eight healthy subjects were given amylose–Ethocel–coated pellets containing 300 mg [13C] glucose together with a capsule that contained amberlite resin labeled with 99mTc to act as a transit marker and outline for the anatomy of the gut. According to γ-scintigraphy, after 7.1 hr all the material was in the colon. Breath 13CO2 data were analyzed by the cusum technique and with total 13CO2 recovery at 25 hr that was found to be 37.5% (Cummings et al. Citation1996).
Epichlorhydrin-treated crosslinked amylose was introduced as a matrix for controlled release of theophylline (Lenaerts, Dumoulin, and Mateescu Citation1991). Amylose ethylcellulose film coating has been investigated for colon targeted drug delivery (Siew, Basit, and Netwon Citation2000). Drug release rate from the films was governed by the concentration of amylose and EC. Drug release rate was inversely proportional to the thickness of the coating and also influenced by the amount of amylose present in the film. These films were shown to be degraded by bacterial enzymes in simulated colonic environment.
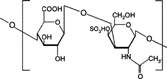
3 Chemical structure of chondroitin sulphate.
CHONDROITIN SULFATE
Chondroitin sulfate is a soluble mucopolysaccharide (Wastenson Citation1971; Toledo and Dietrich Citation1977) that is used as a substrate by the bacteroid inhabitants of the large intestine mainly by Bacteroides thetaiotaomicron and B. ovatus (Salyers Citation1979) (). Periplasmic enzymes are probably responsible for chondroitin breakdown apparently an outer membrane receptor binds chondroitin sulfate and brings it into contact with enzymes like chondroitin-sulfate-lyase (Salyers and O'Brien Citation1980).
Chondroitin sulfate could be used as carrier for colon targeted delivery of bioactive agents. However, use would depend on its persistence as a solid dosage form in the physiological environment of the stomach and small intestine. Since natural chondroitin sulfate is readily water-soluble, it might not protect its load successfully. However, crosslinked chondroitin sulfate would be less hydrophilic and thus would provide a better shield.
Chondroitin sulphate was crosslinked with 1,12 diaminododecane using dicyclohexylcarbodiimide as a catalyst and formulated in a matrix with indomethacin as a drug marker. The indomethacin release kinetics from the various formulations was analyzed in PBS with and without rat caecal content at 37°C under carbon dioxide atmosphere. It was concluded that release of indomethacin depended upon the biodegradation action of the caecal content (Rubinstein, Nakar, and Sintov Citation1992a, Citation1992b). The extent of crosslinking was assessed by the amount of methylene blue that adsorbed as a result of cation exchange. The crosslinked chondroitin dry powder was sieved and mixed with indomethacin for the preparation of matrix tablets. The indomethacin release kinetics from the various formulations was analyzed in PBS with and without rat caecal content at 37°C under a carbon dioxide environment. In separate experiments, the caecal content was sonicated to cause the lysis of the bacterial cell membranes. The drug release profiles indicated a constant rate of biodegradation in the presence of rat caecal content, and diffusion-driven release from the matrix's surface area in PBS control. Prolonged incubation in PBS with rat caecal content increased drug release, and after 2 hr the released indomethacin levels were significantly higher than those in the PBS controls. The maximal cumulative percent drug release values for the PBS controls were 30.1 ± 10.0, 19.7 ± 15.0, and 9.0 ± 4.1 for the three levels of crosslinkage of the chondroitin sulfate carriers, respectively, whereas those for the caecal content media were 71.0 ± 19.0, 48.0 ± 35.0, and 22.5 ± 7.9, respectively. The linear correlation between the degree of crosslinking and the amount of drug released in the presence of caecal content suggests that drug release in the colon can be controlled by adjusting the relative amounts of the variously crosslinked chondroitin sulfate formulations in the matrices.
Sintov, Di-Capua, and Rubinstein (Citation1995) crosslinked chondroitin sulphate with 1,12-diaminododecane to give a series of crosslinked products with reduced water solubility. Indomethacin tablets were prepared using two types of crosslinked polymers: very low water soluble and relatively high water soluble and analyzed for their water uptake and drug release characteristics.
INULIN
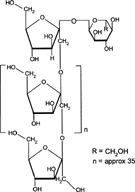
Inulin is a naturally occurring storage polysaccharide found in many plants such as onion, garlic, artichoke, and chicory. Chemically, it belongs to the glucofructans and consists of a mixture of oligomers and polymers containing 2 to 60 (or more) β 2-1 linked D-fructose molecules (). Most of these fructose chains have a glucose unit as the initial moiety. It is not hydrolyzed by the endogenous secretions of the human digestive tract (Dysseler and Hoffen Citation1995). However, bacteria harboring in the colon and more specifically Bifidobacteria are able to ferment inulin (McKeller and Modler Citation1989; Wang and Gibson Citation1993; Gibson and Roberfroid Citation1995).
4 Chemical structure of inulin.
The inulin has been incorporated into Eudragit RS films for preparation of mixed films that resisted degradation in the upper GIT but digested in human fecal medium by the action of Bifidobacteria and Bacteroids (Vervoort and Kinget Citation1996). Various inulin hydrogels have been developed that serve as potential carriers for the introduction of drugs into the colon (Vervoort et al. Citation1997, Citation1998a, Citation1998b; Maris et al. Citation2001). Vinyl groups were introduced in inulin chains to form hydrogels by free radical polymerization. Inulin was reacted with glycidyl methacrylated in N,N-dimethylformamide in the presence of 4-dimethylaminopyridine as catalyst. 1H and 13C NMR spectroscopy were used for the characterization of the obtained reaction product and revealed the conversion of the incorporated vinyl groups into covalent crosslinks upon free radical polymerization of aqueous solutions of the derivatized inulin (Vervoort et al. Citation1997). Maris et al. (Citation2001) synthesized and characterized new hydrogel systems composed of methacrylated inulin, copolymerized with the aromatic azo agent BMAAB and HEMA or MA. The gels were assessed by studying the influence of various parameters on the dynamic and equilibrium degree of swelling.
Inulin hydrogels have been developed and characterized for dynamic and equilibrium swelling properties and in vitro degradation study (Vervoort et al. Citation1998a, Citation1998b). The rate of water transport into the inulin hydrogels was quite high (mean swelling time < 1.2 hr) and the hydrogels exhibited anomalous dynamic swelling behavior. The equilibrium swelling of the hydrogels was influenced by the degree of substitution and feed concentration of methacrylated inulin, by the initiator concentration, by the ionic strength, and by an acidic pH of the swelling solvent. The enzymatic digestibility of the prepared inulin hydrogels was assessed by performing an in vitro study using an inulinase preparation derived from Aspergillus niger. The amount of fructose liberated from the inulin hydrogels by the action of inulinase was quantified using the anthrone method. The equilibrium swelling ratio as well as the mechanical strength of the hydrogels were studied before and after incubation in inulinase solutions. The data obtained by these different methods indicate that enzymatic digestion of the inulin hydrogels appeared to be enhanced by a prolonged degradation time, a higher inulinase concentration, and a lower degree of substitution and feed concentration of the hydrogel polymer. The inulin hydrogels exhibited an increase in equilibrium swelling after degradation compared with the swelling before degradation, suggesting that inulinase enzymes diffuse into the inulin hydrogel networks causing bulk degradation.
Stubbe et al. (Citation2001) developed azo-containing polysaccharide gels more specifically azo-inulin and azo-dextran gels. Compared with azo-hydrogels that can only be degraded by reduction of the azo groups, the goal of the study was to evaluate whether, in vitro, azo-polysaccharide gels could be degraded through both reduction of the azo-groups in the crosslinks as well as enzymatic breakdown of the polysaccharide backbone. The azo-polysaccharide gels were synthesized by radical crosslinking of a mixture of methacrylated inulin or methacrylated dextran and BMAAB and were characterized by dynamic mechanical analysis and swelling measurements. Azo-dextran gels could be obtained from methacrylated dextran having low degree of substitution but not from lowly substituted methacrylated inulin. Increasing the amount of BMAAB resulted in denser azo-inulin and azo-dextran networks. Compared with their swelling in dimethylformamide, all azo-dextran gels became more swollen in water while azo-inulin gels shrank upon exposure to water, indicating a more hydrophobic character of the azo-inulin gels. Breakdown of the inulin and dextran chains in the azo-polysaccharide gels by inulinase and dextranase, respectively, was observed. However, the degradation of azo-dextran gels by dextranase seemed more pronounced than the degradation of the azo-inulin gels by inulinase. In rat caecal content medium, reduction of the azo function in azo-inulin gels was not observed. This may be attributed to a low partitioning of nicotinamide-adenine dinucleotide phosphate in the gels.
GUAR GUM
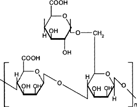
Guar gum, obtained from the ground endosperms of Cyamposis tetragonolobus, consists chiefly of high molecular weight hydrocolloidal polysaccharide, composed of galactan and mannan units combined through glycosidic linkages and shows degradation in the large intestine due the presence of microbial enzymes (Tomolin, Taylor, and Read Citation1989; Bayliss and Houston Citation1986; Macfarlane et al. Citation1990). The structure of guar gum is a linear chain of β-D-mannopyranosyl units linked (1→4) with single-member α-D-galactopyranosyl units occurring as side branches (Goldsten, Alter, and Seaman Citation1992) (). Guar gum has a molecular weight of approximately 1 million, giving it a high viscosity in solution. This galactomannan is soluble in cold water, hydrating quickly to produce viscous pseudoplastic solutions that although shear-thinning generally have greater low-shear viscosity than other hydrocolloids (Johnson and Gee Citation1981). This gelling property retards release of the drug from the dosage form, and it is susceptible to degradation in the colonic environment. Homogenized and diluted feces from human source were incubated with the guar gum to investigate the degradation of polysaccharide by intestinal microflora. It produced a rapid decrease in viscosity and fall in pH while no such results were observed when it was incubated with autoclaved fecal homogenates (Tomlin et al. Citation1986).
5 Chemical structure of guar gum.
The guar gum was reacted with increasing amount of borax, and the properties of the new polymers were characterized by viscosity measurement prior to drying, atomic adsorption, FT-IR, water solubility measurements, and equilibrium water uptake measurements. It was found that despite the reaction with borax, solid guar gum (either in film form or compressed tablets) maintained its ability to degrade by galactomannanase. The modified products were found to possess better swelling properties and, as a result, better enzymatic degradation. The increase in the polymer biodegradation was directly proportional to the increase in their fluid uptake characteristics (Rubinstein and Gilko-Kabir Citation1995). To reduce the swelling properties of the guar gum, it was reacted with glutaraldehyde under acidic conditions to obtain different products with increasing crosslinking densities. The products were characterized by measuring their swelling properties in simulated gastric and intestinal fluids and their crosslinking densities. The release kinetics of three different drugs—sodium salicylate, indomethacin, and budesonide—was assessed from the crosslinked products into buffer solutions with or without a mixture of galactomannanase and α-galactosidase, and their in vivo degradation in the caecum of conscious rats with and without antibiotic treatment. It was found that while there was significant reduction in swelling, the different products were able to degrade in the biological milieu. It was concluded that new biodegradable hydrogels could be utilized for colonic delivery of poorly water-soluble drugs, such as indomethacin or budesonide but not highly water soluble drugs, such as sodium salicylate (Gilko-Kabir et al. Citation1998).
6 Crosslinking reaction of guar gum with STMP (A) and glutaraldehyde (B and C).
In another attempt to reduce the solubility of guar gum, CitationGilko-Kabir et al. (2000a, 2000b) reacted it with sodium trimeta phosphate (STMP). Swelling of guar gum in artificial gastrointestinal fluids was reduced from 100–120-fold to 30–35-fold, depending on the amount of crosslinker used, showing a bell-shape dependency. The reaction with STMP caused guar gum to loose its nonionic nature (). This was verified by the dependency found between the extent of swelling of the crosslinked polymer and the amount of sodium chloride (ionic strength) in the solution. An increase in sodium chloride concentration caused a decrease in the fluid uptake, which is attributed to the counterion shield effect on charged functional groups like phosphates. The increase in the ionic strength of the solution decreases the osmotic pressure inside the charged gel and its swelling is thus reduced. The release kinetics of preloaded hydrocortisone was carried out from formulated hydrogels into buffer solutions with or without addition of α-galactosidase and β-mannanase. Colon-specificity was also assessed by direct measurement of the polymer degradation in the caecum of conscious rats and it was found that the product that was crosslinked with 0.1 equivalent of trisodium trimetaphosphate was able to prevent the release of 80% of its hydrocortisone load for at least 6 hr in PBS (pH 6.4). When a mixture of α-galactosidase and β-mannanase was added to the buffer solution, an enhanced release was observed. In vivo degradation studies in the rat caecum showed that despite the chemical modification of guar gum, it retained its enzyme-degrading properties in a crosslinker concentration-dependent manner (Gliko-Kabir et al. 2000b).
Matrix tablets of guar gum with dexamethasone and budesonide have been investigated for colon targeted drug delivery (Wong et al. Citation1997). Enhanced release of drug was observed when release studies were conducted in simulated colonic fluid than in simulated gastric fluid and simulated intestinal fluid where negligible amount of drug was released. The extent of acceleration in drug dissolution depend on the concentration of galactomannanase. Galactomannanase (> 0.01 mg/ml) accelerated dissolution of both dexamethasone and budesonide form matrix tablets in simulated colonic fluid. Rama Prasad, Krishnaiah, and Satyanarayana (Citation1998) formulated guar gum matrix tablets incorporating indomethacin as model drug for colonic delivery. Drug release rate studies were carried out in 0.1 M HCl for 2 hr and then in Sorensen's phosphate buffer for 3 hr. To assess the susceptibility of guar gum by colonic bacteria, release rate studies were performed in the presence of rat caecal contents. To induce the enzymes that specifically degrade guar gum, the rats were given 1 ml of aqueous dispersion of guar gum (2% w/w). Only 29.21 ± 0.64% of drug was released after 21 hr in PBS without rat caecal contents at pH 6.8, while it was 49.70 ± 3.88% in concentration of 2% caecal matter that further increased to 59.64 ± 4.05% in case of 4% caecal matter.
Drug release studies after inducting of enzymes for 3 days revealed that at 2% w/w caecal content level 60.19 ± 1.44% drug was released after 21 hr while it was 78.54 ± 3.08% with 4% caecal matter. Release of the drug was further increased when the studies were carried out in presence of caecal matter obtained after 7 days induction of enzymes. A linear relationship was obtained between the percent drug released and the time of induction. Further guar gum matrix tablets were evaluated by conducting gamma scintigraphic studies after incorporating 99m-Tc-DTPA as tracer, in 6 healthy male human volunteers (Krishnaiah et al. Citation1998). Scintigraphs taken at regular intervals showed that some amount of tracer present on the surface of the tablets was released in stomach and small intestine and the bulk of the tracer present in the tablet mass delivered to the colon. The colonic arrival of the tablets was found to be 2 to 4 hr. On entering the colon, the tablets were found to be degraded in 5 of 6 volunteers thereby releasing a larger amount of the tracer. Kenyon et al. (Citation1997) developed four formulations of dexamethasone using guar gum in which one was designed for rapid release while the others were aimed at delayed release of drug. Formulations were administered to human volunteers after labeling with radioactive 153Sm and scintigraphs were taken from time to time. It was revealed from the serum concentration profile and scintigraphs that rapid release tablet disintegrated in the stomach. One of the delayed release formulations started to disintegrate in the small intestine while the other two exhibited disintegration time of 3.6 ± 1.6 and 5.8 ± 2.3 hr. Colon targeted release was endorsed based on the observations that all three formulations disintegrated completely in the colon releasing 72–82% of drug.
Guar gum compressed tablets using 5-ASA as drug core has been investigated for colon-specific delivery (Krishnaiah, Satyanarayana, and Rama Prasad Citation1999). Drug release rate studies for tablets coated with 300, 200, and 150 mg guar gum revealed that 5.98, 8.67, and 12.09% of drug was released after 26 hr while tablets formulated with 125 mg guar gum took only 5 min to disintegrate in simulated gastric fluid. Formulations prepared using thick coat of guar gum (200 and 300 mg) could cause release of reasonable amount of drug in presence of rat caecal contents even in 26 hr. Formulations prepared with 150 mg of guar gum as coating showed optimum release (95.51%) of 5-ASA after 26 hr in presence of rat caecal contents. The drug release studies suggest that the thickness of guar gum coating in the range of 0.61–0.91 mm was sufficient for colon-specific delivery of 5-ASA. Krishnaiah et al. (Citation2001) investigated the influence of metronidazole and tinidazole on the usefulness of guar gum as a carrier for colon-specific delivery of albendazole (as model drug) from guar gum matrix tablets. The in vitro drug release studies were performed in simulated colonic fluids (4% w/v of rat caecal contents) obtained after oral treatment of rats for 7 days either with varying doses of metronidazole/tinidazole and 1 ml of 2% w/v of guar gum or with 1 ml of 2% w/v of guar gum alone as control after completing the dissolution study in 0.1 M HCl (2 hr) and pH 7.4 Sorensen's phosphate buffer (3 hr). The guar gum matrix tablets were degraded by colonic bacteria of rat caecal contents and released about 44% of albendazole in simulated colonic fluids (control study) at the end of 24 hr indicating the susceptibility of the guar gum formulations to the rat caecal contents. The results of the study showed that concomitant administration of either metronidazole or tinidazole with guar gum based colon-specific drug delivery systems may interfere with the targeting of drugs to colon.
Matrix tablets containing various proportions of guar gum were prepared by wet granulation technique using starch paste as a binder (Krishnaiah et al. Citation2002a). The tablets were evaluated for hardness and drug content and were subjected to in vitro drug release studies. The results of the in vitro release study revealed that in simulated gastric and small intestinal conditions the release of celecoxib was much less (2–4%) depending on the proportion of guar gum used in the formulation. When the dissolution study was continued in simulated colonic fluids in presence of rat caecal content, the release of drug was increased substantially indicating the susceptibility of the guar gum formulations to the rat caecal contents. With the aim of reducing the systemic side effects associated with intravenous administration of 5-fluorouracil, Krishnaiah et al. (Citation2002b) developed novel tablet formulations for site-specific delivery of 5-fluorouracil to the colon without the drug being released in the stomach or small intestine using guar gum as a carrier. Fast-disintegrating 5-fluorouracil core tablets were compression-coated with different proportion of guar gum and were subjected to in vitro drug release studies. Guar gum compression-coated tablets released only 2.5–4% of the drug in simulated GI fluids. When the dissolution study was continued in simulated colonic fluids (4% w/v rat caecal content medium), tablets compression-coated with 60, 70, and 80% of guar gum released another 70, 55, and 41% of the drug, respectively. Thimma and Tammishetti (Citation2001) reported barium chloride crosslinked carboxymethyl guar gum beads for GI drug delivery.
DEXTRANS
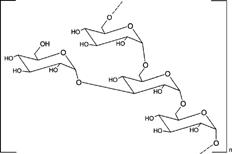
This class of polysaccharide consists of linear chains of α-D-glucose molecules; 95% of the chains consists of 1:6-α-linked linear glucose units while the side chains consist of 1:3-α-linked moieties (). They are obtained from microorganisms of the family of Lactobacillus (Leuconostoc mesenteroides). Dextrans are colloidal, hydrophilic, and water-soluble substances, inert in biological system, and do not affect cell viability. Dextranases are the enzymes that hydrolyze glycosidic linkages of the dextrans. Dextranase activity of the colon is shown by anaerobic Gram-negative intestinal bacteria especially the Bacteroides (Hehre and Sery Citation1952). Dextran also has been found to be degraded in human feces due to bacterial action (Aberg Citation1953).
7 Chemical structure of dextran.
Dextrans of various molecular weights have been used as drug carriers. Various dextran ester prodrugs have been prepared and evaluated for their efficacy to deliver the drug to the target organ, i.e., colon. Harboe, Johansen, and Larsen (Citation1988; Harboe et al. Citation1989a) synthesized dextran ester prodrug. The bioavailability of naproxen after oral administration of aqueous solutions of a dextran T-70-naproxen ester was compared with that of an oral solution of an equivalent dose of naproxen in pigs. The bioavailability was close to 100% in comparison to the oral dose of parent drug, and drug activation took place in the lower part of the GIT. It was established that several features of the prodrug indicated that naproxen was released from the prodrug prior to systemic absorption and that drug activation involved the action of one or more enzyme systems located in the GIT. In rabbits, the plasma concentration-time curves for the conjugate were characterized by an initial lag time of about 2–3 hr, whereas naproxen was detected in plasma immediately after per oral administration of the drug compound per se. The distribution of the prodrug along the GIT at various times after conjugate administration was assessed by HPLC analysis of conjugated and free naproxen in various segments of the GIT. From these experiments, drug regeneration was effective in the bowel below the ileum.
Dextran ester prodrugs of ketoprofen and naproxen were prepared using dextran with molecular weight 10,000–50,0000 which showed degradation in the colon. In vitro release revealed that release of naproxen from prodrug was several folds higher in caecum homogenates than in control medium or homogenates of the small intestine of pig (Larsen et al. Citation1989a, Citation1989b, Citation1991b; Harboe et al. Citation1989b).
The side effects of steroid therapy, which are used in the treatment of chronic colitis, may be decreased by selectively delivering drug to the colon (Friend et al. Citation1991; Tozer et al. Citation1991). One potential way of achieving this is to attach the drug to dextran, a polar macromolecule that prevents drug absorption in the small intestine. Previous reports on colon-specific delivery using dextran prodrugs have focused on non-steroidal anti-inflammatory agents, which are directly coupled to dextran using the carboxylic acid groups of the drugs (Harboe et al. Citation1988; Larsen and Johansen Citation1989). Experiments with these conjugates have demonstrated that dextranases and esterases in the colon degrade the conjugate and liberate drug at this site (Harboe et al. Citation1989b; Larsen, Jensen, and Olesen Citation1991a).
Glucocorticoids-dextran ester prodrugs have been prepared and proved efficacious in delivering drugs to colon (McLeod, Friend, and Tozer Citation1993; McLeod et al. Citation1994a, Citation1994b). Methylprednisolone and dexamethasone were attached to dextran with molecular weight 72,600 by a succinate linker and, in addition, dexamethasone was attached by glutaric acid to investigate the effect of a linker molecules on hydrolysis kinetics. The kinetics of degradation of the hemiester and corresponding dextran conjugates were measured as a function of pH and temperature. Intermolecular migration of the linker molecule from the 21- to the 17-position on the glucocorticoid occurred in all three hemiester, although to a greater extent in methylprednisolone-hemiester. The dextran conjugates also were incubated at 37°C, pH 6.8, and the chemical degradation half-lives was determined.
Dextran hydrogels have been shown to be the promising carrier for the delivery of drugs to colon. Bronsted, Hovgaard, and Simonsen (1995a, 1995b) prepared dextran hydrogels utilizing diidocyanate as crosslinker, which were shown to be degradable in vitro and in vivo in rat caecum. Simonsen et al. (Citation1995) reported the degradability of dextran hydrogels in the human colonic fermentation model; however, the fermentation of gels started after 24 hr which would cause delay in drug release.
Hovgaard and Brondsted (Citation1995) have studied the suitability of dextran hydrogels for colon targeted delivery of hydrocortisone. The hydrogels were characterized by equilibrium degree of swelling and mechanical strength, and the degradation of hydrogels was studied in vitro using dextranase, in vivo in rats, and in human fermentation model. They found that by changing the chemical composition of the hydrogels, it is possible to control the equilibrium degree of swelling, mechanical strength, and biodegradability. The dextran hydrogels were degraded in vivo in the caecum of rats but not the stomach. Furthermore, the dextran hydrogels were degraded in a human colonic fermentation model, indicating that dextranses are indeed present in human colonic contents. Finally, release of hydrocortisone from the hydrogels was found to depend on the presence of dextranases in the release medium. pH-sensitive dextran hydrogels were prepared by activation of dextran (T-70) with 4-nitrophenyl chloroformate, followed by conjugation of the activated dextran with 4-aminobutyric acid and crosslinking with 1,10-diaminodecane. The crosslinking efficiencies determined by mechanical measurements were in the range of 52–63%. Incorporation of carboxylpropyl groups in dextran hydrogels led to a higher equilibrium and faster swelling under high pH conditions. The swelling reversibility of hydrogels also was observed after repeated changes in buffers between pH 2.0 and 7.4.
The slow rates of swelling and deswelling in response to changes in pH were attributed to the hydrophilic nature of dextran and formation of hydrogen bonds between the hydroxyl groups of dextran with water molecules. The pronounced effect of carboxylic acid content on degradation of hydrogels was observed after 4 hr incubation with dextranase, and the influence significantly decreased after exposure to the enzyme for 8 hr. The mechanism of bulk degradation of hydrogels under high swelling extent was substantiated using Coomassie blue protein assay. The release rate of bovine serum albumin from hydrogels was primarily determined by the swelling extent. The release rate was further enhanced by addition of dextranase in buffer solutions (Chiu et al. Citation1999).
Bauer and Kesselhut (Citation1995) synthesized dextran fatty acid ester and showed that lauroyl dextran esters with molecular weight of approximately 250,000 and degree of substitution ranging from 0.11 to 0.3 were suitable for colon targeted drug delivery as film coatings. In vitro studies with lauroyl dextrans esters bearing theophylline were carried out and Hirsch et al. (Citation1997) showed that addition of dextranase accelerated the drug release. However, further studies negated the utility of dispersion of lauroyl dextrans as coating material as they did not exhibit ideal zero-order dissolution before and quick disintegration after enzyme addition (Hirsch et al. Citation1999). The dextran capsules appear to be excellent candidates for colon-specific drug delivery of almost any drug molecules as the polymer composition governs the specific release pattern (Bronsted, Andersen, and Hovgaard 1998). A reaction mixture containing dextran, magnesium chloride, glutaraldehyde, and polyethyleneglycol 400 in water was applied onto molding pins of nylon producing capsule caps and bodies. The capsule materials were characterized by measuring the mechanical strength in compression and equilibrium degree of swelling.
Based on these results, an optimal composition for the capsule material was selected. The dextran capsules were loaded with hydrocortisone and subsequently drug release was studied. The release was found to be about 10% during the initial 3 hr in a buffer solution. Over a period of 24 hr the release was about 35%. However, when the dextran capsules were challenged with a dextranase solution, simulating the arrival of the drug delivery system to the colon, the capsules quickly broke and the drug was released as a dose dump.
Dextran ester prodrugs of metronidazole have been prepared and characterized for colon-specific drug release (Johansen and Larsen Citation1984, Citation1985; Vermeersch et al. Citation1985). The kinetics of hydrolysis of metronidazole monosuccinate dextran ester conjugates in aqueous solution over the pH range 6.61–7.80 revealed that the degradation of dextran conjugates proceeds through parallel formation of metronidazole and metronidazole monosuccinate, respectively. The regeneration rates of the latter compounds followed first-order kinetics. Dextran ester prodrugs of 5-ASA were prepared and drug release rate study revealed that no drug was released in the presence of contents of small intestine of rats while the presence of diluted caecal contents accelerated drug release (Jung et al. Citation1998). Dextran-nalidixic acid ester (dextran-NA) with a varied degree of substitution was synthesized as a colon-specific prodrug of nalidixic acid (Lee et al. Citation2001). No drug was detected during the 6 hr of the incubation period of prodrug in buffer of pH 1.2 and 6.8, which indicated that dextran-NA might be chemically stable during the transit through the GIT. When dextran-NA was incubated with caecal contents of rats at 37°C, the extent of drug released in 24 hr was considerably increased. NA was not liberated from the incubation of dextran-NA with the homogenate of tissue and contents of the small intestine.
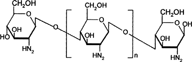
8 Chemical structure of chitosan.
CHITOSAN
Chitosan is a functional linear polymer derived from chitin, the most abundant natural polysaccharide next to cellulose, and it is not digested in the upper part of the GIT by human digestive enzymes (Furda Citation1983; Omrod, Holmes, and Miller Citation1998). Chitosan is a copolymer consisting of 2-amino-2-deoxyD-glucose units linked with β-(1→4) bonds (). It should be susceptible to glycosidic hydrolysis by microbial enzymes in the colon because it possesses glycosidic linkages similar to those of other enzymatically depolymerized polysaccharides.
CitationTozaki et al. (1997b, 2002) developed colon-specific insulin delivery with chitosan capsules. In vitro drug release experiments from chitosan capsules containing 5(6)-carboxyfluorescein (CF) were carried out by a rotating basket method with slight modifications. The intestinal absorption of insulin was evaluated by measuring the plasma insulin levels and its hypoglycemic effects after oral administration of the chitosan capsules containing insulin and additives. Little release of CF from the capsules was observed in an artificial gastric juice (pH 1) and in an artificial intestinal juice (pH 7). However, the release of CF was markedly increased in the presence of rat caecal contents. A marked absorption of insulin and a corresponding decrease in plasma glucose levels were observed following the oral administration of these capsules that contain 20 IU of insulin and sodium glycocholate as compared with the capsules containing only lactose or only 20 IU of insulin. The hypoglycemic effect started at 8 hr after the administration of chitosan capsules when the capsules entered the colon, as evaluated by the transit time experiments with chitosan capsules. These findings suggest that chitosan capsules may serve as useful carriers for the colon-specific delivery of peptides including insulin.
The naturally occurring polymer chitosan was reacted separately with succinate and phthalic anhydrides and resulted in the formation of semisynthetic derivatives, chitosan succinate and chitosan phthalate. The matrices bearing diclofenac sodium as model drug were prepared and incorporated into tablets. The in vitro studies revealed very slight release of drug under acidic conditions while improved drug release profiles were observed under basic conditions suggesting suitability for colon targeted drug delivery (Aiedeh et al. Citation1999).
Tozaki, Fujita, and Odoriba (Citation1999) used rats to study the colon specificity of chitosan capsules bearing R-68070, a thromboxane synthetase inhibitor used for the treatment of chemically induced ulcerative colitis. Insulin is degraded by the digestive action of enzymes of upper gastrointestinal tract and hence Tozaki and co-workers (Tozaki et al. Citation1997a) incorporated insulin into chitosan capsules to deliver insulin in the colon It was found that these capsules were stable in the stomach and small intestine but degraded by microorganism in rat caecal contents upon entering into the colon, proving their utility as carriers for colon targeted delivery of peptide and nonpeptide drugs. A pH-sensitive drug delivery carrier also has been reported for chitosan-based hydrogels (Vandelli et al. Citation1996; Shu, Zhu, and Song Citation2001). Suzuki et al. (Citation1998) prepared hard capsules of chitosan with enteric polymers for colon targeted drug delivery.
Chitosan has favorable biological properties such as nontoxicity, biocompatibility, and biodegradability. In addition, it has the advantage of being widely approved as a food ingredient, which suggests its acceptability as a new excipient for oral administration (Weiner Citation1993). Despite these promising characteristics, a limitation of chitosan is its rapid dissolution in the gastric cavity. Chemical crosslinking with aldehydes has been, so far, a way of overcoming this problem (Berthold, Cremer, and Kreuter Citation1996; Thanoo, Sunny, and Jaykrishnan Citation1992; Akbuga and Durmaz Citation1994). Nevertheless, the toxicity of aldehydes enormously limits the exploitation of these crosslinked microcapsules. Furthermore, this crosslinking process is not totally effective in preventing the release of the encapsulated drug. A multiparticulate system of chitosan would be the desired dosage form for colon-specific drug delivery. Lorenzo-Lamosa et al. (Citation1998) investigated a microparticulate system that combined specific biodegradability and a pH-dependent release profile. It consists of chitosan microcores bearing diclofenac sodium entrapped within acrylic microspheres. Chitosan microcores were prepared using spray drying and then microencapsulated into Eudragit® L-100 and Eudragit® S-100 using an oil-in-oil solvent evaporation method. Release of the drug from chitosan multireservoir system was adjusted by changing the chitosan molecular weight or the type of chitosan salt. Furthermore, by coating the chitosan microcores with Eudragit®, perfect pH-dependent release profiles were attained. In addition, it was observed from the IR spectra of the Eudragit coated chitosan microcores that chitosan was ionically crosslinked with the Eudragit which further controlled the release of the drug in the alkaline pH of the small intestine.
Similarly, a multiparticulate system of chitosan hydrogel beads has been investigated for colon-specific delivery of macromolecules using fluorescein isothiocanate-labeled bovine serum albumin as a model protein. The hydrogel beads were formed by polyelectrolyte complexation of chitosan with its counterion, tripolyphosphate. The protein release experiments were carried out in vitro under different conditions to simulate the pH and times likely to be encountered during intestinal transit to the colon. The results showed that the hydrogel beads were degraded by rat caecal and colonic enzymes, resulting in a marked acceleration in the release of protein (Zhang, Ibrahim, and Neau Citation2002).
A chitosan-dispersed drug delivery system, composed of active ingredient reservoir and the outer drug release regulating layer dispersing chitosan powder in hydrophobic polymer, was newly developed for colon-specific drug delivery (Shimono et al. Citation2002). Chitosan was dissolved in aqueous solutions containing aspartic, glutamic, hydrochloric, lactic, and citric acids to obtain different chitosan salts (Orienti et al. Citation2002). Chitosan salts were collected from the solutions by spray drying and the powders obtained were mixed with sodium diclofenac, taken as a model antiinflammatory drug. The authors investigated the suitability of chitosan salified with different acids in colon-specific drug delivery based on the possibility of minimizing release during transit in the upper part of the GIT and enhancing polymer hydrolysis by β-glucosidase once in the colon. The in vitro release studies were performed using a diffusion cell in which compartments were separated by dialysis membrane. The release studies were also carried out at pH 7.0 buffer in presence of β-glucosidase. It was concluded that chitosan salified with aspartic, glutamic, hydrochlori, and lactic acids is a suitable supporting material for the preparation of colon-specific drug delivery systems. Physical mixtures of the chitosan salts with diclofenac sodium (1:1) provided slower drug release with respect to the pure drug both at acidic and alkaline pHs. Their interactions with β-glucosidase at pH 7.0 enhanced the release rate due to the enzyme-catalyzed hydrolysis of chitosan. Among the different salts analyzed, aspartate and glutamate provided the lowest release at the acidic pHs and the highest at alkaline pHs. Moreover, in the presence of β-glucosidase the release rate increase was more pronounced due to their solubility characteristics favoring enzyme-polymer interactions.
9 Chemical structure of cyclodextrins.
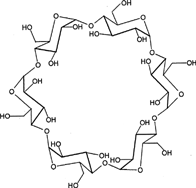
CYCLODEXTRINS
Cyclodextrins (CyDs) are cyclic oligosaccharides consisting of six to eight glucose units joined through α-1,4 glucosidic bonds (). CyDs remain intact during their passage through stomach and small intestine. However, in the colon, they undergo fermentation from the presence of vast colonic microflora into small saccharides and thus absorbed from these regions (Antenucci and Palmer Citation1984; Gerloczy et al. Citation1985; Flourie et al. Citation1993). CyDs form inclusion complexes with drug molecules because the interior of the molecule is relatively lipophilic while the exterior is hydrophilic (Uekama, Hiramaya, and Irie Citation1991; Stella and Rajewski Citation1997; Andersen et al. Citation1983). It has been investigated through a study in healthy human volunteers that β CyDs are degraded to a very small extent in the small intestine but are completely digested in large intestine. Most bacterial strains that are isolated from human beings are capable of degrading CyDs. This has been proved by their ability to grow on cyclodextrins by utilizing them as the sole carbon source and by the stimulation of cyclodextrinase activity as low as 2–4 hr of exposure to CyDs. This property of the drug may be exploited for the formation of colon targeted drug delivery systems. Several CyD conjugates have been prepared and the enantioselective hydrolysis has been described (Karunaratne, Farmer, and Hancock Citation1993; Tanaka, Kominato, and Yamamoto Citation1994; Coates et al. Citation1994).
Formulation of prodrug of CyDs with drug molecules can provide a versatile means for construction of not only colon targeted delivery systems, but also delayed release systems. Biphenylacetic acid (BPAA), an anti-inflammatory drug, was conjugated with α-, β-and γ-CyDs and in vivo release pattern was investigated in rat after oral administration. The CyD prodrug maintained the physical integrity in stomach and small intestine which is reflected from the observations that the absorption of prodrugs from upper GIT was negligible, and after 6 hr most of the prodrug approached the caecum and colon. The α- and γ-CyD amide prodrugs were hydrolyzed to the maltose conjugate in the colon while the α- and γ-CyD ester prodrugs produced BPAA in the caecum and colon. The anti-inflammatory effect of the α- and γ-CyD ester prodrugs was assessed and compared with those of BPAA alone and the drug CyD complex prepared by the kneading method in a molar ratio of 1:1. In the case of β-CyD complex, a rapid anti-inflammatory response was observed from the small intestine after a fast dissolution of the complex. In sharp contrast, the γ-CyD ester prodrug required a fairly long lag time to exhibit the drug activity, because BPAA was produced after the prodrug had reached the caecum and colon (Minami, Hiramaya, and Uekama Citation1998; Uekama, Minami, and Hirayama Citation1997).
10 Chemical structure of alginates.
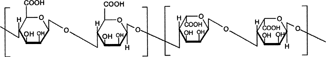
Hiramaya, Minami, and Uekama et al. (Citation1996) prepared two CyD conjugates: ester and amide conjugates. They showed that ester conjugate released the drug preferentially when incubated with the contents of caecum or colon, whereas no appreciable drug release was observed on incubation with the contents of either stomach or intestine in intestinal or liver homogenates or in rat blood. Systemic side effects of prednisolone were significantly reduced when conjugated with α-CyD (Yano et al. Citation2002). The lower side effect of the conjugate was attributed to passage of the conjugate through the stomach and small intestine without significant degradation or absorption, followed by the degradation of the conjugate site-specifically in the large intestine.
ALGINATES
Alginates, natural hydrophilic polysaccharide derived from seaweed, consist of 1→4, linked D-mannuronic acid and L-glucuronic acid residues (). Alginates are easily gelled in presence of a divalent cation as calcium ion. The gelation or crosslinking is due to the stacking of the glucuronic acid blocks of alginate chains.
Calcium alginates beads can be prepared by drop-wise addition of the solution of sodium alginate into the solution of calcium chloride. The alginate beads have the advantage of being nontoxic, and dried alginate beads reswell in presence of dissolution media and can act as controlled release systems. Calcium alginate beads were prepared as cores and 5-ASA was spray-coated on them (Shun and Ayres Citation1992). Different enteric as well as sustained release polymers were applied as coat on calcium alginate beads. A system was prepared by coating calcium alginate beads with Aquacoat® that is a pH-independent polymer followed by 2% w/w coating of Eudragit L-30D. Being enteric polymer, Eudragit® resisted the release of drug in acidic media and drug release was triggered at alkaline pH and controlled by thickness of Aquacoat®. When drug-loaded calcium alginate beads swell sufficiently (osmotic gradient) to exceed the strength of outer sustained released coat, the film bursts to release the drug. Such a system delivers drug to the distal intestine with minimal initial leak and provides sustained release in the colon. Kiyoung, Kun, and Yueim (Citation1999) prepared alginate beads and coated them with dextran acetate. In the absence of dextranase, minimal drug release occurred whereas it was significantly improved in the presence of dextranase.
LOCUST BEAN GUM
It is also called carob gum, as it is derived from carob (Ceratonia siliqua) seeds. Locust bean gum has an irregularly shaped molecule with branched beta-1,4-D-galactomannan units. This neutral polymer is only slightly soluble in cold water, it requires heat to achieve full hydration and maximum viscosity.
The colon-specific drug delivery systems based on polysaccharides, locust bean gum and chitosan in the ratio of 2:3, 3:2 and 4:1, were evaluated using in vitro and in vivo methods (Raghvan et al. 2002). Core tablets containing mesalazine with average weight of 80 mg were prepared by compressing the materials using 6-mm round, flat, and plain punches on a single station tablet machine. The formulated core tablets were compression-coated with different quantities of locust bean gum and chitosan. The in vitro studies in pH 6.8 phosphate buffer containing 2% w/v rat caecal contents showed that the cumulative percentage release of mesalazine after 26 hr was 31.25 ± 0.56, 46.25 ± 0.96, and 97.5 ± 0.26, respectively. The in vivo studies conducted in 9 healthy male human volunteers for the various formulations showed that the drug release was initiated only after 5 hr, which is the transit time of small intestine. In vitro drug release studies and in vivo studies revealed that the locust bean gum and chitosan as a coating material applied over the core tablet was capable of protecting the drug from being released in the physiological environment of stomach and small intestine and was susceptible to colonic bacterial enzymatic actions with resultant drug release in the colon. Hirsch et al. (Citation1999) carried out dissolution studies on theophylline tablets coated with crosslinked locust bean gum and showed the mechanical instability of these coatings in the dissolution media, thereby suggesting the nonsuitability of such films as colon carriers.
SUMMARY
The growing interest of researchers toward oral drug delivery to colon is justified by the need of such systems, which would effectively target the drug release to the colon circumventing the hostile environment of upper GIT. Conventional drug delivery systems suffer from the disadvantages of being nonspecific in nature and consequently release maximum proportion of drug in stomach and small intestine. This causes unwanted absorption of drugs from these regions, which results in generation of toxic effects as well as increase in the cost of therapy. Administration of peptides and proteinous drugs through conventional dosage forms lead to degradation of activity, and those drugs, which are optimally absorbed from large intestine, have low bioavailability. Targeted drug delivery to the colon has advantages in treatment of diseases associated with the colon. It also is beneficial for those drugs, which are unstable in upper GIT, such as peptides and proteins. Several strategies are available to cause the drug release at the target site, i.e., colon. The approach is based on the formation of prodrug and involves covalent linkage between drug and carrier. The type of linkage that is formed between drug and carrier would decide the triggering mechanism for the release of drug in colon. The presence of azo-reductase enzymes plays a pivotal role in the release of drug from azo bond prodrugs while glycosidase activity of the colonic microflora is responsible for liberation of drugs from glycosidic prodrugs.
The most common approach utilizes enteric polymers, that resist degradation in acidic environment and release drug in alkaline media due to formation of salt. However, this approach may not produce intended results due to variation of GI pH. This variation in pH is brought about by pathological conditions, diet composition, or because of inter- and intra-subject variation. The strategy, which depends on colonic microflora for liberation of entrapped drug is, seems most suitable. The rationale is the ability to maintain the integrity of polysaccharide matrix in the hostile environment of stomach and small intestine and afterward release of drug in colon. This release is affected due to the existence of vast colonic microflora having enzymes, that digest polysaccharides.
The most favorable property of these materials is that they are already approved as pharmaceutical excipients. A large number of polysaccharides such as amylose, guar gum, pectin, chitosan, inulin, cyclodextrins, chondroitin sulphate, dextrans, and locust bean gum have been investigated for their use in colon targeted drug delivery systems. The most important fact in the development of polysaccharide derivatives for colon targeted drug delivery is the selection of a suitable biodegradable polysaccharide. As these polysaccharides are usually soluble in water, they must be made water-insoluble by crosslinking or hydrophobic dramatization. Very important is an optimal proportional of the hydrophobic and hydrophilic parts, respectively, and the number of free hydroxy groups in the polymeric molecule.
M. K. Chourasia is thankful to the Council of Scientific and Industrial Research, New Delhi, India, for providing financial assistance to carry out project on colon targeting of drugs.
REFERENCES
- Aberg B.. 1953. Breakdown of dextran by human feces. Scand. J. Clin. Lab. Invest.. 5: 37–38. [PUBMED], [INFOTRIEVE]
- Adkin A. D., Kenyon C. J., Lerner E. I., Landau I., Strausss E., Carbon D., Penhasi A., Rubinstein A., Wilding I. R.. 1997. The use of scintigraphy to provide proof of concept for novel polysaccharide preparation designed for colonic drug delivery. Pharm. Res.. 14: 103–107. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Ahrabi S. F., Heinamaki J., Sande S. A., Graffner C.. 2000a. Development of pectin matrix tablets for colonic delivery of model ropivacaine. Eur. J. Pharm. Sci.. 10: 43–52. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Ahrabi S. F., Heinamaki J., Sande S. A., Graffner C.. 2000b. Influence of neutron activation factors on matrix tablets for site specific delivery to the colon. Eur. J. Pharm. Sci.. 10: 225–235. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Aiedeh K., Taha M. O.. 1999. Synthesis of chitosan succinate and chitosan phthalate and their evaluation as suggested matrices in orally administered colon-specific drug delivery systems. Arch. Pharm. (Weinheim.). 332: 103–107. [CROSSREF]
- Akbuga J., Durmaz D.. 1994. Preparation and evaluation of crosslinked chitosan microspheres containing furosemide. Int. J. Pharm.. 11: 217–222. [CROSSREF]
- Andersen G. H., Robbins F. M., Domingues F. J., Morres R. G., Long C. L.. 1983. The utilisation of schardinger dextrins by the rat. Toxicol. Appl. Pharmacol.. 5: 257–266. [CROSSREF]
- Antenucci R., Palmer J. K.. 1984. Enzymatic degradation of α and β-cyclodextrins by bacteroids of the human colon. J. Agric. Food Chem.. 32: 1316–1321
- Ashford M., Fell J. T., Attwood D., Sharma H., Woodhead P.. 1993. An evaluation of pectin as a carrier for drug targeting to colon. J. Control. Rel.. 26: 213–220. [CROSSREF]
- Ashford M., Fell J. T., Attwood D., Sharma H., Woodhead P.. 1994. Studies on pectin formulations for colonic drug delivery. J. Control. Rel.. 30: 225–232. [CROSSREF]
- Azad Khan A. K., Truelove S. C., Aronseq J. K.. 1982. The disposition and metabolism of sulphasalazine (salicylazosulphapyridine) in man. Br. J. Clin. Pharmacol.. 13: 523–528
- Bauer K. H., Kesselhut J. F.. 1995. Novel pharmaceutical excipients for colon targeting. STP Pharm. Sci.. 5: 54–59
- Bayliss C. E., Houston A. P.. 1986. Degradation of guar gum by faecal bacteria. Appl. Environ. Microbiol.. 48: 626–632
- Berthold A., Cremer K., Kreuter J.. 1996. Influence of crosslinking on the acid stability and physicochemical properties of chitosan microspheres. STP Pharm. Sci.. 6: 358–364
- Brondsted H., Andersen C., Hovgaard L.. 1998. Crosslinked dextran—a new capsule material for colon targeting of drugs. J. Control. Rel.. 53(1–3)7–13. [CROSSREF]
- Brondsted H., Hovgaard L., Simonsen J.. 1995a. Dextran hydrogels for colon-specific drug delivery. III., In vitro and in vivo degradation. STP Pharm. Sci.. 5: 60–64
- Brondsted H., Hovgaard L., Simonsen J.. 1995b. Dextran hydrogels for colon-specific drug delivery. IV. Comparative release mechanism of hydrocortisone and prednisolone phosphate. STP Pharm. Sci.. 5: 65–69
- Chan R. P., Pope D. J., Gilbert A. P., Baron J. H., Lennard-Jones J. P.. 1983. Studies of two novel sulfaslazine analogue: ipsalazine and balsalazine. Dig. Dis. Sci.. 28: 609–615. [PUBMED], [INFOTRIEVE]
- Chickering D. E., Mathiowitz E.. 1995. Bioadhesive microspheres. I. A novel electrobalance-based method to study adhesive interactions between individual microspheres and intestinal mucosa. J. Control. Rel.. 34: 251–262. [CROSSREF]
- Chickering D. E., Jacob J. S., Mathiowitz E.. 1995. Bioadhesive microspheres. 2. Characterisation and evaluation of bioadhesion involving hard, bioerodible polymers and soft-tissue. Reacting Polymers. 25: 189–206. [CROSSREF]
- Chiu H. C., Hsiue G. H., Lee Y. P., Huang L. W.. 1999. Synthesis and characterization of pH-sensitive dextran hydrogels as a potential colon-specific drug delivery system. J. Biomater. Sci. Polym. Ed.. 10: 591–608. [PUBMED], [INFOTRIEVE]
- Coates J. H., Easton C. J., Fryer N. L., Lincolin S. F.. 1994. Complementary diastereo selectivity in the synthesis and hydrolysis of acylated cyclodextrins. Chem. Lett.. 1153–1156
- Cui N., Friend D. R., Fedorak R. N.. 1994. A budesonide prodrug accelerates treatment of colitis in rats. Gut. 35: 1439–1446. [PUBMED], [INFOTRIEVE]
- Cummings J. H., Englyst H. N.. 1987. Fermentation in the human large intestine and available substrates. Am. J. Clin. Nutr.. 45: 1243–1247. [PUBMED], [INFOTRIEVE]
- Cummings J. H., Southgate D. A. T., Branch W. J., Wiggins H. S., Houston H., Jenkins D. J. A., Jirraj T., Hill M. J.. 1979. The digestion of pectin in human gut and its effect on calcium absorption and large bowel function. Br. J. Nutr.. 41: 477–485. [PUBMED], [INFOTRIEVE]
- Cummings J. H., Milojevic S., Harding M., Coward W. A., Gibson G. R., Botham R. L., Ring S. G., Wraight E. P., Stockham M. A., Allwood M. C., Newton J. M.. 1996. In vivo studies of amylose- and ethylcellulose-coated [13C]glucose microspheres as a model for drug delivery to the colon. J. Control. Rel.. 40: 123–131. [CROSSREF]
- Dysseler B. S., Hoffen M. J.. 1995. Inulin, an alternative dietary fiber. Properties and quantitative analysis. Eur. J. Clin. Nutr.. 49: S145–S152. [PUBMED], [INFOTRIEVE]
- El-Gibaly I.. 2002. Oral delayed-release system based on Zn-pectinate gel (ZPG) microparticles as an alternative carrier to calcium pectinate beads for colonic drug delivery. Int. J. Pharm.. 232: 199–211. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Englyst H. N.. 1987. Digestion of the polysaccharides of potato in the small intestine of man. Am. J. Clin. Nutr.. 45: 423–431. [PUBMED], [INFOTRIEVE]
- Englyst H. N., Cummings J. H.. 1985. Digestion of the polysaccharides of some cereal foods in the human small intestine. Am. J. Clin. Nutr.. 42: 778–787. [PUBMED], [INFOTRIEVE]
- Englyst H. N., Cummings J. H.. 1986. Digestion of the polysaccharides of banana (Musa paradisinca sapietum) in man. Am. J. Clin. Nutr.. 44: 42–50. [PUBMED], [INFOTRIEVE]
- Englyst H. N., Wiggins H. S., Cummings J. H.. 1982. Determination of nonstarch polysaccharides in plant foods by gas liquid chromatography of constituent sugars as alditol acetates. Analyst. 107: 307–318. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Englyst H. N., Hay S., MacFarlane G. T.. 1987. Polysaccharide breakdown by mixed populations of human faecal bacteria. FEMS Microbiol. Ecol.. 95: 163–171
- Englyst H. N., Kingman S. M., Cummings J. H.. 1992. Classification and measurement of nutritionally important starch fractions. Eur. J. Clin. Nutr.. 46: S33–S50. [PUBMED], [INFOTRIEVE]
- Fernandez-Hervas M. J., Fell J. T.. 1998. Pectin/chitosan mixtures as coatings for colon-specific drug delivery: An in vitro evaluation. Int. J. Pharm.. 169: 115–119. [CROSSREF]
- Flourie B., Molis C., Achour L., Dupas H., Hatat C., Rambaud J. C.. 1993. Fate of β-cyclodextrin in the human intestine. J. Nutr.. 123: 676–680. [PUBMED], [INFOTRIEVE]
- Friend D. R.. 1992. Glycosides in colonic drug delivery. Oral Colon Specific Drug Delivery, D. R., Friend, 153–187, Boca Raton, FL, CRC Press
- Friend D. R., Chang G. W.. 1984. A colon-specific drug delivery based on the drug glycosidases of colonic bacteria. J. Med. Chem.. 27: 261–266. [PUBMED], [INFOTRIEVE]
- Friend D. R., Phillips S., McLeod A., Tozer T. N.. 1991. Relative anti-inflammatory effect of oral dexamethasone-β-D-glucoside and dexamethasone in experimental IBD. Proc. Int. Control. Bioact. Mater.. 18: 612–613
- Fukui E., Miyamura M., Kobayashi M.. 2001. An in vitro investigation of the suitability of press-coated tablets with hydroxypropylmethylcellulose acetate succinate (HPMCAS) and hydrophobic additives in the outer shell for colon targeting. J. Control. Rel.. 70: 97–107. [CROSSREF]
- Furda I.. 1983. Aminopolysaccharides—their potential as dietary fiber. Unconventional Sources of Dietary Fiber, Physiological and In Vitro Functional Properties, I., Furda, 105–122, Washington, DC, American Chemical Society
- Gazzaniga A., Bussetti C., Moro L., Sangali M. E., Giordano F.. 1995. Time dependent oral delivery system for colon targeting. STP Pharm. Sci.. 5: 83–88
- Gazzaniga A., Iamartino P., Maffione G., Sangalli M. E.. 1994. Oral delayed release system for colon specific drug delivery. Int. J. Pharm.. 108: 77–83. [CROSSREF]
- Gerloczy A., Fonagy A., Keresztes P., Perlaky L., Szejtli J.. 1985. Absorption, distribution, excretion and metabolism of orally administered 14C-β-cyclodextrin in rat. Arzneim Forsch/Drug Res.. 35: 1042–1047
- Gibson G. R., Roberfroid M. R.. 1995. Dietary modulation of the human colonic microflora: introducing the concept of prebiotics. J. Nutr.. 125: 1401–1414. [PUBMED], [INFOTRIEVE]
- Gilko-Kabir I., Yagen B., Penhasi A., Rubinstein A.. 1998. Low swelling crosslinked guar and its potential use as colon-specific drug carrier. Pharm. Res.. 15: 1019–1025. [CROSSREF]
- Gilko-Kabir I., Yagen B., Penhasi A., Rubinstein A.. 2000a. Phosphated crosslinked guar for colon-specific drug delivery. I. Preparation and physicochemical characterization. J. Control. Rel.. 63: 121–127. [CROSSREF]
- Gilko-Kabir I., Yagen B., Baluom M., Rubinstein A.. 2000b. Phosphated crosslinked guar for colon-specific drug delivery. II In vitro and in vivo evaluation. J. Control. Rel.. 63: 129–134. [CROSSREF]
- Goldsten A. M., Alter E. N., Seaman J. K.. 1992. Guar gum. Oral Colon-Specific Drug Delivery and Their Derivatives, R. L., Whistler, 1–43, , NY, Plenum Press
- Gupta V. K., Beckert T. E., Price J. C.. 2001a. A novel pH- and time-based multi-unit potential colonic drug delivery system. I. Development. Int. J. Pharm.. 213: 83–91. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Gupta V. K., Assmus M. W., Beckert T. E., Price J. C.. 2001b. A novel pH- and time-based multi-unit potential colonic drug delivery system. II. Optimization of multiple response variables. Int. J. Pharm.. 213: 93–102. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Harboe E., Larsen C., Johansen M., Olesen H. P.. 1989a. Macromoleculer prodrugs. XIV. Absorption characteristics of naproxen after oral administration of a dextran T-70-naproxen ester prodrugs in pigs. Int. J. Pharm.. 53: 157–165. [CROSSREF]
- Harboe E., Larsen C., Johansen M., Olesen H. P.. 1989b. Macromoleculer prodrugs. XV. Colon targeted delivery. bioavailability of naproxen from orally administered dextran-naproxen ester prodrugs varying in molecular size in the pig. Pharm. Res.. 6: 919–923. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Harboe E., Johansen M., Larsen C.. 1988. Macromolecular prodrugs. VI. Coupling of the highly lipophilic agent naproxen to dextrans and in vitro characterization of the conjugates. Farmaci. Sci. Ed.. 16: 73–85
- Hardy J. G., Evans D. F., Zaki I., Clark A. G., Tonnesen H. H., Gamst O. N.. 1987. Evaluation of an enteric coated naproxen tablet using gamma scintigraphy and pH monitoring. Int. J. Pharm.. 37: 245–250
- Hehre E. J., Sery T. W.. 1952. Dextran splitting anaerobic bacteria from the human intestine. J. Bacteriol.. 63: 424–426
- Hergenrother R. W., Wabers H. D., Cooper S. L.. 1992. The effect of chain extenders and stabilizers on the in vivo stability of polyurethanes. J. Appl. Biomat.. 3: 17–22
- Hiramaya F., Minami K., Uekama K.. 1996. In vitro evaluation of biphenyl acetic acid-beta-cyclodextrin conjugates as colon-targeting prodrugs: drug release behaviour in rat biological media. J. Pharm. Pharmacol.. 48: 27–31
- Hirsch S., Binder V., Kolter K., Kesselhut J. F., Bauer K. H.. 1997. Lauroyldextran as a coating material for site-specific drug delivery to the colon: in-vitro dissolution of coated-tablets. Proc. Int. Symp. Control. Rel. Bioact. Mater.. 24: 379–380
- Hirsch S., Binder V., Kolter K., Kesselhut J. F., Bauer K. H.. 1999. Lauroyldextran and cross-linked galactomannan as coating materials for site-specific drug delivery to the colon. Eur. J. Pharm. Biopharm.. 47: 61–71. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Hovgaard L., Brondsted H.. 1995. Dextran hydrogels for colon-specific drug delivery. J. Control. Rel.. 36: 159–166. [CROSSREF]
- Huang S. L., Bansleben D. A., Knox J. R.. 1979. Biodegradable polymers: chymotrypsin degradation of low molecular weight poly(ester-urea) containing phenylalaline. J. Appl. Polym. Sci.. 23: 429–437. [CROSSREF]
- Jain, Nitin K., Chourasia M. K., Jain S., Jain S. K., Jain N. K.. 2003. Development and characterization of engineered commensal bacteria for the delivery of enzymes. Ind. J. Pharm. Sci.. 65: 113–121
- Johansen M., Larsen C.. 1984. Stability kinetics and hydrolysis of metronidazole monosuccinate in aqueous solution and in plasma. Int. J. Pharm.. 21: 201–209. [CROSSREF]
- Johansen M., Larsen C.. 1985. A comparison of the chemical stability and the enzymatic hydrolysis of a series of aliphatic and aromatic ester derivatives of metronidazole. Int. J. Pharm.. 27: 219–231. [CROSSREF]
- Johnson J. C., Gee J. M.. 1981. Effect of gel forming gum on the intestinal unstirred layer and sugar transport in vitro. Gut. 22: 398–403. [PUBMED], [INFOTRIEVE]
- Jung Y. J., Lee J. S., Kim H. H., Kim Y. K., Kim Y. M.. 1998. Synthesis and properties of dextran-5-amino salicylic acid ester as a potential colon-specific prodrug of 5-amino salicylic acid. Arch. Pharm. Res.. 21: 179–186. [PUBMED], [INFOTRIEVE]
- Karunaratne D. N., Farmer S., Hancock R. E. W.. 1993. Synthesis of bulky beta-lactams for inhibition of cell surface beta-lactamase activity. Bioconjug. Chem.. 4: 434–439. [PUBMED], [INFOTRIEVE]
- Kenyon C. J., Nardi R. V., Wong D., Hooper G., Wilding I. R., Friend D. R.. 1997. Colonic delivery of dexamethasone: a pharmacoscintigraphic evaluation. Aliment. Pharmacol. Ther.. 11: 205–213. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Khan M. Z., Prebeg Z., Kurjakovic N.. 1999. A pH-dependent colon targeted oral drug delivery system using methacrylic acid copolymers I. Manipulation of drug release using Eudragit® L100–55 and Eudragit® S100 combinations. J. Control. Rel.. 58: 215–222. [CROSSREF]
- Kiyoung L., Kun N., Yueim K.. 1999. Polysaccharides as a drug coating polymer. Polym. Prep.. 40: 359–360
- Krishnaiah Y. S. R., Satyanarayana S., Rama Prasad Y. V., Narasimha Rao S.. 1998. Gamma scintigraphic studies on guar gum matrix tablets for colonic drug delivery in healthy subjects. J. Control. Rel.. 55: 245–252. [CROSSREF]
- Krishnaiah Y. S. R., Satyanarayana S., Rama Prasad Y. V.. 1999. Studies of guar gum compression coated 5-aminosalicylic acid tablets for colon-specific delivery. Drug Dev. Ind. Pharm.. 25: 651–657. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Krishnaiah Y. S. R., Veer Raju P., Dinesh Kumar B., Bhaskar P., Satyanarayana V.. 2001. Development of colon targeted drug delivery systems for mebendazole. J. Control. Rel.. 77: 87–95. [CROSSREF]
- Krishnaiah Y. S. R., Satyanarayana V., Dinesh Kumar B., Karthikeyan R. S.. 2002a. Studies on the development of colon-targeted delivery systems for celecoxib in the prevention of colorectal cancer. J. Drug Target.. 10: 247–254. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Krishnaiah Y. S., Satyanarayana V., Dinesh Kumar B., Karthikeyan R. S.. 2002b. In vitro drug release studies on guar gum-based colon targeted oral drug delivery systems of 5-fluorouracil. Eur. J. Pharm. Sci.. 16: 185–92. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Larsen C., Johansen M.. 1989. Macromolecular prodrugs XI. Regeneration rates of various NSAID compounds from their corresponding dextran ester prodrugs in aqueous buffer and in different biological media. Acta Pharm. Nord.. 2: 57–66
- Larsen C., Jensen B. H., Olesen H. P.. 1991a. Stability of ketoprofen-dextran ester prodrugs in homogenates of various segments of the pig GI tract. Acta Pharm. Nord.. 3: 41–44. [PUBMED], [INFOTRIEVE]
- Larsen C., Jensen B. H., Johansen M., Olesen H. P.. 1991b. Bioavailability of ketoprofen from orally administered ketoprofen dextran ester prodrug in the pig. Acta. Pharm. Nord.. 3: 71–76. [PUBMED], [INFOTRIEVE]
- Larsen C., Harboe E., Johansen M., Olesen H. P.. 1989a. Macromolecular prodrugs. XVI. Colon targeted delivery. Comparison of the rate of release of naproxen from dextran ester prodrug in homogenates of various segments of the pig gastrointestinal tract. Pharm. Res.. 6: 995–999. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Larsen C., Harboe E., Johansen M., Olesen H. P.. 1989b. Macromoleculer prodrugs. XV. Colon targeted delivery. Bioavailability of naproxen from orally administered dextran ester prodrugs varying molecular size in the pig. Pharm. Res.. 6: 919–923. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Lee J. S., Jung Y. J., Doh M. J., Kim Y. M.. 2001. Synthesis and properties of dextran-nalidixic acid ester as a colon-specific prodrug of nalidixic acid. Drug Dev. Ind. Pharm.. 27: 331–336. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Lenaerts V., Dumoulin Y., Mateescu M. A.. 1991. Controlled release of theophylline from crosslinked amylose tablets. J. Control. Rel.. 15: 39–46. [CROSSREF]
- Levitt M. D., Hirsh P., Fetzer C. A., Sherman M., Levine A. S.. 1987. H2 excretion after ingestion of complex carbohydrates. Gastroenterology. 92: 383–386. [PUBMED], [INFOTRIEVE]
- Lorenzo-Lamosa M. L., Remunan-Lopez C., Vila-Jato J. L., Alonso M. J.. 1998. Design of microencapsulated chitosan microspheres for colonic drug delivery. J. Control. Rel.. 52: 109–118. [CROSSREF]
- Macfarlane G. T., Hay S., Macfarlane S., Gibson G. R.. 1990. Effect of different carbohydrates on growth polysaccharidases and glycosidase production of Bacteroides ovatus in batch and continuous culture. J. Appl. Bacteriol.. 68: 179–187. [PUBMED], [INFOTRIEVE]
- Macleod G. S., Fell J. T., Collett J. H.. 1997. Studies on the physical properties of mixed pectin/ethylcellulose films intended for colonic drug delivery. Int. J. Pharm.. 157: 53–60. [CROSSREF]
- Macleod G. S., Fell J. T., Collett J. H., Sharma J. L., Smith A. M.. 1999. Selective drug delivery to the colon using pectin: chitosan: hydroxypropyl methylcellulose film coated tablets. Int. J. Pharm.. 187: 251–257. [CROSSREF], [PUBMED], [INFOTRIEVE]
- MacNeil M. E., Rashid A., Stevens H. N. F.. 1990. Dispensing device. World Patent, WO 9009168
- Maris B., Verheyden L., Reeth K. V., Samyn C., Augustijns P., Kinget R., Van den Mooter G.. 2001. Synthesis and characterisation of inulin-azo hydrogels designed for colon targeting. Int J. Pharm.. 213: 143–152. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Markus, Rudolph W., Klein S., Beckert T. E., Petereit H., Dressman J. B.. 2001. A new 5-aminosalicylic acid multi-unit dosage form for the therapy of ulcerative colitis. Eur. J. Pharm. Biopharm.. 51: 183–190. [CROSSREF]
- McKeller R. C., Modler H. W.. 1989. Metabolism of fructooligosaccharides by Biffidobacterium spp. Appl. Microbiol. Biotechnol.. 31: 537–541. [CROSSREF]
- McLeod A. D., Friend D. R., Tozer T. N.. 1993. Synthesis and chemical stability of glucocorticoid-dextran ester: potential prodrugs for colon-specific drug delivery. Int. J. Pharm.. 92: 105–114. [CROSSREF]
- McLeod A. D., Fedorak R. N., Friend D. R., Tozer T. N., Cui N.. 1994a. A glucocorticoid prodrug facilitates normal mucosal function in rat colitis without adrenal suppression. Gastroenterology. 106: 405–413. [PUBMED], [INFOTRIEVE]
- McLeod A. D., Tolentino L., Tozer T. N.. 1994b. Glucocorticoid-dextran ester conjugates as potential prodrugs for colon-specific drug delivery: steady-state pharmacokinetics in rat. Biopharm. Drug Dispos.. 15: 151–164. [PUBMED], [INFOTRIEVE]
- Meshali M. M., Gabar K. E.. 1993. Effect of interpolymer complex formation of chitosan with pectin or acacia on the release behaviour of chlorpromazine HCI. Int. J. Pharm.. 89: 177–181. [CROSSREF]
- Milojevic S., Newton J. M., Cummings J. H., Gibson G. R., Botham R. L., Ring S. C., Stockham M., Allwood M. C.. 1996a. Amylose as a coating for drug delivery to the colon: preparation and in vitro evaluation using 5-aminosalicylic acid pellets. J. Control. Rel.. 38: 75–84. [CROSSREF]
- Milojevic S., Newton J. M., Cummings J. H., Gibson G. R., Botham R. L., Ring S. C., Stockham M., Allwood M. C.. 1996b. Amylose as a coating for drug delivery to the colon: preparation and in vitro evaluation using glucose pellets. J. Control. Rel.. 38: 85–94. [CROSSREF]
- Minami K., Hiramaya F., Uekama K.. 1998. Colon-specific drug delivery based on a cyclodextrin prodrug: release behaviour of biphenyl acetic acid from its cyclodextrin conjugates in rat intestinal tracts after oral administration. J. Pharm. Sci.. 87: 715–720. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Moore W. E. C., Holdeman L. V.. 1975. Discussion of current bacteriological investigations of the relationship between intestinal flora, diet and colon cancer. Cancer Res.. 35: 3418–3420
- Morishita I., Morishita M., Takayama K., Machida Y., Nagai T.. 1993. Enteral insulin delivery by microspheres in three different formulations using Eudragit L100 and S100. Int. J. Pharm.. 91: 29–37. [CROSSREF]
- Munjeri O., Collett J. H., Fell J. T.. 1997. Hydrogel beads based on amidated pectins for colon-specific drug delivery: the role of chitosan in modifying drug release. J. Control. Rel.. 46: 273–278. [CROSSREF]
- Muraoka M., Hu Z., Shimokawa T., Sekino S., Kurogoshi R., Kuboi Y., Yoshikawa Y., Takada K.. 1998. Evaluation of intestinal pressure-controlled colon delivery capsule containing caffeine as a model drug in human volunteers. J. Control. Rel.. 52: 119–129. [CROSSREF]
- Omrod D. J., Holmes C. C., Miller T. E.. 1998. Dietary chitosan inhibits hypercholesterolaemia and antherogensis in the apoliprotein E-deficient mouse model of atherosclerosis. Atherosclerosis. 138: 329–334. [CROSSREF]
- Orienti I., Cerchiara T., Luppi B., Bigucci F., Zuccari G., Zecchi V.. 2002. Influence of different chitosan salt on the release of sodium diclofenac in colon-specific delivery. Int. J. Pharm.. 238: 51–59. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Park K., Shalaby S. W. W., Park H.. 1993. Biodegradation. Biodegradable Hydrogels for Drug Delivery, J., Guillet, 13–34, Lancaster, PA, Technomic Publishing
- Potts J. E., Clendianaings R. A., Ackard B.. 1973. The Biodegradability of Synthetic Polymers. Polymer Science and Technology. vol. 3. ed., J., Guillet, 61–79, New York, Plenum Press
- Raghavan C. V., Muthulingam C., Leno Jenita J. A. J., Ravi T. K.. 2002. An in vitro and in vivo investigation into the suitability of bacterially triggered delivery system for colon targeting. Int. J. Pharm.. 50: 892–895
- Rama Prasad Y. V., Krishnaiah Y. S. R., Satyanarayana S.. 1998. In vitro evaluation of guar gum as a carrier for colon-specific drug delivery. J. Control. Rel.. 51: 281–287
- Ratner B. D., Gladhill K. W., Hobert T. A.. 1988. Analysis of in vitro enzymatic and oxidative degradation polyurethanes. J. Biomed. Mater. Res.. 22: 509–527. [PUBMED], [INFOTRIEVE]
- Rihova B.. 1995. Bioadhesive polymers for oral drug delivery. Adv. Exp. Med. Biol.. 371B: 1491–1494. [PUBMED], [INFOTRIEVE]
- Rodriguez M., Antunez J. A., Taboada C., Seijo B., Torres D.. 2001. Colon-specific delivery of budesonide from microencapsulated cellulosic cores: evaluation of the efficacy against colonic inflammation in rats. J. Pharm. Pharmacol.. 53: 1207–1215. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Rodriguez M., Vila-Jato J. L., Torres D.. 1998. Design of a new multiparticulate system for potential site-specific and controlled drug delivery to the colonic region. J. Control. Rel.. 55: 67–77. [CROSSREF]
- Rubinstein A.. 1990. Microbially controlled drug delivery to the colon. Biopharm Drug Dispos.. 11: 465–475. [PUBMED], [INFOTRIEVE]
- Rubinstein A., Radai R.. 1991. Pectic salt as a colonic drug delivery system. Proc. Int. Symp. Control. Rel. Bioact. Mater.. 18: 221–222
- Rubinstein A., Radai R.. 1995. In vitro and in vivo analysis of colon specifically of calcium pectinate formulations. Eur. J. Pharm. Biopharm.. 41: 291–295
- Rubinstein A., Gilko-Kabir I.. 1995. Synthesis and swelling-dependent enzymatic degradation of borax-modified guar gum for colonic delivery purposes. STP Pharm. Sci.. 51: 41–46
- Rubinstein A., Nakar D., Sintov A.. 1992a. Colonic drug delivery: enhanced release of indomethacin from cross linked chondroitin matrix in rat caecal content. Pharm. Res.. 9: 276–278. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Rubinstein A., Nakar D., Sintov A.. 1992b. Chondroitin sulphate: a potential prodrug for colon-specific drug delivery. Int. J. Pharm.. 84: 141–150. [CROSSREF]
- Rubinstein A., Radai R., Ezra M., Pathak S., Rokem J. M.. 1993. In vitro evaluation of calcium pectinate: a potential colon-specific drug delivery carrier. Pharm. Res.. 10: 258–263. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Rubinstein A., Pathak S., Friendman M., Rokem J. S. R.. 1990. In vitro method for drug release analysis for microbially controlled drug delivery. Proc. Int. Symp. Control. Rel. Bioact. Mater.. 17: 466–467
- Saffran M., Bedra C., Kumar G. S., Neckers D. C.. 1988. Vasopressin: A new model for the study of effects of additives on the oral and rectal administration of peptide drugs. J. Pharm. Sci.. 77: 33–38. [PUBMED], [INFOTRIEVE]
- Saffran M., Kumar G. S., Savariar C., Burnham J. C., Williams F., Neckers D. C.. 1986. A new approach to the oral administration of insulin and other peptide drugs. Science. 233: 1081–1084. [PUBMED], [INFOTRIEVE]
- Salyers A. A.. 1979. Energy sources of major intestinal fermentative anaerobes. Am. J. Clin. Nutr.. 32: 158–163. [PUBMED], [INFOTRIEVE]
- Salyers A. A., O'Brien M.. 1980. Cellular location of enzymes involved in chondroitin sulfate breakdown by Bacteroides thetaiotaomicron. J. Bacteriol.. 143: 772–780. [PUBMED], [INFOTRIEVE]
- Schacht E., Gevaert A., Kenawy E. R.. 1996. Polymers for colon specific drug delivery. J. Control. Rel.. 39: 327–328. [CROSSREF]
- Schacht E., Callant D., Verstraete W.. 1991. Synthesis and evaluation of polymeric prodrugs of 5-aminosalicylic acid. Proc. Int. Symp. Contr. Rel. Bioact. Mater.. 18: 686–687
- Scheline R. R.. 1973. Metabolism of foreign compounds by gastrointestinal microorganisms. Pharmacol. Res.. 25: 451–523
- Semde R., Amighi K., Moes A. J.. 1998. Epichlorhydrin cross-linked pectins for colonic drug delivery. Proc. Int. Symp. Control. Rel. Bioact. Mater.. 25: 806–807
- Semde R., Amighi K., Devleeschouwer M. J., Moes A. J.. 2000a. Effect of pectinolytic enzymes on the theophylline release from pellets coated with water insoluble polymers containing pectin HM or calcium pectinate. Int. J. Pharm.. 197: 169–179. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Semde R., Amighi K., Devleeschouwer M. J., Moes A. J.. 2000b. Studies of pectin HM/Eudragit RL/Eudragit NE film-coating formulations intended for colonic drug delivery. Int. J. Pharm.. 197: 181–192. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Shibata N., Ohno T., Shimokawa T., Hu Z., Yoshikawa Y., Koga K., Murakami M., Takada K.. 2001. Application of pressure-controlled colon delivery capsules to oral administration of glycyrrizin in dogs. J. Pharm. Pharmacol.. 53: 441–447. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Shimono N., Takatori T., Masumi T., Ueda M., Mori M., Higashi Y., Nakamura Y.. 2002. Chitosan dispersed system for colon-specific drug delivery. Int. J. Pharm.. 245: 45–54. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Shu X. Z., Zhu K. J., Song W.. 2001. Novel pH-sensitive citrate crosslinked chitosan film for drug controlled release. Int. J. Pharm.. 212: 19–28. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Shun Y. L., Ayres J. W.. 1992. Calcium alginate beads as core carriers of 5-aminosalicylic acid. Pharm. Res.. 9: 714–790
- Siew L. F., Basit A. W., Netwon J. M.. 2000. The properties of amylose-ethyl cellulose films cast from organic based solvents as potential coating colonic drug delivery. Eur. J. Pharm. Sci.. 11: 133–139. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Simonsen L., Hovgaard L., Mortensen P. B., Bronsted H.. 1995. Dextran hydrogels for colon-specific drug delivery. V. Degradation in human intestinal incubation models. Eur. J. Pharm. Sci.. 3: 329–337. [CROSSREF]
- Sintov A., Di-Capua N., Rubinstein A.. 1995. Cross-linked chondroitin sulphate: characterization for drug delivery purposes. Biomaterials. 16: 473–478. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Spitael J., Kinget R.. 1977. Factors affecting the dissolution rate of enteric coatings. Pharm. Ind.. 39: 502–505
- Spitael J., Kinget R.. 1979. Solubility and dissolution rate of enteric polymers. Acta Pharm. Technol.. 25: 163–168
- Spitael J., Kinget R.. 1980. Influence of solvent composition upon film-coating. Pharm. Acta Helv.. 55: 157–160. [PUBMED], [INFOTRIEVE]
- Spitael J., Kinget R., Naessens K.. 1980. Dissolution rate of cellulose acetate phthalate and Bronsted catalysis. Pharm. Ind.. 42: 846–849
- Sriamornsak P., Nunthanid J.. 1998. Calcium pectinate gel beads for controlled release drug delivery: I. Preparation and in vitro release studies. Int. J. Pharm.. 160: 207–212. [CROSSREF]
- Steggerda F. R.. 1968. Gastroinestinal gas following food consumption. Ann. N.Y. Acad Sci.. 150: 57–61. [PUBMED], [INFOTRIEVE]
- Stella V. J., Rajewski R. A.. 1997. Cyclodextrins: their future in drug formulation and delivery. Pharm. Res.. 14: 556–567. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Stubbe B., Maris B., Van den Mooter G., Demeester J.. 2001. The in vitro evaluation of azo containing polysaccharide gels' for colon delivery. J. Control. Rel.. 75: 103–114. [CROSSREF]
- Suzuki T., Matsumoto T., Hagino Y., Tozaki H., Yamamoto A., Muranishi S.. 1998. Biodegradation and medical applications of chitosan hard capsules. Sci. Technol. Polym. Adv. Mater., Prasad P., 567–571., 4th ed., New York, Plenum Press
- Swanson D., Barclay B., Wong P., Theeuwes F.. 1987. Nifedipine gastro-intestinal, therapeutics system. Am. J. Med.. 83: 3–5. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Swift G.. 1992. Biodegradable polymers in the environment: are they really biodegradable?. Proc. ACS Div. Polym. Mat. Sci. Eng.. 66: 403–404
- Takaya T., Niwa K., Muraoka M., Ogita I., Nagai N., Yano R., Kimura G., Yoshikawa Y., Yoshikawa H., Takada K.. 1998. Importance of dissolution process on systemic availability of drugs delivered by colon delivery system. J. Control. Rel.. 50: 111–122. [CROSSREF]
- Tanaka H., Kominato K., Yamamoto R.. 1994. Synthesis of doxorubicin-cyclodextrin conjugates. J. Antibiot.. 47: 1025–1029. [PUBMED], [INFOTRIEVE]
- Thanoo B. C., Sunny M. C., Jaykrishnan A.. 1992. Crosslinked chitosan microspheres: preparation and evaluation as a matrix for the controlled release of pharmaceuticals. J. Pharm. Pharmacol.. 44: 283–286. [PUBMED], [INFOTRIEVE]
- Theeuwes F., Guittard G., Wong P.. 1990. Delivery of drugs to colon by oral dosage forms. U.S. Patent 4904474
- Thimma R. T., Tammishetti S.. 2001. Barium chloride crosslinked carboxymethyl guar gum beads for gastrointestinal drug delivery. J. Appl. Poly. Sci.. 82: 3084–3090. [CROSSREF]
- Toledo O. M. S., Dietrich C. P.. 1977. Tissue specific distributions of sulfated mucopolysaccharides in mammals. Biochim. Biophys. Acta. 498: 114–122. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Tomlin B. J., Read N. W., Edwards C. A., Duerden B. J.. 1986. The degradation of guar gum by fecal incubation system. Brit. J. Nutr.. 55: 481–486. [PUBMED], [INFOTRIEVE]
- Tomolin J., Taylor J. S., Read N. W.. 1989. The effect of mixed faecal bacteria on a selection of viscous polysaccharide in vitro. Nutr. Rep. Int.. 39: 121–135
- Touitou E., Rubinstein A.. 1986. Targeted eternal delivery of insulin to rats. Int. J. Pharm.. 30: 95–99. [CROSSREF]
- Towle G. A., Christensen O.. 1973. Pectin. Industrial Gums and Their Derivatives, R. L., Whistler, J. N., BeMiller, 429–461, New York, Academic Press
- Tozaki M., Emi Y., Horisaka E., Fujita T., Yamamoto A., Muranishi S.. 1997a. Degradation of insulin and calcitonin and their protection by various protease inhibitors in rat caecal contents: implications in peptide delivery to the colon. J. Pharm. Pharmacol.. 49: 164–168. [PUBMED], [INFOTRIEVE]
- Tozaki H., Komoike J., Tada C., Maruyama T., Terabe A., Suzuki T., Yamamoto A., Muranishi S.. 1997b. Chitosan capsules for colon-specific drug delivery: improvement of insulin absorption from the rat colon. J. Pharm. Sci.. 86: 1016–1021. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Tozaki M., Fujita T., Odoriba T.. 1999. Colon specific delivery of R68070, a new thromboxane synthetase inhibitor, using chitosan capsules: therapeutic effects against 2,4,6-trinitrobenzene sulfonic acid-induced ulcerative colitis in rats. Life Sci.. 64: 1155–1162. [PUBMED], [INFOTRIEVE]
- Tozaki H., Odoriba T., Okada N., Fujita T., Terabe A., Suzuki T., Okabe S., Murnishi S., Yamamoto A.. 2002. Chitosan capsules for colon-specific drug delivery: enhanced localization of 5-aminosalicylic acid in the large intestine accelerates healing of TNBS-induced colitis in rats. J. Control. Rel.. 82: 51–61
- Tozer T. N., Rigod J., McLeod A. D., Gungon R., Hong M. K., Friend D. R.. 1991. Colon-specific delivery of dexamethasone from a glucoside prodrug in the guinea pig. Pharm. Res.. 8: 445–454. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Turkoglu M., Ugurlu T.. 2002. In vitro evaluation of pectin-HPMC compression coated 5-aminosalicylic acid tablets for colonic delivery. Eur. J. Pharm. Biopharm.. 53: 65–73. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Uekama K., Hiramaya F., Irie T.. 1991. Pharmaceutical uses of cyclodextrin derivatives. High Performance Biomaterials, M., Szycher, 789–786, Lancaster, PA, Technomic Publishing
- Uekama K., Minami K., Hirayama F.. 1997. 6A-O-[(4-biphenylyl)acetyl]-alpha-, -beta- and -gamma-cyclodextrins and 6A-deoxy-6A-[[(4-biphenyl)acetyl]amino]-alpha-, -beta-, and -gamma-cyclodextrins: potential prodrugs for colon-specific delivery. J. Med. Chem.. 40: 2755–2761. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Van den Mooter G., Samyn C., Kinget R.. 1992. Azo polymers for colon-specific drug delivery. Int. J. Pharm.. 87: 37–46. [CROSSREF]
- Van den Mooter G., Samyn C., Kinget R.. 1993. Azo polymers for colon-specific drug delivery. Part II: influence of the type of azo polymer on the degradation by intestinal microflora. Int. J. Pharm.. 97: 133–139. [CROSSREF]
- Vandelli M. A., Leo E., Forni F., Bernatei M. T.. 1996. In vitro evaluation of a potential colonic drug delivery system that releases drug after a controllable lag-time. Eur. J. Pharm. Biopharm.. 43: 148–151
- Van Soest P. J.. 1978. Dietary fibers: their definition and nutritional properties. Am. J. Clin. Nut.. 31: S12
- Vermeersch J., Vandoorne F., Permentier D., Schacht E.. 1985. Macromoleculer prodrugs of metronidazole I. Esterification of hydroxyl containing polymers with metronidazole monosuccinate. Bull. Soc. Chim. Belg.. 94: 591–596
- Vervoort L., Kinget R.. 1996. In vitro degradation by colonic bacteria of inulin HP incorporated in Eudragit films. Int J. Pharm.. 129: 185–190. [CROSSREF]
- Vervoort L., Van den Mooter G., Augustijns P., Busson R., Toppet S., Kinget R.. 1997. Inulin hydrogels as carrier for colonic drug targeting. I. Synthesis and characterization of methacrylated inulin and hydrogel formation. Pharm. Res.. 14: 1730–1736. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Vervoort L., Rombaut P., Van den Mooter G., Augustijns P., Kinget R.. 1998a. Inulin hydrogels. I. Dynamic and equilibrium swelling properties. Int. J. Pharm.. 172: 127–135. [CROSSREF]
- Vervoort L., Rombaut P., Van den Mooter G., Augustijns P., Kinget R.. 1998b. Inulin hydrogels. II. In vitro degradation study. Int. J. Pharm.. 172: 137–145. [CROSSREF]
- Wakerly Z., Fell J. T., Attwood D., Parkins D.. 1996. Pectin/ethyl cellulose film coating formulations for colonic drug delivery. Pharm. Res.. 13: 1210–1212. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Wakerly Z., Fell J. T., Attwood D., Parkins D.. 1997. Studies on drug release from pectin/ethylcellulose film-coated tablets: a potential colonic delivery system. Int. J. Pharm.. 153: 219–224. [CROSSREF]
- Wang X., Gibson G. R.. 1993. Effects of the in vitro fermentation of oligofructose and inulin by bacteria growing in the human large intestine. J. Appl. Bacterol.. 75: 373–380
- Wastenson A.. 1971. Properties of fractionated chondroitin sulfate form ox nasal septa. Biochem. J.. 122: 477–785
- Weiner M. L.. 1993. An overview of the regulatory status and of the safety of chitin and chitosan as food and pharmaceutical ingredients. Advances in Chitin and Chitosan., Brine C. J., P. A., Sandford, J. P., Zikakis, 663–672, London, Elsevier Applied Sciences
- Willoughby C. P., Aronson J. K., Agback H., Bodin N. O., Anderson E., Truelove S. C.. 1981. Disposition in normal volunteers of sodium azodisalicylate, a potential therapeutic agent in inflammatory bowl disease. Gut. 22: A431
- Willoughby C. P., Aronson J. K., Agback H., Bodin N. O., Anderson E., Truelove S. C.. 1982. Disposition in normal volunteers of sodium azodisalicylate, a potential therapeutic agent in ulcerative colitis. Gut. 23: 1081–1087. [PUBMED], [INFOTRIEVE]
- Wong D., Larrabeo S., Clitford K., Tremblay J., Friend D. R.. 1997. USP dissolution apparatus III (reciprocating cylinder) for screening of guar-based colonic delivery formulation. J. Control. Rel.. 47: 173–179. [CROSSREF]
- Yano H., Hirayama F., Kamada M., Arima H., Uekama K., Uekama K.. 2002. Colon-specific delivery of prednisolone-appended alpha-cyclodextrin conjugate: alleviation of systemic side effect after oral administration. J. Control. Rel.. 79: 103–112. [CROSSREF]
- Yehe P. Y., Berenson M. M., Samowitz W. S., Kopeckova P., Kopecek J.. 1995. Site-specific drug delivery and penetration enhancement in the gastrointestinal tract. J. Control. Rel.. 36: 109–124. [CROSSREF]
- Zhang H., Ibrahim A. A., Neau S. H.. 2002. An in vitro evaluation of a chitosan-containing multiparticulate system for macromolecule delivery to the colon. Int. J. Pharm.. 239: 197–205. [CROSSREF], [PUBMED], [INFOTRIEVE]