Abstract
The solid dispersions with poloxamer 188 (P188) and solid solutions with polyvinylpyrrolidone K30 (PVPK30) were evaluated and compared in an effort to improve aqueous solubility and bioavailability of a model hydrophobic drug. All preparations were characterized by differential scanning calorimetry, powder X-ray diffraction, intrinsic dissolution rates, and contact angle measurements. Accelerated stability studies also were conducted to determine the effects of aging on the stability of various formulations. The selected solid dispersion and solid solution formulations were further evaluated in beagle dogs for in vivo testing. Solid dispersions were characterized to show that the drug retains its crystallinity and forms a two-phase system. Solid solutions were characterized to be an amorphous monophasic system with transition of crystalline drug to amorphous state. The evaluation of the intrinsic dissolution rates of various preparations indicated that the solid solutions have higher initial dissolution rates compared with solid dispersions. However, after storage at accelerated conditions, the dissolution rates of solid solutions were lower due to partial reversion to crystalline form. The drug in solid dispersion showed better bioavailability in comparison to solid solution. Therefore, considering physical stability and in vivo study results, the solid dispersion was the most suitable choice to improve dissolution rates and hence the bioavailability of the poorly water soluble drug.
Due to the advent of high throughput screening in drug discovery process the resultant compounds are often high molecular weight and highly lipophilic and show poor solubility in water and in turn poor bioavailability (Lipinski, et al. 1999). The solubility and bioavailability enhancement of such a poorly soluble drug pose one of the most challenging aspects of drug development.
Today among the many methods available to improve bioavailability, formulation of solid dispersions and solid solutions has gained enormous attention. By definition, solid dispersions and solid solutions can be differentiated based on the molecular state of the drug in the carrier matrix. If the drug is converted to amorphous form and forms one phase system with polymer, it can be classified as a solid solution, whereas if the drug exists as microcrystalline dispersion, i.e., forms two-phase system, it is generally referred to as a solid dispersion (Goldberg, Gibaldi, and Kanig Citation1965; Sekiguchi and Obi Citation1961).
The improvement of bioavailability with these systems is based primarily on improving dissolution rates. In case of solid dispersion this is achieved by improvement in the wetting behavior of hydrophobic drug as well as deagglomeration and micellaization of the drug with hydrophillic polymers. In solid solution, the improvement in dissolution rate is due to the high energy, amorphous nature of the drug.
Thermodynamically, solid solutions are unstable compared with solid dispersions because in the solid solutions the drug exists in a high energy amorphous form (Goldberg et al. Citation1966), that is prone to precipitation or crystallization under environmental stress such as moisture and heat, especially during processing and storage of the drug products. Solid dispersions on the other hand may provide the advantages of the solid solution while having fewer physical stability problems. The key empirical distinction of a solid dispersion is that drug retains its unique physical state after processing and hence the reversion to more stable crystalline form is not a major concern (Goldberg, Gibaldi, and Kanig Citation1966b; Leuner and Dressman 1000).
A selected model drug under study has poor water solubility and therefore limited oral bioavailability. The primary objective of the study was to select the optimal physical system based on physical stability, improved solubility and dissolution rates, ease of manufacturing, and in vivo bioavailability. To achieve this goal, solid dispersions and solid solutions of the model drug were formulated using selected hydrophilic carriers, poloxamer 188 (P188) and polyvinylpyrrolidone K30 (PVPK30), respectively. Characterization of the solid dispersions with P188 and solid solutions with PVPK30 was carried out and the release profiles were compared with corresponding physical mixtures. In addition, the physical stability of the drug in solid dispersions and solid solutions was monitored at accelerated stress conditions. In vivo studies were conducted to evaluate the performance of the solid dispersion and solid solution in beagle dogs.
MATERIALS AND METHODS
The drug was provided by Hoffmann-La Roche (Nutley, NJ, USA). Poloxamer 188 (P188 NF®) and PVPK30 (polyvinylpyrrolidone K30) were purchased from BASF Corporation (Mount Olive, NJ, USA). All chemicals used were of analytical grade.
Preparation of Physical Mixtures with P188 and PVPK30
The drug and carriers were mixed thoroughly in a mortar with pestle and passed through a 60-mesh (250 μ m) screen. This mixture was further mixed in Turbula mixer for an additional 15 min. The different ratios of drug and carrier prepared for the study were: 20:80, 30:70, 50:50, 60:40, and 80:20.
Preparation of Solid Dispersion with P188 using the Fusion Method
P188 was heated at 60°C in an oven, until it melted completely. The drug was added to the molten polymer at 60°C and mixed thoroughly in mortar with pestle. The mixture was cooled to ambient conditions, milled, and passed through a 40-mesh (425 μ m) screen. Different ratios of drug and P188 prepared were 20:80, 30:70, 50:50, and 80:20.
Preparation of Solid Solution with PVPK30 using Solvent Method
The drug and PVPK30 were dissolved in ethyl alcohol and then the solvent was evaporated in rotavap (Buchi Rotavapor, Buchi Laboratories, Switzerland) at 60°C under vacuum. The dried powder was then mixed in mortar with pestle and passed through a 40-mesh (425 μ m) screen.
Characterization of Various Formulations
Powder X-Ray Diffraction (XRD)
The various samples were analyzed by powder X-ray diffraction (Scintag Inc., CA, USA) using Cu Kα radiation to determine the crystalline state of the drug in the solid dispersions and solid solutions. The XRD pattern was collected in the angular range of 1 < 2θ < 40° in a step scan mode (step width 0.02°, scan rate 1 deg/min).
Thermal Analysis
Thermal analysis was carried out using SII 5200 differential scanning calorimeter (DSC) (TA Instruments, NJ, USA) equipped with a liquid nitrogen cooling accessory. Samples (5–10 mg) were prepared in sealed pans. The samples were scanned at a heating rate of 5°C/min. To determine glass transition temperature (Tg), samples were subjected to heat-cool-heat cycle. The heat-cool-heat cycle helped in erasing sample history and obtaining accurate Tg. Data were analyzed using the DSC 5200 disk station analysis program.
Solubility Measurements
Solubility of the drug was determined as a function of carrier concentration. P188 and PVPK30 were separately dissolved in water to prepare aqueous solution of carriers (10%, 20%, 30% w/v). An excess amount of the drug was added to 10 mL of purified water or to an aqueous solution of carriers. The samples were allowed to shake for 48 hr at room temperature. The solution was filtered through a 0.45 μ pore size filter. The solution concentration was measured by HPLC analysis.
Measurement of Contact Angle
To determine wettability of the substrates, a contact angle goniometer (Ramé-Hart Inc., NJ, USA) was used. The pellets were prepared for various formulations (100 mg) using Carver press with 5/16” flat surface punches at 4000 psi pressure and a dwell time of 60 sec. A drop of water (approx 2 μ mL) was placed on the pellet with a micropipette and the contact angle was measured with the help of the microscope assembly with an overall system magnification of 23x.
Dissolution Studies
The intrinsic dissolution studies were carried out to evaluate the dissolution rate of various formulations. Intrinsic dissolution or constant surface area dissolution was studied using a Wood's apparatus. About 100 mg of the powder was compressed in Wood's apparatus die (∼diameter 0.8 cm) using a Carver press at 3000 psi pressure and a dwell time of 60 sec.
Dissolution was performed using a USP dissolution apparatus II (Distek dissolution system model 2100, NJ, USA) connected to HP UV-VIS spectrophotometer (Model 8452 A diode array spectrophotometer, NJ, USA) with a 500 mL of simulated gastric fluid without pepsin at pH 1.5 as a dissolution medium at 37°C and a paddle speed of 25 rpm.
Stability Studies
The stability studies were conducted to determine the effect of aging on the physical stability of the drug in various formulations. The solid dispersion with P188 and solid solution with PVPK30 and the drug “as is” were stored at 40°C/75%RH and at room temperature for 1 month in open glass vials. Various analytical methods such as DSC, X-ray diffraction pattern, and intrinsic dissolution studies were carried out and compared with initial formulations.
In Vivo Bioavailability Studies
The single dose pharamcokinetic profiles were evaluated in fasted beagle dogs at ∼10 mg/kg dose using various formulations with the drug as solid dispersion and drug as solid solution. The blood samples were collected at regular time intervals and the drug-plasma concentrations were determined using HPLC. The HPLC method found the column at phenomenex prodigy 5, 4.6 × 250, or equivalent; the mobil phase was acetonitrile/ water/ trimethylamine/acetic acid in 40: 60: 0.2: 0.1 v/v compositions. Detection was UV at 254 nm and the injection volume at 10 μ L.
RESULTS AND DISCUSSION
Powder X-Ray Diffraction Studies
Powder X-ray diffraction (PXRD) is an important tool in investigating the crystalline nature of the drug in solid dispersions and solid solutions. shows the comparison of PXRD of pure drug, pure P188, physical mixture, and solid dispersion. The X-ray diffraction pattern for the model drug showed distinctive peaks at 9, 10, 11, 12, 15, 16, 17, 19, 20, 21, 22, 26, and 27° indicating its crystalline nature. In the solid dispersion and physical mixture with P188, the drug retained its original characteristic peaks confirming the crystalline nature of the drug. P188 is crystalline in nature and gives two characteristic peaks: one starts at 19° and the broader one between 22° and 23°. In the case of solid solution with PVPK30, absence of drug peaks indicated the amorphous nature of the drug; however, the drug retained its characteristic peaks in physical mixture with PVPK30 ().
FIG. 1 Powder x-ray diffraction pattern of pure drug, physical mixture, and solid dispersion with P188.
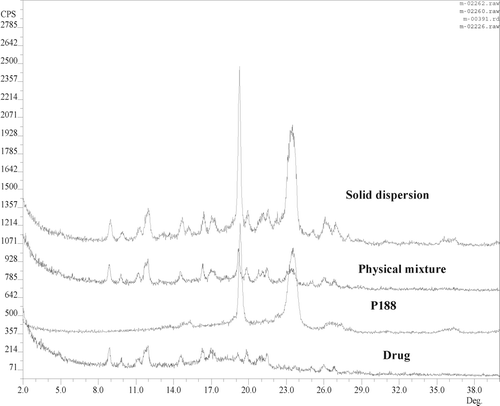
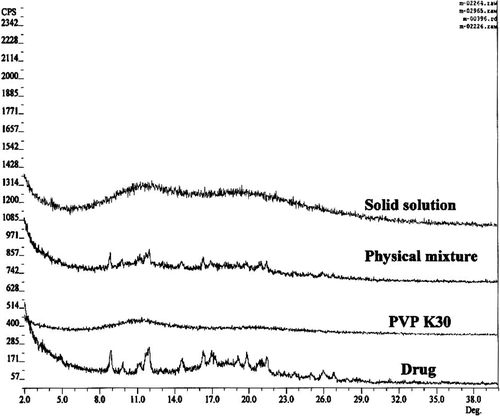
FIG. 2 Powder x-ray diffraction pattern of pure drug, physical mixture, and solid solution with PVP K30.
Thermal Analysis
According to Van't Hoff equation (Martin Citation1993), if the drug and polymer form an eutectic mixture, the melting point of the drug in carrier will decrease as the mole fraction of polymer increases. The melting endotherm of the pure drug was obtained at 238.6 ± 0.3°C and for P188 was at 51.6 ± 0.4°C ().
FIG. 3 Representative thermograms show distinct melting endotherms for drug and P188 in solid dispersion.
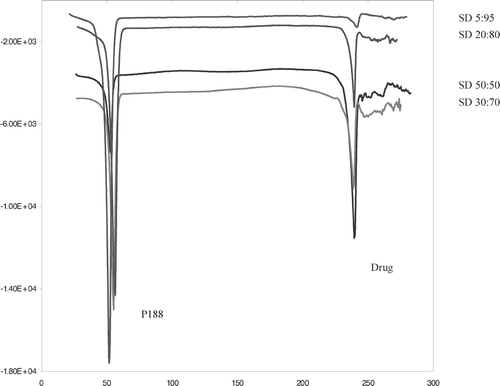
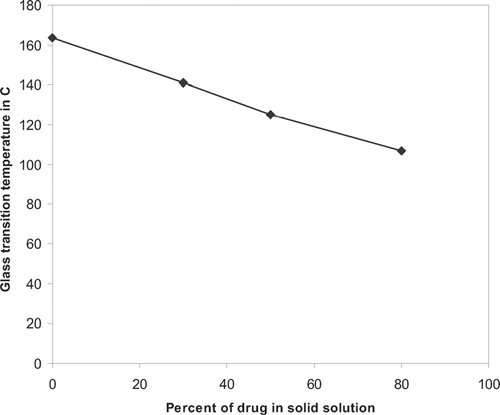
The DSC studies were performed on physical mixtures and solid dispersions with various amounts of drug and P188. There was no change in drug or polymer melting endotherm with increasing amount of P188 in the formulations (). This was further shown by plotting melting temperature and heat of melting (Δ H) of drug versus percentage of P188 (). This plot indicated that the melting temperature and Δ H of the drug remained unchanged with increase in P188 concentration, suggesting that the drug did not form a eutectic mixture with the carrier. The PVPK30 showed a glass transition at 163.5 ± 0.2°C. DSC thermogram of physical mixtures with PVPK30 showed the presence of a drug endotherm and a glass transition of the PVPK30 ().
FIG. 4 Melting temperature and Δ H of the drug in solid dispersions as a function of P188 concentrations.
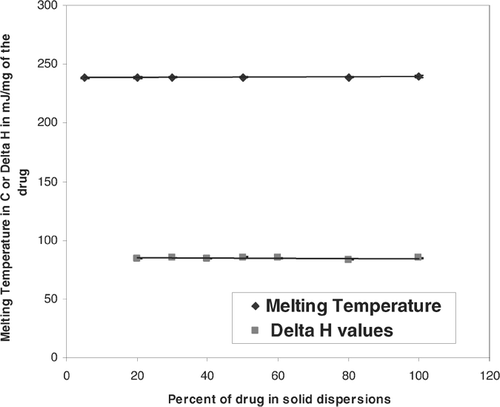
TABLE 1 Glass transition temperatures of various formulations
During the first heating cycle of the physical mixture, water evaporation peak at 100°C and drug melting endotherm at ∼238°C was observed. During the second heating run only single glass transition was observed suggesting that during the first run, the drug formed one phase system with PVPK30 and depressed the glass transition temperature of the PVPK30 ().
DSC analysis of solid solutions ( and ) showed single glass transition temperature. The glass transition temperature of PVPK30 was decreased with increase in drug concentration in solid solutions. These results indicated the presence of one phase system. Thermal analysis data were further confirmed by the powder X-ray diffraction results that showed an amorphous form of the drug in presence of the PVPK30.
FIG. 5 Change in glass transition with increase in drug concentration in solid solution.
Solubility and Contact Angle Studies
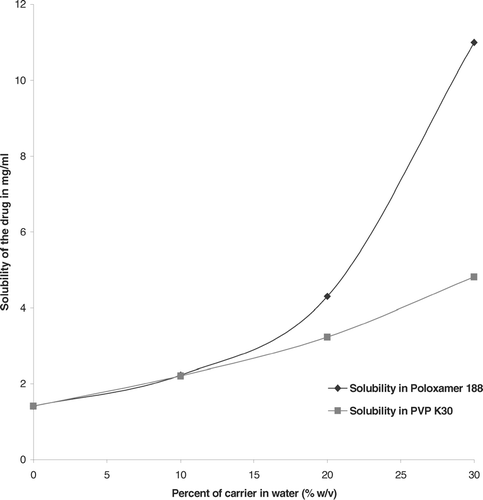
As shown in , the solubility of the drug increased with an increase in polymer concentrations. The pure drug had water solubility of 1.41 mg/mL. The solubility of drug was increased 7-fold in poloxamer aqueous solution (30% w/v) whereas only a 3-fold increase was observed with PVPK30 aqueous solution (30%w/v). The increase in the solubility of drug with P188 may be attributed to micellar solubilization (Sekikawa, Nakano, and Arita Citation1978). The increase in solubility was further supported by evaluating the wetting effect of polymers.
FIG. 6 Solubility of drug as a function of P188 and PVP K30 concentrations.
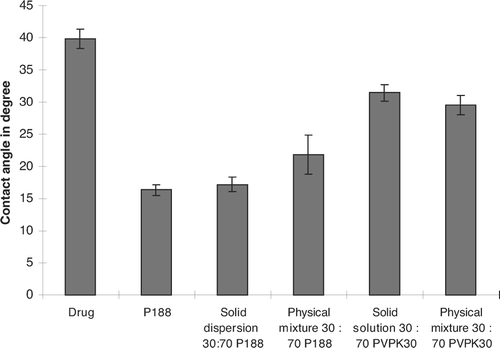
The contact angle results are presented in . The contact angle for the drug was observed to be 39.8°, which indicated the hydrophobic nature of the drug. The phsical mixture and solid dispersion showed reduction in the contact angle of the drug in presence of the P188. The physical mixture showed ∼2-fold reduction while that of the solid dispersion showed ∼2.5-fold reduction in contact angle when compared with the pure drug. The physical mixture and solid solution with PVPK30 showed only slight decrease in contact angle compared with the pure drug. The contact angle studies suggest that the P188 helped improve wettability of the hydrophobic drug compared to with PVPK30.
FIG. 7 Contact angle studies compare drug, physical mixtures (P188 and PVP K30), to solid dispersion and solid solution.
Dissolution Studies
The intrinsic dissolution rates for various formulations are summarized in . Comparing the drug, physical mixtures, and solid dispersions, the solid dispersions showed highest intrinsic dissolution rate. For example, the solid dispersion at drug:P188 ratio of 30:70 showed a 2.5-fold increase in intrinsic dissolution rate compared with physical mixture and a 100-fold increase in dissolution rate compared with the pure drug ( and ). Dissolution profile of physical mixtures and corresponding solid dispersions showed an increase in intrinsic drug dissolution rate with increase in concentration of P188 (). The dissolution rate of solid dispersion at drug:P188 ratio of 20:80 was 4-fold higher than the dissolution rate of the solid dispersion with ratio 80:20 ().
FIG. 8 Comparison of dissolutions profiles of drug, physical mixture, (a) solid dispersion, and (b) solid solution.
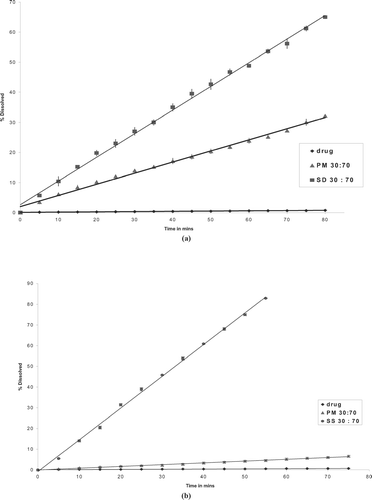
FIG. 9 Dissolution profiles of various solid dispersions with P188.
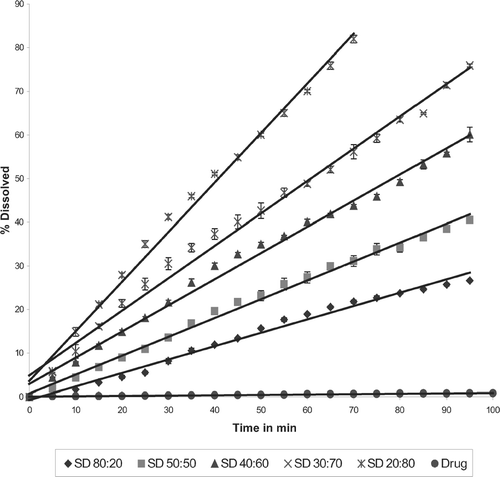
TABLE 2 Intrinsic dissolution rates of various formulations
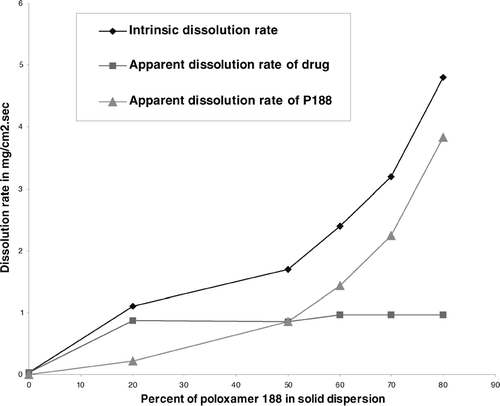
To understand further the role of polymer in improving the dissolution rate, the intrinsic dissolution rates were normalized with respect to concentration of the drug and polymer to obtain apparent dissolution rate for drug and polymer. The apparent dissolution rates of drug and polymer suggested that in solid dispersions, P188 increased the intrinsic solubility of the drug by 27-fold that was achieved at 20% polymer concentration. Further increase in concentration of polymer did not help in improving intrinsic dissolution of the drug. The observed increase in intrinsic dissolution rates could be attributed to faster erosion of the matrix that is primarily polymer controlled ( and ).
FIG. 10 Change of dissolution rates of the drug in solid dispersions with change in concentration of P188.
The intrinsic dissolution rate of the solid solution at drug:PVPK30 ratio of 30:70 was 8-fold higher compared with the dissolution rate of corresponding physical mixture, and it was 180-fold higher than the pure drug ( and ). The solid solutions with increasing amounts of PVPK30 showed increase in intrinsic drug dissolution rates ( and ). However, the apparent dissolution rate of drug in solid solution did remain constant with increase in PVPK30 concentration. The enhancement in intrinsic dissolution rate was attributed to amorphous nature of the drug which increased intrinsic solubility of the drug by 54-fold.
FIG. 11 Dissolution profile of various solid solutions with PVP K30.
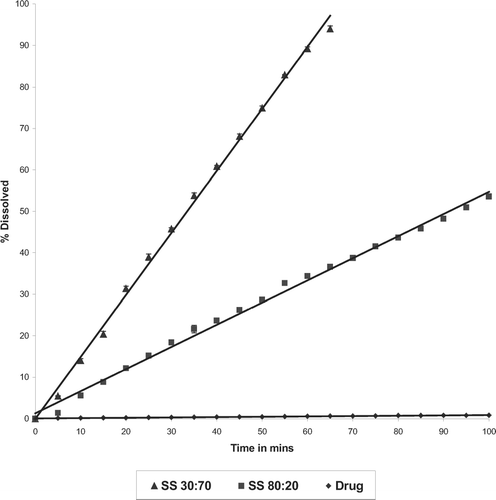
The physical mixtures prepared by P188 showed higher drug release rate compared with physical mixture with PVPK30, which can be attributed to the improved wetting of the drug (). Comparing the intrinsic dissolution rates of solid dispersion and solid solution, (drug: polymer ratio of 30:70) the solid solution showed 2-fold higher dissolution rate ().
Stability Studies
The accelerated stability studies were conducted to determine the effect of aging on the physical stability of various formulations under stress storage conditions. The solid dispersion with P188 and solid solution with PVPK 30 were stored at 40°C and 75% RH in open glass vials for 1 month. The chemical stability was found to be satisfactory for various formulations after storage at such accelerated conditions. The physical stability of the drug was studied using various analytical techniques such as DSC, powder X-ray diffraction, and intrinsic rate dissolution method.
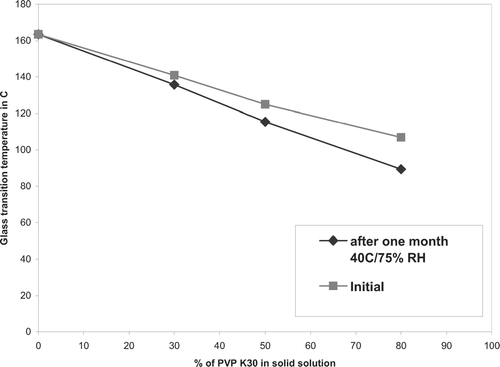
This solid dispersion showed similar DSC thermogram and X-ray diffraction patterns when compared with initial solid dispersion stored at room temperature. In solid solution (drug:PVPK30 ratio of 80:20), the DSC showed presence of drug endotherm during first heating cycle, and the glass transition was depressed to 96.3°C compared with initial glass transition of 106.9°C indicating higher probability of reversion to crystalline in presence of moisture and heat ().
FIG. 12 Decrease in glass transition temperature after storing for 1 month at 40°C/75%RH.
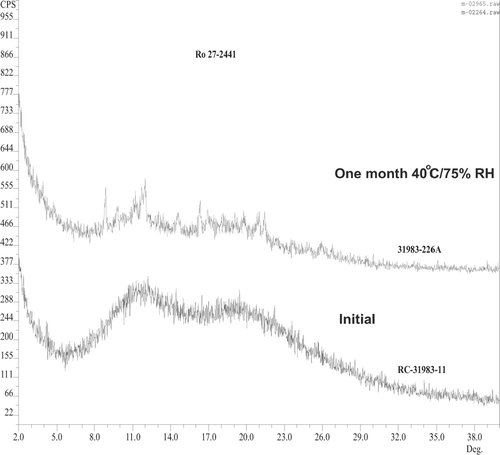
The powder X-ray diffraction for this solid solution kept at 40°C/75%RH for 1 month showed an appearance of distinct drug peaks especially at 11° and 22° and small peak at 17° and 20°, which confirms the recrystallization of the drug in the solid solution with PVPK30 ().
FIG. 13 XRD pattern compares solid solution with ratio of 80:20 before and after storing at 40°C/75% RH for 1 month.
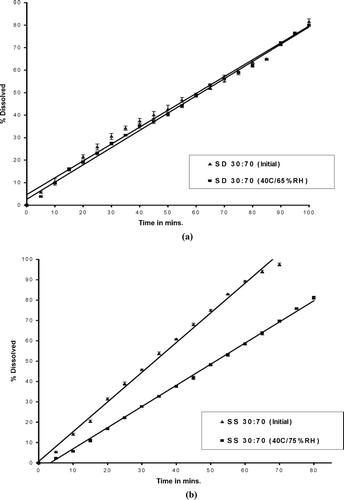
The solid solution at drug:PVPK30 ratio of 30:70 after storage did not show any change in DSC and powder X-ray diffraction pattern when compared with initial formulation. The explanation can be the high amount of PVPK30 helped in maintaining the drug in amorphous form (Simonelli, Metha, and Higuchi Citation1970; Hammad and Muller Citation1998). The intrinsic dissolution rates studies showed that the solid dispersion kept at 40°C/75%RH for 1 month had similar dissolution rate compared with initial formulations (). In with solid solution, however, after 1 month slower dissolution rate was obtained compared with the initial samples. The solid solution at drug: PVPK30 ratio of 30:70 showed 1.4-fold decrease in dissolution rate after storing at 40°C/75%RH for 1 month compared with initial formulation ().
FIG. 14 Dissolution rate profiles compare (a) solid dispersion and (b) solid solution (before and after storage of 1 month at 40°C/75%RH).
Data from different analytical techniques together indicated that in solid dispersion, the drug did not show any physical change after storage at 40°C/75%RH for 1 month. While in solid solution, the drug partially recrystallizes from amorphous form, which negatively affected the dissolution rate of the drug. Based on these studies solid dispersion showed superior stability under stress conditions compared with solid solution.
In Vivo Bioavailability Studies
In vivo studies were carried out in 3 male beagle dogs in fasted condition for each formulation. The pharmacokinetic parameters are tabulated in . Solid dispersion with P188 showed higher Cmax and AUC compared with solid solution with PVPK30.
TABLE 3 In vivo parameters for the solid dispersion and solid solution
In vivo studies do not correlate well with in vitro dissolution results with respect to solid dispersion and solid solution. The difference between in vivo and in vitro performances of these two formulations can be attributed to the following:
The reversion of amorphous form in solid solution to stable crystalline form in vivo.
The uncontrollable crystallization of solid solution in vivo may yield large particles of the drug, thus further lowering its dissolution and thus bioavailability.
In solid dispersion P188 acts as wetting agent and the drug maintains its microcrystalline dispersion with polymer, which improves solubility, intrinsic dissolution, and thereby bioavailability of the drug.
SUMMARY AND CONCLUSION
The solid dispersions with P188 and the solid solutions with PVPK30 were prepared and were characterized by various analytical techniques. The thermal analysis of the physical mixtures and solid dispersions with P188 suggested that the drug does not form eutectic mixture with carrier. Thermal analysis in combination with powder X-ray diffraction studies indicated that in solid dispersions, the drug retained its crystalline form and the drug forms two phase system with P188. However, the solid solutions with PVPK30 showed amorphous nature of the drug and single glass transition temperature indicating one phase system with PVPK30.
In solid dispersion, P188 improved the wetting of the drug, increased the intrinsic solubility, and thus improved intrinsic dissolution rate of the drug. The contact angle studies were well correlated to the dissolution studies for the physical mixture and solid dispersion. Solid dispersion showed higher drug release compared with physical mixtures with P188. Solid solutions showed higher intrinsic dissolution rates compared with solid dispersions that was attributed to high energy amorphous nature of the drug.
The stability studies suggested that in the solid dispersion with P188, the drug retains its crystalline form and showed identical dissolution profiles after storing at high temperature and humidity conditions. In the solid solution with PVPK30, the drug partially converted back to stable crystalline form under stress storage conditions, which negatively affected the dissolution rate of the solid solutions. In vivo studies further confirmed the role of P188 in improving the bioavailability of the drug in solid dispersion compared with solid solution with PVPK30.
In conclusion, the model hydrophobic drug showed increased dissolution rates in presence of hydrophilic carriers like P188 and PVPK30. In solid dispersion the drug retained its crystalline form and P188 improved wetting of hydrophobic drug and minimized agglomeration, which resulted in microcrystalline uniform dispersion of drug in the carrier matrix. Initially solid solutions with PVPK30 showed better dissolution rate due to the conversion of drug to amorphous form. However, stability of the amorphous drug limited its use for formulation development. Based on solubility studies, dissolution rate studies, stability studies, and in vivo studies, the solid dispersion with P188 proved to be the method of choice.
REFERENCES
- Goldberg A. H., Gibaldi M., Kanig J. L. Increasing dissolution rates and gastrointestinal absorption of drugs via solid solutions and eutectic mixtures. I-theoretical considerations and discussion of the literature. J. Pharmaceut. Sci. 1965; 54: 1145–1148
- Goldberg A. H., Gibaldi M., Kanig J. L. Increasing dissolution rates and gastrointestinal absorption of drugs via solid solutions and eutectic mixtures. II-experimental evaluation of eutectic mixtures: urea-acetoaminophen system. J. Pharmaceut. Sci. 1966a; 55: 482–487
- Goldberg A. H., Gibaldi M., Kanig J. L. Increasing dissolution rates and gastrointestinal absorption of drugs via solid solutions and eutectic mixtures. III-Experimental evaluation of griseofulvin-succinic acid solution. J. Pharmaceut. Sci. 1966b; 55: 487–492
- Hammad M. A., Muller B. W. Solubility and stability of clonazepam in mixed Micelles. Int. J. Pharmaceut. 1998; 169: 55–64
- Leuner C., Dressman J. Improving drug solubility for oral delivery using solid solutions. Eur. J. Pharmaceut. Biopharmaceut. 2000; 50: 47–60
- Lipinski C. A., Lombardo F., Dominyl B. W., Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev. 1997; 23: 3–25
- Martin A. Thermal analysis. Physical Pharmacy. Lea & Febiger, Philadelphia 1993, (8′)-88
- Sekiguchi K., Obi N. Studies on absorption of eutectic mixture. I. A comparison of the behavior of eutectic mixture of sulfathiazole and that of ordinary sulfathiazole in man. Chem. Pharmaceut. Bull. 1961; 9: 866–872
- Sekikawa H., Nakano M., Arita T. Inhibitory effect polyvinylpyrrolidone on the crystallization of drugs. Chem. Pharmaceut. Bull. 1978; 26: 118–126
- Simonelli A. P., Metha S. C., Higuchi W. I. Inhibition of sulfathaizole crystal growth by poluvinylpyrrolidone (PVP). J. Pharmaceut. Sci. 1970; 59: 633–637