Abstract
To improve its oral absorption, rapidly dissolving ibuprofen solid dispersions (SD) were prepared in a relatively easy, simple, quick, inexpensive, and reproducible manner, characterized by scanning electron microscopy (SEM), differential scanning calorimetry (DSC), and Fourier transform infrared spectroscopy (FTIR). They were evaluated for solubility, in vitro release, and oral bioavailability of ibuprofen in rats. Loss of individual surface properties during melting and resolidification as revealed by SEM indicated the formation of effective SDs. Absence or shifting toward the lower melting temperature of the drug peak in SDs and physical mixtures in DSC study indicated the possibilities of drug-polymer interactions. However, no such interactions in the solid state were confirmed by FTIR spectra that showed the presence of drug crystalline in SDs. Quicker release of ibuprofen from SDs in rat intestine resulted in a significant increase in AUC and Cmax, and a significant decrease in Tmax over pure ibuprofen. Preliminary results from this study suggested that the preparation of fast-dissolving ibuprofen SDs by low temperature melting method using PEG 6000 as a meltable hydrophilic polymer carrier could be a promising approach to improve solubility, dissolution, and absorption rate of ibuprofen.
Ibuprofen () is a nonsteroidal anti-inflammatory drug that has been widely used in the treatment of mild to moderate pain and fever. As its serum concentrations and analgesic effect are correlated, rapid ibuprofen absorption could be a prerequisite for the quick onset of its action. Because of high membrane permeability, extent of ibuprofen absorption approaches up to 100%. Dissolution thus becomes the rate-limiting step for absorption, and the quick release of ibuprofen in the gastrointestinal tract following oral administration is desirable (Laska et al. Citation1986). Various formulations such as prodrugs (Murtha and Ando Citation1994), inclusion complexes (Ghorab and Adeyeye Citation2001), and microcapsules (Adeyeye and Price Citation1994), of ibuprofen were developed. However, the dissolution rate and the oral bioavailability of ibuprofen from these formulations differed widely; methods were time; consuming and costly; and some formulations were bulky with poor flow characteristics and handling difficulties.
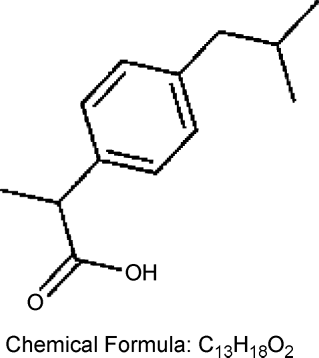
FIG. 1 Structure of ibuprofen.
Solid dispersions (SDs) of poorly water soluble drugs in hydrophilic carrier matrix have been reported to improve their solubility and dissolution rate (Serajuddin Citation1999; Passerini et al. Citation2002; Seo et al. Citation2003). Moreover, they also are proven to enhance their bioavailability by increasing their saturation solubility in gastrointestinal fluids. However, ibuprofen SDs using solvent or solvent-melting method could be problematic, because it might not be always easy to find a common solvent, and large volumes of solvents and long duration of heating might be necessary to enable complete dissolution of both components. Also, the common methods such as vacuum drying, spray-drying, spraying on sugar beads using a fluidized bed coating system, and lyophilization used for the removal of organic solvents from SDs could make the process relatively more complicated, tedious, and costly.
In addition, they might also associate with the solvent related environmental problems (Seo et al. Citation2003). SDs by melting could be problematic (for drugs with higher melting temperature) because of the possible thermal instability of the components, and the hardening of melts resulting into difficulties in the pulverization for subsequent formulation. With ibuprofen because of its low melting temperature, melting at lower temperature using meltable hydrophilic polymers might be feasible. However, the traditional melting methods have been reported to be associated with many processing difficulties such as the temperature and shear rate control, reproducibility, and scalability. For many drugs including ibuprofen, SDs by melt agglomerations in high shear mixers, using a hot solution of meltable hydrophilic carriers as a binding solution, have been claimed to be advantageous industrially (Passerini et al. Citation2002; Seo et al. Citation2003; Vilhelmsen et al. Citation2005). They also were associated with many disadvantages, e.g., separate melting of polymer with or without drug was an extra step that could make the process complicated and costly; the yield in many cases was low because of the polymer/drug loss while pouring into the powder mix; and the processes themselves were very much similar to the wet granulation method used in tablet manufacturing process. This makes them relatively more demanding in terms of time and technology. Although the drying was not needed, in many cases, the improvement in the drug dissolution was lower compared with the SDs of equivalent composition prepared by melting method. In addition, use of such inert fillers as lactose might increase the bulk and the price of these formulations (Passerini et al. Citation2002; Seo et al. Citation2003; Vilhelmsen et al. Citation2005). Therefore, it would be an advantage if the formation of ibuprofen SDs could be achieved using a rapid, less expensive, controllable, and reproducible process (Vilhelmsen et al. Citation2005).
Polyethylene Glycols (PEGs) are semicrystalline polymers used extensively in the SDs preparation for their wetting, solubilizing, and surface active properties (Craig Citation1990). They have been reported to enhance the solubility, dissolution, and bioavailability of many poorly water soluble drugs using various techniques including melting agglomeration and melting. In this study, low temperature melting method was used to prepare fast releasing ibuprofen-PEG 6000 SDs, which were evaluated for their in vitro and in vivo performances. Polyethylene glycol 6000 (PEG 6000) was empirically selected as a meltable polymer for its low melting point, surfactant properties and oral safety (Serajuddin Citation1999; Passerini Citation2002; Seo Citation2003; Vilhelmsen Citation2005; Craig Citation1990).
MATERIALS AND METHODS
Ibuprofen was supplied by Yuhan Research Institute (South Korea), and PEG 6000 was purchased from Fluka Biochemika (Germany). All other chemicals were of reagent grade and used without further purification.
Solubility of Ibuprofen in Molten Polymer
First, 50g PEG 6000 were melted (59–60°C) in a glass beaker, second, 5 g ibuprofen at a time was added into it. The mixture was continuously stirred and the dissolution of ibuprofen in molten PEG 6000 was visually recorded. When the earlier sample was completely dissolved, another 5 g ibuprofen were added and the procedure was repeated until any undissolved ibuprofen was visible.
Preparation of Solid Dispersions and Determination of Drug Content and Percent Yield
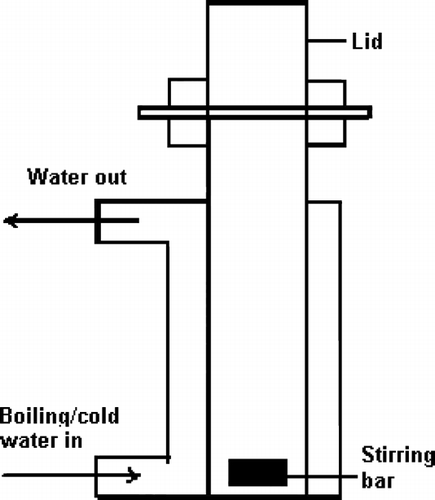
Ibuprofen and PEG 6000 in 4:1, 2:1, 1:1, 1:3, 1:5, 1:7, and 1:10 weight ratios were mixed in a mortar-and-pestle to obtain a homogeneous physical mixture that was sieved through 40 mesh screen and transferred into a locally designed ointment formulation vessel (). Hot water (90–95°C) was continuously circulated using a temperature controlled circulating water bath and the resulting clear molten solution was magnetically stirred at 700 rpm. After 10–15 min, the clear solution was cooled by circulating cold water (< 4°C) for 1 hr and the solidified SDs were then ground by using a mortar-and-pestle, sieved through a 40 mesh screen, and stored in a screw capped vial at room temperature for further use. Drug content was calculated by dissolving SDs equivalent to 20 mg ibuprofen in a suitable quantity of methanol, filtering (0.20 μ m), suitably diluting with methanol, and analyzing by HPLC. Similarly, the percentage yield of each formulation was determined according to the total recoverable final weight of SDs and the total original weights of ibuprofen and PEG 6000 used.
FIG. 2 Locally designed ointment formulation vessel.
Scanning Electron Microscopy
The surface morphology of Ibuprofen, PEG 6000, physical mixtures; and SDs were examined using a SEM (S-4100, Hitachi, Japan). The powders were fixed on a brass stub using double-sided adhesive tape and made electrically conductive by coating in a vacuum (6Pa) with platinum (6 nm/min) using Hitachi Ion Sputter (E-1030) for 240°Cs at 15 mA.
Determination of Solubility
Ibuprofen, physical mixtures, or SDs equivalent to 250 mg of ibuprofen were added to 10 ml phosphate buffer pH 6.8 (PB) in test tubes, vortexed for 2 min, and shaken at 25°C (shaking water bath KMC 12055 WI) for 24 hr. Resultant samples containing undissolved SDs suspended in the test medium were centrifuged at 10000 rpm for 5 min and the clear supernatants obtained were filtered (0.20 μ m), suitably diluted with PB of 25°C and analyzed by HPLC.
In Vitro Ibuprofen Release
Many dissolution studies concerning ibuprofen have been performed using dissolution mediums containing a small amount of acids or surfactants that may accelerate its dissolution rate by their wetting, micellar solubilization, and/or deflocculation properties. Hence, the conclusion of its increased dissolution from improved formulations may not always be justified until its dissolution in water is carried out as a control. It also has been reported that a biowaiver for immediate-release ibuprofen solid oral dosage form is scientifically justified, provided that the dosage form is rapidly dissolving (85% in 30 min or less) in pH 6.8 buffer (Potthast et al. Citation2005). Hence, the solubility and dissolution tests were performed in PB pH 6.8 without any exogenous surfactants.
Based on these facts, dissolution tests of ibuprofen, physical mixture and SDs (equivalent to 10 mg ibuprofen) were performed in 500 ml PB pH 6.8 (37± 0.5°C) devoid of surfactant; and acids as the dissolution medium using USP model digital tablet dissolution test apparatus (Shinseang Instrument; South Korea) at the paddle rotation speed of 50 rpm. At the specified times, 5 ml samples were withdrawn, filtered, and assayed for ibuprofen content by HPLC. Equivalent amount of fresh medium prewarmed to 37 ± 0.5°C was replaced after each sampling. Dissolution tests also were performed on samples under real time stability test (23–27°C and 60–65% relative humidity) after 6 months of storage.
Solubility of Ibuprofen in Aqueous Polymer Solutions
To 10 ml of each of 5, 10, 20, 40, 60, 80, and 100 mg/ml solutions of PEG 6000 in PB in test tubes, 250 mg ibuprofen was added, vortexed for 2 min, and shaken at 25°C in a temperature controlled water bath for 120 hr. This time was determined previously to achieve equilibrium. Resultant samples containing undissolved ibuprofen suspended in the test medium were centrifuged at 10000 rpm for 5 min and the clear supernatants obtained were filtered (0.20 μ m), suitably diluted with corresponding polymer solutions of 25°C and analyzed by HPLC.
Differential Scanning Calorimetry
The DSC measurements were performed on a differential scanning calorimeter (DSC-6100, Seiko Instruments, Japan) with a thermal analyzer. Under nitrogen flow of 25 ml/min, ∼ 2 mg of ibuprofen, PEG 6000, their physical mixture, or SDs were placed in a sealed aluminum pan, and heated at a scanning rate of 5°C/min from 20°C to 100°C. An empty aluminum pan was used as reference.
Fourier Transform Infrared Spectroscopy
FTIR spectra were obtained using FTIR spectrometer-430 (Jasco, Japan). The samples (ibuprofen, PEG 6000, physical mixtures, or SDs) were previously ground and mixed thoroughly with potassium bromide, at 1:5 (sample: potassium bromide) weight ratio. The potassium bromide discs were prepared by compressing the powders at a pressure of 5 tons for 5 min in a hydraulic press. Scans were obtained at a resolution of 2 cm− 1, from 4000 to 500 cm− 1.
Pharmacokinetic Study
Animals care and procedures were conducted according to the guidelines for animal use in toxicology (Society of Toxicology USP 1989) and the study protocol was approved by the Animal Care and Use Committee of the College of Pharmacy, Yeungnam University. A total of 20 male Sprague-Dawley rats (average weight 250 ± 20 g) were divided into 4 groups; right femoral artery was cannulated under light ether anesthesia and hard gelatin capsules (Suheung Capsule, Seoul, South Korea) of ibuprofen powder, physical mixture, or SDs equivalent to 25 mg/kg ibuprofen were administered orally. At predetermined time intervals, 0.2 ml of blood was collected and the plasma was separated by centrifuging at 3000 rpm for 10 min (5415C, Eppendorf, USA) (Geisslinger et al. Citation1989).
Plasma (0.05 ml) was mixed with 0.4 ml of acetonitrile solution containing flufenamic acid (5 μ g/ml) as an internal standard, centrifuged at 3000 rpm for 10 min to precipitate the proteins, and the supernatant layer (0.4 ml) was evaporated in a rotary centrifugal vacuum evaporator. The residue was reconstituted in 50 μ l mobile phase and 20 μ l of the resulting solution was analyzed by HPLC (Canaparo et al. Citation2000). The noncompartmental pharmacokinetic parameters were calculated using the Winnonlin (version 1.1, Scientific Consulting NC, USA) software program. The data from different formulations were compared for statistical significance by one-way analysis of variance (ANOVA). The statistical significance of means among different formulations was then compared by multiple range method of least significant difference.
Drug Analysis
Ibuprofen concentrations were analyzed by Jasco P987 HPLC system equipped with a Jasco UV detector (UV-975). Separation was performed with 50 injection volume (pharmacokinetic study-20 μl) on a reverse-phase C18 column (Inertsil GL Science column.5μm particle size, 4.6?150 mm). The mobile phase was acetonitrile:phosphate buffer (pH 3.5) (6:4 v/v). The eluent was monitored at 220 nm with a flow rate of 1.2 ml/min (Canaparo et al. Citation2000).
RESULTS AND DISCUSSION
Preparation of Solid Dispersions and Determination of Drug Content and Percent Yield
SD preparation was relatively simple and the cooled masses of SDs were frangible enough to be ground easily. Ibuprofen assay in all SDs was almost 100% and the percentage yield was greater than 98% (data not shown). This method was relatively more feasible to prepare ibuprofen-PEG 6000 SDs because of their low melting points, the ease in controlling the processing variables such as temperature and shearing rate, and the short duration of preparation (∼ 1–2 hr). In addition, the results were reproducible with relatively higher percentage yields. Drug content analysis indicated that the ibuprofen was uniformly distributed in SDs and the higher yield showed relatively lower process loss.
Scanning Electron Microscopy
In scanning electron micrographs (), ibuprofen appeared as smooth-surfaced rectangular crystalline structures (A) and PEG 6000 as smooth-surfaced irregular particles (B). Physical mixtures contained individual ibuprofen and PEG 6000 particles (C) and 1:1 w/w SD (D) and 1:10 w/w SD (E) appeared as smooth scaly surfaced uniform and homogeneously mixed mass. The surface morphology of SDs almost resembled to that of pure PEG 6000, indicating that ibuprofen was adsorbed into the PEG 6000 and homogeneously dispersed into the polymer. SEM pictures suggested that the individual surface properties of PEG 6000 and ibuprofen were lost during melting and solidification indicating the formation of effective SD systems.
FIG. 3 Scanning electron micrographs. (A). Ibuprofen, (B). Polyethylene glycol 6000, (C). 1:1 w/w physical mixtures. (D). 1:1 w/w solid dispersions, and (E). 1:10 w/w solid dispersions.

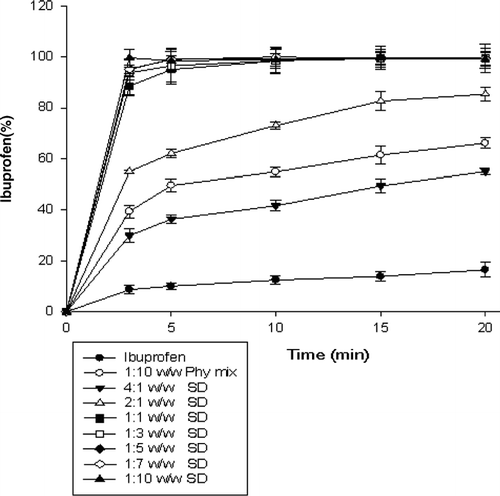
Solubility, Dissolution, and Phase Solubility
Solubility and dissolution of ibuprofen increased with the increment in the ratio of PEG 6000 in SDs ( and ). Phase solubility study showed that the solubility of ibuprofen almost linearly increased as the concentration of PEG 6000 increased (R2 = 0.89) (). Enhanced solubility and dissolution of ibuprofen from physical mixtures could be related to the surface activity, wetting effect that may lead to reduced agglomeration and hence increased surface area and solubilizing effect of PEG 6000 (Seo et al. Citation2003; Vilhelmsen et al. Citation2005; Craig Citation1990; Sudha et al. Citation2007; Law et al. Citation2003; Zerrouk et al. Citation2002; Craig Citation2002; Mura et al. Citation1996; Corrigan Citation1985; Verheyen et al. Citation2002). In the dry state, drug particles were in close contact or adhered to the polymer particles as a result of mixing (shown by SEM). When the mixture came in contact with water, the polymer particles might have hydrated rapidly (because of the high hydrophilic potency of PEG 6000) into polymer solution. This hydration contributed to the increased wettability of the drug particles and to the local enhancement of the drug solubility at the diffusion layer surrounding the particles and subsequently releasing the drug into the medium (Craig Citation2002; Mura and Ando Citation1996). This also could explain the higher solubility of drug in phase solubility study where the ibuprofen particles were dispersed in aqueous polymer solutions.
FIG. 4 Solubility of solid dispersions in phosphate buffer (pH 6.8). Data are expressed as mean ± SD (n = 3).
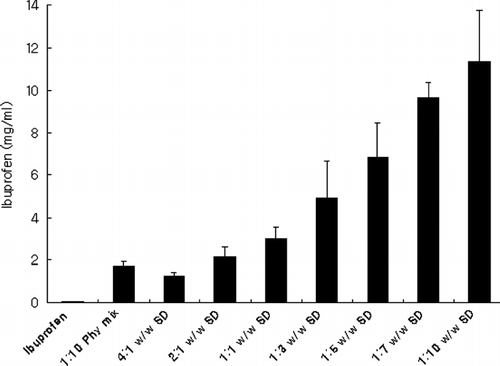
FIG. 5 Dissolution profiles of solid dispersions in phosphate buffer (pH 6.8). Data are expressed as mean ± SD (n = 3).
Differential Scanning Calorimetry and Fourier Transform Infrared Spectroscopy
To understand the possible mechanisms of improved dissolution, SDs were characterized by DSC and FTIR. The DSC thermograms () of ibuprofen showed apparent endothermic peak at 74.86°C with enthalpy of fusion (Δ H) 129.0 J/g. Similarly, the melting peak of PEG 6000 was observed at 59.41°C with enthalpy of fusion (Δ H) 197.83 J/g. Sharp ibuprofen as well as PEG 6000 peaks were lost in SDs and physical mixtures. In 1:1 w/w SDs, a sharp peak was found at 49.19°C with enthalpy of fusion (Δ H) 139.4 J/g. Similarly, another broad reduced endotherm was observed at 57.39°C. In 1:10 w/w SDs, a sharp small peak was observed at 47.83°C and another sharp peak was observed at 57.73°C with enthalpy of fusion (Δ H) 176.8 J/g. The 1:1 w/w physical mixtures showed a sharp peak at 49.02°C with an enthalpy of fusion (Δ H) 155.8 J/g and another broad endotherm at 64.94°C.
FIG. 6 Phase solubility behavior of ibuprofen at 25°C in polyethylene glycol 6000 solutions in phosphate buffer (pH 6.8). Data are expressed as mean ± SD (n = 3).
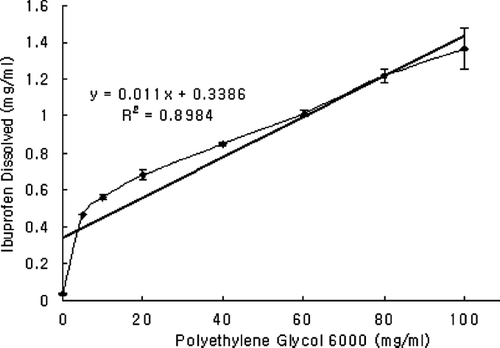
FIG. 7 Differential scanning calorimetric thermograms of ibuprofen, polyethylene glycol 6000, physical mixtures, and solid dispersions.
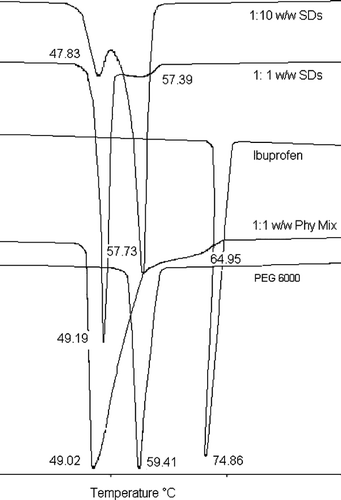
Absence or shifting toward the lower melting temperature of the drug peak in SDs and physical mixtures in DSC study indicated the possibilities of interactions between ibuprofen and PEG 6000. In a binary solid system such as a crystalline drug and a crystalline polymer, if the drug is soluble in the molten polymer at the melting temperature of the polymer or vice versa, then the systems could be eutectic (Carstensen Citation2001). To have a high solubility in the molten polymer, the drug molecule should have a weak affinity to the crystalline drug and/or a strong affinity to the molten polymer. Solids with low melting temperatures have weak affinity in solid state. Therefore a drug having low melting temperatures will have high solubility in the molten polymer. Consequently, this drug and the polymer will form an eutectic system (Sudha et al. Citation2007). PEGs are known to form eutectic systems with negligible solid solution (Law et al. Citation2003, Citation2002, Citation2001; Anastasiadou et al. Citation1983; Ford and Rubenstein Citation1978; Zerrouk et al. Citation2002). However, the amount of drug at eutectic composition differed widely. For example, the concentration of drug at eutectic composition with PEGs was 25% fenofibrate with PEG 8000 (Law et al. Citation2001), 17% for diazepam with PEG 4000 (Anastasiadou et al. Citation1983), 15% for indomethacin with PEG 6000 (Ford and Rubeinstein Citation1978), 35 and 33% for flurbiprofen with PEG 4000 and 6000 (Lacoulonche et al. Citation1998), and 15% for tamazepam with PEG 6000 (Moter et al. Citation1998).
PEG-drug eutectics belong to a category that exhibits complete miscibility in the liquid state and complete immiscibility in the solid state (Vasil'ev Citation1964). In this category, the liquid phase interactions between unlike components are expected to be stronger than those between like components. The lattice mismatch between PEGs and small organic molecules makes the formation of PEG–drug solid solution difficult and the liquid phase PEG–drug miscibility leads to simple binary eutectic systems (Law et al. Citation2001). Since the melted PEG 6000 solution was viscous, stirring might have caused a thorough mixing and ibuprofen was soluble in molten PEG 6000 up to 50% w/w. However, they were miscible at any proportion at the temperature used in the SD preparation (90–95°C).
Ibuprofen has been reported to form eutectics with PEG 8000 and its concentration at eutectic composition was 35% w/w (Law et al. Citation2002) Earlier reports showed that the PEG-drug phase diagrams are not sensitive to the PEG molecular weight (Lacoulonche et al. Citation1998, Citation1996). Because of low melting temperature, ibuprofen was highly soluble in molten PEG 6000 at any given temperature and thus is expected to form a strong eutectic relative to other compounds having higher melting temperature (Law et al. Citation2002).
When the melts with eutectic composition are cooled, the two phases begin to crystallize spontaneously, and the eutectic crystallization rate accelerates at the contact between the two phases, and proceeds with the minor phase of the eutectic growing in the interstitial spaces of the primary phase. This process leads to a marked reduction in particle size of the minor component. According to Tamman's rule, the lower melting component (PEG) is the major phase in the eutectic mixture and PEG–drug eutectic crystallization will have a well-defined microstructure with a reduction in drug particle size (Podolinsky and Taran Citation1981; Law et al. Citation2003). The complete miscibility of ibuprofen and PEG in the liquid phase indicates polymer–drug interaction at elevated temperatures, and the recognition that there would be negligible polymer–drug interaction in the solid state (as showed by FTIR) due to lattice mismatch that imposes a significant constraint on drug loading at the solid solution limit (Law et al. Citation2003).
As the melt of any composition other than that corresponding to the eutectic is cooled, one component will progressively solidify, thereby rendering the remaining liquor richer in the other component until the eutectic composition is reached. At that point, the remaining liquid will solidify as a fine dispersion. According to Tamman's rule, PEG–ibuprofen eutectic crystallization might have well defined microstructure with a reduction in drug particle size (Passerini et al. Citation2002; Law et al. Citation2003). As long as the amount of ibuprofen in the dispersion is less than the eutectic composition e.g., 1:10 w/w SDs, all of it would be present as a eutectic mixture (the small peak at 47.83°C). The consistent dissolution profile obtained above or below the eutectic point (in 1:1 to 1:10 w/w SDs) implies that the eutectic point may not determine the upper limit for drug loading in ibuprofen-PEG 6000 SDs prepared by this method (Law et al. Citation2002).
The DSCs of SDs and physical mixtures (PMs) exhibited two endothermic events. The peaks at 49.19°C of 1:1 w/w SD, at 49.02°C of PMs and at 47.83°C of 1:10 w/w SDs were possibly the melting endotherm of eutectic. After the eutectic has melted, the solid phase suspended in the liquid melt might possibly be ibuprofen (whose concentration might have been above the eutectic composition in 1:1 w/w SDs and PMs), which melted to produce the second peak, the reduced broad fusion peaks at 57.39°C in 1:1w/w SD and 64.95°C in PMs. Similarly, in 1: 10 w/w SDs, the second peaks at 57.73°C might represent the melting of PEG 6000 (Vasil'ev et al. Citation1964; Bowden Citation1938).
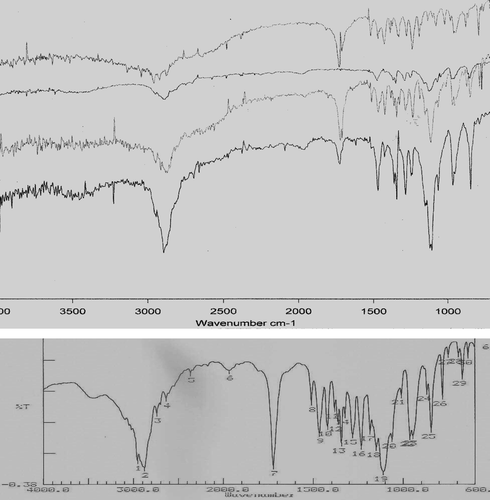
In FTIR analysis (), the spectrum of pure ibuprofen showed an intense, well-defined infrared band at around 1721 cm− 1 (carbonyl-stretching of isopropionic acid group) and another spectrum at ∼ 3000 cm− 1, whereas the PEG 6000 showed a characteristic spectra of C–H stretching of OC2H5 groups from 2800 to 2900 cm− 1 and C–O stretching from 1000 to 1200 cm− 1. A decrease in the intensity of ibuprofen peak was observed with the increasing amount of PEG 6000 in SDs. The solid dispersions showed IR spectra almost similar to those of their corresponding physical mixtures. Presence of the stretching vibration of ibuprofen carbonyl peak in SDs and physical mixtures indicates that the drug crystalline form may not be altered during solid dispersion formation and its attenuated intensities were thought to be due to the lower drug content. This observation fits well with the previous studies in which ibuprofen was found to be primarily crystalline (Passerini et al. Citation2002; Vilhelmsen et al. Citation2005).
FIG. 8 Fourier transform infrared absorption spectra of ibuprofen, (top to bottom). 1:1 w/w solid dispersions, 1:10 w/w solid dispersions, polyethylene glycol 6000, and 1:1 w/w physical mixtures.
The complete miscibility of ibuprofen and PEG 6000 in the liquid phase indicates polymer–drug interaction at elevated temperatures and the ibuprofen can act as a hydrogen bond acceptor or donor.
However, the absence of major shift in the peak positions, retention of drug peak, and the almost equivalent addition spectra (of ibuprofen and PEG 6000) for SDs and physical mixtures suggested the absence of interactions in the solid state between PEG 6000 and the ibuprofen.
Since the principal aim of our work was to formulate fast dissolving ibuprofen SDs and to evaluate their in vitro and in vivo performances, DSC or FTIR study was not performed and compared with other SDs and physical mixtures. The phase diagrams were not constructed to understand the mechanism of eutectic formation. The fact that ibuprofen-PEG 6000 SD systems were completely miscible in the liquid state and immiscible in the solid state indicates that they might have crystallized out simultaneously as microfine crystals from the molten mixture. This effect results in increased ibuprofen surface area that might have played an important role for enhanced dissolution rate (Passerini et al. Citation2002; Seo et al. Citation2003). Thus so, the enhancement of dissolution from the solid dispersions may be attributed partly to the reduction in particle size in ibuprofen crystalline due to the formation of eutectic system with PEG 6000. The similarity in the dissolution profiles of 1:1 to 1: 10 w/w SDs could indicate that along with eutectic formation, a combination of other factors such as surface activity, wetting, and solubilization effect of PEG 6000 might have affected ibuprofen solubilization and dissolution. In case of SDs where the drug concentration exceeded the eutectic composition, and improved solubility and dissolution might be partly due to the enhanced dissolution of the noneutectic portion of drug in the solid dispersion—through mechanisms including reduced agglomeration, increased solubility, and melting point depression of drug by the polymer.
Particle size reduction and improved wetting may lead to reduced agglomeration and hence increased surface area (Zerrouk et al. Citation2002). Since the physical mixture of the same composition dissolved more slowly than the solid dispersions, the intrinsic effect of PEG 6000 also might have had an important role to increase the dissolution rate of ibuprofen. Such differences in the dissolution pattern of PMs and the corresponding SDs might be attributed to a significant reduction of the drug particle size in the carrier matrix (Rabasco et al. Citation1991).
Pharmacokinetic Study
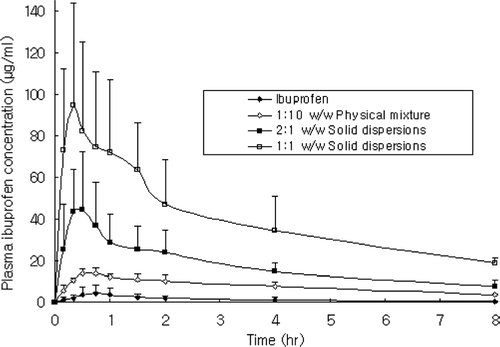
The total plasma concentrations of ibuprofen in SDs and physical mixtures were significantly higher compared with those in ibuprofen powder (p < 0.0034) ( and ). Unlike SDs (p < 0.009), the Tmaxvalue physical mixtures (p > 0.18) were not significantly different than that of ibuprofen (p > 0.17). But the AUC and Cmax of ibuprofen from physical mixtures and SDs were significantly increased (p < 0.0005). However, the elimination rate constant (Kel) and half-life (T1/2) values of ibuprofen from physical mixtures and SDs were not significantly different compared with ibuprofen powder (p > 0.05). The significantly higher AUC and Cmax, and the earlier Tmax for ibuprofen from SDs indicated the higher extent of absorption for SDs because of their improved dissolution rate in rat intestine. In summary, the SDs resulted in much higher bioavailability compared with ibuprofen as reflected by both AUC and Cmax values. These results showed that the ibuprofen was more readily available from SDs than from pure ibuprofen or a simple physical mixture containing high proportion of PEG 6000. Taken together, the fast and complete dissolution resulting from improved solubility of the ibuprofen was responsible for its enhanced oral absorption.
TABLE 1 Pharmacokinetic parameters of ibuprofen delivered by ibuprofen powder, ibuprofen-polyethylene glycol 6000 solid dispersions, and physical mixtures
FIG. 9 Plasma concentration-time profiles of ibuprofen after oral administration of ibuprofen powder, physical mixtures, and solid dispersions equivalent to 25 mg/kg ibuprofen in rats. Data are expressed as mean ± SD (n = 5).
CONCLUSION
Rapid release of ibuprofen was achieved in a relatively easy, simple, quick, inexpensive, and reproducible manner. Quicker dissolution of SDs in rat intestine resulted into its rapid absorption and improved bioavailability compared with pure ibuprofen. Preliminary results from this work suggested that the preparation of ibuprofen solid dispersion using PEG 6000 as a meltable hydrophilic polymer carrier could be a promising approach to improve its solubility, dissolution, and bioavailability.
This research was supported by the Regional R & D Cluster Project designated by the Ministry of Science and Technology & the Ministry of Commerce, Industry, and Energy (2007) and financially supported by the Ministry of Science and Technology (F104AA010008-06A0101-00810) and by Korea Ministry of Commerce, Industry, and Energy grant (10006577, 2002) in South Korea.
REFERENCES
- Adeyeye C. M., Price J. C. Development and evaluation of sustained-release ibuprofen-wax microspheres. II. In vitro dissolution studies. Pharm. Res. 1994; 11: 575–579
- Anastasiadou C., Henry S., Legendre B., Souleau C., Duchene D. Solid dispersions: comparison of prepared melts and coprecipitates of diazepam and polyoxyethylene glycol 4000. Drug Dev. Ind. Pharm. 1983; 9: 103–115
- Bowden S. T. The Phase Rule and Phase Reactions:Theoretical and Practical. MacMillan Press, London 1938
- Canaparo R., Muntoni E., Zara G. P., Della P. C., Berno E., Costa M., Eandi M. Determination of ibuprofen in human plasma by high-performance liquid chromatography: validation and application in pharmacokinetic study. Biomed. Chromatogr. 2000; 14: 219–226
- Carstensen J. T. Melting point diagrams and eutectics. Advanced Pharmaceutical Solids. Marcel Dekker, New York 2001; 170–189
- Corrigan O. I. Mechanisms of dissolution of fast release solid dispersions. Drug Dev. Ind. Pharm. 1985; 11: 697–724
- Craig D. Q. M. The mechanisms of drug release from solid dispersions in water-soluble polymers. Int. J. Pharm. 2002; 231: 131–144
- Craig D. Q. M. Polyethylene glycols and drug release. Drug Dev. Ind. Pharm. 1990; 16: 2501–2506
- Ford J. L., Rubeinstein M. J. H. Phase equlibria and dissolution rates of indomethacin-polyethylene glycol 6000 solid dispersions. Pharm. Acta Helv. 1978; 53: 93–98
- Geisslinger G., Dietzel K., Bezler H., Nuernberg B., Brune K. Therapeutically relevant differences in the pharmacokinetical and pharmaceutical behavior of ibuprofen lysinate as compared to ibuprofen acid. Int. J. Clin. Pharmacol. Ther. Tox. 1989; 27: 324–328
- Ghorab M. K., Adeyeye M. C. Enhancement of ibuprofen, dissolution via wet granulation with beta-cyclodextrin. Pharm. Dev. Tech. 2001; 6: 305–314
- Lacoulonche F., Chauvet A., Masse J., Egea M. A., Garcia M. L. An investigation of FB interactions with poly(ethylene glycol) 6000, poly(ethylene glycol) 4000 and poly-epsilon-caprolactone by thermoanalytical and spectroscopic methods and modeling. J. Pharm. Sci. 1998; 87: 543–551
- Lacoulonche F., Manderioli A., Bramanti G., Ceccarelli L. Properties of solid dispersions of naproxen in various polyethylene glycols. Drug Dev. Ind. Pharm. 1996; 22: 909–916
- Laska E. M., Sunshine A., Marrero I., Olson N., Siegel C., McCormick N. The correlation between blood levels of ibuprofen and clinical analgesic response. Clin. Pharmacol. Ther. 1986; 40: 1–7
- Law D., Wang W., Schmitt E. A., Qiu Y., Steven L. K., Fort J. J. Properties of rapidly dissolving eutectic mixtures of poly(ethylene glycol) and fenofibrate: the eutectic microstructure. J. Pharm. Sci. 2003; 92: 505–515
- Law D., Wang W., Schmitt E. A., Michelle A. L. Prediction of poly (ethylene) glycol-drug eutectic compositions using an index based on the van't Hoff equation. Pharm. Res. 2002; 19: 315–321
- Law D., Krill S. L., Schmitt E. A., Fort J. J., Qiu Y., Wang W., Porter W. R. Physicochemical considerations in the preparation of amorphous ritonavir/PEG 8000 solid dispersions. J. Pharm. Sci. 2001; 90: 1015–1025
- Moter G. V., Augustijns P., Blaton N., Kinget R. Physicochemical characterization of solid dispersions of temazepam with polyethylene glycol 6000 and PVP K30. Int. J. Pharm. 1998; 164: 67–80
- Mura P., Manderioli A., Bramanti G., Ceccarelli L. Properties of solid dispersions of naproxen in various polyethylene glycols. Drug Dev. Ind. Pharm. 1996; 22: 909–916
- Murtha J. L., Ando H. Y. Synthesis of the cholesteryl ester prodrugs cholesteryl ibuprofen and cholesteryl flufenamate and their formulation into phospholipid microemulsions. J. Pharm. Sci. 1994; 83: 1222–1228
- Passerini N., Gonzalez-Rodriguez M. L., Cavallari C., Rodriguez L., Albertini B. Preparation and characterisation of ibuprofen-poloxamer 188 granules obtained by melt granulation. Eur. J. Pharm. Sci. 2002; 15: 71–78
- Podolinsky V. V., Taran Y. Influence of adsorption on structure formation in eutectic systems. J. Cryst. Growth. 1981; 52: 82–87
- Potthast H., Dressman J. B., Junginger H. E., Midha K. K., Oeser H., Shah V. P., Vogelpoel H., Barends D. M. Biowaiver monographs for immediate release solid oral dosage forms: ibuprofen. J. Pharm. Sci. 2005; 94: 2121–2131
- Rabasco A. M., Ginés J. M., Fernández-Arévalo M., Holgado M. A. Dissolution rate of diazepam from polyethylene glycol 6000 solid dispersions. Int. J. Pharm. 1991; 67: 201–205
- Seo A., Holm P., Kristensen H. G., Schæfer T. The preparation of agglomerates containing solid dispersions of diazepam by melt agglomeration in a high shear mixer. Int. J. Pharm. 2003; 259: 161–171
- Serajuddin A. T. M. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999; 88: 1058–1066
- Sudha R. V., Zeren W., Stefanie H., Steven L. K. Factors affecting the formation of eutectic solid dispersions and their dissolution behavior. J. Pharm. Sci. 2007; 96: 294–304
- Vasil'ev M. E. Classification of the phase diagrams of binary metallic systems according to atomic interaction forces. Russ. Phys. Chem. 1964; 38: 473–476
- Verheyen S., Blaton N., Kinget R., Mooter G. V. Mechanism of increased dissolution of diazepam and temazepam from polyethylene glycol 6000 solid dispersions. Int. J. Pharm. 2002; 249: 45–58
- Vilhelmsen T., Eliasen H., Schæfer T. Effect of a melt agglomeration process on agglomerates containing solid dispersions. Int. J. Pharm. 2005; 303: 132–142
- Zerrouk N., Chemtob C., Arnaud P., Toscani S., Dugue J. In vitro and in vivo evaluation of carbamazepine-PEG 6000 solid dispersions. Int. J. Pharm. 2002; 225: 49–62