
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Several dehydroabietylamine derivatives containing heterocyclic moieties such as thiophene and pyrazine ring were successfully synthesized. The antiproliferative activities of these thiophene-based Schiff-bases, thiophene amides, and pyrazine amides were investigated in vitro against Hela (cervix), MCF-7 (breast), A549 (lung), HepG2 (liver), and HUVEC (umbilical vein) cells by MTT assay. The toxicity of L1−L10 (IC50 = 5.92− >100 μM) was lower than L0 (1.27 μM) and DOX (4.40 μM) in every case. Compound L1 had higher anti-HepG2 (0.66 μM), anti-MCF-7 (5.33 μM), and anti-A549 (2.11 μM) and compound L3 had higher anti-HepG2 (1.63 μM) and anti-MCF-7 (2.65 μM) activities. Both of these compounds were recognized with high efficiency in apoptosis induction in HepG2 cells and intercalated binding modes with DNA. Moreover, with average IC50 values of 0.66 and 5.98 μM, L1 was nine times more effective at suppressing cultured HepG2 cells viability than normal cells (SI = 9). The relative tumor proliferation rate (T/C) was 38.6%, the tumor inhibition rate was up to 61.2%, which indicated that L1 had no significant toxicity but high anti-HepG2 activity in vivo. Thus, it may be a potential antiproliferation drug with nontoxic side effects.
1. Introduction
Currently, various chemotherapy drugs have been developed for the treatment of different cancers; however, undesirable side effects may greatly impede their use in clinical progression. Among a variety of cancer cures, the antiproliferative approach plays a crucial role in controlling this deadly disease (Xin et al., Citation2018; Lei et al., Citation2019b). Up to now, numerous molecules containing heterocyclic rings have showed great antiproliferative potential, particularly those with thiophene and pyrazine rings.
Thiophene derivatives are ubiquitous in nature and can be found in the structure of various drugs and medicines, produced by combustion of fossil fuels or by the general cooking process (Dyreborg et al., Citation1996; Dalvie et al., Citation2002; Medower et al., Citation2008). Thiophenes bear extensive pharmacological properties such as analgesic, antipyretic, and antiandrogenic activities (Hana et al., Citation2008; Huang et al., Citation2012; Iványi et al., Citation2012). Furthermore, a great number of thiophene derivatives are used as antitumor agents (Lesyk et al., Citation2006; Kulandasamy et al., Citation2009; Khalil et al., Citation2010; Ye et al., Citation2010). For instance, OSI-930 is an investigational anticancer agent which contains thiophene moiety (Petti et al., Citation2005; Garton et al., Citation2006). Ghorab et al. (Citation2016) reported a series of thiophene derivatives with high anti-MCF-7 (breast adenocarcinoma) cancer activity (half maximal inhibitory concentration [IC50] = 33.1–66.3 μM). Mohareb and Al-Omran (Citation2012) have studied three thiophene derivatives exhibited much higher inhibitory effects toward three tumor cell lines, MCF-7, NCI-H460 (non-small cell lung cancer), and SF-268 (CNS cancer) with GC50 value in the range of 0.01–16.2 μM than the reference drug, doxorubicin (DOX).
Recently, nitrogen heterocycles have also been reported to exhibit therapeutics anticancer (Azuine et al., Citation2004; Lei et al., Citation2019a,Citationc) and anti-microbial (Nomiya et al., Citation2000; Mathew et al., Citation2006) activities. Among them, the pyrazine heterocycles have widespread application in food science, materials, and medicinal chemistry (Mondal et al., Citation2010; Saito et al., Citation2010; Badrinarayanan & Sperry, Citation2011; Zitko et al., Citation2011). For example, pyrazinamide, a pyrazine derivative, is an antimicrobial agent that is most commonly used for treatment of active tuberculosis during the initial phase of therapy in combination with other agents. Quinoxaline compounds have been reported to possess a wide range of interesting biological properties such as anticancer, antiviral, antimicrobial, antifungal, antitubercular, anti-inflammatory, and anti-angiogenesis agents (Seitz et al., Citation2002; Smits et al., Citation2008; Vicente et al., Citation2009; Lee et al., Citation2010; Sridevi et al., Citation2010; Ingle et al., Citation2013; Aissi et al., Citation2014; Soozani et al., Citation2018), containing pyrazine motif. Ahmed et al. (Citation2018) have synthesized several compounds and evaluated anticancer effects against three cancer lines (HCT-116, MCF-7, and HepG2), and the results revealed that pyrazine derivatives were the most active compounds with IC50 value of 1.89 and 2.05 Μm. Another pyrazine derivative, pyrazin-2(1H)-one, has attracted considerable attention due to its biological activities, such as anti-viral, antibacterial, anti-inflammatory, and anticancer (colon cancer therapies) activities (Lindsley et al., Citation2005).
Recently, dehydroabietylamine (L0), which is one of the most vital modified products of rosin, has attracted considerable attention due to the broad spectrum of biological properties (Singh et al., Citation2014; Lin et al., Citation2015; Auxiliadora et al., Citation2016; Bahekar et al., Citation2016; Fei et al., Citation2016; Liu et al., Citation2016; Wang et al., Citation2016; Huang et al., Citation2017; Liu et al., Citation2017). In general, a focus of research on dehydroabietylamine derivatives with their anticancer, antibacterial, antifungal, and cytotoxic activities has been paid their attention in forest chemistry too. Rao et al. (Citation2012) screened a series of imines, amides, and ureas with a dehydroabietyl skeleton for their anticancer activities against SMMC7721 (liver), A549 (lung), C6 (glioma), and MCF-7 cancer cell lines with smallest IC50 values of 6.65, 0.75, 0.81, and 10.65 μM. Lately, our group has found that several Schiff-bases and amide compounds displayed highly potent inhibitory activities against HepG2 (liver), MCF-7 and A549 cells with smallest IC50 values of 0.14, 0.24, 2.58, and 3.17 μM (Zhao et al., Citation2018).
Up until this day, finding new molecules with more effective, less toxic, and target-specific DNA binding properties is one of the most important interest in medicinal chemistry. Cisplatin, a well-known active anticancer drug, can covalently bind to DNA, but its usage is limited with side effects and acquired cellular resistance (Rajendiran et al., Citation2007). The aforementioned thiophene, pyrazine, and dehydroabietylamine analogs are all recognized as excellent antiproliferative agents; thus, our group was interested in the further investigation of these derivatives. Our goal was to achieve high anticancer activity, low toxicity, and target-specific DNA binding properties with the dehydroabietylamine derivatives including thiophene Schiff-bases (L1−L3), thiophene amides (L4−L6), and pyrazine amides (L7−L10). These new compounds were screened for antiproliferative activities against Hela (cervix), MCF-7, A549, HepG2 human cancer cell lines in vitro by MTT assay, in addition to L1 in vivo. We have verified that several compounds owned high antiproliferative activities against these cancer cells and some of them exhibited more potent antiproliferative activities as compared to dehydroabietylamine. Subsequently, the induction of apoptosis effect on L1 and L3 with HepG2 cells was also investigated and the result suggested they could inhibit cell proliferation by inducing apoptosis. In this report, we hope to display a simple but effective strategy that may make contributions to the exploration of future anticancer drugs.
2. Results and discussion
2.1. Chemistry
All compounds were synthesized by the facile and efficient synthetic route from the commercially available (+)-Dehydroabietylamine, [α]20 D = +55.1 (c 2.4 pyridine) (Scheme 1). To explore the relationship between the compound’s structures and biological activities, three thiophene aldehydes with different functional groups, three carboxylic acids, and four pyrazine carboxylic acids with various substituents were designed to react with dehydroabietylamine to prepare thiophene Schiff-bases (L1−L3), thiophene amides (L4−L6), and pyrazine amides (L7−L10). L1−L10 were obtained under neutral to slightly alkaline conditions (pH = 7.0–7.4) during the experimental process.
Scheme 1. Synthesis of L1−L10.
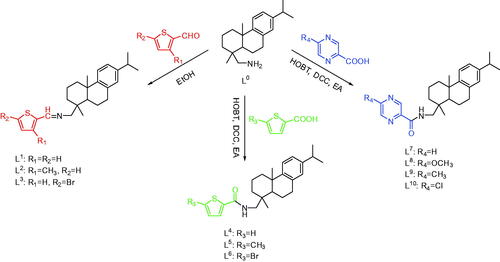
Both of the thiophene Schiff-bases L3 and thiophene amides L4 owned novel structural features with two aromatic rings (thiophene ring and benzene ring) and two aliphatic rings from dehydroabietylamine. For L3, its faint yellow block-shaped single crystal was found to be a monoclinic crystal in a chiral space group P21 with a Flack parameter of 0.009(8). The thiophene ring with C5, N1, and C6 was coplanar (). The molecules are stably connected by slightly weak C1 − H1⋅⋅⋅S1# hydrogen bond to assemble an infinite one-dimensional chain structure along b axis (). The distance of H1⋅⋅⋅S1 (2.8019(18) Å) and C1⋅⋅⋅S1 (3.6459(64) Å) is corresponding to Shi’s report (H16⋅⋅⋅S2 2.95 Å and C16⋅⋅⋅S2 3.61 Å), which is slightly shorter than the sum of van der Waals radii, proving that a weak interaction existed between H1⋅⋅⋅S1 (Shi & Wen, Citation1998). The length of the new imine double bond C5 − N1 (1.2520(7) Å) is in accordance with the report from Lu (C10 − N2 1.2913(3) Å) (Lu et al., Citation2016) and our previous result of C7 − N1 1.2690(4) Å (Zhao et al., Citation2018). For L4, its colorless block-shaped single crystal was obtained as orthorhombic crystal system in a chiral space group P21 with a Flack parameter of 0.08(5). The molecules are connected by N1 − H1⋅⋅⋅O1# hydrogen bond to assemble an infinite one-dimensional chain structure along b axis (). The hydrogen-bond parameters of L3 and L4 are shown in . Selected bond lengths and angles of L3 and L4 are shown in . The crystallographic data are shown in Table S1 in Supplementary Material.
Figure 1. (a) Molecular structure of L3 (hydrogen atoms omitted for clarity). (b) The 1D chain structure formed by intermolecular hydrogen bonds.
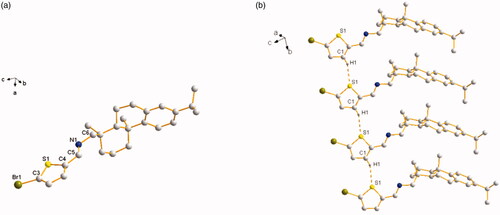
Figure 2. (a) Molecular structure of L4 (hydrogen atoms omitted for clarity). (b) The 1D chain structure formed by intermolecular hydrogen bonds.
Table 1. Hydrogen bonds of L3 and L4.
Table 2. Selected bond lengths (Å) and angles (deg) for L3 and L4.
2.2. Biological evaluation
2.2.1. Antiproliferative activities
Compounds L1−L10 were evaluated against human cancer cell lines Hela, HepG2, MCF-7, A549, and the normal cell HUVEC with the antiproliferative activities in vitro by MTT assay, and results are summarized as IC50 values in . DMSO was used as negative control and DOX (Doxorubicin) as positive control, which is a common chemotherapy medication used to cure cancer (Wang et al., Citation2004).
Table 3. Cytotoxicity of L1−L10 and DOX against certain axenic cancer cells and normal cell.
A histogram was drawn more distinctly to compare the antiproliferative activity of L0−L10 against axenic cancer cells and cytotoxicity based on IC50 values (). We used DOX as positive control, L0 also as control to compare the antiproliferative activity among its modified products for the reasoned that the antiproliferative activity of L0 was slightly lower than DOX (except Hela cells), whereas that was better than a variety of pharmaceutical products. The values of p > 0.05 were considered that the antiproliferative activity of L0 and DOX in statistics difference were insignificant, except for MCF-7 cells. The result is shown in .
Figure 3. The comparison of antiproliferative effects and cytotoxicity of DOX, L0−L10.
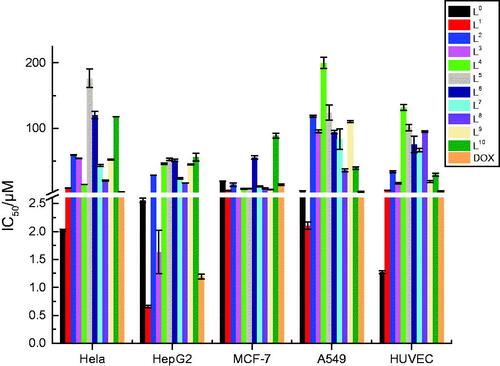
From , the result showed quite clearly that L1 and L3 had higher antiproliferative activity against HepG2 cells while L2 had higher antiproliferative activity against A549 cells. L1−L10 were low toxic as their toxicity (IC50 = 5.92− >100 μM) was all lower than L0 (1.27 μM) and DOX (4.40 μM) for HUVEC cells. For further observation, for HepG2 cells, the IC50 values of L1 (0.66 μM) and L3 (1.63 μM) were much smaller than those of L0. Moreover, with average IC50 values of 0.66 and 5.92 μM, L1 was nine times more effective at suppressing cultured HepG2 cells viability than normal cells, and L3 as well with average IC50 values of 1.63 and 16.65 μM. For MCF-7 cells, most compounds had higher anti-MCF-7 activity than both L0 and DOX with their smaller IC50 value compared to L0 (excluding L6 and L10), particularly, L1 (5.33 μM) and L3 (2.65 μM). For A549 cells, the IC50 value of L1 (2.11 μM) was smaller than L0, which meant L1 had higher anti-A549 activity than L0; in especial, the IC50 value of L1 was smaller than DOX. The security index (SI) value of L1 is 9.0 and L3 is 10.2, which suggested they had higher anticancer activity and lower toxicity compared with L0(0.5)and DOX (3.7). Actually, among these investigated compounds, L1 had the smallest IC50 value of 0.66 μM against HepG2 cells and lower toxicity, which shows that L1 may be a promising anticancer drug.
Furthermore, by contrast with the structure and cytotoxicity in vitro on the cancer cell lines of L1−L10, compounds owned − CH3 electron-donating group (L2, L5, L9) had relatively lower antiproliferative activity than those without electron-donating group (L1, L4, L7). As reported by Hande, introduction of a polar group containing a hydrogen-bond donor resulted to enhanced anticancer activity (Hande, Citation1998). Although the correspondingly better solubility of Schiff-bases and the special structure of Schiff-base (−CH = N−), which can conjugate with the thiophene ring and grant it with higher stability and bioactivity, these Schiff-base compounds (L1−L3) had higher antiproliferative activity against HepG2 and MCF-7 cells. As a whole, all these synthetic compounds could inhibit the proliferation of cancer cells by penetrating into cytoplasm with endocytosis, we reasoned that the strong chelating ability of ‘S’ and ‘N’ may lead to the easy formation of hydrogen bonds with carboxyl and amino groups in the cancer cell lines through their unique lone pair electrons.
2.2.2. Induction of apoptosis
As a routine chemotherapeutic agent for a broad range of malignancies, DOX can prevent tumor proliferation via inducing apoptosis. Apoptosis, known as programed cell death, is a crucial process related to the regulation of development and homeostasis (Arjmand & Aziz, Citation2009; Qi et al., Citation2019). It plays an important role in cancer, as its induction in cancer cells is significant to a successful therapy, thereby carrying out apoptosis assay can afford meaningful information to the study of the mode of action. In this paper, we have estimated the potential mechanism of cell proliferation inhibitory activity of L1 and L3 through the assay apoptosis with Annexin V-FITC/PI and flow cytometry in HepG2 cells. The flow-cytometric analysis is shown in .
Figure 4. Apoptosis assay of HepG2 cells treated with L1 and L3. C was DMSO (negative control), others were L1 at 0.26, 2.6, 13 μM, and L3 at 0.22, 2.2, 11 μM, respectively.
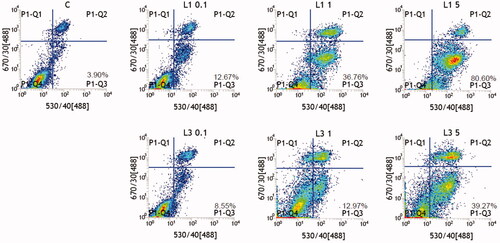
According to , after L1 and L3 incubated with HepG2 cells with the concentration range from 0.22 to 13 μM, respectively, live cells reduced and apoptotic cells increased; herein, they obviously exhibited the dosage-dependent manner. When the concentrations of L1 were 0.26, 2.6, and 13 μM, the apoptotic ratios of L1 were 12.67%, 36.76%, and 80.60%, respectively. Simultaneously, when the concentrations of L1 were 0.22, 2.2, and 11 μM, the apoptotic ratios of L3 were 8.55%, 12.97%, and 39.21%, respectively. Compared to the apoptotic ratio of these two compounds, L1 had obviously higher ability to induce apoptosis at the similar concentration. During the induction of apoptosis process, live cells trend to develop toward apoptotic cells with the enhanced concentration. Herein the reported results distinctly illustrated that L1 and L3 could inhibit cell proliferation by inducing apoptosis.
For further investigation, the logP values of all compounds were calculated by hyperchem 8.0 (). Compared with DOX (1.50), the values of logP of L1 (0.98), L2 (1.17) and L3 (1.32) were smaller with stronger hydrophily, which implied they had better solubility and may be in favor of excellent cytotoxic activities. According to a report, the ideal drug potency and satisfied pharmacokinetic profiles required good water solubility to achieve the desired therapeutic efficacy (Li et al., Citation2014). Any drug to be absorbed should exist in the form of solution at the site of absorption (Muhammad & Bashir, Citation2017). The value of logP of L4−L10 (>3) was larger, which suggested they had stronger lipophicity and its slightly poor solubility might lead to relatively worse effects on cancer cells. All compounds herein had been next examined with pKb in the range of 9.00–14.96; thus, they seemed no regular effect on cytotoxic activities.
Table 4. Physicochemical data (logP and pKb) of L1−L10, DOX.
2.2.3. DNA binding modes
Intercalation is well known to strongly influence the properties of the DNA and has been reported as a preliminary step in mutagenesis. It was reported that DOX had the ability to remain inside nucleated cells because of its lipophilic characteristics and DNA intercalating or binding properties (Arjmand & Aziz, Citation2009; Sun et al., Citation2019). We further investigated whether L1 and L3 had similar activity to DOX; herein we studied DNA binding properties of them. The DNA (Salmon Sperm DNA) binding modes were evaluated by ethidium bromide (EB) fluorescence displacement experiments. Actually, EB had no perceptible emission in buffer solution; after adding DNA, the fluorescence intensity improved obviously, which was considered of its strongly intercalation with DNA base pairs. The intercalation of the compound with the base pairs of DNA can be confirmed when the DNA − EB emission can be decreased or quenched upon adding a compound (Li et al., Citation1996; Gao et al., Citation2010). As expected, the emission intensity apparently reduced (shown in ) by adding L1 and L3 to DNA-EB, which exhibits that L1 and L3 can bind to DNA at the sites occupied by EB, and it can interact with DNA by intercalation.
Figure 5. Emission spectra of DNA − EB in the absence and presence of increasing amounts of L1 and L3 at room temperature, respectively ([EB] = 2 × 10−5 M, [DNA] = 1 × 10−4 M, and [L1, L3] = 1.5 × 10−5 M).
![Figure 5. Emission spectra of DNA − EB in the absence and presence of increasing amounts of L1 and L3 at room temperature, respectively ([EB] = 2 × 10−5 M, [DNA] = 1 × 10−4 M, and [L1, L3] = 1.5 × 10−5 M).](/cms/asset/dfa5840a-9477-4372-b81e-c8855d9cb15c/idrd_a_1716879_f0005_c.jpg)
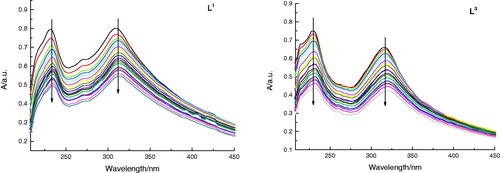
The DNA binding modes of L1 and L3 were detected by the application of Absorption Spectral as well (shown in ). Normally, a compound binding to DNA can generate hypochromism and bathochromism by intercalation. The absorption spectra exhibited a hypochromic shift after promoting added amounts of DNA to solution of L3, which illustrated an intercalative binding mode. The result was in coincidence with that of fluorescence studies. To sum up, the absorption and fluorescence spectral all verified that L1 and L3 with the same as DOX could bind with DNA through intercalation.
Figure 6. Absorption spectra of L1 and L3 (5 × 10−5 M) in the absence and presence of increasing amounts of DNA (5 × 10−5 M to 10−3 M) at room temperature in Tris-NaCl-HCl buffer (pH = 7.3). The arrow shows the absorbance change when increasing the DNA concentration.
In view of aforementioned findings, these synthetic dehydroabietylamine derivatives had relatively high potential antiproliferative activity and low toxicity, some of them also could induce apoptosis at low concentration. Moreover, to understand the cytotoxicity and the DNA binding modes was significative for designing new and potential drugs.
2.2.4. Antiproliferation activity in vivo
Herein, we have intravenously injected compound L1(dose: 0.6 mg/kg) into the mice with HepG2 cells during 25 days with every 3 days in vivo experiment for further investigation.
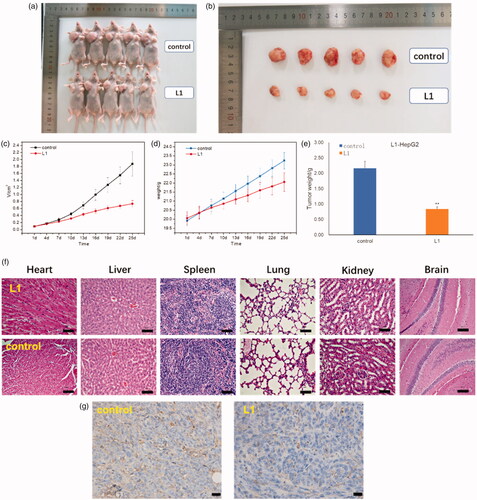
As shown in , it is obvious that the volume and weight of tumor mice were decreased after injected with compound L1 (0.6 mg/kg) as compared with PBS control. The average volume of tumor (HepG2) was 0.739 cm3 when the PBS control was 1.876 cm3. In addition, the weight of tumor was 0.84 g compared with PBS control was 2.16 g. The relative tumor proliferation rate (T/C) was 38.6% and the tumor inhibition rate was up to 61.2%. Moreover, no obvious toxicity was also observed in the heart, liver, spleen, lung, kidney, and brain tissues of the mice injected with compound L1 in , which exhibited no significant changes in morphology of these organs. In , CD31 immumohistochemical staining with mice was taken on the 25th day after an intravenous injection of compound L1 (0.6 mg/kg) showed tumor angiogenesis rate was decreased compared with PBS control, which demonstrated compound L1 could suppress tumor growth. In general, these findings suggested that L1 had high anti-HepG2 activity both in vitro and in vivo, and L1 had great promising future as nontoxic side effects and effective antiproliferation drug.
Figure 7. (a) Whole appearance and (b) the volume of tumor mice injected with PBS (control) and compound L1 after 25 days. (c) Change in tumor volume of mice injected with compound L1 (0.6 mg/kg) compared with PBS control. (d) Change in body weight of mice injected with compound L1 (0.6 mg/kg) and PBS. (e) The tumor weight of mice injected with compound L1 (0.6 mg/kg) and PBS control after 25 days. (f) H&E staining of the brain, heart, liver, spleen, lung, and kidney tissues collected from mice on the 25th day after an intravenous injection of compound L1 (0.6 mg/kg) and PBS control. (g) CD31 immumohistochemical staining with mice on the 25th day after an intravenous injection of compound L1 (0.6 mg/kg) and PBS control. Scale bar = 20 μm; error bars are based on standard errors of the mean (n = 5).
3. Conclusions
In conclusion, our formulation has shown thiophene Schiff-bases (L1−L3), thiophene amides (L4−L6) and pyrazine amides (L7−L10) with high antiproliferative activity, relatively low toxicity and DNA binding modes. The toxicity of L1−L10 (IC50 = 5.92− >100 μM) was all lower than L0 (1.27 μM) and DOX (4.40 μM). Compound L1 had higher anti-HepG2 (0.66 μM), anti-MCF-7 (5.33 μM) and anti-A549 (2.11 μM) activity. Compound L3 had higher anti-HepG2 (1.63 μM) and anti-MCF-7 (2.65 μM) activity. Additionally, L1 and L3 were verified with high efficiency apoptosis induction in HepG2 cells and intercalated modes of binding with DNA. L1 had no significant toxicity but high anti-HepG2 activity both in vitro and in vivo; it may be an effective antiproliferation drug with nontoxic side effects.
These findings can provide a convenient procedure of the rational design in new and potential cellular targeting compounds, which may help to explore the future selective anticancer drugs directly toward cellular targets. However, the possible cellular and structural mechanisms are still unclear and its investigation is currently ongoing.
4. Experimental section
4.1. General experimental procedures
All reagents and solvents were analytical reagent (AR) grade and used as received unless otherwise indicated. The IR spectra were recorded on a Bruker Vectex 80 FT − IR spectrometer with KBr discs in the 4000–500 cm−1 range. The 1 H NMR spectra were measured on a Bruker Avance III 600 MHz NMR spectrometer (Billerica, MA, USA). 1 H and 13 C {1 H} NMR spectra were recorded in CDCl3 as solvent unless otherwise stated. Chemical shifts (δ) were given as parts per million (ppm) relative to the NMR solvent signals (CDCl3 7.26 and 77.00 ppm for 1 H and 13 C{1H} NMR, respectively). J values were given in Hz. HRMS were measured to determine purity of all tested compounds by LTQ Orbitrap XL mass spectrometer (Thermo Electron, USA). Reactions were monitored by TLC using silica gel 60 F–254 in 0.25 mm thick plates. Compounds on TLC plates were detected under UV light at 254 nm. Purifications were performed by flash chromatography on silica gel (300–400 mesh). The DNA binding modes were investigated by Lambda 950 (Perkin Elmer, USA) and LS55 Fluorescence Spectrophotometer (Perkin Elmer, USA).
4.2. Synthesis of compound L1−L10
Thiophene Schiff-bases (L1−L3), thiophene amides (L4−L6), and pyrazine amides (L7−L10) were synthesized by the following methods.
4.2.1. Synthesis of 2-thiophene-dehydroabietylamine-Schiff-base (L1)
L0 (1.43 g, 5.0 mmol), 2-thiopehne-formaldehyde (0.56 g 5.0 mmol) with acetic acid as the catalyst was dissolved in ethanol (50 mL) and refluxed for 24 h. When reaction mixture was cooled to the room temperature, lots of white needlelike crystal precipitation appeared. Then white needlelike crystals were obtained by recrystallization from ethanol solution. (1.58 g, 83%), mp: 83.6-85.4 °C; IR (neat) νmax/cm−1 3423, 2929, 2861, 1649, 1598, 1450, 1375, 1036, 977, 747, 637; 1 H-NMR (CDCl3, 600 MHz) δ 1.04 (3 H, s), 1.21-1.23 (9 H, t, J = 7.2 Hz), 1.30-1.50 (3 H, m), 1.60-1.66 (2 H, m), 1.71–1.79 (2 H, m), 1.88–1.90 (1 H, m), 2.24–2.26 (1 H, d, J = 12 Hz); 2.79–2.81 (2 H, m), 2.81–2.86 (1 H, m), 3.40 (2 H, m), 6.86 (1 H, s), 6.98 (1 H, d, J = 7.8 Hz), 7.02 (1 H, dd, J = 3.6 Hz), 7.17 (1 H, d, J = 8.4 Hz), 7.25 (1 H, d, J = 3.0 Hz), 7.31–7.32 (1 H, m), 8.30 (1 H, s); 13 C{1H} (CDCl3, 151 MHz) δ 18.95, 19.45, 24.04, 24.05, 25.72, 30.58, 33.46, 36.72, 37.73, 38.27, 38.48, 46.11, 123.83, 124.48, 126.88, 127.26, 128.52, 129.70, 135.01, 143.02, 145.39, 147.51, 153.95; MS [M + H]+ m/z 380.2419 (calcd for C25H33NS 379.2334). Anal. calcd for C25H33NS: C, 79.10; H, 8.76; N, 3.69; S, 8.45. Found: C, 79.21; H, 8.62; N, 3.81; S, 8.36.
4.2.2. Synthesis of 3-methyl-2-thiophene-dehydroabietylamine-Schiff-base (L2)
When the mixture was cooled to the room temperature, removed the solvent by reduced pressure distillation, and received the brown oil compound. Finally, the brown products were obtained by recrystallization from methanol solution and dried in vacuum. (1.34 g, 68%), mp: 38.2–39.1 °C; IR (neat) νmax/cm−1 3428, 2929, 2868, 1629, 1447, 1378, 1207, 1050, 825, 716, 620; 1 H–NMR (CDCl3, 600 MHz) δ 1.02 (3 H, s), 1.21–1.24 (9 H, t, J = 7.2 Hz), 1.38–1.52 (3 H, m), 1.60–1.66 (2 H, m), 1.69–1.80 (2 H, m), 1.91–1.95 (1 H, m), 2.25–2.27 (1 H, d, J = 11.4 Hz), 2.37(3 H, s), 2.80–2.87 (3 H, m), 3.41 (2 H, m), 6.83 (1 H, s), 6.98 (1 H, d, J = 7.2 Hz), 7.17 (1 H, dd, J = 8.4 Hz), 7.22 (1 H, d, J = 4.8 Hz), 7.24–7.25 (1 H, m), 8.36 (1 H, s); 13 C{1H} ((CD3)2SO, 151 MHz) δ 15.79, 18.50, 18.69, 24.37, 24.45, 25.45, 29.46, 33.38, 35.02, 36.08, 37.40, 38.01, 40.41, 44.88, 50.04, 123.96, 124.36, 126.03, 126.81, 129.54, 134.89, 141.97, 142.87, 145.45, 147.28, 165.57; MS [M + H]+ m/z 394.5419 (calcd for C26H35NS 393.2490). Anal. calcd for C26H35NS: C, 79.33; H, 8.96; N, 3.56; S, 8.15. Found: C, 79.02; H, 8.69; N, 3.75; S, 8.54.
4.2.3. Synthesis of 5-bromine-2-thiophene-dehydroabietylamine-Schiff-base (L3)
After the reaction mixture cooling to the room temperature, lots of pale yellow needlelike crystal precipitation appeared. Then faint yellow block-shaped single crystals were obtained by recrystallization from ethanol solution. (1.99 g, 87%), mp: 132.9–134.3 °C; IR (neat) νmax/cm−1 3394, 2977, 2922, 1630, 1427, 1378, 1085, 1044, 879, 797, 620; 1 H–NMR (CDCl3, 600 MHz) δ 1.04 (3 H, s), 1.21–1.23 (9 H, t, J = 7.2 Hz), 1.30–1.50 (3 H, m), 1.60–1.66 (2 H, m), 1.71–1.79 (2 H, m),1.88–1.90 (1 H, m), 2.24–2.26 (1 H, d, J = 12 Hz); 2.79–2.81 (2 H, m), 2.81–2.86 (1 H, m), 3.40 (2 H, dd, J = 12 Hz), 6.89 (1 H, s), 6.99–7.01 (3 H, m), 7.19–7.20 (1 H, d, J = 8.4 Hz), 8.19 (1 H, s); 13 C{1H} (CDCl3, 151 MHz) δ 18.92, 18.95, 19.40, 24.00, 25.63, 30.54, 33.45, 36.74, 37.70, 38.24, 38.49, 46.15, 72.94, 116.61, 123.83, 124.43, 126.86, 129.52, 130.19, 134.89, 144.72, 145.39, 147.42, 153.02; MS [M + H]+ m/z 458.1526 (calcd for C25H32BrNS 457.1439). Anal. calcd for C25H32BrNS: C, 65.49; H, 7.03; N, 3.05; S, 6.99. Found: C, 65.34; H, 7.12; N, 3.11; S, 6.90.
4.2.4. Synthesis of 2–thiophene-acyl-dehydroabietylamine (L4)
Equal 2-thiophene-carboxylic acid (0.64 g, 5.0 mmol) and HOBT (0.68 g, 5.0 mmol) were dissolved in ethyl acetate (40 mL), stirred for 0.5 h at 0 °C. Then DCC (1.03 g, 5.0 mmol) was slowly added, stirred for 2.5 h at 0 °C. The ethyl acetate solution (10 mL) of L0 (1.43 g, 5.0 mmol) was added to the reaction system slowly, the mixture was stirred at room temperature for 8 h. Filter to remove DCU, filtrate was diluted to 200 mL and washed with 5% NaHCO3 (3*20 mL), 10% citric acid (2*20 mL) and saturated salt water. Ethyl acetate layer was dried with anhydrous Na2SO4 for 2 h, and solvents were evaporated to get milky white powders. Then colorless block-shaped single crystals were obtained by recrystallization from ethanol solution. (1.74 g, 88%), mp: 153.7–156.0 °C; IR (neat) νmax/cm−1 3428, 2922, 2860, 1638, 1561, 1439, 1371, 1044, 976, 771, 606; 1 H NMR (CDCl3, 600 MHz): δ 1.05 (3 H, s), 1.21–1.24 (9 H, s), 1.38–1.44 (2 H, m), 1.56–1.57 (1 H, m), 1.60–1.67 (2 H, m), 1.71–1.81 (2 H, m), 1.89–1.92 (1 H, m), 2.25–2.30 (1 H, m), 2.76–2.91 (3 H, m), 3.42–3.61 (2 H, dd, J = 12 Hz), 6.87 (1 H, s), 6.98 (1 H, dd, J = 7.2 Hz), 7.14 (1 H, d, J = 7.2 Hz), 7.18 (1 H, d, J = 8.4 Hz), 7.29–7.30 (1 H, m), 7.69–7.72 (1 H, m), 8.01–8.05 (1 H, m), 8.34 (1 H, s), 8.61(1 H, d, J = 3.6 Hz); 13 C{1H} NMR (CDCl3, 151 MHz): δ 18.76, 18.90, 19.64, 23.96, 25.64, 30.48, 33.40, 36.60, 37.66, 38.20, 38.42, 45.63, 58.45, 72.91, 121.16, 123.80, 124.41, 124.61, 126.87, 135.00, 136.55, 145.39, 147.47, 149.18, 154.68, 161.83; MS [M + H]+ m/z 375.2800 (calcd for C26H34N2 374.2722). Anal. calcd for C26H34N2: C, 83.37; H, 9.15; N, 7.48. Found: C, 83.43; H, 9.28; N, 7.29.
4.2.5. Synthesis of 5-methyl-2-thiophene-acyl-dehydroabietylamine (L5)
The condensation reaction used HOBT and DCC like the method of L4. The solution was dried with anhydrous Na2SO4 and evaporated to get milky white powders. (1.72 g, 84%), mp: 169.4–171.7 °C; IR (neat) νmax/cm−1 3413, 2922, 2854, 1629, 1553, 1453, 1290, 1050, 1046, 811, 743, 606; 1 H–NMR ((CD3)2SO, 600 MHz) δ 0.90 (3 H, s), 1.13–1.15 (9 H, t, J = 7.2 Hz), 1.32–1.36(2H, m), 1.43–1.45 (1 H, m), 1.56–1.61(2H, m), 1.69–1.71 (2 H, m), 1.96–1.99 (1 H, m), 2.24 (1 H, d, J = 12.6 Hz), 2.42–2.44 (3 H, s), 2.73–2.78 (3 H, m), 2.90–3.40 (2 H, dd, J = 7.2 Hz), 6.77 (1 H, s), 6.82 (1 H, d, J = 1.8 Hz), 6.92 (1 H, dd, J = 8.4 Hz), 7.12 (1 H, d, J = 8.4 Hz), 7.60 (1 H, d, J = 3.6 Hz), 8.09 (1 H, s, –NH); 13 C{1H} NMR ((CD3)2SO, 151 MHz) δ 15.61, 18.74, 19.13, 19.30, 24.35, 24.40, 25.82, 30.37, 33.35, 36.35, 37.48, 38.32, 45.04, 49.72, 123.93, 124.46, 126.64, 126.88, 128.64, 135.00, 138.10, 144.73, 145.28, 147.44, 161.88; MS [M + H]+ m/z 410.2527, MS [M + Na]+ m/z 432.2346 (calcd for C26H35NOS 409.2439). Anal. calcd for C26H35NOS: C, 76.24; H, 8.61; N, 3.42; S, 7.83. Found: C, 76.19; H, 8.72; N, 3.36; S, 7.74.
4.2.6. Synthesis of 5-bromine-2-thiophene-acyl-dehydroabietylamine (L6)
HOBT and DCC were used in the condensation reaction (same as L4). Ethyl acetate was dried with and evaporated to get milky white powders. (1.92 g, 81%), mp: 170.7–173.2 °C; IR (neat) νmax/cm−1 3414, 2922, 2849, 1629, 1550, 1413, 1282, 1077, 805, 736, 620; 1 H–NMR ((CD3)2SO, 600 MHz) δ 0.90 (3 H, s), 1.13–1.15 (9 H, t, J = 6.6 Hz), 1.32–1.35 (2 H, m), 1.42–1.46 (1 H, m), 1.57–1.61(2H, m), 1.62–1.73 (2 H, m), 1.95–1.99 (1 H, m), 2.24 (1 H, d, J = 12.6 Hz), 2.72–2.79 (3 H, m), 2.92–3.41 (2 H, m), 6.82 (1 H, s), 6.92 (1 H, dd, J = 7.8 Hz), 7.12 (1 H, d, J = 7.8 Hz), 7.22 (1 H, d, J = 3.6 Hz), 7.68 (1 H, d, J = 4.2 Hz), 8.33 (1 H, s, –NH); 13 C{1H} NMR ((CD3)2SO, 151 MHz) δ 18.71, 19.12, 19.26, 24.35, 24.40, 25.81, 30.34, 33.34, 36.35, 37.48, 38.34, 40.42, 45.78, 45.08, 49.87, 117.02, 123.94, 124.46, 126.89, 129.26, 131.81, 134.95, 142.47, 145.29, 147.40, 160.88; MS [M + H]+ m/z 475.3262 (calcd for C25H32BrNOS 474.3138). Anal. calcd for C25H32BrNOS: C, 63.28; H, 6.80; N, 2.95; S, 6.76. Found: C, 63.19; H, 6.87; N, 2.83; S, 6.82.
4.2.7. Synthesis of 2-pyrazine-acyl-dehydroabietylamine (L7)
After the condensation reaction of 2-pyrazine-carboxylic acid (0.62 g, 5.0 mmol) and dehydroabietylamine (1.43 g, 5.0 mmol) used HOBT and DCC, which was same as L4, solvents were evaporated to get yellow powders. (1.53 g, 78%), mp: 159.8–162.3 °C; IR (neat) νmax/cm−1 3401, 2929, 2854, 1653, 1524, 1392, 1153, 1016, 873, 819, 620; 1 H–NMR (CDCl3, 600 MHz) δ 1.02 (3 H, s), 1.21–1.23 (9 H, t, J = 7.2 Hz), 1.38–1.41 (1 H, m), 1.51–1.53 (2 H, m), 1.66–1.71(1H, m), 1.77–1.79 (2 H, m), 1.91–2.04 (2 H, m), 2.29 (1 H, d, J = 12.6 Hz), 2.80–2.89 (2 H, m), 2.90–2.95(1H, m), 3.26–3.54 (2 H, dd, J = 7.2 Hz), 6.89 (1 H, s), 6.98 (1 H, dd, J = 8.4 Hz), 7.16 (1 H, d, J = 7.8 Hz), 7.91 (1 H, s, –NH), 8.49 (1 H, d, J = 1.2 Hz), 8.72 (1 H, d, J = 2.4 Hz), 9.39 (1 H, s); 13 C{1H} NMR ((CD3)2SO, 151 MHz) δ 18.43, 18.68, 24.38, 24.44, 25.43, 29.41, 33.36, 35.02, 36.04, 37.40, 37.99, 44.89, 50.15, 123.99, 124.37, 126.79, 134.85, 143.90, 144.75, 145.50, 145.57, 147.26, 151.88, 167.45; MS [M + H]+ m/z 392.2721, [M + Na]+ m/z 414.2539 (calcd for C25H33N3O 391.2624). Anal. calcd for C25H33N3O: C, 76.69; H, 8.49; N, 10.73. Found: C, 76.57; H, 8.56; N, 10.64.
4.2.8. Synthesis of 5-methoxyl-2-pyrazine-acyl-dehydroabietylamine (L8)
A mixture of Dehydroabietylamine (L0) (10.0 mmol), 5-chloro-pyrazine-2-carboxylic acid methyl ester (10.0 mmol) was dissolved in EtOH (150 mL) and refluxed for 24 h. Then the mixture was cooled to the room temperature, removed the solvent by reduced pressure distillation, and received the orange powders. Finally, the orange powders were obtained by recrystallization from ethanol solution. (1.37 g, 65%), mp: 117.6–119.8 °C; IR (neat) νmax/cm−1 3422, 2929, 2868, 1714, 1593, 1433, 1276, 1125, 1016, 825, 620; 1 H–NMR ((CD3)2SO, 600 MHz) δ 0.92 (3 H, s), 1.11–1.21 (9 H, t, J = 7.2 Hz), 1.22–1.26 (2 H, m), 1.38–1.49 (2 H, m), 1.52–1.64 (2 H, m), 1.64–1.72 (1 H, m), 1.88–1.92 (1 H, m), 2.23 (1 H, d, J = 12.0 Hz), 2.63–2.79 (3 H, m), 3.16–3.53 (2 H, m), 3.79 (3 H, s), 6.79 (1 H, s), 6.92 (1 H, dd, J = 7.8 Hz), 7.11 (1 H, d, J = 7.8 Hz), 7.80 (1 H, s, –NH), 8.08 (1 H, s), 8.59 (1 H, s); 13 C{1H} NMR ((CD3)2SO, 151 MHz) δ 14.49, 18.73, 19.00, 19.25, 21.13, 24.31, 25.58, 29.97, 33.35, 36.02, 37.44, 44.35, 50.62, 51.86, 60.18, 123.92, 124.40, 126.82, 129.62, 134.04, 134.78, 145.32, 147.40, 157.09, 165.16; MS [M + H]+ m/z 422.2804, [M + Na]+ m/z 444.2620 (calcd for C26H35N3O2 421.2729). Anal. calcd for C26H35N3O2: C, 74.07; H, 8.37; N, 9.97. Found: C, 74.01; H, 8.46; N, 9.89.
4.2.9. Synthesis of 5–methyl-2-pyrazine-acyl-dehydroabietylamine (L9)
L9 was synthesized according to the method used for L4. Obtained L9 is pale yellow powders. (1.66 g, 82%), mp: 130.7–133.4 °C; IR (neat) νmax/cm−1 3387, 2922, 2854, 1664, 1534, 1439, 1378, 1290, 1167, 1030, 819, 634; 1 H–NMR (CDCl3, 600 MHz) δ 1.02 (3 H, s), 1.21–1.23 (9 H, t, J = 6.6 Hz), 1.38–1.42 (2 H, m), 1.51–1.53 (2 H, m), 1.67–1.73 (1 H, m), 1.70–1.81 (2 H, m),1.97–2.01 (1 H, m), 2.28 (1 H, d, J = 12.0 Hz), 2.63 (3 H, s), 2.81–2.94 (3 H, m), 3.25–3.49 (2 H, dd, J = 7.2 Hz), 6.88 (1 H, s), 6.98 (1 H, dd, J = 1.8 Hz), 7.16 (1 H, d, J = 7.2 Hz), 7.85(1 H, s, –NH), 8.34(1 H, d, J = 3.0 Hz), 9.25 (1 H, d, J = 1.8 Hz); 13 C{1H} NMR (CDCl3, 151 MHz) δ 18.92, 19.12, 21.77, 23.94, 24.92, 25.47, 30.43, 33.91, 36.31, 37.58, 37.83, 38.28, 45.42, 49.75, 123.87, 124.24, 126.95, 134.84, 141.80, 142.22, 143.39, 145.61, 147.07, 156.91, 163.28; MS [M + H]+ m/z 406.2879, [M + Na]+ m/z 428.2698 (calcd for C26H35N3O 405.2780). Anal. calcd for C26H35N3O: C, 77.00; H, 8.70; N, 10.36. Found: C, 76.92; H, 8.62; N, 10.44.
4.2.10. Synthesis of 5-chloro-2-pyrazine-acyl-dehydroabietylamine (L10)
The condensation reaction yielded pale yellow powders, which was synthesized according to the method used for L4. (1.59 g, 75%), mp: 152.4–154.5 °C; IR (neat) νmax/cm−1 3387, 2929, 2860, 1664, 1529, 1447, 1282, 1167, 1125, 1022, 825, 620; 1 H–NMR ((CD3)2SO, 600 MHz) δ 0.90 (3 H, s), 1.12–1.18 (9 H, t, J = 7.2 Hz), 1.32–1.42 (3 H, m), 1.57–1.76 (4 H, m), 2.01–2.04 (1 H, m), 2.24 (1 H, d, J = 13.8 Hz), 2.73–2.81 (3 H, m), 3.09–3.44 (2 H, dd, J = 6.6 Hz), 6.83 (1 H, s), 6.92 (1 H, dd, J = 7.8 Hz), 7.11 (1 H, d, J = 7.8 Hz), 7.12 (1 H, d, J = 8.4 Hz), 7.60 (1 H, d, J = 3.6 Hz), 8.60 (1 H, s, –NH), 8.83 (1 H, s), 9.00 (1 H, s); 13 C{1H} NMR ((CD3)2SO, 151 MHz) δ 18.66, 19.08, 19.20, 24.30, 25.66, 30.18, 33.27, 36.31, 37.53, 38.36, 38.47, 40.62, 45.39, 49.76, 123.94, 124.36, 126.86, 134.91, 143.47, 143.83, 144.14, 145.35, 147.44, 151.13, 162.78; MS [M + Na]+ m/z 448.2146 (calcd for C25H32ClN3O 425.2234). Anal. calcd for C25H32ClN3O: C, 70.49; H, 7.57; N, 9.86. Found: C, 70.37; H, 7.48; N, 9.94.
4.3. Biological assays
4.3.1. Cell culture, antiproliferative activities and cytotoxicity assay
Reagents and compounds Dulbecco’s Modified Eagle’s Medium (DMEM) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), fetal bovine serum (FBS) and penicillin/streptomycin were all commercially purchased.
Hela, HepG2, MCF-7, A549, and HUVEC cells were used in the antiproliferative activity assay. Cancer cells were seeded in 96-well plates with a density of 104 cells per well, after 12 hours of incubation at 5% CO2 and 37 °C, the culture media was removed and cells were incubated with L1−L10 dissolved in DMEM at different concentrations (each concentration repeated 3 times) for 36 h at 5% CO2 and 37 °C. Subsequently, removed the culture media and the new culture medium containing MTT (1 mg/mL) was added, followed by incubating for 4 h to allow the formation of formazan dye (Xia et al., Citation2015; Zhao et al., Citation2018). After removing the medium, 200 μL DMSO was added to each well to dissolve the formazan crystals. Absorbance was measured at 595 nm in a microplate photometer. Cell viability values were determined (at least three times) according to the following formulae: cell viability (%) = the absorbance of experimental group/the absorbance of blank control group ×100%.
4.3.2. Induction of apoptosis by flow-cytometric analysis
Induction apoptosis assay was operated by Becton Dickinson Ultra-high speed separation flow-cytometry instrument and Annexin V-FITC/PI was purchased from Nanjing Keygen Biotech Co. Ltd.
We further investigated whether L1 and L3 could induce apoptosis; DMSO was used as negative control. HepG2 cells (1 × 106) were cultured in 35 mm dishes and incubated at 37 °C for 24 h. After incubation with DMSO at 5 μg/mL, L1 at 1, 2, 5 μg/mL, and L3 at 0.1, 1, 5 μg/mL for 24 h (each concentration repeated 3 times, the incubation time is optimum), the treated cells were washed, trypsinized (non-EDTA), and centrifuged (2000 rpm/min). Then the cells were collected and resuspended in 500 μL of buffer solutions loaded with Annexin V-FITC apoptosis detection reagent (with 5 μL Annexin V-FITC and 5 μL PI). The Annexin V-FITC-stained cells were incubated for 5–15 min in the dark, and approximate 104 cells were collected for flow-cytometry analysis with a single 488 nm argon laser.
4.3.3. In vivo experiment
In vivo experiment was taken by Nanjing Keygen Biotech Co. Ltd. For developing the tumor model, 1 × 106 HepG2 cells were subcutaneously injected into the right armpit of every Balb/C nude mouse. And then two groups of HepG2-tumor-bearing mice with five mice per group were randomly chosen in our experiment: (1) PBS (as a control) and (2) compound L1. After the size of tumors reached 80 mm3, all agents including PBS, compound L1 solutions were administrated via an intravenous injection (dose = 0.6 mg/kg), respectively. During the next 25 days, the tumor size of every mouse in our experiments was measured by a vernier caliper every 3 days. Moreover, to accurately evaluate the growth inhibition of tumors, the mice were sacrificed after 25 days, and then their tumors were collected, photographed, and weighed. In addition, the sections of tumor, heart, kidney, liver, lung, and spleen tissues of different groups harvested on the 25th day were observed using H&E staining, and then examined by a pathologist. The tumor size was calculated as the volume = 0.5 × (tumor length) × (tumor width)2. The inhibition efficiency of tumor growth was calculated according to the following equation:
CD31 immumohistochemical staining with mice was conducted on the 25th day after an intravenous injection of compound L1 (0.6 mg/kg) and PBS control.
Supplemental Material
Download ()Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Ahmed HEA, Ihmaid SK, Omar AM, et al. (2018). Design, synthesis, molecular docking of new lipophilic acetamide derivatives affording potential anticancer and antimicrobial agents. Bioorg Chem 76:332–42.
- Aissi RE, Liu J, Besse S, et al. (2014). Synthesis and biological evaluation of new quinoxaline derivatives of ICF01012 as melanoma-targeting probes. ACS Med Chem Lett 5:468–73.
- Arjmand F, Aziz M. (2009). Synthesis and characterization of dinuclear macrocyclic cobalt(II), copper(II) and zinc(II) complexes derived from 2, 2, 2′, 2′, S, S[bis(bis-N, N-2-thiobenzimidazolyloxalato-1, 2-ethane)]: DNA binding and cleavage studies. Eur J Med Chem 44:834–44.
- Auxiliadora MDA, Pablo BR, Francisco BF, Miguel AG. (2016). Synthesis and antileishmanial activity of C7- and C12-functionalized dehydroabietylamine derivatives. Eur J Med Chem 121:445–50.
- Azuine MA, Tokuda H, Tokuda H, et al. (2004). Cancer chemopreventive effect of phenothiazines and related tri-heterocyclic analogues in the 12-O-tetradecanoylphorbol-13-acetate promoted Epstein-Barr virus early antigen activation and the mouse skin two-stage carcinogenesis models. Pharmacol Res 49:161–9.
- Badrinarayanan S, Sperry J. (2011). Biomimetic synthesis of 2,5-bis(indol-3-ylmethyl)pyrazine via intermolecular amino aldehyde cyclization. Synlett 162:2339–42.
- Bahekar SP, Hande SV, Agrawal NR, et al. (2016). Sulfonamide chalcones: synthesis and in vitro exploration for therapeutic potential Against Brugia Malayi. Eur J Med Chem 124:262–9.
- Dalvie DK, Kalgutkar AS, Khojasteh-Bakht SC, et al. (2002). Biotransformation reactions of five membered aromatic heterocyclic rings. Chem Res Toxicol 15:269–99.
- Dyreborg S, Arvin E, Broholm K. (1996). Effects of creosote compounds on the aerobic bio-degradation of benzene. Biodegradation 7:191–201.
- Fei BL, Yin B, Li DD, Xu WS, et al. (2016). Enantiopure copper(II) complex of natural product rosin derivative: DNA binding, DNA cleavage and cytotoxicity. J Biol Inorg Chem 21:987–96.
- Gao EJ, Wang KH, Zhu MC, Liu L. (2010). Hairpin-shaped tetranuclear palladium(II) complex: synthesis, crystal structure, DNA binding and cytotoxicity activity studies. Eur J Med Chem 45:2784–90.
- Garton AJ, Crew APA, Franklin M, et al. (2006). OSI-930: a novel selective inhibitor of kit and kinase insert domain receptor tyrosine kinases with antitumor activity in mouse Xenograft models. Cancer Res 66:1015–24.
- Ghorab MM, Alsaid MS, Al-Dosari MS, et al. (2016). Novel quinolines carrying pyridine, thienopyridine, isoquinoline, thiazolidine, thiazole and thiophene moieties as potential anticancer agents. Acta Pharm 66:155–71.
- Hana HY, Khalil WKB, Elmakawy AI, Elmegeed GA. (2008). Androgenic profile and genotoxicity evaluation of testosterone propionate and novel synthesized heterocyclic steroids. J Steroid Biochem Mol Biol 110:284–94.
- Hande KR. (1998). Clinical applications of anticancer drugs targeted to topoisomerase II. Biochem Biophys Acta 1400:173–84.
- Huang XC, Huang RZ, Li LX, et al. (2017). Synthesis and biological evaluation of novel chalcone derivatives as a new class of microtubule destabilizing agents. Eur J Med Chem 132:11–25.
- Huang LH, Zheng YF, Song CJ, et al. (2012). Synthesis of novel D-ring fused 70-aryl-androstano[17,16-d][1,2,4] triazolo[1,5-a]pyrimidines. Steroids 77:367–74.
- Ingle R, Marathe R, Magar D, et al. (2013). Sulphonamidoquinoxalines: search for anticancer agent. Eur J Med Chem 65:168–86.
- Iványi Z, Szabó N, Huber J, et al. (2012). Synthesis of dring-substituted (50R)- and (50S) 17b-pyrazolinylandrostene epimers and comparison of their potential anticancer activities. Steroids 77:566–74.
- Khalil AM, Berghot MA, Ghada EA, Gouda MA. (2010). Synthesis and antimicrobial evaluation of some new thiophene derivatives. Eur J Med Chem 40:1658–69.
- Kulandasamy R, Adhikari AV, Stables JP. (2009). A new class of anticonvulsants possessing 6 Hz activity: 3,4-dialkyloxy thiophene bishydrazones. Eur J Med Chem 44:4376–84.
- Lee SB, Park YI, Dong MS, Gong YD. (2010). Identification of 2,3,6-trisubstituted quinoxaline derivatives as a Wnt2/beta-catenin pathway inhibitor in nonsmall-cell lung cancer cell lines. Bioorg Med Chem Lett 20:5900–4.
- Lei M, Miao H, Wang XY, et al. (2019a). Trifluoromethyl aryl sulfonates (TFMS): an applicable trifluoromethoxylation reagent. Tetrahedron Lett 60:1389–92.
- Lei M, Sha SJ, Wang XY, et al. (2019b). Co-delivery of paclitaxel and gemcitabine via a self-assembling nanoparticle for targeted treatment of breast cancer. RSC Adv 9:5512–20.
- Lei M, Zhang HY, Miao H, et al. (2019c). Preparation and biological evaluation of soluble tetrapeptide epoxyketone proteasome inhibitors. Bioorg & Med Chem 27:4151–62.
- Lesyk R, Zimenkovsky B, Atamanyuk D, et al. (2006). Anticancer thiopyrano[2,3-d][1,3]thiazol-2-ones with norbornane moiety. synthesis, cytotoxicity, physico-chemical properties and computational studies. Bioorg Med Chem 14:5230–40.
- Li Z, Wang J, Zhou Y, Liu H. (2014). Lead compound optimization strategy (3)-structure modification strategies for improving water solubility. Acta Pharm Sin 49:1238–47. −
- Li Q, Chu DTW, Claiborne A, et al. (1996). Synthesis and structure-activity relationships of 2-pyridones: a novel series of potent DNA gyrase inhibitors as antibacterial agents. J Med Chem 39:3070–88.
- Lin TT, González MA, Carboni MG, et al. (2015). (+)-Dehydroabietylamine derivatives target triple-negative breast cancer. Eur J Med Chem 102:9–13.
- Lindsley CW, Zhao ZJ, Leister WH, et al. (2005). Discovery of 2,3,5-trisubstituted pyridine derivatives as potent Akt1 and Akt2 dual inhibitors. Bioorg Med Chem Lett 15:761–4.
- Liu CX, Lin ZX, Zhou AM. (2016). Design, synthesis, cytotoxicities and DNA cleavage activities of dibenzoxepine and isoquinoline derivatives starting from dehydroabietylamine. J Asian Nat Prod Res 18:1169–77.
- Liu XY, Zhang RH, Li TQ, et al. (2017). Novel fully biobased benzoxazines from rosin: synthesis and properties. ACS Sustainable Chem Eng 5:10682–92.
- Lu W, Yang SL, Xu L, et al. (2016). Crystal structure and magnetic properties of salicylaldehyde schiff base binuclear copper(II) complex. Chemical Reagents 38:716–20.
- Mathew V, Keshavayya J, Vaidya VP. (2006). Heterocyclic system containing bridgehead nitrogen atom: synthesis and pharmacological activities of some substituted 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles. Eur J Med Chem 41:1048–58.
- Medower C, Wen L, Johnson WW. (2008). Cytochrome P450 oxidation of the thiophene-containing anticancer drug 3-[(Quinolin-4-ylmethyl)-amino]-thiophene-2-carboxylic acid (4-trifluoromethoxy-phenyl)-amide to an electrophilic intermediate. Chem Res Toxicol 21:1570–7.
- Mohareb RM, Al-Omran F. (2012). Reaction of pregnenolone with cyanoacetylhydrazine: novel synthesis of hydrazide-hydrazone, pyrazole, pyridine, thiazole, thiophene derivatives and their cytotoxicity evaluations. Steroids 77:1551–9.
- Mondal R, Ko S, Bao Z. (2010). Fused aromatic thienopyrazines: structure, properties and function. J Mater Chem 20:10568–76.
- Muhammad SR, Bashir S. (2017). Application of various polymers and polymers based techniques used to improve solubility of water soluble drugs: a review. Acta Pol Pharm 74:347–56.
- Nomiya K, Noguchi R, Ohsawa K, et al. (2000). Synthesis, crystal structure and antimicrobial activities of two isomeric Gold(I) complexes with nitrogen containing heterocycle and triphenylphosphine ligands, [Au(L)(PPh3)] (HL = pyrazole and imidazole. J Inorg Biochem 78:363–70. ).
- Petti F, Thelemann A, Kahler J, et al. (2005). Temporal quantitation of mutant kit tyrosine kinase signaling attenuated by a novel thiophene kinase inhibitor OSI-930. Mol. Cancer Ther 4:1186–97.
- Qi S, Guo L, Yan S, et al. (2019). Hypocrellin A-based photodynamic action induces apoptosis in A549 cells through ROS-mediated mitochondrial signaling pathway. Acta Pharm Sin B 9:279–93.
- Rajendiran V, Karthik R, Palaniandavar M, et al. (2007). Mixed-ligand copper(II)-phenolate complexes: effect of coligand on enhanced DNA and protein binding, DNA cleavage, and anticancer activity. Inorg Chem 46:8208–21.
- Rao XP, Huang XZ, He L, et al. (2012). Antitumor activity and structure-activity relationship of diterpenoids with a dehydroabietyl skeleton. Cchts 15:840–4.
- Saito R, Matsumura Y, Suzuki S, Okazaki N. (2010). Intensely blue-fluorescent 2,5-bis(benzoimidazol-2-yl)pyrazine dyes with improved solubility: their synthesis, fluorescent properties, and application as microenvironment polarity probes. Tetrahedron 66:8273–9.
- Seitz LE, Suling WJ, Reynolds RC. (2002). Synthesis and antimycobacterial activity of pyrazine and quinoxaline derivatives. J Med Chem 45:5604–6.
- Shi JC, Wen TF. (1998). Intramolecular hydrogen bond between aromatic hydrogen and coordinated sulfur atom (sp2 C − H⋅⋅⋅S). Chem J Chin Univ 19:1650–3.
- Singh N, Mishra BB, Bajpai S, Singh RK, et al. (2014). Natural product based leads to fight leishmaniasis. Bioorg Med Chem 22:18–45.
- Smits RA, Lim HD, Hanzer A, et al. (2008). Fragment based design of new h4 receptor-ligands with anti-inflammatory properties in vivo. J Med Chem 51:2457–67.
- Soozani A, Keivanloo A, Bakherad M. (2018). One-pot palladium-catalyzed synthesis of functionalized 10H-pyrido [1,2-a]quinoxalin-10-ones under copper-free conditions. Tetrahedron 74:150–6.
- Sridevi CH, Balaji K, Naidu A, Sudhakaran R. (2010). Synthesis of some phenylpyrazolo benzimidazolo quinoxaline derivatives as potent antihistaminic agents. Eur J Chem 7:234–8.
- Sun J, Ye C, Bai E, et al. (2019). Co-delivery nanoparticles of doxorubicin and chloroquine for improving the anti-cancer effect in vitro. Nanotechnology 30:085101–26.
- Vicente E, Duchowicz PR, Castro EA, Monge A. (2009). QSAR analysis for quinoxaline-2-carboxylate1,4-di-N-oxides as anti-mycobacterial agents. J Mol Graph Model 28:28–36.
- Wang YY, He Y, Yang LF, Peng SH, et al. (2016). Synthesis of novel diterpenoid analogs with in-vivo antitumor activity. Eur J Med Chem 120:13–25.
- Wang SW, Konorev EA, Kotamraju S, et al. (2004). Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms: intermediacy of H2O2- and p53-dependent pathways. J Biol Chem 279:25535–43.
- Xia B, Wang B, Zhang WY, Shi JS. (2015). High loading of doxorubicin into styrene terminated porous silicon nanoparticles via p-stacking for cancer treatments in vitro. RSC Adv 5:44660–5.
- Xin JC, Li N, Ma QS, et al. (2018). Synthesis and antitumor activity of 1-Phenyl-4-substituted phthalazine derivatives. Chin J Org Chem 38:451–6.
- Ye D, Zhang Y, Wang F, et al. (2010). Novel thiophene derivatives as PTP1B inhibitors with selectivity and cellular activity. Bioorg Med Chem 18:1773–82.
- Zhao FY, Lu W, Su F, et al. (2018). Synthesis and potential antineoplastic activity of dehydroabietylamine imidazole derivatives. Med Chem Commun 9:2091–9.
- Zhao FY, Wang WF, Lu W, Xu L, et al. (2018). High anticancer potency on tumor cells of dehydroabietylamine Schiff-base derivatives and a copper(II) complex. Eur J Med Chem 146:451–9.
- Zitko J, Dolezal M, Svobodova M, et al. (2011). Synthesis and antimycobacterial properties of N-substituted 6-amino-5-cyanopyrazine-2-carboxamides. Bioorg Med Chem 19:1471–6.