
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The novel curcumin analog C086, previously identified as an oral novel heat shock protein 90 (Hsp90) inhibitor, was found to exhibit anti-hepatoma activity in vitro and in vivo. However, owing to its limited aqueous solubility, the usage of C086 in the clinical application was restricted. This research focused on the increase of the aqueous solubility and bioavailability of C086 via a solid dispersion preparation to improve its accumulation in the liver, which accordingly enhanced anti-hepatoma activity. C086-solid dispersion (C086-SD) was successfully prepared by using solvent evaporation technology. As compared with bulk compound, aqueous solubility obtained with the optimal formulation (C086/PVP k30:1/6 (w/w)) was increased by 1.741 million-fold, and in the following oral administration experiment, bioavailability was found to be improved by an approximately 28-fold relative to C086-Suspension and accumulate preferably in the liver. Accordingly, C086-SD exhibited stronger potent anti-proliferative effects against liver cancer cell line (i.e. HepG2) than pure C086. Moreover, C086-SD was found to have an enhanced anti-hepatoma effect using the orthotopic hepatocellular carcinoma xenograft in BALB/C nude mice. The results above suggested the potential application of C086-SD in the treatment of liver cancer.
Introduction
Hepatocellular carcinoma (HCC) is now considered one of the leading causes of cancer-related deaths all over the world. Even after surgical procedures, the overall 5-year survival rate of HCC patients remains very poor. Adjuvant drug therapy is an effective method to improve the survival of patients with liver cancer. Therefore, it is essential to find effective compounds against liver cancer. Curcumin (a polyphenolic compound derived from turmeric) is proved as a safe and cost-effective natural compound with potential preventive and therapeutic effects on various tumors (e.g. gastric cancer, prostate cancer, breast cancer, and liver cancer) (Kelloff et al., Citation1996). In addition, curcumin was identified as a heat shock protein 90 (Hsp90) inhibitor by our lab (Wu et al., Citation2006), which may be a potential therapeutic agent for multiple oncogene-addicted cancer types (Garcia-Carbonero et al., Citation2013). However, the application of curcumin was limited by its poor bioavailability when orally administered owing to its poor aqueous solubility and rapid system elimination (Anand et al., Citation2007; Yang et al., Citation2007; Liu et al., Citation2016; Ma et al., Citation2019).
To address this issue, our lab has made much effort on curcumin modification (Chen et al., Citation2011; Liu et al., Citation2014; Wu et al., Citation2015; Zheng et al., Citation2016; Fan et al., Citation2017; Ye et al., Citation2017; Fan et al., Citation2018; Wang et al., Citation2019) and formulation development (Huang et al., Citation2008; Wu et al., Citation2011) for improving its pharmacokinetic properties and solubility since 2006. A series of curcumin analogs via using structure-based drug design has been developed. Consequently, their druggability (e.g. affinity with Hsp90, pharmacological effect, pharmacokinetic behaviors, and stability) have also been evaluated. Fortunately, 4-(4-hydroxy-3-methoxyphenyl) curcumin, namely C086 (), was found to be a promising preclinical candidate (Chen et al., Citation2011; Liu et al., Citation2014; Wu et al., Citation2015; Wang et al., Citation2019). This compound showed a higher affinity with Hsp90 in comparison to Hsp90 N terminal inhibitor NVP-AUY922 (Soga et al., Citation2013) and was further proved as a potential Hsp90 inhibitor (Wu et al., Citation2015). C086 exhibited more potent anti-cancer activities (e.g. colon cancer, chronic myeloid leukemia, and lung cancer) in vitro and in vivo than curcumin (Chen et al., Citation2011; Liu et al., Citation2014; Wu et al., Citation2015). Notably, C086 inhibited not only Hsp90 chaperone function but also Bcr-Abl kinase activity; and therefore, C086 overcame imatinib resistance in chronic myelogenous leukemia cells from either mutation or amplification of Abl kinase (Wu et al., Citation2015). Furthermore, C086 inhibited the growth of human leukemia progenitor/stem cells (Wang et al., Citation2019). The preliminary experiments demonstrated that C086 could improve pharmacokinetic characteristics such as enhanced oral bioavailability (17.8-fold), extended terminal elimination half-life (2.72-fold) with respect to curcumin using the same formulation (suspension) in a rat model (data shown in Supplementary Table S1). Despite the potential bioactivities, pilot experiments showed that C086 was found to belong to biopharmaceutics classification system II (BCS II) with its poor aqueous solubility (less than 1 µg/mL). This solubility may lead to incomplete absorption in the gastrointestinal fluids, thereby appearing a low absolute bioavailability (1.32%). Therefore, it is imperative to develop new formulations to enhance the water-solubility and accordingly to improve the bioavailability of C086.
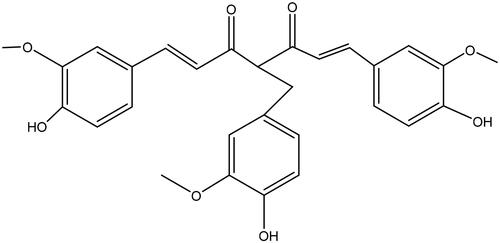
Figure 1. The chemical structure of C086.
Currently, diverse drug delivery systems and formulation strategies (solid dispersion technique, nano-emulsion, complexation, and lipid-based formulations) have been used to tackle the low bioavailability due to the limited solubility (Ghadi & Dand, Citation2017; Williams et al., Citation2013). Among techniques mentioned above, solid dispersion was widely employed to reach supersaturation, which provided a high rate of transport of the drug across a membrane (Wilson et al., Citation2018) and then enhanced oral bioavailability. The solid dispersions have become extensively used to deliver poorly soluble drugs. Recently, the solid dispersion technology has been successfully utilized in 27 commercially available formulations, including three orally dosed oncolytics (vemurafenib, regorafenib, and everolimus) (Sawicki et al., Citation2016; Tran et al., Citation2019). Water-solubility and dissolution for these formulations were tremendously improved compared with their conventional formulations of the respective drugs. For example, the dissolution of vemurafenib solid dispersion (Zelboraf®, Roche) was even 30-fold higher compared to its crude powder. Meanwhile, the solid dispersions can be further formulated into other solid formulations (capsule, granule, and tablet), which provide conveniences for the patients.
The curcumin-solid dispersion (Curcumin-SD) was previously developed by our lab (Huang et al., Citation2008), with increased aqueous solubility (69.2 mg/mL). This formulation enhanced oral bioavailability by 23-fold and increased the maximum plasma (Cmax) by 3-fold in comparison with curcumin suspension (Supplementary Figure S4 and Table S1). Based on the data obtained, it was found that the solid dispersion could be an effective approach to improve the solubility and bioavailability for a curcumin-like molecule (C086). Consequently, in this research, C086-solid dispersion (C086-SD) was prepared and characterized by X-ray powder diffraction (XRPD), differential scanning calorimetry (DSC), Fourier transform infrared (FTIR) spectrometer, in vitro dissolution, and stability study. In order to evaluate the enhancement of solid dispersion technology on the aqueous solubility and bioavailability for C086 (a poorly water-soluble compound), oral bioavailability, and anti-cancer activity of C086 and C086-SD were compared. Moreover, the anti-cancer effect of this formulation on orthotopic hepatocellular carcinoma xenograft model in BALB/c nude mice was evaluated.
Materials and methods
Materials and animals
C086, with a purity: 98.2%, was synthesized by the Institute of Clinical Pharmacology, Fujian Medical University (Fuzhou, China). Curcumin was purchased from Sinopharm Chemical reagent Co.Ltd (China, lot: 20151223). Triptolide was purchased from the National Institutes for Food and Drug Control (purity > 99%, Beijing, China, lot: 111567–200502). Polyvinylpyrrolidone 30 (PVP K30) was purchased from Sigma Chemicals (San Francisco, USA). Acetonitrile and methanol of chromatographic grade were obtained from Fisher Scientific (Toronto, Canada). Ethyl acetate and ethyl tertbutyl ether of the chromatographic grade were provided by Tianjin Concord Science and Technology Co., Ltd., (Tianjin, China) and Merck Co., Inc. (Darmstadt, Germany). Primary antibodies against β-actin, phospho-MEK, and phospho-ERK (p-ERK) were obtained from Cell Signaling Technology. The Annexin V: fluorescein isothiocyanate (FITC). HepG2 was purchased from Shanghai Cell Institute of the Chinese Academy of Sciences. (Shanghai, China). Unless otherwise specified, all materials were of analytical grade.
Male Sprague-Dawley rats (180–230 g) and ICR mice (18–22 g) were obtained from the Experimental Animal Center of the Fujian Medical University, with Permit Numbers SCXK (Min) 2012-0001 and SCXK (Min) 2016-0002, respectively. BALB/C-nude mice (Specific Pathogen Free,18–22 g) were purchased from Shanghai SLAC Laboratory Animal Co., Ltd. [SCXK (hu)2012-0002]. The research adhered to the Principles of Laboratory Animal Care (NIH publication #85-23, revised in 1985). All care and handling of animals were performed following the Institutional Authority for the Fujian Medical University. All animal experiments were evaluated and approved by the Animal and Ethics Review Committee of Faculty of the Fujian Medical University.
Preparation of C086-solid dispersion (C086-SD)
C086-SD with different weight ratios of drug/Polyvinylpyrrolidone (PVP) K30 (1:3, 1:4, 1:6, w/w) was prepared using a solvent evaporation method. In brief, PVP K30 was dissolved in an appropriate amount of ethanol, and C086 was dissolved in acetone. The resultant polymer solution was then added to the drug solution and mixed well. Subsequently, the solvent was removed at 50 °C in a water bath. The solid was dried for 24 hours under vacuum and passed through a 100-mesh sieve (0.149 mm). The obtained C086-SD was stored in a desiccator at room temperature for further characterization.
High-performance liquid chromatography (HPLC) analysis in vitro
The quantification of C086 was conducted using an Agilent HPLC system (1260 InfinityII) with a diode-array detector (Agilent Technologies Inc., USA) set at 440 nm. The mobile phase was 5% acetic acid (v/v) in water/acetonitrile (50/50, v/v). An Eclipse XDB-C18 column (4.6 mm × 250 mm, 5 µm, Agilent Technologies Inc., USA) was used with a flow rate of 1.0 mL/min. The injection volume was 10 μL. The chromatograms were analyzed using ChemStation software. Adequate selectivity was demonstrated in Supplementary Figure S1. Adequate linearity was shown in the concentration range of 0.4 to 100 µg/mL (r = 0.9999). The concentration range showed adequate inter- and intra-day precision (RSD (%) < 2.0).
Solubility, drug loading, dissolution and stability studies
The solubility of C086 was tested for the prepared C086-SD with different weight ratios of (C086: PVP K30;1:3,1:4, and 1:6) and C086 raw powder. Briefly, an excess amount of the samples was added into stoppered conical flasks containing 4 mL ultrapure water. The resulting mixtures were incubated in a water shaker bath with an agitation speed of 100 rpm at 37 ± 0.5 °C for 72 h. The drug loading of C086-SD was calculated based on the actual content of C086 in solid dispersion. Approximately 100 mg of the solid dispersions were weighed into a 10 mL volumetric flask. Ultrapure water was added, vortexed until thoroughly dissolved and then to the desired volume. The samples obtained were filtered (cellulose 0.45 μm syringe filter) and further diluted with the mobile phase and then analyzed by HPLC described above.
In vitro dissolution testing of coarse C086, the prepared C086-SD with different weight ratios of (C086:PVP K30;1:3,1:4, and 1:6) was carried out in 900 mL of distilled water containing 0.1% (w/v) sodium lauryl sulfate (SLS) using the paddle method at 37 ± 0.5 °C, with a rotation speed of 100 rpm in a ZRS-8G dissolution apparatus (Tianjin, China). The amount of each sample was equivalent to 10 mg of C086. At predetermined time intervals (0.5, 2.5, 10, 20, 30, 45, 60 min), l mL of release medium was withdrawn and replenished with fresh medium. The samples were filtered (cellulose 0.45 μm syringe filter) and analyzed by HPLC.
Three batches of C086-SD filled in the hydroxypropyl methylcellulose (HPMC) capsule (size 0) were stored at 25 °C ± 2 °C/60% RH ± 5% RH for 12 months (ICH Q1A (R2) 2003). The C086 assay was analyzed in 0, 1, 2, 3, 6, 9, 12 months. In addition, the stability during storage of C086-SD in normal saline solution at 4 °C and 25 °C was investigated. C086 was then analyzed in 0,12,24, and 48 h.
Physicochemical characterization
Solid state characterization of the obtained solid dispersions and physical mixtures of C086 and PVP K 30 were characterized by XRPD, DSC and FTIR. The tested samples included C086 pure powder, PVP K30, physical mixture (C086: PVP K30;1:6,w/w), and C086-SD (C086:PVP K30;1:6,w/w).
XRPD data were collected using an Empyrean diffractometer (Panalytical, Netherlands) with graphite monochromated Cu Kα radiation (40 kV and 40 mA). Each diffraction pattern was obtained with a step size of 0.02 °C in the 2θ range of 5°–60°.
DSC was performed on the NETZSCH DSC 214 (Gerätebau GmbH, Selb, Germany). The scans were carried out under the nitrogen gas at a flow rate of 20 mL/min, and a heating rate of 20 °C/min was used from 20 to 250 °C.
The FTIR spectra of samples were measured using the Nicolet Avatar 330 FTIR Spectrometer (IS50, ThermoFisher Scientific, USA). The samples were mixed with KBr, ground and pressed into thin slices and scanned in the range of 4000–400 cm−1. For each spectrum, 32 scans were conducted with a resolution of four cm−1. The obtained data were analyzed using the Omnic software (Version 8.2.0.387).
Pharmacokinetic and biodistribution study of C086 in vivo
Liquid chromatography-tandem mass spectrometry (LC-MS) analysis of C086 in vivo
A bioanalytical method for C086 in plasma samples or tissue homogenates was developed to investigate the pharmacokinetic profiles and biodistribution of C086 in vivo. C086 was extracted from either plasma samples or tissue homogenates using ethyl acetate-methyl tert butyl ether by the liquid-liquid extraction method. Triptolide was used as an internal standard (IS). Briefly, the IS solution (10 μg/mL,10 μL) was added into 100 μL of plasma samples or tissue homogenates. Then ethyl acetate-methyl tert butyl ether (1:1, v/v,900 μL) was added into the plasma samples or the tissue homogenate samples and vortex-mixed for 3 min followed by centrifugation at 8,000 rpm (4 °C) for 10 min. The supernatants were transferred to the polypropylene centrifuge tubes and dried using a Martin Christ rotational vacuum concentrator (RVC2-33IR, Gefriertrocknungsanlagen GmbH, Germany) at 40 °C. The dry residues were reconstituted in 100 μL mobile phase (acetonitrile/water: 45/55 (v/v)) following by centrifugation at 15,000 rpm (4 °C) for 10 min. The resulting supernatants (5 μL) were used for LC-MS analysis.
The LC-MS system consisted of an Agilent 1260 Infinity LC system and an Agilent 6410 Triple quadrupole Mass Spectrometer with electrospray ionization (ESI) ion source. Chromatographic separation was performed on an Eclipse XDB-C18 (4.6 × 150 mm, 3.5 μm, Agilent Technologies Inc., USA) through an isocratic mobile phase (acetonitrile/water:45/55 (v/v)) at 30 °C. The flow rate was 0.20 mL/min. The following MS detection parameters were used: 3500 V capillary voltage; 25 psi nebulizer gas pressure (N2); 10 L/min drying gas flow (N2); 320 °C gas temperature. The voltages were set at 120 V for both C086 and triptolide. Detection of ions was conducted in the negative-ion selected reaction monitoring mode with the following transitions in selected-ion monitoring (SIM) mode: m/z 503.2 (C086), and m/z 359.3 (triptolide). The data acquisition was ascertained by MassHunter software Version B.03.01 (Agilent, USA).
The developed LC-MS method was validated for selectivity (Supplementary Figure S2, S3), linearity, accuracy, precision, recovery, matrix effect, and stability following the FDA Guidelines for Industry: Bioanalytical Method Validation (Health U D o, & Services H., Citation2018).
In vivo bioavailability of C086 in rats
In vivo bioavailability of C086 was investigated using a rat model. Briefly, male Sprague-Dawley rats (180–230 g) were randomly assigned to two groups (n = 6 per group) and treated with the following formulation: (1) C086-SD (dose:140 mg/kg); (2) C086-Suspension (dose:450 mg/kg). C086-Suspension was prepared by dispersing the coarse drug powder in carboxymethylcellulose sodium (0.5%, w/v). C086-SD was dissolved in normal saline solution (0.9%,w/v).At predetermined time points (0.167, 0.417, 0.75, 1, 1.5, 2.0, 2.5, 4.0, 6.0, and 8.0 h) , blood samples were collected by retro-orbital plexus puncture into heparinized tubes following oral administration. The blood was centrifuged at 8,000 rpm for 5 min to obtain plasma. The plasma samples were stored at −80 °C until assay. The plasma concentration of C086 was determined by the LC-MS method described above. All pharmacokinetic parameters were analyzed using the non-compartmental method by DAS V3.2.1 software. The results were expressed as a mean ± standard deviation. The relative bioavailability was calculated by EquationEquation (1)(1)
(1) .
(1)
(1)
Frel and D represent relative bioavailability and dose, respectively. Reference and test represent reference formulation and test formulation, respectively. AUC0-t represents the area under the plasma concentration-time curve from time zero to t of C086.
Biodistribution of C086-SD in mice
Drug biodistribution of C086-SD was conducted in mice. Twenty-one ICR mice were randomly divided into seven groups, with three mice in each group. Mice were intragastrically administered with C086-SD (dose:140 mg/kg). All animals were sacrificed at predetermined time points (1.5, 3, 4, 5, 6, 7, 8 h) by removal of circulating blood from organs before organ collection. Organs, including heart, liver, lung, kidney, and spleen, were collected and then washed with phosphate-buffered saline. These samples were drained by filter papers, weighed, and then stored at −80 °C until further analysis. Before analysis, tissue samples were thawed to room temperature and homogenized by adding appropriate normal saline at the ratio of 1:4 (w/v). The concentration of C086 was determined using the LC-MS method described above.
Anti-hepatoma effect of C086 in vitro and in vivo
Anti-proliferation assay
Anti-proliferative effects of C086 were tested using the HCC cell line (HepG2). Briefly, cells were plated in 96-well plates at a density of 40,000 viable cells per well and then treated with a series of doses of C086 solution (0.1% (v/v) dimethyl sulfoxide, DMSO) and C086-SD at the concentration of 1.7, 3.4, 6.8, 13.6, 20.4, and 27.2 μmol/L. The control groups were treated with PVP K30 at a concentration that was equivalent to C086-SD, as well as DMSO at a concentration equal to the C086 solution. Cell viability was evaluated with a methyl thiazol tetrazolium (MTT) assay (Beyotime Biotechnology, Shanghai) according to the manufacturer's instructions. The absorbance of the plate was measured at 570 nm using a Bio-Rad microplate reader 450 (Hercules, CA, USA). The cell inhibition rate was calculated based on EquationEquation (2)(2)
(2) . Half maximal inhibitory concentration (IC50) was calculated by Graphpad Prism (Version 7.0).
(2)
(2)
Orthotopic hepatocellular carcinoma xenograft in BALB/C nude mice
HepG2 cells (1 × 107/mL cells suspension) in 50 μL Dulbecco's modified eagle medium (DEME) containing 50% Matrigel were injected into the left lobe of the liver in BALB/C nude mice (Feng et al., Citation2011; Reiberger et al., Citation2015). After one week of inoculation, the tumor-bearing mice were randomly divided into three groups (n = 6 for each group). The tumor-bearing mice were intragastrically administered with: (1) normal saline (Control); (2) C086-Suspension (dose:100 mg/kg);and (3)C086-SD (dose:100 mg/kg), once a day for seven consecutive days. The body weight was recorded every other day. At the end of the experiments, mice were weighed and then sacrificed, the liver tumors were excised carefully and weighted. After tumor suppression study, the tumor tissues from different groups were collected for further analysis.
Western blotting analysis
HepG2 cells and whole cells were collected, and then the whole-cell lysates were prepared by using radioimmunoprecipitation (RIPA) lysis buffer. Tumor tissues (about 100 mg per mouse) were minced and homogenized on an ice bath in 0.5 mL of ice-cold RIPA lysis buffer. The detailed procedure was carried out as previously described (Chen et al., Citation2011).
Statistical analysis
Statistical analysis of the data was performed with ANOVA and unpaired student t-test with p < .05 as the minimal level of significance.
Results and discussion
Physiochemical characterization of C086-SD
Solid dispersion can be performed by several process methods such as solvent evaporation (most widely used), melting, precipitation, and electro-spinning. Practically, the solvent-evaporation and melting method are extensively and easily scaled-up in industrial products. PVP K30 is generally recognized as safe and has strong hydrophilic properties and physiological tolerance. Therefore, PVP K30 is preferentially selected as the carrier because of its extensive usage in the development of solid dispersions (Sethia & Squillante, Citation2004; Sharma & Jain, Citation2010).
Solubility, dissolution, drug loading and stability
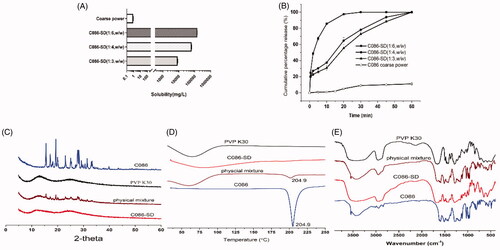
The solubility of C086 in the prepared C086-SD with three mass ratios (C086: PVP K30;1:3, 1:4, 1:6, w/w) was approximately 8.57, 64.01, and 143.65 mg/mL, respectively (). C086-SD obtained (C086: PVP K30;1:6,w/w) displayed a 1.741 × 105-fold increase in solubility compared with free C086. The dissolution profile was plotted on cumulative percentage release (%) vs time (min). As shown in , the cumulative release of C086-SD is approximately 100% in 20 mins, whereas the release of unformulated C086 was low with less than 10% within 60 mins, revealing that C086-SD increased the dissolution rate by 17-fold in comparison with free C086.The solubility and dissolution rate of C086 was greatly enhanced with the increasing proportion of PVP K30 in the solid dispersion formulations. These results were consistent with the previous literature (Kaewnopparat et al., Citation2009). Finally, C086-SD (C086: PVP K30;1:6, w/w) was chosen in further study, and the drug loading of C086 in this optimum formulation was 15.02 ± 0.98% (w/w).
Figure 2. Physicochemical characterization of C086-SD. (A) Aqueous solubility. (B) Dissolution. (C) XRPD patterns, (D) DSC thermograms, and (E) FTIR spectra of pure C086, PVP K30, physical mixture (C086/PVP K30:1/6 (w/w)), and C086-SD (C086/PVP K30:1/6 (w/w)).
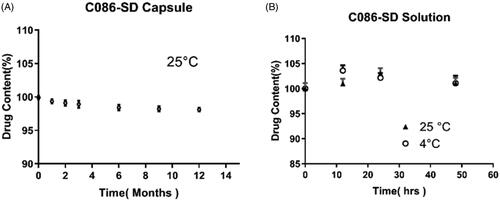
As well recognized, this solid dispersion was sensitive to temperature and humidity during storage on account of the hygroscopicity of PVP K30 and this solid dispersion's thermodynamic instability. The stability of these formulations based on the solid dispersions can be enhanced by using the HPMC capsule due to featured characteristics (low water content). Therefore, the HPMC capsule (0) was used for the prepared C086-SD (Al-Tabakha, Citation2010). For the C086-SD capsule, no obvious change was observed during 12-month storage (), suggesting that the HPMC capsule was a good promising form for these formulations developed on the solid dispersion. Likewise, the C086-SD solution in normal saline was stable for 48 hours at 4 °C and 25 °C with no significant change in the assay, with RSD(%) 3.04% and 3.38%, respectively (). This formulation provided the basis for drug preparation and storage in pharmacokinetics and pharmacology experiments in vivo.
Figure 3. The stability of C086-SD. (A) Long-term stability of C086-SD Capsule at 25 °C ± 2 °C/60% RH ± 5% RH for 12 months. (B) Short-term stability of C086-SD in normal saline at 4 °C and 25 °C for 48 hours.
Physiochemical characterization of C086-SD
XRPD, DSC and FTIR were utilized to characterize the solid state of C086 in the solid dispersions obtained (C086-SD (C086/PVP K30:1/6 (w/w)) and the corresponding physical mixtures.
In the XRPD patterns (), the featured crystal diffraction peaks of C086 appeared at a diffraction angle of 2θ at 15°,19°,27°, etc., presenting a crystalline form, while PVP K30 is an amorphous powder having no crystalline structure. These crystalline peaks attributable to C086 have completely disappeared in C086-SD, hinting that C086 was dispersed homogeneously in PVP K30 in the amorphous or molecular states. As for the corresponding physical mixture, various characteristic crystal peaks of C086 could be traced with low intensity, and no new peaks were observed. The result suggests the solid state of C086 was not changed and no chemical interaction between the drug and the carrier existed.
In the DSC thermograms (), the single sharp endothermic peak of C086 was at 204.9 °C, suggesting the crystal melting point. There was a broad endotherm peak ranging from 45 to 90 °C for amorphous PVP K30 because of the presence of residual moisture. The characteristic melting peak of C086 was almost nonexistent in the C086-SD, whereas the physical mixture exhibited an endothermic peak at 204.9 °C, which was lower than that of pure C086.The results indicated that the C086 crystal was transformed into the amorphous state in the solid dispersion formulation.
The FTIR spectra () were carried out to elucidate further the interaction between C086 and PVP K30 in the solid state. As for C086, the peak 3,545 cm−1 indicated the presence of phenolic OH. The sharp absorption bands at 1,591 cm−1 (stretching vibrations of the benzene ring), 1,617,1,510 cm−1 (C = O and C = C vibration), 1,429 cm−1 (olefinic C–H bending vibration), 1,285 cm−1 (aromatic C–O stretching vibrations), and 1,031 cm−1 (C–O–C stretching vibrations) were noted. The characteristic absorption peaks of PVP K30 around 2953 cm−1,1655 cm−1 and 1290 cm−1 were attributed to C–H, C = O, and C–N stretching vibration. The FTIR spectra of the corresponding physical mixtures were similar to the synthetic spectra producing by the addition of C086 and PVP K30, indicating the absence of any drug-carrier chemical interaction. This result was consistent with the results of XRPD. In particular, the phenolic O-H stretching vibration at 3545 cm−1 of C086-SD disappeared, revealing the intermolecular hydrogen bonding between C086 and PVP K30. The possible reason is that PVP K30 and C086 form hydrogen bonds at the hydroxyl position of phenol, and the carbonyl group of PVP K30 forms positive and negative ionic bonds with the C086 bicarbonyl group potentially (He et al., Citation2019). Therefore, this hydrogen bonding might prevent the association of C086 molecules to form the crystal nucleus during the preparation of solid dispersion, thereby inhibiting the crystal growth (Sekikawa et al., Citation1978; Cong et al., Citation2014).
In a word, according to these results of XRPD, DSC, FTIR, the solid state of C086 appeared to be no changed in the physical mixture, revealing that C086 was compatible with PVP K30.C086 (C086-SD) was uniformly dispersed into the PVP K30 carriers in the amorphous form, leading to the enhanced drug solubility and dissolution rate.
Pharmacokinetic and biodistribution study of C086 in vivo
Liquid chromatography-tandem mass spectrometry (LC-MS) analysis of C086 in vivo
The validated bioanalytical method for the quantitative evaluation of C086 in plasma or tissue homogenate is critical for pharmacokinetic studies in vivo. Based on the previous results (data not shown), C086 was extracted from plasma or tissue homogenates using liquid-liquid extraction according to the sensitivity of C086 quantification via LC-MS. Representative chromatograms of rat plasma samples (i.e. Blank rat plasma, blank rat plasma spiked with triptolide (1) and C086 (2), and actual samples) and mice tissue homogenates (e.g. lung) were shown in Supplementary Figure S2 and S3, respectively. No interference peaks were observed in either plasma samples or tissue homogenates, suggesting that the assay was free of potentially interfering substances, which included endogenous matrix components, metabolites, and anticipated concomitant medications. Adequate linearity was obtained for both plasma and tissue homogenate calibration curves (r > 0.99). The mean matrix effects and extraction recoveries of C086 at three levels of the low, medium, and high concentrations in both plasma and tissue samples were within the acceptable criteria. All stability conditions tested (i.e. room temperature for 24 hours, in three freeze-thaw cycles, and storage at −80 °C for 15 days) had an acceptable criterion of relative error difference (<15%). All these results indicated that the developed LC-MS method was reliable for the quantitative analysis of C086 pharmacokinetics and biodistribution in vivo.
In vivo bioavailability of C086 in rats
Pharmacokinetic studies of C086-SD and C086-Suspension were conducted in rats to investigate whether the solid dispersion could improve oral bioavailability of C086 in vivo. The plasma concentration-time curve and primary pharmacokinetic parameters C086-SD and C086-Suspension were given in and , respectively. The values of Cmax and the area under the curve (AUC) were normalized by dividing the value of the dose (AUC/D and Cmax/D, respectively). As listed in , normalized AUC0-t of C086-SD (46.357 ± 3.736 g/L·h) was approximately 29 times (p < .05) as high as that of the C086-Suspension (1.558 ± 0.178 g/L·h), suggesting the relative bioavailability of C086-SD was approximately 2976%. Moreover, C086-SD demonstrated a 47-fold increase in normalized Cmax concentration and a 2-fold increase in time to maximum plasma concentration observed (Tmax) respectively compared with unformulated C086. These results suggested that the solid dispersion formulation could greatly improve oral bioavailability. There were the following reasons: (1) PVP K30 as a carrier could prolong the circulation lifetime in the body (Kaneda et al., Citation2004); (2) Solid dispersions could increase the dissolution rate and the adhesiveness of gut lumen, resulting in rapid absorption of the C086 molecules across the gastrointestinal membrane (Chen et al., Citation2018). Interestingly, the mean residence time (MRT0-∞) and terminal elimination half-life (T1/2) of C086-Suspension appeared to be approximately 3.2 times and 2.5 times as much as that of C086-SD, respectively. These results might be explained that the native C086 powder in C086-Suspension could not dissolve in the gastrointestinal tract, hence prolonging the gastric residence and extent of the release. The extended time in the body might be one reason that C086-Suspension (dose:100 mg/kg) could exhibit pharmacological effects because of adequate drug plasma levels sustained. In a word, C086-SD significantly improved bioavailability.
Figure 4. Mean plasma concentration-time curves of C086-Suspension and C086-SD following oral administration in rats (n = 6).
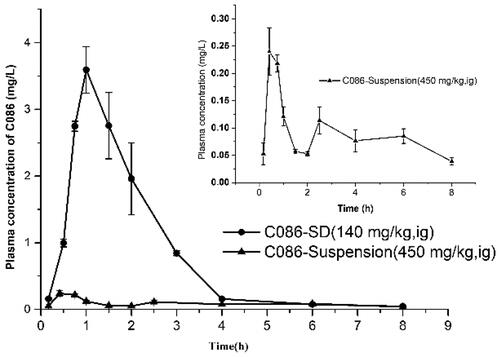
Table 1. Pharmacokinetic parameters of C086 following oral administration in rats (Mean ± SD, n = 6).
Biodistribution of C086-SD in mice
To better identify target organs and anticipate the safety and efficacy of C086-SD, drug biodistribution of C086-SD were investigated in ICR mice. The C086 tissue concentrations of the prepared C086-SD following oral administration were shown in , suggesting that C086 underwent a wide distribution to the entire desired tissues. Of all these tissues, the liver, heart, and lung reached a peak in three hours, whereas spleen and kidney in five hours. Interestingly, there appeared a steep increase in the liver after 6 hours, and the liver maintained the level of C086 (>0.546 µg/g) for 8 hours, which explained this formulation might preferably be accumulated in the liver. AUC0-t in the liver was the highest distribution, and the rank order of AUC0-t in the other tissues was heart > lung > spleen > kidney (). As seen from , MRT0-t in each tissue was between 3.67 and 4.20 h (p > .05). This result was consistent with the MRT0-t obtained from in vivo bioavailability. To elucidate the selectivity of the formulation to the tissues, targeting efficiency (T) was calculated according to EquationEquation (3)(3)
(3) : where T and NT represent target tissue and not target tissue, respectively, AUC0-t is the area under tissue concentration-time curve from time zero to t of C086. The value of T in the liver reached up to 1.872 (>1), while the value of T in the heart, spleen, kidney, and lung was less than 1, revealing that C086-SD mainly accumulated in the liver (). It may be speculated that this formulation might be potential use as a therapeutic agent against liver cancer. Thus, the inhibitory effects of liver cancer needed to be further investigated.
(3)
(3)
Figure 5. The tissue distribution of C086-SD following a single oral administration (140 mg/kg) using as a mice model (n = 3). (A) C086 concentration (µg/g) in key tissues at different times. (B) AUC0-t (µg/g*h) in key tissues. (C) MRT0-t (h) in key tissues.
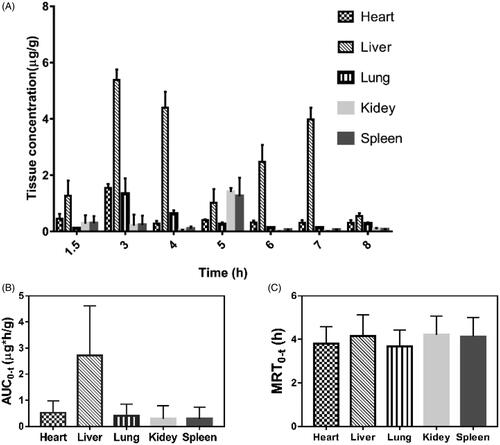
Table 2. The targeting efficiency of C086-SD following oral administration in mice (Mean ± SD, n = 3).
T and NT represent target tissue and not target tissue, respectively. AUC0-t represents the area under the tissue concentration-time curve from time zero to t of C086.
Anti-hepatoma effect of C086 in vitro and in vivo
Given that C086-SD had high exposure in the liver, the potential therapeutic effect of C086 on liver cancer was explored by conducting the anti-hepatoma effect of C086 via the HCC cell line (HepG2). The IC50 of C086-SD and C086 was 7.82 μmol/L and 6.74 μmol/L, respectively, suggesting C086-SD had a more potent inhibition on HepG2 cells than that of C086 (). Since HepG2 was sensitive to C086, and the distribution of this compound was high in the liver, an orthotopic hepatocellular carcinoma xenograft model by using orthotopic liver injection HepG2 cells in BALB/c nude mice was established to investigate the anti-hepatoma activity in vivo. At the end of the experiment, excised tumors were weighed and photographed (). The tumor inhibition rate was calculated based on Equation (4). As seen from , the tumor inhibition rate of C086-SD and C086-Suspension was 56.25% and 39.06%, respectively as compared with the control group. Moreover, all animals in C086 treatment groups (C086-Suspension and C086-SD) survived without appreciable weight loss during the experiment (p > .05, data not shown). These results suggested that C086-SD showed stronger anti-hepatoma effect than C086-Suspension on the orthotopic hepatoma xenograft model in vivo without observed adverse effect.
(4)
(4)
Figure 6. The anti-proliferative effects of C086 in vitro (n = 3). (A) The inhibition rate in HepG2 cells treated with indicated concentrations of C086-SD and C086 for 48 h and then assessed by an MTT assay, *p < .05 vs C086 at the same concentration. (B) The relative expression of phospho-MEK (p-MEK), MEK, phospho-ERK (p-ERK) and ERK in HepG2 cells were detected by using Western Blotting. (C) Histogram of the relative expression of proteins (density ratio of protein/β-actin): *p < .05 vs either control or C086.
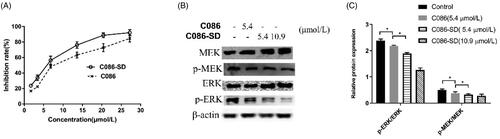
Figure 7. In vivo anti-cancer evaluation effect in the orthotopic hepatocellular carcinoma xenograft in BALB/C nude mice. (A) Photograph of HepG2 orthotopic tumors harvested on the last day of the experiment from nude mice (six per each group). (B) The weight of the harvested tumor: **p < .01 vs control and *p < .05 vs C086. (C) The expression of phospho-MEK (p-MEK) and phospho-ERK (p-ERK) in tumor tissue were detected by using Western Blotting. (D) Histogram of the relative expression of proteins (density ratio of protein/β-actin): *p < .05 and **p < .01 vs either control or C086. C086 = C086-Suspension.
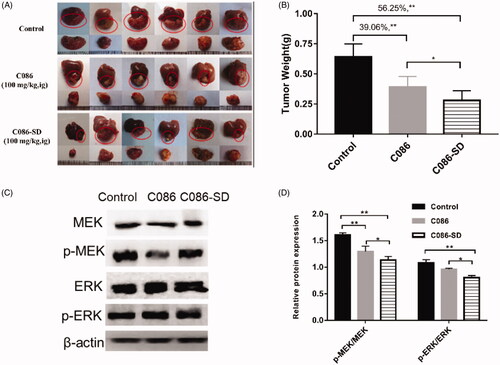
To further understand the underlying mechanism of the anti-hepatoma effect of C086, the proliferation effect associated with HCC was investigated. Also, one of the primary signaling pathway that promoted proliferation and apoptosis-resistance of HCC was the Raf/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) cascade (Samatar & Poulikakos, Citation2014; Yu et al., Citation2015; Yang & Liu, Citation2017). Raf was a client protein for Hsp90, and in turn, Hsp90 inhibitors (C086) could down-regulate the Raf/MEK/ERK pathway by the degradation of the client proteins Raf (Dou et al., Citation2005). Accordingly, the ERK and MER protein levels, as well as their phosphorylated forms (phosphorylated-ERK (p-ERK)) and phosphorylated-MER (p-MEK)), were determined using western blot analysis in HepG2 cells and tumor tissue. In the HepG2 cells and HepG2-derived tumor obtained in orthotopic hepatoma xenograft mice, western blot analysis showed that the total MEK, ERK, and their phosphorylated forms were significantly reduced in the C086-treated groups compared with the control group ( and ). Importantly, C086-SD (either cell line or tumor) appeared to have a more significant down-regulation effect on the activated MEK and ERK relative to C086 (p < .05) ( and ), as well as C086-SD-treated in HepG2 cells demonstrated a concentration-dependent manner ().In a word, the results above revealed that C086 was able to inhibit the Raf/MEK/ERK pathway in either HepG2-derived tumors or HepG2 cell line and accordingly could inhibit the proliferation in the HCC and showed anti-hepatoma activity. Therefore, it was consistent with our lab's previous results that C086 was proved to be an Hsp90 inhibitor.
Collectively, C086-SD was proved to effectively improve the anti-hepatoma effect of C086 in vitro and in vivo, suggesting that the solid dispersion formulation improved the solubility and transmembrane transport, increased the bioavailability of C086, and in turn enhanced its anti-hepatoma effect. The present results indicate that C086-SD had a potential application in the treatment of liver cancer.
Conclusions
In this study, solid dispersion technology was used to prepare C086-SD. The results showed that, compared with C086 alone, C086-SD improved the solubility of C086 by 1.741 × 105-fold and bioavailability by 28-fold, and in turn, promoted the preferential accumulation of C086 in the liver. Excitingly, for the first time, it was found that C086-SD enhanced the anti-cancer effect on orthotopic human hepatoma xenograft model in nude mice, and its anti-hepatoma effect may be related to its inhibition of Raf/MEK/ERK pathway, thus inhibiting the proliferation of HCC cells. In conclusion, this study shows that solid dispersion technology effectively improved the oral bioavailability of C086 and its anti-hepatoma activity in vivo, and C086-SD has potential applications in the treatment of liver cancer.
Supplemental Material
Download ()Acknowledgements
We would like to thank Dr. Joan Lausier for doing the proofreading for this manuscript. We also thank the public technology center of Fujian Medical University, for providing liquid chromatography-mass spectrometry instruments.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Related Research Data
References
- Al-Tabakha MM. (2010). HPMC capsules: current status and future prospects. J Pharm Pharm Sci 13:428–42.
- Anand P, Kunnumakkara AB, Newman RA, et al. (2007). Bioavailability of curcumin: problems and promises. Mol Pharm 4:807–18.
- Chen C, Liu Y, Chen Y, et al. (2011). C086, a novel analog of curcumin, induces growth inhibition and down-regulation of NFκB in colon cancer cells and xenograft tumors. Cancer Biol Ther 12:797–807.
- Chen Y, Liu Y, Xu J, et al. (2018). Design and evaluation of nanocomposite microparticles to enhance dissolution and oral bioavailability of andrographolide. Powder Technol 323:219–29.
- Cong W, Shen L, Xu D, et al. (2014). Solid dispersion tablets of breviscapine with polyvinylpyrrolidone K30 for improved dissolution and bioavailability to commercial breviscapine tablets in beagle dogs. Eur J Drug Metab Pharmacokinet 39:203–10.
- Dou F, Yuan LD, Zhu JJ. (2005). Heat shock protein 90 indirectly regulates ERK activity by affecting Raf protein metabolism. Acta Biochim Biophys Sin (Shanghai) 37:501–5.
- Fan YJ, Liu Y, Zhang LR, et al. (2017). C0818, a novel curcumin derivative, interacts with Hsp90 and inhibits Hsp90 ATPase activity. Acta Pharm Sin B 7:91–6.
- Fan YJ, Zhou YX, Zhang LR, et al. (2018). C1206, a novel curcumin derivative, potently inhibits Hsp90 and human chronic myeloid leukemia cells in vitro. Acta Pharmacol Sin 39:649–58.
- Feng YX, Wang T, Deng YZ, et al. (2011). Sorafenib suppresses postsurgical recurrence and metastasis of hepatocellular carcinoma in an orthotopic mouse model. Hepatology 53:483–92.
- Garcia-Carbonero R, Carnero A, Paz-Ares L. (2013). Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol 14:E358–E369.
- Ghadi R, Dand N. (2017). BCS class IV drugs: highly notorious candidates for formulation development. J Control Release 248:71–95.
- He Y, Liu H, Bian W, et al. (2019). Molecular interactions for the curcumin-polymer complex with enhanced anti-inflammatory effects. Pharmaceutics 11:442.
- Health U D o, & Services H. (2018). Bioanalytical method validation, guidance for industry. Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry.
- Huang X, Xu J, Wen C. (2008). Study on preparation and in vitro dissolution of curcumin-plasdone solid dispersion. Chin J Hosp Pharm 21:1819–1822.
- Kaewnopparat N, Kaewnopparat S, Jangwang A, et al. (2009). Increased Solubility, Dissolution and Physicochemical Studies of Curcumin-Polyvinylpyrrolidone K-30 Solid Dispersions. World Acad Sci Eng Technol 55:229–34.
- Kaneda Y, Tsutsumi Y, Yoshioka Y, et al. (2004). The use of PVP as a polymeric carrier to improve the plasma half-life of drugs. Biomaterials 25:3259–66.
- Kelloff GJ, Crowell JA, Hawk ET, et al. (1996). Strategy and planning for chemopreventive drug development: clinical development plans II. J Cell Biochem Suppl 26:54–71.
- Liu W, Zhai Y, Heng X, et al. (2016). Oral bioavailability of curcumin: problems and advancements. J Drug Target 24:694–702.
- Liu Y, Ye M, Wu Q, et al. (2014). Synthesis and evaluation of 4-arylmethyl curcumin analgues as potent Hsp90 inhibitors. Lett Drug Des Discov 11:993–9.
- Ma Z, Wang N, He H, et al. (2019). Pharmaceutical strategies of improving oral systemic bioavailability of curcumin for clinical application. J Control Release 316:359–80.
- Reiberger T, Chen Y, Ramjiawan RR, et al. (2015). An orthotopic mouse model of hepatocellular carcinoma with underlying liver cirrhosis. Nat Protoc 10:1264–74.
- Samatar AA, Poulikakos PI. (2014). Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 13:928–42.
- Sawicki E, Schellens JH, Beijnen JH, et al. (2016). Inventory of oral anticancer agents: pharmaceutical formulation aspects with focus on the solid dispersion technique. Cancer Treat Rev 50:247–63.
- Sekikawa H, Nakano M, Arita T. (1978). Inhibitory effect of polyvinylpyrrolidone on the crystallization of drugs. Chem Pharm Bull 26:118–26.
- Sethia S, Squillante E. (2004). Solid dispersion of carbamazepine in PVP K30 by conventional solvent evaporation and supercritical methods. Int J Pharm 272:1–10.
- Sharma A, Jain CP. (2010). Preparation and characterization of solid dispersions of carvedilol with PVP K30. Res Pharm Sci 5:49–56.
- Soga S, Akinaga S, Shiotsu Y. (2013). Hsp90 inhibitors as anti-cancer agents, from basic discoveries to clinical development. Curr Pharm Des 19:366–76.
- Tran P, Pyo YC, Kim DH, et al. (2019). Overview of the manufacturing methods of solid dispersion technology for improving the solubility of poorly water-soluble drugs and application to anticancer drugs. Pharmaceutics 11:132.
- Wang L, Fan Y, Mei H, et al. (2019). Novel Hsp90 inhibitor C086 potently inhibits non-small cell lung cancer cells as a single agent or in combination with Gefitinib. Cancer Manag Res Volume 11:8937–45.
- Williams HD, Trevaskis NL, Charman SA, et al. (2013). Strategies to address low drug solubility in discovery and development. Pharmacol Rev 65:315–499.
- Wilson V, Lou X, Osterling DJ, et al. (2018). Relationship between amorphous solid dispersion in vivo absorption and in vitro dissolution: phase behavior during dissolution, speciation, and membrane mass transport. J Control Release 292:172–82.
- Wu L, Xu J, Huang X, et al. (2006). Down-regulation of p210 (bcr/abl) by curcumin involves disrupting molecular chaperone functions of Hsp90. Acta Pharmacol Sin 27:694–9.
- Wu L, Yu J, Chen R, et al. (2015). Dual inhibition of Bcr-Abl and Hsp90 by C086 potently inhibits the proliferation of imatinib-resistant CML cells. Clin Cancer Res 21:833–43.
- Wu X, Xu J, Huang X, et al. (2011). Self-microemulsifying drug delivery system improves curcumin dissolution and bioavailability. Drug Dev Ind Pharm 37:15–23.
- Yang K-Y, Lin L-C, Tseng T-Y, et al. (2007). Oral bioavailability of curcumin in rat and the herbal analysis from Curcuma longa by LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci 853:183–9.
- Yang S, Liu G. (2017). Targeting the Ras/Raf/MEK/ERK pathway in hepatocellular carcinoma. Oncol Lett 13:1041–7.
- Ye M, Huang W, Wu WW, et al. (2017). FM807, a curcumin analogue, shows potent antitumor effects in nasopharyngeal carcinoma cells by heat shock protein 90 inhibition. Oncotarget 8:15364–76.
- Yu ZT, Ye SQ, Hu GY, et al. (2015). The RAF-MEK-ERK pathway: targeting ERK to overcome obstacles to effective cancer therapy. Future Med Chem 7:269–89.
- Zheng B, Yang L, Wen C, et al. (2016). Curcumin analog L3 alleviates diabetic atherosclerosis by multiple effects. Eur J Pharmacol 775:22–34.