
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Magnolol, known to have extensive biological activities, is the major bioactive ingredient isolated from the root and stem bark of Magnolia officinalis. However, the clinical application of magnolol is limited by poor aqueous solubility and absorption. The aim of this study is to develop novel mixed micelles and nanosuspensions composed of two biocompatible copolymers, Soluplus® and Poloxamer 188, and to improve the solubility and oral bioavailability of magnolol. The magnolol-loaded mixed micelles (MMs) and magnolol nanosuspensions (MNs) were prepared to use film hydration and antisolvent methods, respectively. The optimal MMs and MNs formulations were prepared to use magnolol, Soluplus®, and Poloxamer 188 in ratios of 1:12:5 and 2:1:1, respectively. The average particle size of MMs was 111.8 ± 14.6, and MNs was 78.53 ± 5.4 nm. The entrapment and drug loading efficiency for MMs were 89.58 ± 2.54% and 5.46 ± 0.65%, correspondingly. The drug loading efficiency of MNs was 42.50 ± 1.57%. In the in vitro release study, MMs showed a slow drug release while that of MNs was fast. The results of the Caco-2 transcellular transport study indicated that both MMs and MNs increased the permeation of magnolol. MMs and MNs markedly promoted gastrointestinal drug absorption by 2.85 and 2.27-fold, respectively, as shown in the pharmacokinetics study. These results indicated that both MMs and MNs formulations prepared with Soluplus® and Poloxamer 188 are promising drug delivery systems for improving the oral absorption of insoluble drugs in the gastrointestinal tract.
1. Introduction
Magnolol, the major bioactive ingredient of Magnolia officinalis, is known to have various activities both in vitro and in vivo, including antioxidant, anti-inflammatory, anticancer, and antidepressant activities. It affects intramolecular hydrogen by hindering hydrogen abstraction from the hydroxyl group, which normally reduces ONOO− and O2 and has antioxidant effects (Chen et al., Citation2011; Zhao & Liu, Citation2011). Magnolol exerts anti-inflammatory properties by downregulating the expression of Toll-like receptor 4 (TLR4), which is upregulated by lipopolysaccharide (LPS). This effect attenuates the TLR4-mediated activation of nuclear factor (NF)-κB and mitogen-activated protein kinase (MAPK) signaling as well as the release of pro-inflammatory cytokines (Fu et al., 2013). Further, magnolol treatment has been shown to induce apoptosis, to suppress cell proliferation through cell cycle arrest and to facilitate pro-apoptotic molecules in human glioma cells (Chen et al., Citation2009; Liang et al., Citation2014; Tsai et al., Citation2020). Recent studies (Zhang et al., Citation2017a) have shown that magnolol also induces autophagy and inhibits the migration and invasion of human PC3 cells. Furthermore, magnolol enhances both phasic and tonic GABAergic neurotransmission in hippocampal dentate granule neurons, which suggests that it may be an effective anxiolytic, sedative, and anticonvulsant drug (Alexeev et al., Citation2012). Unfortunately, poor aqueous solubility and absorption are two major limiting factors in the clinical use of magnolol (Tsai et al., Citation2016; Lee et al., Citation2017). Therefore, developing a novel magnolol formulation with improved oral bioavailability may enhance its therapeutic effects.
Nano-drug carrier systems, including liposomes, micelles, and nanoparticles, can be used to enhance the unfavorable solubility and oral bioavailability of hydrophobic drugs (Zhang et al., Citation2013, Citation2015a; Deng et al., Citation2015; Nekkanti et al., Citation2016; Ma et al., Citation2017). The mixed micelles system is a promising and effective carrier for significantly enhancing the solubility and transmembrane transport of hydrophobic drugs (Zhang et al., Citation2014; Mandal et al., Citation2017; Sandhu et al., Citation2017). Nanosuspensions have also been regarded as potential carriers of hydrophobic and insoluble drugs by improving the apparent solubility with solubilizers and excipients (Zhang et al., Citation2011; Bi et al., Citation2017; Ye et al., Citation2017).
To overcome the challenge of poor solubility and frustrating absorption of magnolol, we tried to develop a novel nano-drug delivery system using drug solubilizers and enhancers of absorption and permeation. Soluplus® and Poloxamer 188 have been used in numerous preparations, including solid dispersions, micelles, liposomes, and nanosuspensions (Lee et al., Citation2016; Medarević et al., Citation2016; Zeng et al., Citation2017; Hu et al., Citation2018; Homayouni et al., Citation2019). Soluplus®, a polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol graft copolymer, is an amphipathic copolymer newly introduced by BASF (Shanghai, China). It has been widely used to increase the aqueous solubility and oral bioavailability of poorly soluble drugs. Given its high fluidity and excellent extrudability, Soluplus® has shown superior performance in forming solid dispersions, especially in the hot-melt extrusion process (Linn et al., Citation2012). As an amphiphilic molecule, Soluplus® also self-assembles into micelles once reaching the critical micelle concentration (Dian et al., Citation2014; Andrade et al., Citation2015). Poloxamer 188 is composed of two polyethylene oxide (PEO) chains separated by a polypropylene oxide (PPO) chain (Yan et al., Citation2010). The Food and Drug Administration (FDA) has recognized Poloxamer 188 as a pharmaceutical excipient, and it is often used as a nanocarrier because of its safety, versatility, and commercial availability (Zhang et al., Citation2015b).
In this study, we prepared magnolol-loaded mixed micelles (MMs) and magnolol nanosuspensions (MNs) by Soluplus® and Poloxamer 188, using their excellent properties such as safety, to increase the aqueous solubility and enhance absorption and permeation of drugs. To evaluate the drug delivery systems, we investigated the physicochemical properties of MMs and MNs, including particle size and zeta potential, surface morphology, encapsulation efficiency and drug content, drug release in vitro, dilution stability. The absorption and efflux characteristics were investigated in Caco-2 cell monolayers. Finally, oral bioavailability was assessed in vivo through pharmacokinetic studies.
2. Materials and methods
2.1. Materials and reagents
Magnolol and honokiol (purity >98%) were purchased from Melonepharma Technology Co., Ltd., (Dalian, China). Poloxamer 188 and Soluplus® were purchased from BASF Ltd., (Shanghai, China). Milli-Q water (Millipore, Bedford, MA) was used throughout the study. Caco-2 cells were purchased from the Shanghai Institute of Cell Bank, China. Chromatography-grade methanol and acetonitrile were used in this study (Tedia Company Inc., Fairfield, OH), and all other reagents were of analytical grade.
2.2. Animals
Male Sprague-Dawley (SD) rats (200 ± 20 g) were purchased from the Shanghai Laboratory Animal Center (SLAC, Shanghai, China). All rats were provided with distilled water ad libitum and maintained at a temperature of 25 °C and relative humidity of 45% for 2 weeks. All animal experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the Jiangsu Provincial Academy of Chinese Medicine.
2.3. Preparation of MMs
MMs were prepared using the film hydration method (Zhang et al., Citation2017b). First, 120 mg of Soluplus® and 50 mg of Poloxamer 188 were co-dissolved in 5 mL of ethanol by gentle agitation at 50 °C, and then 10 mg of magnolol was dissolved in this mixture by gentle agitation until a transparent solution was formed. Subsequently, the solution was rotary evaporated at 50 °C to form a thin film, which was further vacuum-dried for 12 h at −0.1 MPa and 45 °C to remove traces of the remaining ethanol. Then, 5 mL of deionized water was added to hydrate the film, and the MMs were obtained by ultracentrifugation at 14,000 rpm for 15 min.
2.4. Preparation of MNs
MNs were prepared using the anti-solvent precipitation method (Varshosaz et al., Citation2017; Li et al, Citation2020). Briefly, 10 mg each of Soluplus® and Poloxamer 188 were dissolved in 10 mL of deionized water by gentle agitation at 45 °C, and then 20 mg of magnolol in 5 mL of ethanol was added dropwise to this solution. The solution was subsequently rotary evaporated at 45 °C until the ethanol was removed.
2.5. Particle size and zeta potential analyses
The particle size and zeta potential of the MMs and MNs were determined using dynamic light scattering (Malvern ZE S.A., Worcestershire, UK). The results were expressed as the average of three measurements. The polydispersity index (PDI) was then measured to assess the particle size distribution of MMs and MNs.
2.6. Transmission electron microscopy
The morphology of MMs and MNs was evaluated by transmission electron microscopy (TEM) with a JEM-200CX microscope (Japan). A drop of the sample was placed on a carbon-coated copper grid, which was dried under infrared (IR) light for 5 min. The sample was then observed using TEM and photographed using a digital camera.
2.7. Encapsulation efficiency and drug content analysis
In this study, the drug loading and entrapment efficiency (DL% and EE%, respectively) of magnolol in MMs, and the DL% of magnolol in MNs, were determined using high-performance liquid chromatography (HPLC), which consisted a quaternary pump (Waters 2695 separation module, Waters 2489 UV detector) and a Symmetry® C-18 column (5 μm, 250 mm × 4.6 mm). The mobile phase was a mixture of acetonitrile and 0.2% formic acid solution (75/25, v/v). The flow rate was 1 mL/min. The column temperature was maintained at 30 °C. The injection volume was 10 μL. The aforementioned formulations were filtered through a 0.45 µm microporous membrane, followed by a precise drawing of 0.1 mL of the filtrate. An appropriate amount of methanol was added to dilute and disrupt the micelles or the nanosuspensions. DL% and EE% were calculated using the following equations:
2.8. In vitro release study
The in vitro release properties of the MMs and MNs formulations were analyzed using the dialysis method (Jin et al., Citation2016). In brief, groups of samples consisting of 1 mL (2 mg/mL) each of MMs and MNs, or an equivalent amount of magnolol suspension (as the control group), were placed in dialysis membrane bags (molecular weight cutoff [MWCO] = 3500 Da, Greenbird Inc., China). A receptor medium comprising 200 mL of phosphate-buffered saline (PBS, pH 1.2 or 6.8) and 1% (w/v) Tween 80 (Sinopharm Chemical Reagent Co., Ltd) was freshly prepared and incubated at 37 °C under shaking at 100 rpm. Magnolol was completely released under the leakage conditions. Triplicate samples (1 mL) of the release medium were collected at predetermined time points, and equal volumes of fresh release medium were quickly replenished to maintain a constant volume (200 mL). The samples (n = 3) were filtered through 0.22 µm filters, and the amount of magnolol released into the supernatant was evaluated using HPLC.
2.9. Dilution stability
To study the effect of dilution on particle size stability, the MMs and MNs samples were diluted 10, 50, 100, and 200-fold with water (Shen et al., Citation2018), and the particle sizes of the samples were measured using a Malvern zeta sizer as described above. All the experiments were performed in triplicate.
2.10. MTT assay
Caco-2 cell lines were seeded in 96-well microplates at a density of cells/well. After 2 h of incubation, three type formulations (at dose of magnolol) concentrations ranging from 0.25 to 100 μg/mL were added to the cells, and control culture represented cells without treatment. The optical density of treated and untreated wells was measured at 560 nm using a microplate spec-trophotometer (PowerWave XS, Biotek, VT). A graph of percent inhibition versus magnolol concentration was constructed.
2.11. Caco-2 cell culture and transport experiment
The Caco-2 cell culture model, which is a viable model of human intestinal absorption recognized by the FDA, was selected to investigate magnolol absorption. In brief, Caco-2 cells were cultured in an incubator (37 °C, 95% relative humidity, 5% CO2) and used for the transport experiments 21 days post-seeding, during which these cells were sufficiently developed, both in physiological and morphological aspects. Specifically, the transepithelial electrical resistance (TEER) values were >600 Ω cm2. At the start of the experiment, cell monolayers were washed three times with blank Hank’s balanced salt solution (HBSS, pH 7.4); then three types of formulations (free drug, MMs, or MNs) were added to the apical (AP) or basolateral (BL) side at equivalent magnolol concentration (50 μg/mL), and samples were collected from the receiving side after 2 h. The samples were then centrifuged at 14,000 rpm for 5 min at 4 °C, and the supernatant containing magnolol was measured using HPLC. The permeability of magnolol was finally calculated using the following equation:
where V is the volume of the receiver, S is the surface area of the cell monolayer, C is the initial concentration, dC/dt is the rate of concentration change in the receiver side, and dM/dt is the rate of drug transport. The rate of magnolol transport was obtained using linear regression analysis (Zhang et al., Citation2012; Kimura et al., Citation2016; Yano et al., Citation2016).
2.12. Pharmacokinetic studies
Male SD rats (200 ± 20 g) were randomly divided into three groups (n = 6) and fasted overnight prior to the experiment. Groups 1, 2, and 3 were orally administered with magnolol (60 mg/kg in 0.5% HPMC (Sigma, St. Louis, MO)), MMs, and MNs (at dose equivalents of 60 mg/kg magnolol), respectively. At predetermined times (0.167, 0.333, 0.5, 0.75, 1, 2, 4, 6, and 8 h) after administration, blood samples were collected by retro-bulbar puncture into heparinized tubes to determine honokiol concentrations and calculate pharmacokinetic parameters. The blood samples were immediately centrifuged at 6000 rpm for 5 min and the plasma supernatant was collected. Plasma was also placed into tubes containing the internal honokiol standard. The samples were then vortexed for 15 s and methanol was added, followed by vortexing for another 3 min to precipitate the plasma proteins. These samples were then centrifuged at 14,000 rpm for 5 min at 4 °C, and the resulting supernatant was transferred to a separate tube and evaporated to dryness at 20 °C. The residue was dissolved in acetonitrile-water (50:50, v/v) containing 0.2% formic acid, vortexed for 5 min, and centrifuged at 14,000 rpm, and then the samples were subjected to HPLC analysis.
The content of magnolol was determined using HPLC using an Agilent 1260 system (Agilent Co., Ltd.) with a C18 column (250 × 4.6 mm, 5 µm). The mobile phase consisted of an acetonitrile-water mixture (50:50, v/v) containing 0.2% formic acid, and the flow rate was 1 mL/min with a column temperature of 30 °C. The diode array detector (DAD) was set to 290 nm, the injection volume was 20 µL, and the samples were filtered through a 0.22 µm membrane filter (Millipore Corp.).
2.13. Statistical analysis
All values were presented as the mean ± standard deviation. The statistical significance of the results was analyzed using the statistical package for the social sciences (SPSS) software version 19.0 (SPSS Inc., Chicago, IL). The data were analyzed using a t-test and a p < .05 was considered statistically significant.
3. Results
3.1. Characterization of MMs and MNs
3.1.1. Size distribution, zeta potential, and surface morphology
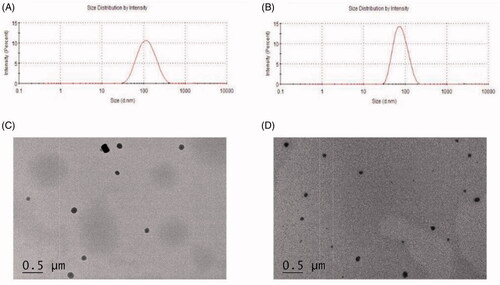
The physicochemical characteristics and drug-loading parameters of MMs and MNs were evaluated. As shown in and , the average particle sizes of MMs and MNs were 111.8 ± 14.6 and 78.53 ± 5.4 nm, with PDIs of 0.17 ± 0.02 and 0.04 ± 0.01, respectively. The zeta potential of MMs and MNs was −1.04 ± 0.12 mV and −24.27 ± 0.22 mV, respectively. showed that the morphology of the MMs and MNs formulations both were spherical shape. The characteristics of the MMs and MNs samples were concordant with those obtained from DLS with appropriate dispersion in both the novel nano-drug delivery systems.
Figure 1. (A) Size distribution of magnolol-loaded mixed micelles(MMs); (B) size distribution of magnolol nanosuspensions(MNs); (C) TEM micrographs and image of MMs, scale bar = 0.5 μm; (D) TEM micrographs and image of MNs, scale bar = 0.5 μm.
3.1.2. Encapsulation efficiency and drug loading
Using HPLC, EE% and DL% were determined to be 89.58 ± 2.54% and 5.46 ± 0.65% in the MMs, respectively, indicating that most of the magnolol was entrapped in the MMs system. The MNs system had a higher drug loading value of 42.50 ± 1.57%, which demonstrates an advantage for MNs over MMs with respect to drug loading.
3.2. In vitro release
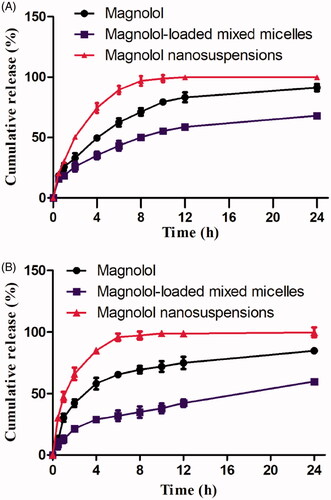
Regarding in-vitro release, showed the release profiles of MMs and MNs compared to free magnolol. As described in the methods section, the in vitro release assay showed that drug released from MMs was slower than from MNs in release media at two different pH levels. For example, at pH 1.2, after 6 h, MNs, free magnolol, and MMs showed a burst release of 90, 65, and 43.15%, respectively. Furthermore, MMs exhibited a sustained release over 24 h (). The percentage of magnolol release from MMs at pH 1.2 and 6.8 was 59.6 and 68.2%, respectively, at 24 h, while free magnolol showed corresponding values of 84.6 and 91.1%, respectively. During the same period, 99.6 and 99.1% of magnolol were released from MNs at a pH of 1.2 and 6.8, respectively, which was considerably faster than the release from MMs and that of free drug. These results demonstrate that the developed MMs and MNs systems efficiently solubilize magnolol and have the potential for being used as oral bioavailability enhancers.
Figure 2. (A) In vitro release profiles of magnolol in phosphate buffer, pH 1.2; (B) in vitro release profiles of magnolol in phosphate buffer, pH 6.8.
3.3. Dilution stability
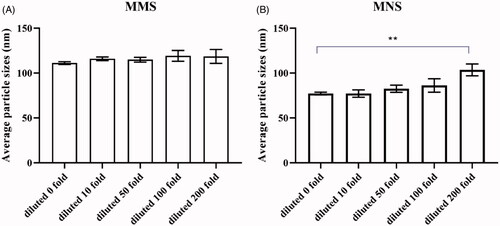
To prove both formulations could keep the size of the particle constant when they were orally administered to the body undergo strong dilution in physiological fluids, the dilution stability of MMs and MNs was investigated. Particle sizes of MMs showed no significant change (), suggesting that MMs had a stabilizing effect against dilution. MNs showed relative stability in 200 fold diluted (). This effect possibly occurred because magnolol was spontaneously incorporated into the micelle cores from the aqueous environment. Hydrophobic micelles were likely to exhibit a higher solubilization capacity than hydrophilic forms do, and the increased hydrophobicity of the MMs cores enhanced their stability following dilution. Furthermore, in MNs, Soluplus® and Poloxamer 188 likely adsorbed onto the surface of the nanoparticles more strongly than the other stabilizers did, thereby protecting the drug against dilution (Deng et al., Citation2010; Zong et al., Citation2017). However, when a large amount of liquid dilution occurs, the concentration of stabilizer in MNs decreases, and the stabilizer cannot maintain adsorption on the surface of magnolol. magnolol particles aggregate and settle with each other, resulting in larger particle size.
Figure 3. (A) Diluted stability of MMs (diluted 10, 50, 100, 200-fold); (B) diluted stability of MNs (diluted 10, 50, 100, 200-fold). **p < .01.
3.4. MTT assay
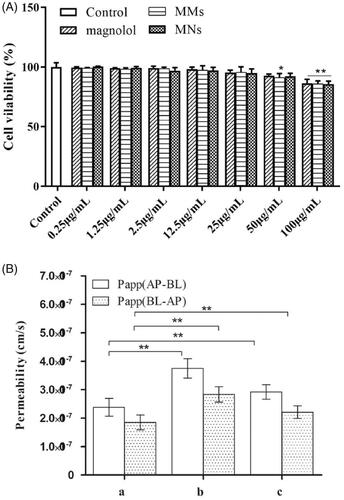
Caco-2 cells were treated with different concentrations of magnolol, MMs, and MNs (0.25 to 100 μg/mL) for 2 h, and individual cell viability was shown in . The results demonstrated nanosuspensions and mixed-micelles exhibited enhanced anti-proliferative activity in comparison to free magnolol in a dose-dependent manner. The IC50 value of magnolol, MMs, and MNs were 153.528, 135.282, and 143.89 μg/mL, respectively.
Figure 4. (A) Caco-2 cells were treated with various concentrations of magnolol for 2 h. Cell viability was measured by MTT assay. Data are presented as mean ± standard deviation; n = 3; (B) AP-BL and BL-AP permeability of magnolol, MMs, and MNs across the Caco-2 cell monolayer after 2 h incubation at 37 °C (mean ± standard deviation; n = 3). *p < .5; **p < .01.
3.5. Transport of magnolol, MMs, and MNs across Caco-2 cell monolayers
Caco-2 monolayers were used to investigate the transport of magnolol and the corresponding formulations through the intestinal epithelial barrier. As shown in , the transport of magnolol (50 µg/mL), MMs, and MNs from the AP to BL direction and vice versa, was investigated. For AP-BL transport, the PappAB value of magnolol was 2.38 ± 0.31 × 10−7 cm/s. The MMs and MNs formulations significantly (MMs, p < .01; MNs, p < .05) increased the absorptive directional (AP-BL) transport of magnolol (3.75 ± 0.34 × 10−7 cm/s and 2.92 ± 0.25 × 10−7 cm/s, respectively). For BL-AP transport, the permeability (Papp) of MMs (2.83 ± 0.27 × 10−7 cm/s) and MNs (2.21 ± 0.22 × 10−7 cm/s) increased to a small and statistically insignificant degree compared with that of magnolol (1.85 ± 0.26 × 10−7 cm/s). These results indicate that MMs and MNs increased the absorption of magnolol.
3.6. Pharmacokinetic studies
The plasma drug concentration-time curve of magnolol, MMs, and MNs after a single oral administration in rats (n = 6) is shown in and . The average maximum plasma concentration (Cmax) values were 0.305 ± 0.031, 0.587 ± 0.048, and 0.65 ± 0.125 mg/L in the free magnolol, MMs, and MNs groups, respectively. This result indicates that the formation of micelles and nanosuspensions markedly increases the Cmax of magnolol in the blood. The Cmax value of MNs was 2.13 and 1.11-fold higher than those of free magnolol and MMs, respectively. The notable Cmax enhancement in MNs was attributed to the rapid release of magnolol. The time to reach Cmax (Tmax) values after oral administration of free magnolol, MMs, and MNs were 0.708 ± 0.102, 0.792 ± 0.102, and 0.5 h, respectively. The Tmax showed no obvious difference between the magnolol and MMs groups, whereas their half-life (t1/2) values were significantly different (p < .01). MMs and MNs showed prolonged circulatory t1/2 values (3.142 ± 0.285 and 2.776 ± 0.417 h, respectively), which increased 4.12 and 3.64-fold relative to that of the free drug. The average area under the curve from time 0 to infinity (AUC0–∞) values for free magnolol, MMs, and MNs were 0.837 ± 0.084, 2.904 ± 0.465, and 2.217 ± 0.332 mg L−1 h−1, respectively. The MMs and MNs groups showed a markedly higher relative bioavailability than that of the free magnolol group (p < .01). In addition, the bioavailability of MMs and MNs (AUC0–∞) relative to that of free magnolol were 346.9 and 234.8%, respectively, demonstrating great potential for clinical use.
Figure 5. The plasma concentration-time curve of magnolol in rats after oral administration of magnolol, MMs and MNs (60 mg/kg, magnolol). Data are presented as mean ± standard deviation; n = 6.
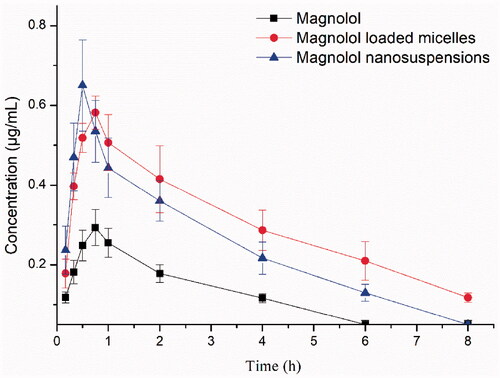
Table 1. Pharmacokinetic parameters of magnolol, magnolol-loaded mixed micelles (MMs) and magnolol nanosuspensions (MNs) (60 mg/kg, magnolol).
4. Discussion
Many drug candidates are highly efficacious but are insoluble in water, which limits their clinical application. Magnolol has numerous pharmacological activities and few associated adverse effects, but its poor oral bioavailability has hindered its clinical efficacy. In our study, we combined the advantages of Soluplus® and Poloxamer 188 to develop novel MMs and MNs formulations with the aim of improving the solubility and oral bioavailability of magnolol. Results of the in vitro drug release study showed that MMs had a slower drug release rate than that of MNs. These results indicated that both MMs and MNs could solubilize magnolol, while MMs may also control the release of magnolol and MNs showed rapid drug release. Both formulations increased the absorption of magnolol in the Caco-2 transcellular transport assay, and both also showed a higher relative bioavailability after oral administration than that of the free drug.
Although both formulations improved the systemic performance of magnolol, they showed marked differences. For example, both MMs and MNs increased drug transport across the Caco-2 cell monolayers, compared to free magnolol, which may be because of smaller particle size of MMs and MNs and also the use of Soluplus® and Poloxamer 188 (Hu et al., Citation2018; Homayouni et al., Citation2019; Lu et al., Citation2020). However, MMs increased drug transport across the Caco-2 cell monolayers relative to that achieved in MNs. This result may be attributed to the high percentage of Soluplus® and Poloxamer 188 in MMs as these solubilizers increase not only the aqueous solubility but also the permeation of magnolol. Compared with free magnolol, MNs substantially decreased the Tmax of magnolol by approximately 30% but the 13% increase observed with MMs. Similarly, MNs and MMs had 2.13 and 1.93-fold (0.65 ± 0.125 and 0.587 ± 0.048, respectively) higher Cmax than did free magnolol. MMs prolonged the circulatory t1/2 by 4.12-fold (3.142 ± 0.285 h) relative to that of MNs, which was 3.64-fold (2.776 ± 0.417 h) greater than that of the free drug. In addition, both formulations improved oral bioavailability; compared with that of the free drug, the oral bioavailability of MMs was 3.47-fold (2.904 ± 0.465) higher than that of MNs, which was 2.65-fold (2.217 ± 0.332) higher than that of free magnolol. These results suggested that it would be possible to formulate mixed micelle and nanosuspensions based on free magnolol with a decreased therapeutic dose (Fares et al., Citation2018; Tang et al., Citation2019). These results supported a significant increase in the bioavailability of magnolol by MMs and MNs. The increase in bioavailability can be explained for various reasons. First, MMs and MNs increase the dissolution by decreasing particle size, providing better absorption and permeation of magnolol. In addition, the nanosized particles can improve paracellular and intracellular transports, thereby increasing the intestinal absorption and protecting against drug degradation by enteric enzymes (Yun et al., Citation2013; Na et al., Citation2020). MNs provides an immediate release of drug contributed to smaller Tmax and high Cmax. The improved bioavailability of magnolol in MNs can be ascribed to the presence of surfactants and stabilizers, which increases the apparent solubility, as well as to particle size reduction leading to amplified surface area (Mori et al., Citation2016; Nagaraj et al., Citation2017). This induces immediate drug release, making more drugs available at the absorption site. Furthermore, a small number of MNs may pass through the Peyer’s patch of the small intestine and then be directly absorbed in nanoparticle form (Han et al., Citation2014). MMs may prolong the drug retention time in the gastrointestinal tract and increase passive drug absorption due to high adhesion to the intestinal wall, and further enhances intracellular uptake by enterocytes. Additionally, endocytosis is thought to be the main gastrointestinal tract absorption mechanism of micelles which gives rise to a long t1/2 (Mathot et al., Citation2007; Alqahtani et al., Citation2017).
5. Conclusion
The developed novel Soluplus® and Poloxamer 188-based MMs and MNs markedly increased the solubility and oral absorption of magnolol. In the Caco-2 transport study and pharmacokinetic study, MMs showed better absorption and higher AUC for magnolol than MNs did. In contrast, MNs exhibited a higher Cmax for magnolol than MMs did. Both the MNs and MNs formulations markedly improved the oral bioavailability of magnolol and, therefore having immense clinical application prospects.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Alexeev M, Grosenbaugh DK, Mott DD, Fisher JL. (2012). The natural products magnolol and honokiol are positive allosteric modulators of both synaptic and extra-synaptic GABA(A) receptors. Neuropharmacology 62:2507–14.
- Alqahtani MS, Islam MS, Podaralla S, et al. (2017). Food protein based core-shell nanocarriers for oral drug delivery: effect of shell composition on in vitro and in vivo functional performance of zein nanocarriers. Mol Pharm 14:757–69.
- Andrade F, das Neves J, Gener P, et al. (2015). Biological assessment of self-assembled polymeric micelles for pulmonary administration of insulin. Nanomedicine 11:1621–31.
- Bi C, Miao X, Chow S, et al. (2017). Particle size effect of curcumin nanosuspensions on cytotoxicity, cellular internalization, in vivo pharmacokinetics and biodistribution. Nanomedicine 13:943–53.
- Chen LC, Liu YC, Liang YC, et al. (2009). Magnolol inhibits human glioblastoma cell proliferation through upregulation of p21/Cip1. J Agric Food Chem 57:7331–7.
- Chen YH, Huang PH, Lin FY, et al. (2011). Magnolol: a multifunctional compound isolated from the Chinese medicinal plant Magnolia officinalis. Eur J Integr Med 3:e317–24.
- Deng J, Huang L, Liu F. (2010). Understanding the structure and stability of paclitaxel nanocrystals. Int J Pharm 390:242–9.
- Deng J, Zhang Z, Liu C, et al. (2015). The studies of N-octyl-N-arginine-chitosan coated liposome as an oral delivery system of cyclosporine A. J Pharm Pharmacol 67:1363–70.
- Dian L, Yu E, Chen X, et al. (2014). Enhancing oral bioavailability of quercetin using novel soluplus polymeric micelles. Nanoscale Res Lett 9:2406.
- Fares AR, Elmeshad AN, Kassem M. (2018). Enhancement of dissolution and oral bioavailability of lacidipine via pluronic P123/F127 mixed polymeric micelles: formulation, optimization using central composite design and in vivo bioavailability study. Drug Deliv 25:132–42.
- Fu Y, Liu B, Zhang N, et al. (2013). Magnolol inhibits lipopolysaccharide-induced inflammatory response by interfering with TLR4 mediated NF-κB and MAPKs signaling pathways. J Ethnopharmacol 145:193–9.
- Han M, Yu X, Guo Y, et al. (2014). Honokiol nanosuspensions: preparation, increased oral bioavailability and dramatically enhanced biodistribution in the cardio-cerebro-vascular system. Coll Surf B Biointerf 116:114–20.
- Homayouni A, Amini M, Sohrabi M, et al. (2019). Curcumin nanoparticles containing poloxamer or soluplus tailored by high pressure homogenization using antisolvent crystallization. Int J Pharm 562:124–34.
- Hu XY, Lou H, Hageman MJ. (2018). Preparation of lapatinib ditosylate solid dispersions using solvent rotary evaporation and hot melt extrusion for solubility and dissolution enhancement. Int J Pharm 552:154–63.
- Jin X, Zhang Y, Zhang Z, et al. (2016). Juglone loaded Poloxamer 188/phospholipid mixed micelles evaluated in vitro and in vivo in breast cancer. Int J Pharm 515:359–66.
- Kimura O, Fujii Y, Haraguchi K, et al. (2016). Effect of quercetin on the uptake and efflux of aristolochic acid I from Caco-2 cell monolayers. J Pharm Pharmacol 68:883–9.
- Lee CW, Hu SC, Yen FL, et al. (2017). Magnolol nanoparticles exhibit improved water solubility and suppress TNF-α-induced VCAM-1 expression in endothelial cells. J Biomed Nanotechnol 13:255–68.
- Lee SY, Lee JJ, Park JH, et al. (2016). Electrosprayed nanocomposites based on hyaluronic acid derivative and Soluplus for tumor-targeted drug delivery. Coll Surf B Biointerf 145:267–74.
- Li S, Zhang J, Fang Y, et al. (2020). Enhancing betulinic acid dissolution rate and improving antitumor activity via nanosuspension constructed by anti-solvent technique. Drug Des Devel Ther 14:243–56.
- Liang WZ, Chou CT, Chang HT, et al. (2014). The mechanism of honokiol-induced intracellular Ca(2+) rises and apoptosis in human glioblastoma cells. Chem Biol Interact 221:13–23.
- Linn M, Collnot EM, Djuric D, et al. (2012). Soluplus® as an effective absorption enhancer of poorly soluble drugs in vitro and in vivo. Eur J Pharm Sci 45:336–43.
- Lu X, Lu X, Zhang Z, Lv H. (2020). Preparation and characterization of honokiol nanosuspensions and preliminary evaluation of anti-inflammatory effect. AAPS PharmSciTech 1021:62.
- Ma Y, He H, Xia F, et al. (2017). In vivo fate of lipid-silybin conjugate nanoparticles: implications on enhanced oral bioavailability. Nanomedicine 13:2643–54.
- Mandal A, Bisht R, Rupenthal ID, Mitra AK. (2017). Polymeric micelles for ocular drug delivery: from structural frameworks to recent preclinical studies. J Control Release 248:96–116.
- Mathot F, des Rieux A, Ariën A, et al. (2007). Transport mechanisms of mmePEG750P(CL-co-TMC) polymeric micelles across the intestinal barrier. J Control Release 124:134–43.
- Medarević DP, Kleinebudde P, Djuriš J, et al. (2016). Combined application of mixture experimental design and artificial neural networks in the solid dispersion development. Drug Dev Ind Pharm 42:389–402.
- Mori D, Makwana J, Parmar R, et al. (2016). Formulation, evaluation and optimization of the felodipine nanosuspension to be used for direct compression to tablet for in vitro dissolution enhancement. Pak J Pharm Sci 29:1927–36.
- Na YG, Pham TMA, Byeon JJ, et al. (2020). Development and evaluation of TPGS/PVA-based nanosuspension for enhancing dissolution and oral bioavailability of ticagrelor. Int J Pharm 581:119287.
- Nagaraj K, Narendar D, Kishan V. (2017). Development of olmesartan medoxomil optimized nanosuspension using the Box-Behnken design to improve oral bioavailability. Drug Dev Ind Pharm 43:1186–96.
- Nekkanti V, Rueda J, Wang Z, Betageri GV. (2016). Comparative evaluation of proliposomes and self micro-emulsifying drug delivery system for improved oral bioavailability of nisoldipine. Int J Pharm 505:79–88.
- Shen H, Liu S, Ding P, et al. (2018). Enhancement of oral bioavailability of magnolol by encapsulation in mixed micelles containing pluronic F127 and L61. J Pharm Pharmacol 70:498–506.
- Sandhu PS, Kumar R, Katare OP, et al. (2017). Surface-tailored nanomixed micelles containing quercetin-salicylic acid physical complex for enhanced cellular and in vivo activities: a quality by design perspective. Nanomedicine 12:1281–303.
- Tang L, Zhu Z, Xie M, et al. (2019). Effects of β-Cyclodextrin and Hydroxypropyl-β-Cyclodextrin Inclusions on the Degradation of Magnolol by Intestinal Bacteria. AAPS PharmSciTech 20:244.
- Tsai JJ, Chen JH, Chen CH, et al. (2020). Apoptosis induction and ERK/NF-κB inactivation are associated with magnolol-inhibited tumor progression in hepatocellular carcinoma in vivo. Environ Toxicol 35:167–75.
- Tsai T, Kao CY, Chou CL, et al. (2016). Protective effect of magnolol-loaded polyketal microparticles on lipopolysaccharide-induced acute lung injury in rats. J Microencapsul 33:401–11.
- Varshosaz J, Minayian M, Yazdekhasti S. (2017). Physicochemical, pharmacodynamic and pharmacokinetic characterization of soluplus stabilized nanosuspension of tacrolimus. Curr Drug Deliv 14:521–35.
- Yan F, Zhang C, Zheng Y, et al. (2010). The effect of poloxamer 188 on nanoparticle morphology, size, cancer cell uptake, and cytotoxicity. Nanomedicine 6:170–8.
- Yano K, Otsuka K, Kato Y, et al. (2016). Different regulation of P-glycoprotein function between Caco-2 and Caki-1 cells by ezrin, radixin and moesin proteins. J Pharm Pharmacol 68:361–7.
- Ye L, Miao M, Li S, Hao K. (2017). Nanosuspensions of a new compound, ER-β005, for enhanced oral bioavailability and improved analgesic efficacy. Int J Pharm 531:246–56.
- Yun Y, Cho YW, Park K. (2013). Nanoparticles for oral delivery: targeted nanoparticles with peptidic ligands for oral protein delivery. Adv Drug Deliv Rev 65:822–32.
- Zeng YC, Sha LI, Liu C, et al. (2017). Soluplus micelles for improving the oral bioavailability of scopoletin and their hypouricemic effect in vivo. Acta Pharmacol Sin 38:424–33.
- Zhang FH, Ren HY, Shen JX, et al. (2017a). Magnolol suppresses the proliferation and invasion of cholangiocarcinoma cells via inhibiting the NF-κB signaling pathway. Biomed Pharmacother 94:474–80.
- Zhang H, Yang X, Zhao L, et al. (2015a). In vitro and in vivo study of Baicalin-loaded mixed micelles for oral delivery. Drug Deliv 23:1–7.
- Zhang J, Lv H, Jiang K, Gao Y. (2011). Enhanced bioavailability after oral and pulmonary administration of baicalein nanocrystal. Int J Pharm 420:180–8.
- Zhang W, Wang G, See E, et al. (2015b). Post-insertion of poloxamer 188 strengthened liposomal membrane and reduced drug irritancy and in vivo precipitation, superior to PEGylation. J Control Release 203:161–9.
- Zhang Z, Chen Y, Deng J, et al. (2014). Solid dispersion of berberine–phospholipid complex/TPGS 1000/SiO2: preparation, characterization and in vivo studies. Int J Pharm 465:306–16.
- Zhang Z, Cui C, Wei F, Lv H. (2017b). Improved solubility and oral bioavailability of apigenin via Soluplus/Pluronic F127 binary mixed micelles system. Drug Dev Ind Pharm 43:1276–82.
- Zhang Z, Lv H, Jia X, et al. (2012). Influence of vitamin E tocopherol polyethylene glycol succinate 1000 on intestinal absorption of icariside II. Pharmazie 67:59–62.
- Zhang ZH, Wang XP, Ayman WY, et al. (2013). Studies on lactoferrin nanoparticles of gambogic acid for oral delivery. Drug Deliv 20:86–93.
- Zhao C, Liu ZQ. (2011). Comparison of antioxidant abilities of magnolol and honokiol to scavenge radicals and to protect DNA. Biochimie 93:1755–60.
- Zong L, Li X, Wang H, et al. (2017). Formulation and characterization of biocompatible and stable I.V. itraconazole nanosuspensions stabilized by a new stabilizer polyethylene glycol-poly(β-Benzyl-l-aspartate) (PEG-PBLA). Int J Pharm 531:108–17.