
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The combination of chemotherapeutic drug paclitaxel (PTX) and VEGF siRNA could inhibit cancer development with synergistic efficacy. However, efficient and safe delivery systems with high encapsulation efficiency of PTX and a long-time release of drugs are urgently needed. In this study, novel nanoparticles (PTX/siRNA/FALS) were constructed by using tripeptide lipid (L), sucrose laurate (S), and folate-PEG2000-DSPE (FA) to co-deliver PTX and siRNA. The cancer cell targeting nanoparticle carrier (PTX/siRNA/FALS) showed anticipated PTX encapsulation efficiency, siRNA retardation ability, improved cell uptake and sustained and controlled drug release. It led to significant anti-tumor activity in vitro and in vivo by efficient inhibition of VEGF expression and induction of cancer cell apoptosis. Importantly, the biocompatibility of the carriers and low dosage of PTX required for effective therapy greatly reduced the toxicity to mice. The targeting nanoparticles show potential as an effective co-delivery platform for RNAi and chemotherapy drugs, aiming to improve the efficacy of cancer therapy.
1. Introduction
Lung cancer is the leading cause of cancer death, and non-small cell lung cancer (NSCLC) accounts for approximately 85% of all lung cancers (Herbst et al., Citation2018). Despite substantial progress made in developing modalities for lung cancer treatment, including surgical resection, chemotherapy, and radiotherapy, there are still significant challenges in improving the survival rate. In particular, the traditional chemotherapy only provides a dismal 5-year survival rate of less than 15% (Yang et al., Citation2012). One of the major hurdles stems from angiogenesis in the tumor environment, which greatly promotes the growth and metastasis of tumors by providing malignant cells with an adequate supply of oxygen and nutrients (Sausville, Citation2001; Sia et al., Citation2014; Yang et al., Citation2018). Studies demonstrated that selective combination therapy significantly improved cancer treatment compared with single agent therapy (Xu et al., Citation2015). Moreover, anti-angiogenesis therapy and chemotherapy are important therapy regimens against NSCLC (Zhang et al., Citation2013). Integrating anti-angiogenesis therapy with chemotherapy is expected to target the tumor’s vascular endothelial cells and tumor cells simultaneously. Among the growth factor pathways regulating the growth and maintenance of blood vessels, vascular endothelial growth factor (VEGF) is a major driver of tumor angiogenesis (Saharinen et al., Citation2011). Small interfering RNA (siRNA) are known to specifically inhibit the expression of target genes without inflicting significant toxicity (Dykxhoorn & Lieberman, Citation2006; Yang & Zhang, Citation2012). Our expectation is that VEGF siRNA will inhibit VEGF expression (Feng et al., Citation2014) and thereby realize anti-angiogenesis in tumors (Sun et al., Citation2017). Therefore, the simultaneous delivery of chemotherapy drugs and VEGF siRNA for cancer therapy is expected to hold particular promise as a potentially useful dual-modality approach for treating highly vascularized tumors such as NSCLC. Among various chemotherapy drugs, paclitaxel (PTX) is a very suitable candidate, as it has remarkable efficacy against cancers such as lung, ovarian, and breast cancers (Xie et al., Citation2018).
Several studies have reported detailed systems designed for efficient co-delivery of VEGF siRNA and chemotherapy drugs. A targeted core-shell nanoparticle that simultaneously delivered PTX and VEGF siRNA to cells and achieved synergistic effect in cancer therapy was developed by Feng et al. (Feng et al., Citation2014). However, in their study, to reach significant beneficial effects, a high dosage of PTX over 10 mg/kg was required, which was still likely to result in severe side effects. A multifunctional polymeric micelle was designed by Yang et al. (Yang et al., Citation2018) to sequentially deliver PTX and VEGF siRNA. The remarkable pH/redox sensitivity provided a safe and efficient carrier to maximize the synergistic effect of drugs and genes. Unfortunately, although this reported vector was effective for co-delivering VEGF siRNA and chemo drugs at low dosage, the encapsulation efficiency of PTX was less than 75%. This led to difficulties in implementation for clinical applications.
In this study, we explored a novel and simple co-delivery nanoparticle for delivering drug and siRNA through a tripeptide lipid (L) and a sucrose laurate (S). In this delivery vehicle, the novel tripeptide lipid, synthesized by our group, has a carbamate linker, which is stable in blood circulation (pH 7.4). And, a particularly useful feature is that it is susceptible to acid-catalyzed hydrolysis in the acid environment of the endosome (pH 5–6) (Ma, Citation2014; Zhao et al., Citation2014). What’s more, sucrose laurate with sucrose as the hydrophilic head group and fatty acids as lipophilic tail groups is widely used as emulsifiers and solubilizers in pharmaceutical industry as it is natural, nontoxic and biodegradable (Chen et al., Citation2019), which may exert positive effects on both delivery efficiency and toxicity of nanoparticles. For instance, Das et al. (Das et al., Citation2014) explored a stabilized clotrimazole-loaded solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs) for the controlled release of drug by using sucrose laurate as a stabilizer/emulsifier. The effects of sucrose ester structures on liposome-mediated gene delivery were also examined by Zhao et al. (Zhao et al., Citation2018). The results suggested that the incorporation of sucrose laurate into liposomes could increase the efficiency of cationic lipid-based gene delivery systems for gene transfection in vitro and in vivo. Moreover, a review also summarized some research findings and applications of sucrose laurate in nanoparticles (Szűts & Szabó-Révész, Citation2012), which suggested that sucrose laurate is of great potential as pharmaceutical ingredients in novel formulations. In addition, based on the over-expression of folate receptor in NCI-H460 cells, the co-delivery nanoparticles were modified with folate-PEG2000-DSPE (FA) to direct and increase accumulation and uptake of drugs at tumor sites (Cui et al., Citation2016). With the above mentioned design strategy, these co-delivery nanoparticles exhibited a PTX encapsulation efficiency of 97% and was capable of prolonged blood circulation time, improved biodistribution profile and controlled, sustained payload release at tumor sites. As expected, these novel targeting nanocarriers required a low PTX dosage to take full advantage of the synergy between cytotoxic and anti-angiogenesis effects of PTX and anti-VEGF siRNA. Efficient inhibition of tumor growth with very low cytotoxicity to non-cancerous tissues was realized in vitro and in vivo.
2. Materials and methods
2.1. Materials
Paclitaxel (PTX) was purchased from Meilun biology Company (Dalian, China). Paclitaxel injection (free PTX) was produced by Harbin pharmaceutical group bioengineering co., Ltd. (Harbin, China). Carboxyfluorescein-labeled siRNA (FAM-siRNA), VEGF siRNA (5′–3′ sequence: GGAGUACCCUGAUGAGAUCTT - GAUCUCAUCAGGGUACUCCTT), negative control siRNA (siNC) were obtained from GenePharma Co., Ltd. (Shanghai, China). Tripeptide lipids was prepared in our laboratory. Sucrose laurate (1% monoester, HLB = 1) was purchased from the Kaiteki company (Japan). RPMI1640 and fetal bovine serum (FBS) were purchased from Invitrogen Life Technologies (USA). Cell Counting Kit-8 (CCK-8) was purchased from Beyotime (Beijing, China). β-actin antibodies, VEGF antibodies and secondary antibodies were supplied by Proteintech (Wuhan, China). NCI-H460 cells were obtained from the Institute of Biochemistry and Cell Biology (China). BALB/c nude mice (4–6 weeks) were used for all in vivo studies. The Institutional Animal Care and Use Committee of Dalian Medical University approved all experiments performed on the animals.
2.2. Preparation of co-delivery nanoparticles
The thin-film hydration method was used to prepare the PTX-loaded nanoparticles (Zhi et al., Citation2013). Briefly, a solution of the tripeptide lipid and the sucrose laurate (L: S = 4:1, weight ratio) in chloroform containing PTX was evaporated in a vacuum rotary evaporator to form a thin lipid film. Then the PTX-loaded nanoparticles were formed by hydrating the lipid film with 1 mL ultrapure water. Then, the PTX-loaded nanoparticles, PTX/LS (L: S: PTX = 8: 2: 1, weight ratio) and PTX/L (L: PTX = 10: 1, weight ratio) were also prepared by the same method. To construct co-delivery nanoparticles (PTX/siRNA/L and PTX/siRNA/LS), the PTX-loaded nanoparticles and VEGF siRNA were quickly mixed and kept at room temperature for 30 min. For targeted delivery, the co-delivery nanoparticles were mixed with folate-PEG2000-DSPE and kept at 37 °C for 1 h to form the targeted co-delivery nanoparticles (PTX/siRNA/FAL and PTX/siRNA/FALS).
2.3. Drug encapsulation efficiency
The amount of PTX encapsulated in nanoparticles was detected by using UV-spectrophotometer (Li et al., Citation2014b). The unencapsulated PTX was removed from nanoparticles by centrifugation at 1000 rpm for 10 min (Jiang et al., Citation2015). PTX was then measured at a wavelength of 227 nm and quantified by comparing the absorbance with the calibration curve. The encapsulation efficiency and drug loading were obtained by measuring the weight of entrapped PTX. The encapsulation efficiency (EE, %) was calculated by the equation and the drug loading (DL, %) was obtained according to the equation
where W1 represents the weight of entrapped PTX, W0 is the weight of total PTX initially added and WL is the weight of total lipids initially added.
2.4. SiRNA retardation assays
Agarose gel retardation assays was utilized to investigate the complexation of siRNA with cationic nanoparticles. Co-delivery nanoparticles were prepared with the increased lipid to VEGF siRNA N/P ratios from 0.6/1 to 19.2/1. Subsequently, the nanoparticles were subjected to electrophoresis on 1% agarose gels for 40 min at 90 V and observed under ultraviolet imaging system (Syngene, Britain).
2.5. Characterization of co-delivery nanoparticles
Morphological analysis of nanoparticles was characterized by transmission electron microscopy (TEM) (JEOL, Japan). All samples were negatively stained by 2% phosphotungstic acid, and measured at 200 kV. The size and zeta potential of nanoparticles were evaluated by the nano particle analyzer (Horiba, Japan). Briefly, the nanoparticles before measurements were diluted with ultrapure water to 20 μg/mL lipid concentration. The size of nanoparticles was determined under 25 °C at a scattering angle of 90°. The zeta potential of the nanoparticles was measured using the same instrument at 25 °C by the electrophoretic mobility. The results were expressed as the mean ± SD.
2.6. Protein adsorption assay
Protein adsorption assay was performed based on a previous reported method (Jiang et al., Citation2015). Nanoparticles were incubated with bovine serum albumin (BSA) at pH 7.4 (final lipid and protein concentration: 0.15 mg/mL and 0.25 mg/mL) for 2 h at 37 °C to allow protein adsorption. To precipitate the protein-adsorbed aggregate, each sample (300 μL) was centrifuged (13,000 g for 15 min). Protein was quantified by using the BCA Protein Assay Kit (Thermo, USA). Absorbance was monitored at 562 nm with a microplate reader (BioTek, USA).
2.7. Hemolysis assay
One milliliter of blood sample obtained from mice was diluted with 4 mL of phosphate-buffered saline (PBS). Red blood cell (RBC) was harvested after being centrifuged (1800 rpm, 10 min) and washed multiple times with PBS to remove serum. After being diluted with 5 mL PBS to obtain the RBC suspension, 200 µL RBC suspension was mixed with 300 µL co-delivery nanoparticles. The hemolysis of RBC in PBS and in ultrapure water was used as negative and positive control, respectively. After incubation at 37 °C for 6 h, the mixture was centrifuged at 5000 rpm for 10 min to remove RBC. The absorbance of released hemoglobin in the supernatant was subsequently determined at 541 nm. The hemolysis percentage was determined according to the following equation: hemolysis where Atest, Apos, Aneg are the absorbance of the treatment groups, positive control (water) and negative control (PBS), respectively.
2.8. Cellular uptake and intracellular distribution of siRNA
For analyzing quantitatively the cellular uptake of the nanoparticles, NCI-H460 cells were seeded in 24-well plates and incubated for 24 h at 37 °C and 5% CO2 to get a confluence of about 80%. The RPMI1640 medium containing 10% FBS and 1% antibiotics (penicillin and streptomycin) were then replaced by a serum-free RPMI1640 medium. Cells were treated with nanoparticles containing 40 nM FAM-siRNA. Following 2 h, 4 h or 6 h culture, cells were washed with PBS and collected in tube. Finally, the cells were resuspended in RPMI1640 medium containing 10% FBS and 1% antibiotics and immediately analyzed with a flow cytometry (BD Accuri C6, USA). Data were processed using Cflow Plus software (Becton-Dickinson) (Zhao et al., Citation2018).
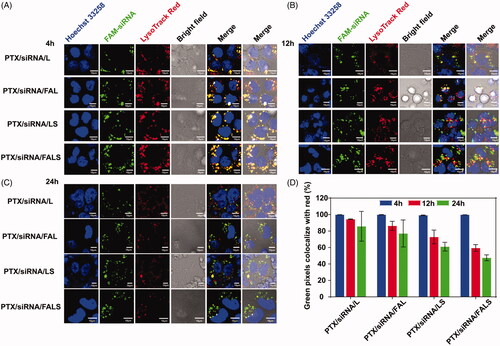
Intracellular distribution of siRNA was observed by confocal laser scanning microscopy (CLSM). NCI-H460 cells were plated onto confocal dishes and incubated under 5% CO2 at 37 °C. After 24 h incubation, the culture medium containing 10% FBS and 1% antibiotics (penicillin and streptomycin) was replaced by serum-free RPMI1640 containing nanoparticles with 40 nM FAM-siRNA. After 6 h culture, the transfection medium was replaced with RPMI1640 containing serum and 1% antibiotics, and the cells were maintained at 37 °C under 5% CO2. After incubation for predetermined time points (4 h, 12 h and 24 h), cells were treated with Lyso Tracker Red (DND-99), and were incubated at 37 °C for 30 min. Cells were then allowed to rinse three times with PBS, followed by fixation with 4% paraformaldehyde at room temperature for 10 min. Nucleus was stained by Hoechst 33258 and cells were observed under a confocal microscope (Olympus, Japan).
2.9. Cytotoxicity and apoptosis of the co-delivery nanoparticles
NCI-H460 cells were seeded at a density of 5000 cells/well in a 96-well plate. After 24 h incubation, the medium was replaced with a serum-free medium containing various concentrations of L, siRNA/L, PTX/L, PTX/siRNA/L, PTX/siRNA/FAL, LS, siRNA/LS, PTX/LS, PTX/siRNA/LS and PTX/siRNA/FALS, respectively. After 6 h culture, the medium was then replaced with RPMI1640 containing serum, and the cells were further incubated for 48 h or 72 h. The cell viability was evaluated by using CCK-8 assay. The absorbance (Abs) at 450 nm was monitored on a plated reader (BioTek Synergy H1, USA). Cell viability was calculated according to the following equation:
NCI-H460 cells were seeded in 24-well plates and incubated at 37 °C under 5% CO2 for 24 h. Then, the medium was replaced with a serum-free medium containing various nanoparticles. After 6 h culture, the medium was replaced with RPMI1640 containing serum and further cultured 12 h or 24 h. Cells were washed with PBS, 200 μL of Hoechst 33258 was added into each well and incubated for 30 min. Cells were washed with PBS for three times. The apoptosis was detected by inverted fluorescence microscope.
2.10. In vitro drug release
The release profiles of PTX from co-delivery nanoparticles were investigated using a dialysis method. Briefly, a dialysis bag (molecular weight cutoff = 1000 Da) with co-delivery nanoparticles was immersed in 40 mL 10% methanol (pH 7.4 and pH 5.5), which was placed in a shaking bed at a stirring speed of 200 rpm at 37 °C. At predetermined time points, 2.0 mL sample was taken from the release medium, followed by supplying a same volume of medium (10% methanol) into the system. The UV spectrophotometer was used to determine the amount of PTX released into 10% methanol.
2.11. In vitro VEGF gene silencing
The Human VEGF immunoassay kit (Proteintech, China) was used to measure the silencing efficacy of VEGF protein. NCI-H460 cells were seeded into 96-well plates and incubated for 24 h. Then, culture medium was removed and serum-free RPMI1640 containing different nanoparticles with 40 nM FAM-siRNA was added. After 6 h culture, the serum-free medium was replaced with fresh complete medium. After 48 h incubation, the medium was collected and analyzed using a human VEGF immunoassay kit according to the manufacturer’s instructions.
2.12. In vivo distribution assay
Approximately 1 × 107 NCI-H460 cells were inoculated into the right armpit of each BALB/c nude mouse. Once the volumes of tumor reached to about 500 mm3, mice were administrated with PTX/siRNA/LS (FAM-siRNA = 0.55 mg/kg, PTX = 8 mg/kg) via tail vein. After the injection, mice were sacrificed at the predetermined intervals. Tumor as well as major organs (liver, lung, heart, spleen, and kidney) were excised and analyzed by a bioluminescence IVIS imaging system (Xenogen, USA).
2.13. In vivo tumor growth inhibition
Approximately 1 × 107 NCI-H460 cells in PBS were subcutaneously injected into the right armpit of BALB/c nude mice to form xenograft. When the xenograft volume reached approximately 250-300 mm3, the mice were randomly divided into eight groups. Different formulations (PTX = 8 mg/kg and/or VEGF siRNA = 0.55 mg/kg) were injected into each mouse once for 2 days by intravenous injection. Mice tumors were measured every two days. The day before the first dose was specified as day 0. A Vernier caliper was used to measure the longest (L) and shortest (S) diameters. Tumor size was calculated according to the formula of V = 0.5LS2. Two days after the last injection, the animals were sacrificed to harvest tumor tissues and fixed in 4% paraformaldehyde, then the tumors were sectioned for TUNEL assay, CD31 immunohistochemistry and hematoxylin and eosin (H&E) stain. All the stained sections were observed by using a fluorescence microscope (Olympus X71, Japan).
2.14. In vivo VEGF downregulation
Western blot was used to determine the expression levels of VEGF, 30 µg of samples were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, USA) (Yoshizawa et al., Citation2008). The membranes were blocked with 5% skimmed milk in tris-buffered saline containing tween-20 (TBST) and then incubated with primary antibodies overnight at 4 °C. Then, PVDF membranes were washed three times with TBST and incubated with horseradish peroxidase-conjugated secondary antibody for 2 h at room temperature. The membrane was washed and developed with BeyoECL Plus reagents and imaged using a BioSpectrum Gel Imaging System (UVP, California, USA).
3. Results and discussion
3.1. Construction and characterization of co-delivery nanoparticles
By rationally optimizing the co-delivery nanoparticles, the strategy for packaging PTX and VEGF siRNA was developed. Firstly, the tripeptide lipid and sucrose laurate were used to encapsulate PTX into liposomes by a thin-film method (). Then, VEGF siRNA was loaded onto PTX-loaded liposome via electrostatic interaction at room temperature () and the targeted functionalized co-delivery nanoparticles were prepared by post-insertion of folate-PEG2000-DSPE (). The detailed characterization of co-delivery nanoparticles was presented in . With the addition of sucrose laurate, the encapsulation efficiency of PTX/LS (green) increased with a significant difference (p < 0.05) compared to PTX/L without sucrose laurate (red) (). Drug loading was also slightly improved. This increased entrapment of PTX was likely due to hydrophobic interactions and steric accommodation (Tai et al., Citation2017) between sucrose laurate and the lipophilic paclitaxel drug (Yan et al., Citation2013; Bnyan et al., Citation2018). In addition, showed that siRNA could be retarded by PTX/LS at an N/P weight ratio of 9.6/1. However, without sucrose laurate the N/P weight ratio increased to 19.2/1 for complete retardation of siRNA. Thus, the addition of sucrose laurate to these nanoparticles markedly increased their interactions with siRNA. Moreover, the interaction of VEGF siRNA with the PTX-loaded nanoparticles did not significantly alter the encapsulation efficiency and drug loading of delivery vehicle. After the insertion of folate-PEG2000-DSPE into the lipid shell, the encapsulation efficiency of PTX/siRNA/FALS was up to 97%, and FA modification did not change the N/P weight ratio for the retardation of siRNA, suggesting that folate-PEG2000-DSPE was a suitable constituent to modify the co-delivery nanoparticles.
Figure 1. Schematic illustration of the construction of co-delivery nanoparticles. The co-delivery nanoparticles were prepared through PTX encapsulating (A), VEGF siRNA loading (B), and folate-PEG2000-DSPE post-inserting (C).
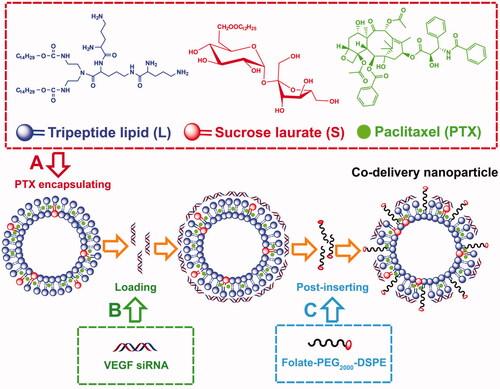
Figure 2. Physicochemical characterization of nanoparticles. (A) Encapsulation efficiency and drug loading of nanoparticles without or with sucrose laurate. (B) Agarose gel electrophoresis of nanoparticles complexed with siRNA; Lane (1): naked siRNA. Lanes (2–7): the weight ratios of lipids to siRNA were (2) 0.6/1, (3) 1.2/1, (4) 2.4/1, (5) 4.8/1, (6) 9.6/1, and (7) 19.2/1. (C) Zeta potentials and (D) sizes of liposomes detected by DLS. (E) TEM images of various nanoparticles. Nanoparticles were negatively stained with 2.0% phosphotungstic acid and observed by transmission electron microscopy. Data are shown as mean ± SD (n = 3).
As important parameters for drug and gene delivery carriers, size and zeta potential of the nanoparticles was measured by Dynamic light scattering (DLS). As shown in , the zeta potentials of L and PTX/L were between 45 mV and 50 mV, and they increased to 55 mV to 60 mV with the addition of sucrose laurate. The slightly higher zeta potential (positive surface charge) of these nanoparticles raised their capacity to form co-delivery nanoparticles due to the increased electrostatic interactions between the cationic nanoparticles and siRNAs. This was consistent with the results of siRNA retardation determined by agarose gel electrophoresis (). Although the zeta potential of the co-delivery nanoparticles decreased with the addition of siRNAs, it remained over 30 mV, leading to a stable colloidal system (Honary & Zahir, Citation2013a). Moreover, nanoparticles with a positive surface charge have the ability to bind to the negative charged cancer cell membrane by a generalized electrostatic interaction, thus yielding a high cellular uptake (Honary & Zahir, 2013 b; Zhao et al., Citation2014). As shown in , the size of the carriers without sucrose laurate (red) was 121.10 nm, 106.47 nm, 125.45 nm and 172.37 nm for L, PTX/L, PTX/siRNA/L and PTX/siRNA/FAL, respectively. And the addition of sucrose laurate (blue) increased their particle size correspondingly. Moreover, due to the interaction between liposomal components and their cargos, the loading of both PTX and VEGF siRNA affected the size of the carrier, but not significantly. We discovered that the sizes of PTX/siRNA/FAL and PTX/siRNA/FALS increased due to the insertion of folate-PEG2000-DSPE on the surface of the lipid shell. Although there are many factors influencing particle size, the co-delivery nanoparticle sizes were found by DLS to lie between 100 nm and 200 nm, which likely ensured the delivery of co-delivery nanoparticles to tumors (Huang et al., Citation2010). The morphology of the nanoparticles was imaged by TEM (), which showed the nanoparticles were approximately spherical in shape, but not entirely consistent with the sizes determined by DLS. The differences were attributed to the fact that the particles measured by the DLS were in a hydrated state in water, while for TEM measurement they were dried after being dropped onto carbon-coated copper grids.
3.2. In vitro simulated blood circulation and release behavior of the nanoparticles
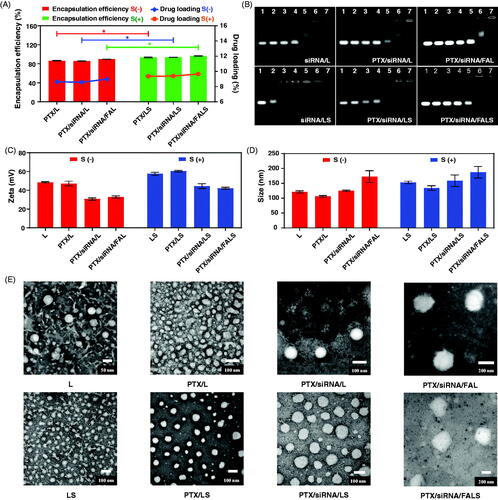
Since the prepared nanoparticles are expected to come into direct contact with blood once systemically administered, a hemolysis test was performed to determine their potential hemolytic toxicity (Dobrovolskaia et al., Citation2008). A 6 h incubation of mouse erythrocytes with the nanoparticles was performed (as shown in ). Very little hemolysis was observed for PTX/siRNA/L, PTX/siRNA/LS, PTX/siRNA/FAL, or PTX/siRNA/FALS samples, with the hemolysis of 1.71%, 1.23%, 0.86% and 0.10%, respectively. Generally, a hemolysis fraction of less than 5% is regarded as being nontoxic and safe (Ramasamy et al., Citation2014). Images of the test samples also showed that after the separation of red cells and supernatant groups, the hemolyzed fractions were significantly different from the positive control (water) group and similar to the negative control (PBS) group (). Moreover, the hemolysis produced by FA modified carriers was significantly (p < 0.05) lower than unmodified co-delivery nanoparticles. This indicated that nanoparticle surface functionalization with PEGylated polyelectrolytes significantly reduced their hemolytic toxicity (Ramasamy et al., Citation2014). In addition, the hemolysis of PTX/siRNA/FALS was significantly (p < 0.05) lower than hemolysis of PTX/siRNA/FAL. This was likely due to the great biocompatibility and less hemolytic of sucrose laurate (Zhao et al., Citation2018). And the addition of the non-ionic surfactant like sucrose laurate resulted in an increased stability of the nanoparticle formulation. Therefore, PTX was well protected and the formulation reduced the direct contact of PTX and cells leading to the decreased incidence of hemolysis (Jeswani et al., Citation2015).
Figure 3. In vitro simulated blood circulation and release behavior of the nanoparticles. (A) Hemolysis assays of nanoparticles after 6 h incubation at 37 °C. (B) Images of the hemolysis of RBCs produced by nanoparticles. a1-a3: negative control (PBS) group, b1–b3: positive control (water) group, c1–c3: PTX/siRNA/L, d1–d3: PTX/siRNA/LS, e1–e3: PTX/siRNA/FAL, f1-f3: PTX/siRNA/FALS. (C) BSA adsorption on nanoparticles after 6 h incubation at 37 °C. (D) In vitro PTX release of nanoparticles at 37 °C (pH = 7.4 and pH = 5.5). (E) Schematic illustration of the targeted delivery and controlled release mechanism of nanoparticles. In a biological environment, nanoparticles were expected to directly contact with red blood cells, leading to the hemolysis (a), and nanoparticles might interact with protein, resulting the protein adsorption of nanoparticles (b). The protein adsorption highly depends on the surface structure of nanoparticles. The high protein adsorption of nanoparticles might prevent exposure of the targeting ligand, resulting a low cell uptake (c). In contrast, a low protein adsorption of nanoparticles would ensure the successful folate receptor-mediated uptake (d), and then nanoparticles could response to the decreased pH in the endosomes, the encapsulated drug would be efficiently released (e).
The surface properties of nanoparticles strongly influence the protein adsorption in blood circulation. Reducing nonspecific protein adsorption of nanoparticles would be expected to prolong their blood circulation time (Noble et al., Citation2014). As shown in , the addition of sucrose laurate significantly decreased protein adsorption to the nanoparticles (p < 0.05). This indicated that the incorporation of sucrose laurate reduced the interaction with negatively charged plasma proteins in the circulation (pH 7.4), resulting in lower non-specific protein absorption. This might be attributed to the competitive adsorption between non-ionic surfactants and proteins (Zhao et al., Citation2014). The lower protein adsorption of targeted co-delivery nanoparticles also benefited from the incorporation of folate-PEG2000-DSPE into the shell, which reduced adsorption of plasma proteins primarily by inhibiting surface interactions, and particularly by avoiding nanoparticle aggregation (Xiao et al., Citation2012). These data showed that the addition of sucrose laurate and folate-PEG2000-DSPE increased the blood circulation time of the nanoparticles and improved their survival as intact drug and gene carriers after intravenous administration.
We next evaluated the degree of protection afforded by nanoparticle encapsulation from release of PTX in the blood circulation system and the maintenance of PTX isolation until controlled release of the drug at the target site. We measured the PTX released from the nanoparticles under simulated physiological conditions in blood (pH 7.4) and its release from simulated endosomal/lysosomal organelles of cancer cells (pH 5.5). As shown in , at pH 7.4 the cumulative PTX release from nanoparticles was low and the cumulative released for all nanoparticles was less than 25% after 6 h. By contrast, at pH 5.5 the PTX release fraction increased significantly (p < 0.05). Both PTX/siRNA/FAL and PTX/siRNA/FALS released more than 88% of PTX after 120 h at pH 5.5. These results indicated that the incorporation of the tripeptide lipid helped to extend stability for efficient delivery from blood circulation and should release PTX in the endosomal/lysosomal environment of cancer cells. We attribute this pH control over release to the presence of carbamate linkers, which are stable at a neutral pH but are susceptible to acid-catalyzed hydrolysis in an acidic environment (Zhi et al., Citation2018). These results also demonstrated that the co-delivery nanoparticles had a sustained release ability, and the release of co-delivery nanoparticles could be further improved by adding sucrose laurates (Szűts & Szabó-Révész, Citation2012) to decrease the surface tension (Youan et al., Citation2003). Sustained release is desirable in anti-cancer therapy, as it increases exposure time to anti-cancer drugs as well as detracts from the possibility that cells adapt combative pathways to thwart toxins and drug resistance (Qu et al., Citation2014). Therefore, we anticipate that the targeted co-delivery nanoparticles could respond to the stimuli of lower pH for controlled drug release, which could contribute to the accumulation of PTX at tumor sites (Qu et al., Citation2014). The targeted delivery and release of nanoparticles were illustrated in , in which the low hemolysis and low protein adsorption of nanoparticles would ensure in vivo safety, successful folate receptor-mediated uptake, and efficient intracellular release of drugs.
3.3. Cellular uptake and intracellular distribution of siRNA
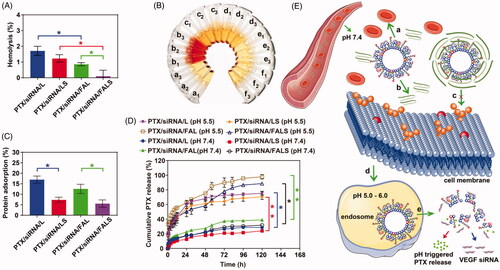
In the case of transfecting siRNA, it is important to obtain to achieve high cellular uptake as well as proper intracellular distribution of siRNA. To measure cellular uptake of co-delivery nanoparticles, we incubated FAM-siRNA-encapsulated co-delivery nanoparticles with NCI-H460 cells and subsequently detected the cells by inverted fluorescence microscopy and flow cytometry. The formulation of FAM-siRNA alone showed minimal fluorescence, while stronger fluorescence was observed in cells that were cultured with co-delivery nanoparticles containing FAM-siRNA. As illustrated in , the fluorescence intensity in cells increased with time, and reached a plateau at 6 h. The strongest mean fluorescence of NCI-H460 cells was observed for the PTX/siRNA/FALS formulation (). Interestingly, the mean fluorescence of PTX/siRNA/LS or PTX/siRNA/FALS was significantly (p < 0.05) stronger than that of PTX/siRNA/L or PTX/siRNA/FAL. This experiment demonstrated that inclusion of sucrose laurate in the nanoparticles notably increased cell uptake of the silencing gene. In particular, compared to PTX/siRNA/FAL, the increased cell uptake of PTX/siRNA/LS likely occurred when the zeta potential increased by the incorporation of sucrose laurate into the nanoparticles (), and thus nanoparticles have stronger interaction with the anionic cell membrane by electrostatic interaction (Honary & Zahir, Citation2013b) to yield the adsorptive-mediated transcytosis (Lakkadwala et al., Citation2019). This increased cell uptake may also benefit from sucrose laurate, as it can enhance the permeation of co-delivery nanoparticles (Kumar and Rajeshwarrao Citation2011). What’s more, the cell uptake of PTX/siRNA/FAL was higher than that of PTX/siRNA/L, similarly, PTX/siRNA/FALS showed higher uptake than PTX/siRNA/LS, suggested that co-delivery nanoparticles modified with FA could increase the cell uptake. The uptake of FA-modified co-delivery nanoparticles (PTX/siRNA/FALS) was believed to result from the combined effect of both folate receptor-mediated and adsorptive-mediated transcytosis that succeeded in increasing cellular uptake and reaching a plateau. The results also indicated that the cell uptake of the formulations in vitro were both lipids and targeted ligand dependent (Lakkadwala et al., Citation2019).
Figure 4. Cell uptake of NCI-H460 cells treated with co-delivery nanoparticles containing FAM-siRNA. (A) Cells were washed with PBS, and observed by inverted fluorescence microscope. Scale bar = 50 μm. (B) Cells were trypsinised and resuspended, and the FAM signal was analyzed by flow cytometer. (C) Mean fluorescence intensity of nanoparticles was calculated. Results are presented as the mean ± SD (n = 3).
After entering the cell, endosomal/lysosomal escape of siRNA is important for subsequent post-transcriptional gene silencing in the cytoplasm (Li et al., Citation2014a). As shown in , at 4 h after transfection most of FAM-siRNA (green fluorescence markers) resided in the endosome/lysosome (red fluorescence markers), as evidenced by colocalization of FAM-siRNA and endosome/lysosome (the co-localization of red and green markers in the merged image generated yellow fluorescence). However, at 12 h after transfection a small portion of green fluorescence separated from the red fluorescence (marked endosome/lysosome) (). As time extended to 24 h, more separation between green and red fluorescence was observed (). The above results suggested FAM-siRNA escaped from the endosomal/lysosomal reservoir and entered the cytoplasm. Moreover, the magnitude of separation of green and red fluorescence can be ranked as PTX/siRNA/L < PTX/siRNA/FAL < PTX/siRNA/LS < PTX/siRNA/FALS (). This triggered escape likely occurred when ion pairs formed between cationic lipids and the anionic lipids in the endosome membrane and thus destabilized the endosomal membrane by excluding surface bound water (Yang et al., Citation2014). Alternatively, these results suggested that with the addition of sucrose laurate, the co-delivery nanoparticles more efficiently escaped from endosomes/lysosomes and released siRNA into the cytoplasm. Their successful escape from the endosome may benefit from sucrose laurate, which enhanced the permeation of co-delivery nanoparticles (Wakabayashi et al., Citation2018) through membranes. Meanwhile, sucrose laurate in formulation might cause the endosomal membrane unstable like others surfactants (Huang et al., Citation2006), helping nanoparticles to destroy endosomes, and then to release siRNA in cytoplasm.
Figure 5. CLSM images of NCI-H460 cells incubated with various nanoparticles containing FAM-siRNA (green) after 4 h (A), 12 h (B) and 24 h (C). Lyso Tracker Red (red) was used to label the endosome/lysosome and Hoechst 33258 (blue) to label nucleus. (D) The ratio of green fluorescence to red fluorescence was quantified with Image-Pro Plus.
3.4. In vitro anti-tumor activity of the co-delivery nanoparticles
The superior uptake efficiency and facile escape from endosomal/lysosomal compartments promotes gene silencing of the non-viral gene vector (Li et al., Citation2014b). The expression levels of VEGF proteins in NCI-H460 cells were measured by the human VEGF ELISA kit after VEGF gene silencing. As shown in , compared to those formulations containing siNC, all nanoparticles with VEGF siRNA exhibited conspicuous VEGF downregulation. The combined delivery of VEGF siRNA and PTX using nanoparticles resulted in significantly lower VEGF expression compared to that by the delivery of VEGF siRNA or PTX alone. Meanwhile, a significantly lower level of VEGF protein was observed after the treatment by PTX/siRNA/LS, compared to PTX/siRNA/L. Additionally, higher silencing effect of VEGF was detected in cells treated with folate targeted co-delivery nanoparticles. This indicated that the FA modification greatly enhanced gene silencing of the co-delivery nanoparticles. The greatest VEGF downregulation in NCI-H460 cells was found in the cells treated with PTX/siRNA/FALS; it reached a silencing efficiency of 92%. From the above results, it is recommended that folate-PEG2000-DSPE and sucrose laurate could be used to increase cellular uptake of PTX/siRNA/FALS, leading to a high gene silencing efficiency.
Figure 6. VEGF protein expression was quantified by the ELISA method. NCI-H460 cells were treated with different nanoparticles after incubation for 48 h. VEGF protein expression of nanoparticles without sucrose laurate (A) and with sucrose laurate (B) was detected. siNC was used as the negative siRNA control. Each bar represents the mean ± SD of at least three experiments. Cell viability of nanoparticles without sucrose laurate (C) and with sucrose laurate (D) was detected by CCK-8 assay. NCI-H460 cells (5000 cells/well) were seeded in a 96-well plate and treated with different formulations for 72 h. Results are presented as the mean ± SD (n = 3). Cell apoptosis of nanoparticles without sucrose laurate (E) and with sucrose laurate (F) was observed by Hoechst 33258 assay. Scale bar = 50 μm.
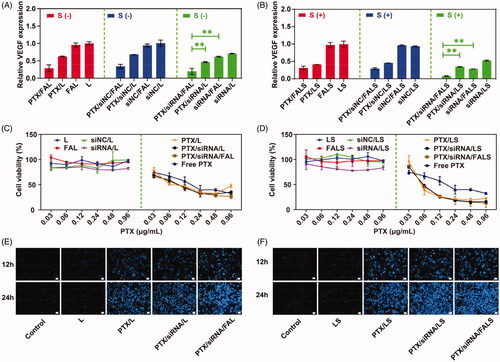
Toxicity against NCI-H460 cells was determined by CCK-8 assay. It could be observed that the cell viability of the blank nanoparticles was maintained in very high levels within the concentration range of experimental samples (Figure S1A & B and ). This indicated that these blank carriers had low cytotoxicity and could be used as a drug carrier. At the same time, in the same experimental concentration range, PTX/L, PTX/LS as well as free PTX showed increased cell inhibition. As listed in , the half-maximal inhibitory concentrations (IC50) values of all nanoparticles loaded with PTX were lower than that of free PTX, indicating that the delivery systems facilitated PTX entry into the cell to some degree and inhibited cell growth better than free PTX. With the addition of sucrose laurate, the IC50 of PTX/LS further decreased to 0.0707 μg/mL, which was 1.9-fold lower than that of PTX/L. The superior uptake efficiency of nanoparticles with sucrose laurate might be attributed to higher levels of PTX in NCI-H460 cells, resulting in more efficient cell inhibition. Moreover, IC50 of PTX/siRNA/LS was slightly lower than that of PTX/LS suggesting that knocking down the VEGF gene could slightly enhance the cell proliferation inhibition of PTX in vitro. The IC50 of FA modified co-delivery nanoparticles was slightly lower than of un-modified co-delivery nanoparticles, implying that the increased cytotoxicity of carriers bearing the FA modification probably benefited from folate receptor-mediated cellular uptake. Additionally, the co-delivery nanoparticles were able to escape from lysosomes, permitting PTX release into cytoplasm, further inhibiting cell growth.
Table 1. In vitro cytotoxicity of nanoparticles against NCI-H460 cells.
The apoptosis indicated that the cells treated with co-delivery nanoparticles presented stronger fluorescence than that treated with the nanoparticles with single PTX or siRNA (). In addition, significantly stronger fluorescence was observed after the treatment by nanoparticles with sucrose laurate () compared to those nanoparticles without sucrose laurate (). It illustrated that with the addition of sucrose laurate, the co-delivery nanoparticles could induce superior cell apoptosis, resulting in more efficient cell growth inhibition ().
3.5. In vivo biodistribution and anti-tumor activity of the co-delivery nanoparticles
To investigate the accumulation of nanoparticles in tumors and other major organs, FAM-siRNA was used to track the distribution of the nanoparticle carriers in an ex vivo evaluation of excised tissues (heart, liver, spleen, lung, and kidney) and tumors. The particles were much likely to accumulate in tumors, and nearly no fluorescence in organs was observed, though PTX/siRNA/LS nanocarriers showed slight fluorescence for liver (). The low nanoparticle accumulation in liver might be due to the great biocompatibility and a low non-specific protein absorption () of the tripeptide lipid nanoparticles, which help them to avoid the recognition and uptake by the mononuclear phagocyte system (MPS)/reticuloendothelial system (RES) (Overchuk & Zheng, Citation2018). What’s more, organic nanoparticles are more rapidly degraded and eliminated by macrophages in liver (Wilhelm et al., Citation2016). Meanwhile, the co-delivery nanoparticle sizes were found by DLS to lie between 100 nm and 200 nm (), which could transport passively through the interendothelial cell gaps across vessel wall (100 − 500 nm) (Wilhelm et al., Citation2016) and accumulate in tumors via the enhanced permeability and retention (EPR) effect (Zamboni et al., Citation2012). In addition, the tumor accumulation of PTX/siRNA/FALS was significantly higher than that of PTX/siRNA/LS, which suggested that the FA modification significantly enhanced the targeting of the nanoparticles to tumors by folate receptor-mediated delivery (Yameen et al., Citation2014). As the tumor accumulation of PTX/siRNA/FALS was significantly higher than of PTX/siRNA/FAL, we also suggested that sucrose laurate significantly improved the directed delivery of siRNA to tumors.
Figure 7. (A) Ex vivo imaging of tumor and organs excised from BALB/c nude mice. Tumor and major organs were collected from the mice at 6 h post-injection, and then the fluorescence signals were detected. (B) Quantitative analysis of the fluorescence intensity of ex vivo images of tumors. (C) Quantitative analysis of the fluorescence intensity of ex vivo images of organs. In vivo anti-tumor efficacy of various nanoparticle formulations against NCI-H460 tumor in mice. (D) Imaging of the tumors collected from mice at the termination of treatment. (E) Tumor growth curve after injection of different nanoparticles for a total five doses. *p < 0.05, **p < 0.01, compared to PTX/siRNA/FALS group. (F) VEGF expression level in tumor was analyzed by Western Blotting. (G) Quantitative analysis of the Western blot bands. The VEGF expression level was normalized to β-actin and analyzed. Data are shown as mean ± SD.
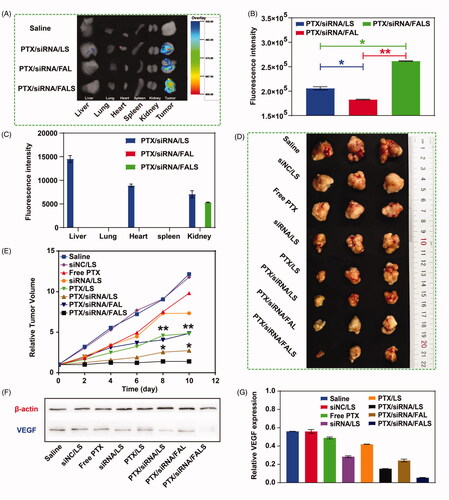
Anti-tumor therapeutic efficacy of nanoparticles was evaluated in BALB/c nude mice bearing NCI-H460 subcutaneous tumors. As shown in , PTX/LS showed a little stronger tumor inhibition than free PTX, due to the improvement of the bioavailability of PTX in vivo. Meanwhile, single VEGF siRNA in the nanoparticles can only slightly retard tumor growth. However, when PTX and VEGF siRNA were co-delivered by the nanoparticles, the anti-tumor activity was improved dramatically via the synergistic effect. What’s more, the relative tumor volume of targeted PTX/siRNA/FALS group was 1.95-fold smaller than that treated with non-targeted PTX/siRNA/LS, demonstrating that FA modified nanoparticle was capable to improve anti-tumor activity by targeted delivery. More interestingly, the tumor inhibition of targeted PTX/siRNA/FALS was 3.46-fold higher than that treated with targeted PTX/siRNA/FAL. It displayed that nanoparticles with the addition of sucrose laurate could lead to more effective tumor inhibition. The above results and the supplement data (Figure S2 A and B) showed that PTX/siRNA/FALS was a promising vector to efficiently co-deliver PTX and siRNA for lung cancer treatment.
3.6. In vivo anti-tumor mechanism of the co-delivery nanoparticles
To study the synergistic mechanism for tumor inhibition afforded by PTX and VEGF siRNA, expression of VEGF protein in the excised tumor tissues was evaluated by Western blotting, and the tumor was also evaluated for TUNEL assay, CD31 immunohistochemistry, and H&E staining. As shown in , mice treated with PTX/siRNA/FALS showed the greatest inhibition of VEGF protein expression. PTX/siRNA/LS also showed significant VEGF downregulation compared to siRNA/LS or PTX/LS. In addition, PTX/siRNA/FALS with sucrose laurate in their delivery vehicles were more potent inhibitors of VEGF expression in tumors than PTX/siRNA/FAL. Thus, the overall results from anti-tumor formulations that were tested showed that the PTX/siRNA/FALS formulation was the most promising vector to efficiently downregulate VEGF expression.
To evaluate the effectiveness of anti-angiogenesis of the above formulations, CD31 antibody (Yang et al., Citation2018), was used to stain endothelial cells in the tumor cross-sections. Compared to saline group, monotherapy performed with siRNA/LS showed slightly lower CD31 staining (), suggesting that this treatment with VEGF siRNA alone produced insignificant anti-angiogenesis. Compared to the monotherapy group, PTX/siRNA/LS, PTX/siRNA/FALS, and PTX/siRNA/FAL all significantly lowered the expression of CD31 markers, indicating apparent inhibition of angiogenesis in these tumors. All corresponding samples also showed similarly low expression of VEGF protein in the above Western blot assays (). It appeared that PTX co-delivered in the combined dual agent formulations PTX/siRNA/LS and PTX/siRNA/FALS sensitized the tumor vasculature to anti-angiogenesis therapy. It has been demonstrated that PTX could lead to a decrease in microvessel density by inducing apoptosis in tumor-associated endothelial cells (Trojan, Citation2009). The targeted nanoparticle PTX/siRNA/FALS formulation had the greatest inhibition of neovascularization and tumor growth based on the greatest accumulation of VEGF siRNA and PTX at tumor sites. These results verified our hypothesis that anti-angiogenesis effectiveness of these nanoparticle formulations was tied to the downregulation of VEGF expression and apoptosis of vascular endothelial cells via the synergistic effect of PTX and VEGF siRNA.
Figure 8. (A) CD31 immunohistochemistry analysis of tumor tissues. (B) TUNEL assay of tumor apoptotic cells in tumor tissues. The tumor sections were stained with fluorescein-dUTP (green) for apoptosis. (C) H&E staining of lung tumors tissues treated with various formulations. Scale bar is 50 µm.
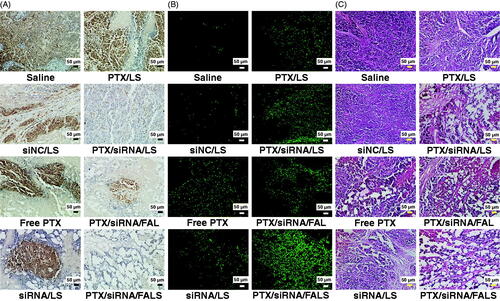
As shown in , no apparent apoptosis was detected in the saline or siNC/LS groups. The monotherapy group vectors siRNA/LS and PTX/LS produced less tumor apoptosis, while a generally more widely extended apoptotic field was seen for the co-delivery nanoparticles. In this research, though tumor apoptosis was primarily attributed to PTX, VEGF expression inhibiton in vivo was also capable of inducing tumor apoptosis. This might be associated with suppressing tumor angiogenesis that could reduce the supply of oxygen and nutrition to tumor tissue. It is important that the greater yield of apoptotic tumor cells was produced by the PTX/siRNA/FALS formulation in comparison to the PTX/siRNA/LS formulation, suggesting that FA modified co-delivery nanoparticles more effectively sensitized cells to the synergistic effects of PTX and VEGF siRNA, perhaps enabled by more efficient targeting. In addition, PTX/siRNA/FALS produced a greater apoptotic outcome in terms of overall cell involvement than PTX/siRNA/FAL. This suggested that the addition of sucrose laurate in targeted delivery nanoparticles further advanced the apoptosis of tumor tissues. At the same time, H&E staining of lung tumors () showed that many mitotic cells could be seen in the tumor tissues of the control group, while all the groups injected with PTX and/or VEGF siRNA exhibited tumor cell contraction and separation from surrounding cells, suggesting that tumor progression was obviously inhibited. The PTX/siRNA/FALS co-delivery nanoparticle demonstrated the greatest proficiency at inhibiting tumor growth. This formulation, comprising tripeptide lipid, sucrose laurate, and folate-PEG2000-DSPE, not only targeted delivery of the dual agents PTX and VEGF siRNA to the tumor site, but also released drugs at the target sites in a controllable manner, leading to the synergistic effect of PTX and VEGF siRNA in tumor tissue.
3.7. In vivo toxicity studies
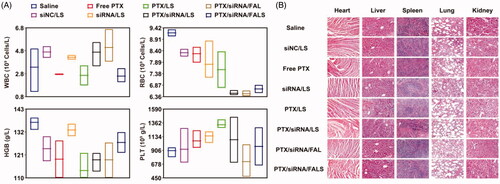
A low level of toxicity and preserving homeostasis in the general cell population is a prerequisite feature for drug delivery systems that are being evaluated for cancer therapy. To investigate the in vivo toxicity of the co-delivery nanoparticles, we measured the body weight change as well as hematology markers and histological markers by H&E staining of excised organs after different treatments. As illustrated in Figure S3, no obvious mouse body weight change occurred in all groups over the duration of the treatments. Representative hematology markers were analyzed including assessment of white blood cell counts (WBC), red blood cell counts (RBC), hemoglobin (HGB) and platelet counts (PLT). The WBC, RBC, HGB and PLT values of all groups were within the normal range at the treatment end point (). The difference in toxicity between therapeutic groups and the saline group was further analyzed by H&E staining of the organs including heart, liver, spleen, lung and kidney. As illustrated in , no apparent abnormality or lesion was observed in the major organs, suggesting that the co-delivery nanoparticles produced little tissue damage. The results of body weight change, hematology markers, and histological markers of these organs confirmed that the co-delivery nanoparticles had little side effects. The low toxicity achieved by co-delivery nanoparticles can be first attributed to the isolation of PTX by its encapsulation, especially during its circulation in blood, and the controlled drug release at the target site. Importantly, the synergistic combination of chemotherapy drugs and siRNA permits one to decrease toxicity and overcome morbidity associated with the high doses of drugs by countering them with a strategy that allows the reduction of dosage of both drug and gene agents while still permitting specific multi-targeting mechanisms to prevail.
Figure 9. (A) Effects of different nanoparticles on hematology of mice were analyzed by SYSMEX XE-2100 Hematology Analyzer and expressed by the floating bars (min to max). Representative hematology markers were analyzed including assessment of white blood cell counts (WBC), red blood cell counts (RBC), hemoglobin (HGB), and platelet counts (PLT). (B) Histological analysis of organs extracted from tumor-bearing mice. Effects of co-delivery nanoparticles on the histopathology of mice organs in toxicity tests. Organ samples were embedded in paraffin, and sections were stained with H&E and viewed under a light microscope at 200× magnification.
4. Conclusion
In this study, a series of nanoparticle formulations were constructed for their effectiveness at co-delivering VEGF siRNA and PTX therapeutic agents. The modified co-delivery nanoparticles (PTX/siRNA/FALS) were found to yield the best performances with the greatest tumor suppression in an in vivo mouse lung tumor model. It preferentially targeted malignant cells expressing the folate receptor and the controlled drug release was triggered by acid-hydrolysis of tripeptide lipids in the nanoparticles. Owing to the exceptional biocompatibility of both tripeptide lipid and sucrose laurate in the delivery vector, as well as the reduced dosage of PTX via the synergistic therapeutic effect, the PTX/siRNA/FALS formulation showed low toxicity to normal tissues in the mouse model. This work provides an efficient and safe approach to the co-delivery of siRNA and PTX via PTX/siRNA/FALS delivery system for tumor targeted therapy.
| Abbreviation | ||
| FA | = | folate-PEG2000-DSPE |
| PTX | = | paclitaxel |
| VEGF siRNA | = | siRNA targeting VEGF gene |
| siNC | = | negative control siRNA |
| L | = | tripeptide lipid nanoparticles |
| FAL | = | FA modified L |
| siNC/L | = | siNC loaded L |
| siRNA/L | = | VEGF siRNA loaded L |
| siNC/FAL | = | siNC loaded FAL |
| siRNA/FAL | = | VEGF siRNA loaded FAL |
| PTX/L | = | PTX loaded L |
| PTX/FAL | = | PTX loaded FAL |
| PTX/siNC/L | = | PTX and siNC co-loaded L |
| PTX/siRNA/L | = | PTX and VEGF siRNA co-loaded L |
| PTX/siNC/FAL | = | PTX and siNC co-loaded FAL |
| PTX/siRNA/FAL | = | PTX and VEGF siRNA co-loaded FAL |
| LS | = | tripeptide lipid/sucrose laurate nanoparticles |
| FALS | = | FA modified LS |
| siNC/LS | = | siNC loaded LS |
| siRNA/LS | = | VEGF siRNA loaded LS |
| siNC/FALS | = | siNC loaded FALS |
| siRNA/FALS | = | VEGF siRNA loaded FALS |
| PTX/LS | = | PTX loaded LS |
| PTX/FALS | = | PTX loaded FALS |
| PTX/siNC/LS | = | PTX and siNC co-loaded LS |
| PTX/siRNA/LS | = | PTX and VEGF siRNA co-loaded LS |
| PTX/siNC/FALS | = | PTX and siNC co-loaded FALS |
| PTX/siRNA/FALS | = | PTX and VEGF siRNA co-loaded FALS |
Supplemental Material
Download ()Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bnyan R, Khan I, Ehtezazi T, et al. (2018). Surfactant effects on lipid-based vesicles properties. J Pharm Sci 107:1237–46.
- Chen S, Hanning S, Falconer J, et al. (2019). Recent advances in non-ionic surfactant vesicles (niosomes): fabrication, characterization, pharmaceutical and cosmetic applications. Eur J Pharm Biopharm 144:18–39.
- Cui SHS, Hui Zhi DFD, Fu Zhao YNY, et al. (2016). Cationic lioposomes with folic acid as targeting ligand for gene delivery. Bioorg Med Chem Lett 26:4025–9.
- Das S, Ng WK, Tan RBH. (2014). Sucrose ester stabilized solid lipid nanoparticles and nanostructured lipid carriers. I. Effect of formulation variables on the physicochemical properties, drug release and stability of clotrimazole-loaded nanoparticles. Nanotechnology 25:105101.
- Dobrovolskaia MA, Aggarwal P, Hall JB, et al. (2008). Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol Pharm 5:487–95.
- Dykxhoorn DM, Lieberman J. (2006). Knocking down disease with siRNAs. Cell 126:231–5.
- Feng Q, Yu M, Wang J, et al. (2014). Synergistic inhibition of breast cancer by co-delivery of VEGF siRNA and paclitaxel via vapreotide-modified core-shell nanoparticles. Biomaterials 35:5028–38.
- Herbst RS, Morgensztern D, Boshoff C. (2018). The biology and management of non-small cell lung cancer. Nature 553:446–54.
- Honary S, Zahir F. (2013a). Effect of zeta potential on the properties of nano-drug delivery systems - a review (Part 2.). Trop. J. Pharm. Res 12:265–73.
- Honary S, Zahir F. (2013b). Effect of zeta potential on the properties of nano-drug delivery systems - a review (Part 1.). Trop. J. Pharm. Res 12:255–64.
- Huang L, Sullenger B, Juliano R. (2010). The role of carrier size in the pharmacodynamics of antisense and siRNA oligonucleotides. J Drug Target 18:567–74.
- Huang Y, Gao J, Chen J, et al. (2006). Cationic liposomes modified with non-ionic surfactants as effective non-viral carrier for gene transfer. Colloids Surf B Biointerfaces 49:158–64.
- Jeswani G, Alexander A, Saraf S, et al. (2015). Recent approaches for reducing hemolytic activity of chemotherapeutic agents. J Control Release 211:10–21.
- Jiang L, Li L, He X, et al. (2015). Overcoming drug-resistant lung cancer by paclitaxel loaded dual-functional liposomes with mitochondria targeting and pH-response. Biomaterials 52:126–39.
- Kumar GP, Rajeshwarrao P. (2011). Nonionic surfactant vesicular systems for effective drug delivery—an overview. Acta Pharm Sin B 1:208–19.
- Lakkadwala S, dos Santos Rodrigues B, Sun C, et al. (2019). Dual functionalized liposomes for efficient co-delivery of anti-cancer chemotherapeutics for the treatment of glioblastoma. J. Control. Release 307:247–60.
- Li Y, Cheng Q, Jiang Q, et al. (2014a). Enhanced endosomal/lysosomal escape by distearoyl phosphoethanolamine-polycarboxybetaine lipid for systemic delivery of siRNA. J Control Release 176:104–14.
- Li Y, Liu R, Yang J, et al. (2014b). Dual sensitive and temporally controlled camptothecin prodrug liposomes codelivery of siRNA for high efficiency tumor therapy. Biomaterials 35:9731–45.
- Ma D. (2014). Enhancing endosomal escape for nanoparticle mediated siRNA delivery. Nanoscale 6:6415–25.
- Noble GT, Stefanick JF, Ashley JD, et al. (2014). Ligand-targeted liposome design: challenges and fundamental considerations. Trends Biotechnol 32:32–45.
- Overchuk M, Zheng G. (2018). Overcoming obstacles in the tumor microenvironment: Recent advancements in nanoparticle delivery for cancer theranostics. Biomaterials 156:217–37.
- Qu M-H, Zeng R-F, Fang S, et al. (2014). Liposome-based co-delivery of siRNA and docetaxel for the synergistic treatment of lung cancer. Int J Pharm 474:112–22.
- Ramasamy T, Haidar ZS, Tran TH, et al. (2014). Layer-by-layer assembly of liposomal nanoparticles with PEGylated polyelectrolytes enhances systemic delivery of multiple anticancer drugs. Acta Biomater 10:5116–27.
- Saharinen P, Eklund L, Pulkki K, et al. (2011). VEGF and angiopoietin signaling in tumor angiogenesis and metastasis. Trends Mol Med 17:347–62.
- Sausville EA. (2001). The challenge of pathway and environment-mediated drug resistance. Cancer Metastasis Rev 20:117–22.
- Sia D, Alsinet C, Newell P, et al. (2014). VEGF signaling in cancer treatment. Curr Pharm Des 20:2834–42.
- Sun W, Wang Y, Cai M, et al. (2017). Codelivery of sorafenib and GPC3 siRNA with PEI-modified liposomes for hepatoma therapy. Biomater Sci 5:2468–79.
- Szűts A, Szabó-Révész P. (2012). Sucrose esters as natural surfactants in drug delivery systems-a mini-review. Int J Pharm 433:1–9.
- Tai K, He X, Yuan X, et al. (2017). A comparison of physicochemical and functional properties of icaritin-loaded liposomes based on different surfactants. Colloids Surf. A 518:218–31.
- Trojan. (2009). Paclitaxel encapsulated in cationic liposomes: a new option for neovascular targeting for the treatment of prostate cancer. Oncol. Rep 25:223–30.
- Wakabayashi R, Sakuragi M, Kozaka S, et al. (2018). Solid-in-oil peptide nanocarriers for transcutaneous cancer vaccine delivery against melanoma. Mol Pharm 15:955–61.
- Wilhelm S, Tavares AJ, Dai Q, et al. (2016). Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater 1:16014.
- Xiao R, Wang R, Zeng Z, et al. (2012). Application of poly(ethylene glycol)-distearoylphosphatidylethanolamine (PEG-DSPE) block copolymers and their derivatives as nanomaterials in drug delivery. Int J Nanomedicine 7:4185–98.
- Xie X, Shao X, Ma W, et al. (2018). Overcoming drug-resistant lung cancer by paclitaxel loaded tetrahedral DNA nanostructures. Nanoscale 10:5457–65.
- Xu X, Ho W, Zhang X, et al. (2015). Cancer nanomedicine: from targeted delivery to combination therapy. Trends Mol Med 21:223–32.
- Yameen B, Choi WI, Vilos C, et al. (2014). Insight into nanoparticle cellular uptake and intracellular targeting. J Control Release 190:485–99.
- Yan F, Li L, Deng Z, et al. (2013). Paclitaxel-liposome-microbubble complexes as ultrasound-triggered therapeutic drug delivery carriers. J Control Release 166:246–55.
- Yang W, Zhang Y. (2012). RNAi-mediated gene silencing in cancer therapy. Expert Opin Biol Ther 12:1495–504.
- Yang Y, Hu Y, Wang Y, et al. (2012). Nanoparticle delivery of pooled siRNA for effective treatment of non-small cell lung cancer. Mol Pharmaceutics 9:2280–9.
- Yang Y, Meng Y, Ye J, et al. (2018). Sequential delivery of VEGF siRNA and paclitaxel for PVN destruction, anti-angiogenesis, and tumor cell apoptosis procedurally via a multi-functional polymer micelle. J Control Release 287:103–20.
- Yang ZZ, Li JQ, Wang ZZ, et al. (2014). Tumor-targeting dual peptides-modified cationic liposomes for delivery of siRNA and docetaxel to gliomas. Biomaterials 35:5226–39.
- Yoshizawa T, Hattori Y, Hakoshima M, et al. (2008). Folate-linked lipid-based nanoparticles for synthetic siRNA delivery in KB tumor xenografts. Eur J Pharm Biopharm 70:718–25.
- Youan BBC, Hussain A, Nguyen NT. (2003). Evaluation of sucrose esters as alternative surfactants in microencapsulation of proteins by the solvent evaporation method. AAPS PharmSci 5:123–31.
- Zamboni WC, Torchilin V, Patri AK, et al. (2012). Best practices in cancer nanotechnology: perspective from NCI nanotechnology alliance. Clin Cancer Res 18:3229–41.
- Zhang Y, Schwerbrock NM, Rogers AB, et al. (2013). Codelivery of VEGF siRNA and gemcitabine monophosphate in a single nanoparticle formulation for effective treatment of NSCLC. Mol Ther 21:1559–69.
- Zhao Q, Liu D, Long Z, et al. (2014). Effect of sucrose ester concentration on the interfacial characteristics and physical properties of sodium caseinate-stabilized oil-in-water emulsions. Food Chem 151:506–13.
- Zhao Y, Liu A, Du Y, et al. (2018). Effects of sucrose ester structures on liposome-mediated gene delivery. Acta Biomater 72:278–86.
- Zhao YN, Qureshi F, Zhang S-B, et al. (2014). Novel Gemini cationic lipids with carbamate groups for gene delivery. J Mater Chem B 2:2920–8.
- Zhi D, Bai Y, Yang J, et al. (2018). A review on cationic lipids with different linkers for gene delivery. Adv Colloid Interface Sci 253:117–40.
- Zhi D, Zhang S, Qureshi F, et al. (2013). Structure-activity relationship of carbamate-linked cationic lipids bearing hydroxyethyl headgroup for gene delivery. Colloids Surf B Biointerf 112:537–41.