
Abstract
Local anesthetics (LAs) have been widely applied in clinic for regional anesthesia, postoperative analgesia, and management of acute and chronic pain. Nanostructured lipid carriers (NLCs) and lipid–polymer hybrid nanoparticles (LPNs) are reported as good choices for LA therapy. Transactivated transcriptional activator (TAT) was reported as a modifier for the topical delivery of drugs. In the present study, TAT modified, levobupivacaine (LEV) and dexmedetomidine (DEX) co-delivered NLCs (TAT-LEV&DEX-NLCs, T-L&D-N) and LPNs (TAT-LEV&DEX-LPNs, T-L&D-L) were designed and compared for the LA therapy. T-L&D-L exhibited better efficiency in improving the skin permeation, analgesic time, and pain control intensity than T-L&D-N both in vitro and in vivo. On the other side, T-L&D-N also improved the therapeutic effect of drugs to a large extent. These two systems both exhibited superiority in some respects. TAT modified LPNs are more promising platform for the long-term local anesthesia.
Introduction
Local anesthetics (LAs) have been widely applied in clinic for regional anesthesia, postoperative analgesia, and management of acute and chronic pain (Andreae & Andreae, Citation2013; de Araújo et al., Citation2019; Levene et al., Citation2019). However, the commercially LAs (lidocaine and bupivacaine) have short-term effects (lasting for hours after a single administration) due to rapid clearance from the injected site, thus resulting repeated injections to control of prolonged, acute or chronic pain (McLure & Rubin, Citation2005; Golembiewski & Dasta, Citation2015). Moreover, systemic toxicity is another clinical limitation including cardiovascular and central nervous systemic toxicity (Brown et al., Citation1995; Shomorony et al., Citation2019). Therefore, there is significant interest in exploiting novel pharmaceutical dosage forms or therapeutic strategies including combination therapy and transdermal drug delivery to prolong the action time and reduce toxicity.
Bupivacaine, a long-acting LA with eight hours of duration of action, has been the most widely used agent during surgical procedures and postoperative pain (Casati & Putzu, Citation2005). Compared to bupivacaine, levobupivacaine (LEV), the pure left-isomer of bupivacaine, provides a similar long-lasting block as racemic bupivacaine, but a greater margin of safety due to less toxic both on cardiovascular system and central nervous system (Casati & Putzu, Citation2005; Cereda et al., Citation2018). The duration of nerve block from bupivacaine or LEV can be prolonged synergistically by co-administration of α2-adrenergic agonist dexmedetomidine (DEX) (Brummett et al., Citation2008; El-Boghdadly et al., Citation2017; Hussain et al., Citation2017; Adel Elmaddawy et al., Citation2018; Rwei et al., Citation2018; El Baz & Farahat, Citation2019; Varshney et al., Citation2019). In our study, we devoted ourselves into designing transdermal drug delivery system (TDDS) to co-deliver LEV and DEX.
Compared with approved injection administration of DEX and bupivacaine, TDDS is a promising alternative way for its following advantages: lower side effects; continuous drug delivery; long-term action and improved patients’ compliance (Blanco et al., Citation2003; Prausnitz & Langer, Citation2008). LEV has a high log P (4.74), which means it is highly soluble in lipids and suitable for delivery by TDDS. Meanwhile, the physicochemical properties of DEX still provide an approach for TDDS including lower molecular weight (200.28 Da) and daily dose (1.2 mg), and proper log P (2.11). For the transdermal delivery of LEV and DEX, the main limitations are to overcome the barrier of the stratum corneum that impedes the penetration of drugs and achieve sustained drug release that prolongs duration of action.
Transactivated transcriptional activator (TAT), one of cell penetrating peptides (CPPs), is a short amino acid sequences able to carry molecules across the barrier of the stratum corneum (Nasrollahi et al., Citation2012; Chu et al., Citation2017; Pescina et al., Citation2018). Recently, TAT has been reported as vehicles for the topical delivery of LAs and achieved some breakthrough results (Wang et al., Citation2013, Citation2016; Chen & You, Citation2017). Nanostructured lipid carriers (NLCs) have been used as drug delivery vehicles for local anesthesia (Ribeiro et al., Citation2016). TAT modified NLCs (TAT-NLCs) were also engineered by Wang et al. for the delivery of lidocaine, achieving enhanced transdermal delivery and anesthesia effect (Wang et al., Citation2016). Other researchers argued that lidocaine-loaded lipid–polymer hybrid nanoparticles (LPNs) are good choice of for LA therapy (Wang et al., Citation2016).
In the present study, TAT modified, LEV and DEX co-delivered NLCs (TAT-LEV&DEX-NLCs, T-L&D-N) and LPNs (TAT-LEV&DEX-LPNs, T-L&D-L) were designed and applied for the LA therapy. They were compared in all respects to determine which one is better for local anesthesia analgesic.
Materials and methods
Materials
Glycerol tripalmitate (GTP), oleic acid (OA), LEV, and DEX were obtained from Sigma-Aldrich (St. Louis, MO). TAT peptide was provided by Chinese Peptide Co., Ltd. (Hangzhou, China). Polyethylene glycol2000 (H2N-PEG-COOH) was provided by Lipoid GmbH (Ludwigshafen, Germany). Poly(d,l-lactic-co-glycolic acid) (PLGA 50:50; MW 0.5–1.5 W) was purchased from Jinan Daigang Biomaterial Co., Ltd. (Jinan, China). All other reagents and solvents were analytical or high performance liquid chromatography (HPLC) grade.
Animals
Sprague-Dawley rats (SD rats, 350–400 g, 10–12 weeks old) and Kunming mice (4–6 weeks old, 18–22 g weight) were purchased from the Medical Animal Test Center of Shandong University and used for experiments after 1 week of acclimatization. Animal experiments were conducted according to the policy of the National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978) and approved by the Medical Ethics Committee of Weifang Medical University (2019SDL098).
Synthesis of TAT conjugated lipid ligand
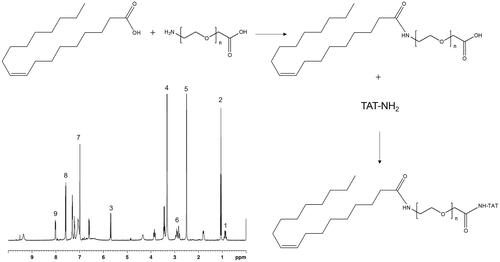
TAT conjugated lipid ligand was synthesized by conjugating TAT with OA, using PEG as a linker (TAT-PEG-OA, ). PEG-OA was first synthesized by dissolving OA, EDC·HCl, and NHS (molar ratio, 1:1.5:1.5) in DMSO under stirring (1 h at room temperature). Then, H2N-PEG-COOH was dissolved in DMSO, added dropwise into the OA mixture under stirring (10 h at room temperature) to achieve PEG-OA (Tan & Wang, Citation2018). EDC·HCl and NHS were then added to PEG-OA (molar ratio, 1.5:1.5:1), followed by adding TAT-NH2 (dissolved in DMSO) dropwise into PEG-OA with gentle stirring (400 rpm at room temperature) temperature for about 24 h (Zhu et al., Citation2014). The solvent was then dialyzed against deionized water for 48 h and lyophilized to get TAT-PEG-OA. The chemical structure of TAT-PEG-OA was confirmed by 1H NMR spectrum (). Peaks 1–3 belong to OA, peak 4 refers to PEG, TAT showed the peaks 5–9.
Figure 1. Synthesis of TAT conjugated lipid ligand: TAT conjugated lipid ligand was synthesized by conjugating TAT with OA, using PEG as a linker (TAT-PEG-OA). The chemical structure of TAT-PEG-OA was confirmed by 1H NMR spectrum.
Preparation of T-L&D-N
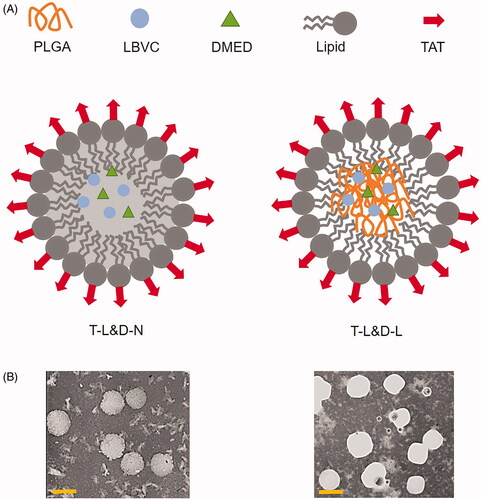
T-L&D-N () was prepared by the hot high pressure homogenization technique (Vitorino et al., Citation2013). Briefly, TAT-PEG-OA (150 mg), GTP (100 mg), LEV (10 mg), and DEX (10 mg) were melted as the lipid phase and then added into the Tween® 80 (0.5%, w/v) hot solution (80 °C), followed by high speed shearing (20,000 rpm for 1 min). The mixture was subjected to a high-pressure homogenization procedure (five cycles at 50 MPa). The T-L&D-N was then cooled and stored at 4 °C. TAT modified, non-drug contained blank NLCs (T-N) were prepared using no drug. Non-TAT modified LEV and DEX co-delivered NLCs (L&D-N) were prepared using PEG-OA instead of TAT-PEG-OA.
Figure 2. Preparation of T-L&D-N and T-L&D-L (A): T-L&D-N was prepared by the hot high pressure homogenization technique and T-L&D-L was prepared by the single-step nanoprecipitation technique. The TEM images of T-L&D-N and T-L&D-L (B).
Preparation of T-L&D-L
T-L&D-L () was prepared by the single-step nanoprecipitation technique (Zhang et al., Citation2008). Briefly, TAT-PEG-OA (100 mg) and lecithin (50 mg) were dissolved in ethanol (4%) heated to 65 °C to ensure all lipids were in liquid phase. PLGA (100 mg), LEV (10 mg), and DEX (10 mg) were dissolved in acetonitrile and added into the preheated liquid phase dropwise under gentle stirring. The mixed solution was vortexed vigorously (3 min) followed by gentle stirring (2 h) at room temperature. The remaining organic solvent and free molecules were removed by washing the NP solution three times using a filter (molecular weight cutoff of 10 kDa). TAT modified, non-drug contained blank LPNs (T-L) were prepared without any drug. Non TAT modified LEV and DEX co-delivered LPNs (L&D-L) were prepared using PEG-OA instead of TAT-PEG-OA. All the formulations were stored at 4 °C before use.
Particle size, zeta potential, and drug entrapment determination
The morphology of NLCs or LPNs was investigated using a transmission electron microscope (TEM, Hitachi, Tokyo, Japan). Average particle size and zeta potential of NLCs or LPNs were determined by photon correlation spectroscopy (Zetasizer 3000, Malvern, Worcestershire, UK) (Tsai et al., Citation2012). The formulations were diluted with double-distilled water before the measurements.
The total concentration of LEV in the nanoparticles was determined by using an HPLC system (Gao & Song, Citation2004). Reversed phase chromatography was performed using a C18 Gemini RP column (150 × 4.6 mm). The flow rate was set at 1 mL/min and the column effluent was monitored at 263 nm. DEX drug loading was determined by HPLC at 214 nm after disrupting the nanoparticles with 100 mM octyl β-d-glucopyranoside (Rwei et al., Citation2018). Drug entrapment efficiency (EE) was calculated using the equation: EE (%)=(Ctotal – Cfree)/Ctotal×100, where Ctotal is the total concentration of drugs in nanoparticles systems and Cfree refers to the concentration of un-entrapped drugs.
Stability of NLCs and LPNs
The long-term stability of T-L&D-N and T-L&D-L was determined after storing the formulations under the condition of 2–8 °C (Zheng et al., Citation2019). At each time point, the mean particle size was measured by the same method in the above section.
In vitro drug release kinetics
In vitro drug release kinetics was performed by dialyzing method (Song et al., Citation2018). The samples were sealed in dialysis bags (molecular weight cutoff of 20 kDa) and immersed in 100 mL of phosphate-buffered saline (PBS) in the presence of 10% FBS placed in a shaking bed (37 °C with a rotation speed of 100 rpm). Samples were collected at predetermined time intervals and replaced with fresh release media. The release solution was subjected to determining the drug content via the method in the ‘Particle size, zeta potential, and drug entrapment determination’ section.
In vitro cytotoxicity assay
In vitro cytotoxicity of NLCs and LPNs was evaluated by MTT assay using Balb/c fibroblasts (3T3 cells) (Ma et al., Citation2017). Cells were seeded in 96-well plates at the density of 1 × 105 cells/well and incubated with T-L&D-N, L&D-N, T-N, T-L&D-L, L&D-L, T-L, and L&D at equivalent drug concentrations for 10 h. The medium was removed and treated with 100 μL of MTT solution (5 mg/mL) for additional 4 h. The absorbance was measured at 570 nm using a microplate reader.
In vitro skin permeation behaviors
In vitro skin permeation behaviors of NLCs and LPNs were examined using the Franz diffusion method (Zhang et al., Citation2016). The skins from the abdominal surface of rats (fur, adherent fat, and subcutaneous tissues were removed) were mounted on the Franz diffusion cells with a surface area of 0.9 cm2 and a receptor volume of 5 mL. Drugs entrapped NLCs, LPNs or free LEV and DEX combination (L&D) (containing 1 mg LEV and 1 mg DEX each) in PBS (pH 7.4) was applied on the donor compartment side of the skin. The receptor medium was withdrawn at determined time intervals and was replaced by the same volume of fresh buffer (pH 7.4) to maintain the sink condition. The receptor medium was subjected to determine the drug content via the method in the ‘Particle size, zeta potential, and drug entrapment determination’ section.
In vivo evaluation of anesthetic effect in mice
To test the anesthetic effects, NLCs and LPNs were monitored by the vocalization response of mice when electrical stimuluses were given (Ma et al., Citation2017). T-L&D-N, L&D-N, T-L&D-L, L&D-L, and L&D (containing 1 mg LEV and 1 mg DEX each) were applied on the lower abdomen of mice. Electrical stimulations (beginning at 1 mA and increasing to a maximum of 8 mA) were applied using a current generator overlying the abdomen at the site of application (Li et al., Citation2019). The enhanced analgesia threshold was recorded in terms of analgesia ratio (AR) at determined time points using the equation: AR (%)=Nnon-vocalization/Ntotal×100, where Nnon-vocalization is the number of non-vocalization response mice and Ntotal refers to the total number of mice tested.
In vivo evaluation of anesthesia antinociception ability in rats
In vivo anesthesia antinociception ability of NLCs and LPNs was assessed in rats using the hot-plate test (Singh et al., Citation2018). After local application of T-L&D-N, L&D-N, T-L&D-L, L&D-L, and L&D (containing 1 mg LEV and 1 mg DEX each) on the planta, rats were placed rats on a hot plate (50 °C). Response time for observed behavioral changes like paw licking, stomping, jumping, and escaping from the hot plate was recorded to examine the normal heat pain threshold before treatment and the pain threshold after treatment. The enhanced pain threshold (EPT) could be recorded using the equation: EPT (%)=(Tafter treatment – Tbefore treatment)/Tbefore treatment×100, where Tafter treatment refers to the time of threshold after treatment and Tbefore treatment is the time of threshold before treatment. The cutoff time for the hot plate test was 15 s.
Statistical analysis
Statistical significance was analyzed by the Student t-test (SPSS 21.0 for Windows; SPSS Inc., Chicago, IL). *p<.05 was considered statistically significant.
Results
Characterization of NLCs or LPNs
The particle size, zeta potential, and EE of NLCs or LPNs are summarized in . Both NLCs and LPNs showed a mean size of about 100 nm. No obvious difference was found in different formulations. Differences in size were not large among the different nanoparticles. Positive zeta potentials were found on both NLCs and LPNs, and TAT modified nanoparticles exhibited higher surface charge. The EE of both drugs is higher than 80% in all the formulations. TEM images of T-L&D-N and T-L&D-L are shown in . During the stability studies, there were no remarkable changes in the particle size of T-L&D-N and T-L&D-L during 4 months of storage at 2–8 °C (), indicating the good stability of these two carriers.
Table 1. The particle size, zeta potential, and EE of NLCs or LPNs.
Table 2. The stability of NLCs or LPNs.
In vitro drug release profiles
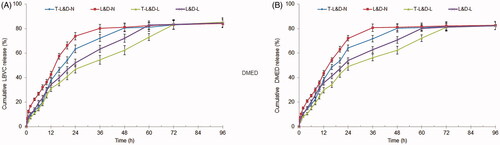
Both NLCs and LPNs illustrated sustained drug release behaviors in . TAT modified nanoparticles showed more sustained drug release than that of non-modified counterparts. Drugs loaded LPNs released drugs more slowly when compared with NLCs. The finishing points of drug release for T-L&D-N and T-L&D-L were 48 and 72 h, respectively.
Figure 3. In vitro LEV (A) and DEX (B) release profiles of NLCs and LPNs: both NLCs and LPNs illustrated sustained drug release behaviors.
In vitro cytotoxicity
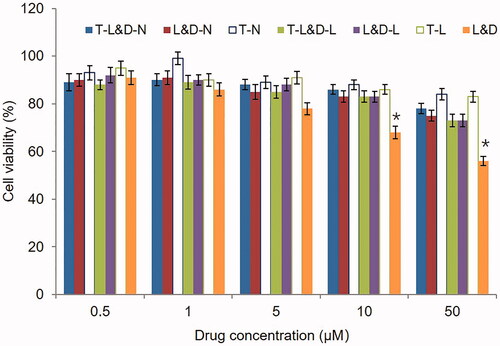
shows that both blank NLCs and LPNs exhibited negligible cytotoxicity. T-L&D-N and T-L&D-L caused lower cytotoxicity than free drugs (L&D) (p<.05). Low toxicity could prove the safety of these two system for drugs delivery.
Figure 4. Cytotoxicity of NLCs and LPNs evaluated on 3T3 cells after 8 h exposure to various concentrations of drugs. *p<.05.
In vitro skin permeation efficiency
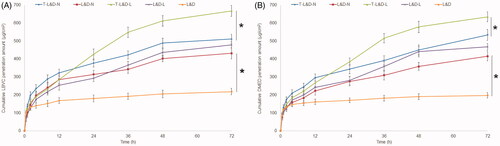
In vitro skin permeation efficiency of NLCs and LPNs was much better than free drugs (L&D) (, p<.05). TAT modified nanoparticles showed enhanced drugs permeation than non-modified ones (p<.05). The skin permeation ability of NLCs and LPNs is different: NLCs showed more permeation amount during the first 24 h, but the next 48 h belongs to LPNs, which gain a more and increased permeation capacity (p<.05). The different permeation behaviors of these two systems may influence the in vivo anesthetic effects.
Figure 5. In vitro LEV (A) and DEX (B) skin permeation efficiency of NLCs and LPNs: in vitro skin permeation efficiency of NLCs and LPNs was much better than free drugs (L&D), TAT modified nanoparticles showed enhanced drugs permeation than non-modified ones. *p<.05.
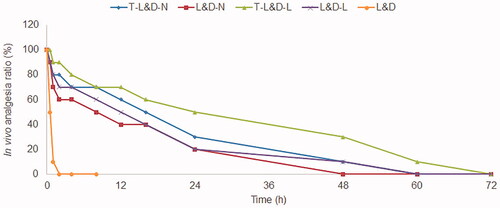
In vivo evaluation of anesthetic effect in mice
The duration rates of mice were summarized after administration of different formulations (). The analgesic response of T-L&D-L was the longest (60 h), longer than that of T-L&D-N (48 h), and other samples (p<.05). T-L&D-N and T-L&D-L illustrated better anesthetic effects compared with their non-modified counterparts (L&D-N and L&D-L, p<.05). The median durations of analgesia for L&D-L and L&D-N were 12 and 8 h, which are significantly longer than that of free L&D (0.5 h, p<.05).
Figure 6. In vivo evaluation of anesthetic effect in mice: the analgesic response of T-L&D-L was the longest, longer than that of T-L&D-N and other samples.
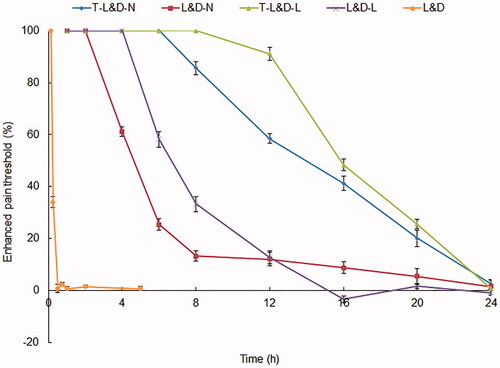
In vivo evaluation of anesthesia antinociception ability in rats
In vivo evaluation of anesthesia antinociception ability in rats was improved by NLCs and LPNs (). The pain threshold of T-L&D-L was the most prominent, which showed continuous effects until 24 h. T-L&D-N also exhibited remarkable ability which is better than non-modified L&D-N, L&D-L, and free L&D (p<.05). These results were in accordance with the in vivo evaluation of anesthetic effect in mice in the above section, which could be the evidence of the outstanding efficiency of the systems.
Figure 7. In vivo evaluation of anesthesia antinociception ability in rats: T-L&D-N also exhibited remarkable ability which is better than non-modified L&D-N, L&D-L and free L&D.
Discussion
Drugs encapsulated by nanoparticles have been reported to increase efficacy and reduce LA toxicity (Yang et al., Citation2019). As discussed in our previous study, nanoparticles having sizes around 100 nm are optimum for drug delivery applications (Pang et al., Citation2020). In the present study, NLCs and LPNs showed a mean size of about 100 nm. NLCs are reported to have the abilities of biocompatibility, modified release kinetics which are suitable for topical drug delivery (Chen et al., Citation2012). LPNs are argued to have controlled release capability, high biocompatibility, and favorable pharmacokinetic profile, while may have an influence on the drug transdermal permeation (Zhang et al., Citation2015; Liu et al., Citation2018).
Drug transdermal permeation included two processes: release from adhesive layer and skin percutaneous permeation (Wang et al., Citation2019). From the results of drug release experiment, it was obvious that the process of drug release may play an important role in drug delivery: Slower release behaviors were found on TAT modified nanoparticles than that of non-modified particles. LPNs also released slower than NLCs. Lipid nanoparticles were reported to reduce the cytotoxicity of drugs (Howell & Chauhan, Citation2009). In this study, biocompatible, low toxic materials were used for the nanocarriers preparation. Hence, the cytotoxicity results indicated that drugs loaded LPNs and NLCs can reduce the cytotoxicity of drugs.
In vitro permeation results illustrated that NLCs formulas good skin permeation capacity, which may be due to their similar property to skin lipids, the high specific surface area for drug absorption because of their smaller size, the existence of a solid matrix, and the biocompatibility makes them suitable for skin administration (Yue et al., Citation2018). LPNs have lipophilic shell and have a natural affinity for skin lipids. This character allows them to facilitate drug transport by favoring the partitioning of the drug from the vehicle into the skin (Hadinoto et al., Citation2013). The skin permeation ability of NLCs and LPNs is different: NLCs showed more permeation amount during the first 24 h, which may be explained by the entrapment of drugs in the inner core of LPNs that hindered the release and permeation. However, after 24 h, more drugs were permeated from LPNs. These behaviors may influence the efficiency of these two systems when used in vivo.
Electrical stimulation testing has long been used as a means of evaluating anesthetic and analgesia effect in animals and humans (Cohen et al., Citation2012). Electrical shock-induced vocalization response suggested that the T-L&D-L prolongs and increases the analgesic properties of the drugs better than T-L&D-N after local administration in mice. TAT modification also illustrated better anesthetic effects compared with their non-modified ones. The penetration of drugs across the skin and their percutaneous delivery are limited by the barrier function of the enormously organized structure of stratum corneum (Alexander et al., Citation2012). TAT is one of the cell-penetrating peptides, which represent amphipathic, arginine-rich, cationic peptides that penetrate and translocate into the cell (Pappalardo et al., Citation2014). TAT peptide could help the carriers to penetrate the skin barrier, enter the dermis and epidermis (Ookubo et al., Citation2014). The results of this section revealed that LPNs may be a better choice for skin penetration and showed better and longer analgesic efficacy.
Hot plate test in rats is one of in vivo analgesic study that assessed by measuring the rats’ pain responses toward thermal stimulus after being put on top of the hot plate (Aziz et al. Citation2019). Rats exposed to NLCs and LPNs were subjected to the hot plate test to determine the effects on pain relief compared with free drugs. The observations suggested that pain thresholds increased by NLCs and LPNs without increasing the dose of the drugs (Yin et al., Citation2019). The most prominent and long-lasting pain threshold of T-L&D-L proved the efficiency of the LPNs and TAT modification. The TAT modified LPNs enhanced the skin permeation of drugs and improved the in vivo efficiency of drugs more remarkably than NLCs counterparts.
Conclusions
T-L&D-L exhibited better efficiency in improving the skin permeation, analgesic time, and pain control intensity than T-L&D-N both in vitro and in vivo. On the other side, T-L&D-N also improved the therapeutic effect of drugs to a large extent. These two systems both exhibited superiority in some respects. In this study, conclusion may be made that TAT modified LPNs are more promising platform for the long-term local anesthesia.
Disclosure statement
The author has no declaration of interest.
References
- Adel Elmaddawy AE, Diab DG, Farag MA. (2018). Levobupivacaine versus levobupivacaine–dexmedetomidine in thoracic paravertebral block for laparoscopic sympathectomy. Anesth Essays Res 12:837–42.
- Alexander A, Dwivedi S, Ajazuddin Giri TK, et al. (2012). Approaches for breaking the barriers of drug permeation through transdermal drug delivery. J Control Release 164:26–40.
- Andreae MH, Andreae DA. (2013). Regional anaesthesia to prevent chronic pain after surgery: a Cochrane systematic review and meta-analysis. Br J Anaesth 111:711–20.
- Aziz ZAA, Nasir HM, Ahmad A, et al. (2019). Enrichment of Eucalyptus oil nanoemulsion by micellar nanotechnology: transdermal analgesic activity using hot plate test in rats' assay. Sci Rep 9:13678.
- Blanco MD, Bernardo MV, Teijón C, et al. (2003). Transdermal application of bupivacaine-loaded poly(acrylamide(A)-co-monomethyl itaconate) hydrogels. Int J Pharm 255:99–107.
- Brown DL, Ransom DM, Hall JA, et al. (1995). Regional anesthesia and local anesthetic-induced systemic toxicity: seizure frequency and accompanying cardiovascular changes. Anesth Analg 81:321–8.
- Brummett CM, Norat MA, Palmisano JM, Lydic R. (2008). Perineural administration of dexmedetomidine in combination with bupivacaine enhances sensory and motor blockade in sciatic nerve block without inducing neurotoxicity in rat. Anesthesiology 109:502–11.
- Casati A, Putzu M. (2005). Bupivacaine, levobupivacaine and ropivacaine: are they clinically different? Best Pract Res Clin Anaesthesiol 19:247–68.
- Cereda CMS, Mecatti DS, Papini JZB, et al. (2018). Bupivacaine in alginate and chitosan nanoparticles: an in vivo evaluation of efficacy, pharmacokinetics, and local toxicity. J Pain Res 11:683–91.
- Chen C, You P. (2017). A novel local anesthetic system: transcriptional transactivator peptide-decorated nanocarriers for skin delivery of ropivacaine. Drug Des Devel Ther 11:1941–9.
- Chen Y, Zhou L, Yuan L, et al. (2012). Formulation, characterization, and evaluation of in vitro skin permeation and in vivo pharmacodynamics of surface-charged tripterine-loaded nanostructured lipid carriers. Int J Nanomedicine 7:3023–32.
- Chu Y, Chen N, Yu H, et al. (2017). Topical ocular delivery to laser-induced choroidal neovascularization by dual internalizing RGD and TAT peptide-modified nanoparticles. Int J Nanomedicine 12:1353–68.
- Cohen R, Kanaan H, Grant GJ, Barenholz Y. (2012). Prolonged analgesia from bupisome and bupigel formulations: from design and fabrication to improved stability. J Control Release 160:346–52.
- de Araújo DR, Ribeiro LNM, de Paula E. (2019). Lipid-based carriers for the delivery of local anesthetics. Expert Opin Drug Deliv 16:701–14.
- El Baz MM, Farahat TEM. (2019). Efficacy of adding dexmedetomidine to intra-articular levobupivacaine on postoperative pain after knee arthroscopy. Anesth Essays Res 13:254–8.
- El-Boghdadly K, Brull R, Sehmbi H, Abdallah FW. (2017). Perineural dexmedetomidine is more effective than clonidine when added to local anesthetic for supraclavicular brachial plexus block: a systematic review and meta-analysis. Anesth Analg 124:2008–20.
- Gao S, Song X. (2004). Determination of bupivacaine hydrochloride in bupivacaine hydrochloride injection by HPLC. Drug Stanoaros China 5:40–1.
- Golembiewski J, Dasta J. (2015). Evolving role of local anesthetics in managing postsurgical analgesia. Clin Ther 37:1354–71.
- Hadinoto K, Sundaresan A, Cheow WS. (2013). Lipid–polymer hybrid nanoparticles as a new generation therapeutic delivery platform: a review. Eur J Pharm Biopharm 85:427–43.
- Howell BA, Chauhan A. (2009). Bupivacaine binding to pegylated liposomes. Anesth Analg 109:678–82.
- Hussain N, Grzywacz VP, Ferreri CA, et al. (2017). Investigating the efficacy of dexmedetomidine as an adjuvant to local anesthesia in brachial plexus block: a systematic review and meta-analysis of 18 randomized controlled trials. Reg Anesth Pain Med 42:184–96.
- Levene JL, Weinstein EJ, Cohen MS, et al. (2019). Local anesthetics and regional anesthesia versus conventional analgesia for preventing persistent postoperative pain in adults and children: a Cochrane systematic review and meta-analysis update. J Clin Anesth 55:116–27.
- Li A, Yang F, Xin J, Bai X. (2019). An efficient and long-acting local anesthetic: ropivacaine-loaded lipid–polymer hybrid nanoparticles for the control of pain. Int J Nanomedicine 14:913–20.
- Liu D, Lian Y, Fang Q, et al. (2018). Hyaluronic-acid-modified lipid–polymer hybrid nanoparticles as an efficient ocular delivery platform for moxifloxacin hydrochloride. Int J Biol Macromol 116:1026–36.
- Ma P, Li T, Xing H, et al. (2017). Local anesthetic effects of bupivacaine loaded lipid–polymer hybrid nanoparticles: in vitro and in vivo evaluation. Biomed Pharmacother 89:689–95.
- McLure HA, Rubin AP. (2005). Review of local anaesthetic agents. Minerva Anestesiol 71:59–74.
- Nasrollahi SA, Taghibiglou C, Azizi E, Farboud ES. (2012). Cell-penetrating peptides as a novel transdermal drug delivery system. Chem Biol Drug Des 80:639–46.
- Ookubo N, Michiue H, Kitamatsu M, et al. (2014). The transdermal inhibition of melanogenesis by a cell-membrane-permeable peptide delivery system based on poly-arginine. Biomaterials 35:4508–16.
- Pang J, Xing H, Sun Y, et al. (2020). Non-small cell lung cancer combination therapy: hyaluronic acid modified, epidermal growth factor receptor targeted, pH sensitive lipid–polymer hybrid nanoparticles for the delivery of erlotinib plus bevacizumab. Biomed Pharmacother 125:109861.
- Pappalardo JS, Langellotti CA, Di Giacomo S, et al. (2014). In vitro transfection of bone marrow-derived dendritic cells with TATp-liposomes. Int J Nanomedicine 9:963–73.
- Pescina S, Ostacolo C, Gomez-Monterrey IM, et al. (2018). Cell penetrating peptides in ocular drug delivery: state of the art. J Control Release 284:84–102.
- Prausnitz MR, Langer R. (2008). Transdermal drug delivery. Nat Biotechnol 26:1261–8.
- Ribeiro LN, Franz-Montan M, Breitkreitz MC, et al. (2016). Nanostructured lipid carriers as robust systems for topical lidocaine-prilocaine release in dentistry. Eur J Pharm Sci 93:192–202.
- Rwei AY, Sherburne RT, Zurakowski D, et al. (2018). Prolonged duration local anesthesia using liposomal bupivacaine combined with liposomal dexamethasone and dexmedetomidine. Anesth Analg 126:1170–5.
- Shomorony A, Santamaria CM, Zhao C, et al. (2019). Prolonged duration local anesthesia by combined delivery of capsaicin- and tetrodotoxin-loaded liposomes. Anesth Analg 129:709–17.
- Singh P, Kongara K, Harding D, et al. (2018). Comparison of electroencephalographic changes in response to acute electrical and thermal stimuli with the tail flick and hot plate test in rats administered with opiorphin. BMC Neurol 18:43.
- Song Z, Shi Y, Han Q, Dai G. (2018). Endothelial growth factor receptor-targeted and reactive oxygen species-responsive lung cancer therapy by docetaxel and resveratrol encapsulated lipid–polymer hybrid nanoparticles. Biomed Pharmacother 105:18–26.
- Tan S, Wang G. (2018). Lung cancer targeted therapy: folate and transferrin dual targeted, glutathione responsive nanocarriers for the delivery of cisplatin. Biomed Pharmacother 102:55–63.
- Tsai MJ, Wu PC, Huang YB, et al. (2012). Baicalein loaded in tocol nanostructured lipid carriers (tocol NLCs) for enhanced stability and brain targeting. Int J Pharm 423:461–70.
- Varshney A, Prabhu M, Periyadka B, et al. (2019). Transversus abdominis plane (TAP) block with levobupivacaine versus levobupivacaine with dexmedetomidine for postoperative analgesia following cesarean delivery. J Anaesthesiol Clin Pharmacol 35:161–4.
- Vitorino C, Almeida J, Gonçalves LM, et al. (2013). Co-encapsulating nanostructured lipid carriers for transdermal application: from experimental design to the molecular detail. J Control Release 167:301–14.
- Wang H, Tian Q, Quan P, et al. (2019). Probing the role of ion-pair strategy in controlling dexmedetomidine penetrate through drug-in-adhesive patch: mechanistic insights based on release and percutaneous absorption process. AAPS PharmSciTech 21:4.
- Wang J, Zhang L, Chi H, Wang S. (2016). An alternative choice of lidocaine-loaded liposomes: lidocaine-loaded lipid–polymer hybrid nanoparticles for local anesthetic therapy. Drug Deliv 23:1254–60.
- Wang Y, Su W, Li Q, et al. (2013). Preparation and evaluation of lidocaine hydrochloride-loaded TAT-conjugated polymeric liposomes for transdermal delivery. Int J Pharm 441:748–56.
- Wang Y, Wang S, Shi P. (2016). Transcriptional transactivator peptide modified lidocaine-loaded nanoparticulate drug delivery system for topical anesthetic therapy. Drug Deliv 23:3193–9.
- Yang Y, Qiu D, Liu Y, Chao L. (2019). Topical anesthetic analgesic therapy using the combination of ropivacaine and dexmedetomidine: hyaluronic acid modified long-acting nanostructured lipid carriers containing a skin penetration enhancer. Drug Des Devel Ther 13:3307–19.
- Yin Q, Hou S, Yin H, et al. (2019). The analgesic and anti-inflammatory effects of zukamu granules, a traditional Chinese medical formulation. Pharm Biol 57:729–35.
- Yue Y, Zhao D, Yin Q. (2018). Hyaluronic acid modified nanostructured lipid carriers for transdermal bupivacaine delivery: in vitro and in vivo anesthesia evaluation. Biomed Pharmacother 98:813–20.
- Zhang L, Chan JM, Gu FX, et al. (2008). Self-assembled lipid–polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano 2:1696–702.
- Zhang L, Wang J, Chi H, Wang S. (2016). Local anesthetic lidocaine delivery system: chitosan and hyaluronic acid-modified layer-by-layer lipid nanoparticles. Drug Deliv 23:3529–37.
- Zhang L, Zhu D, Dong X, et al. (2015). Folate-modified lipid–polymer hybrid nanoparticles for targeted paclitaxel delivery. Int J Nanomedicine 10:2101–14.
- Zheng G, Zheng M, Yang B, et al. (2019). Improving breast cancer therapy using doxorubicin loaded solid lipid nanoparticles: Synthesis of a novel arginine-glycine-aspartic tripeptide conjugated, pH sensitive lipid and evaluation of the nanomedicine in vitro and in vivo. Biomed Pharmacother 116:109006.
- Zhu Y, Cheng L, Cheng L, et al. (2014). Folate and TAT peptide co-modified liposomes exhibit receptor-dependent highly efficient intracellular transport of payload in vitro and in vivo. Pharm Res 31:3289–303.