
Abstract
A LC-MS/MS technique separated the bovine and mouse brain gangliosides monosialotetrahexosylgangliosides (GM1), disialotetrahexosylgangliosides (GD1a), trisialotetrahexosylgangliosides (GT1b) and tetrasialotetrahexosylgangliosides (GQ1b) using a phenyl-hexyl HPLC column and employing a linear methanol gradient in water, which is 0.028% in ammonium hydroxide. The gangliosides were separated according to sialic acid class, and within a particular class, gangliosides having different ceramide carbon chain lengths were also separated. All gangliosides of a particular sialic acid class eluted in characteristic retention time windows in the order of GQ1b, (earliest), GT1b, GD1a, and GM1 (latest). Within each specific retention time window for a particular ganglioside class, gangliosides were separated in the order of increasing ceramide carbon chain length. The phenyl-hexyl column separation of gangliosides is advantageous over established hydrophilic interaction and conventional reversed-phase chromatography techniques, in that the former separates gangliosides according to sialic acid class but not ceramide composition and the latter distributes all the sialic acid ganglioside classes throughout the entire chromatogram. The mechanism of separation of the ganglioside sialic acid classes is proposed to be a π-electron repulsion of negatively-charged gangliosides by the column phenyl moiety.
Graphical Abstract
Introduction
Gangliosides are a heterogeneous group of molecules classified as glycosphingolipids. Its structure consists of a polar oligosaccharide portion that is covalently linked to a non-polar ceramide moiety. The ceramide structure consists of a fatty acid group attached via an amide bond to a long-chain amino alcohol sphingoid base, also known as a long-chain base, which predominantly has a trans double bond between the C4 and C5 carbon atoms. The oligosaccharide portion consists of multi-linked saccharide units, of which one or more are sialic acid (N-acetylneuraminic acid).
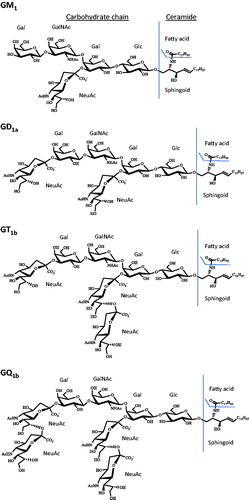
The core chain of the oligosaccharide group of mammalian brain gangliosides are predominantly the tetrasaccharide core of the ganglio series, 95% being GM1, GD1a, GD1b, GT1b, and GQ1b.[Citation1] The nomenclature for gangliosides is as follows:[Citation2] number 1 indicates the tetrasaccharide ganglio oligosaccharide core sequence of neutral saccharides attached to the ceramide, being galactose β1-3 → N-acetylgalactosamine → β1-4 galactose ⟶ β1-4 glucose β1-1′ → ceramide. The letters M, D, T, and Q, indicate mono-, di-, tri- and tetra- number of sialic acid groups, respectively, that are bonded to the neutral core ganglio sequence, either as single sialic acid moieties attached at one or more sites and/or through a multiple sialic acid chain. The lower case letter indicates a particular isomer with respect to the location of the sialic acid groups. The convention used in this manuscript identifies the tetrasaccharide ganglio class, followed by the number of carbons in the ceramide, followed by a colon, followed by the number of double bonds in the ceramide. A “d” indicates the dihydroxysphingosine form of the sphingoid base. Thus GM1 d38:1 is the GM1 class that has 38 carbons in the ceramide having one double bond. As previously determined for bovine brain GM1 gangliosides, the most prevalent GM1 d38:1 ceramide is GM1 d20:1-18:0, having a ceramide with a 20-carbon dihydroxysphingosine sphingoid base that has one double bond and a saturated 18-carbon fatty acid group.[Citation3] However, GM1 d38:1 can also include other associated structures, such as GM1 d18:1-20:0. Structures of each of the sialic acid ganglioside classes used in this study are given in .
Figure 1. Chemical structures of GM1, GD1a, GT1b, GQ1b gangliosides. Gangliosides with d36:1 ceramides. Gal is galactose, GalNAc is N-acetylgalactosamine, Glc is glucose and NeuAc is N-acetylneuraminic acid (sialic acid).
The fatty acid component in the ceramide for mammalian brain gangliosides is predominately a saturated 18-carbon chain (80−92%), with the remaining fatty acids being mostly C16, C20, C22, and C24.[Citation4] The most prevalent sphingoid base components of the ceramide are the dihydroxylated d18:1 (18 carbons with one double bond) and d20:1 sphingosines, totaling 74–96% for any particular oligosaccharide class of gangliosides present in the human brain,[Citation5] followed by the second most prevalent sphingoid base components, being d18:0 and d20:0 sphinganines, totaling 5–10% for brain gangliosides of various mammals.[Citation6]
Significant ganglioside heterogeneity results from substantial oligosaccharide diversity, in which over 180 different structures have been identified.[Citation7] There is also diversity in the fatty acid component, in which carbon chain lengths from 10 to 34 have been identified, as well as the reported presence of unsaturated and 2-hydroxy fatty acids.[Citation8–15] Finally, there is diversity in the sphingoid base in terms of chain length (16–20 carbons),[Citation16–18] presence of saturated and unsaturated carbon chains,[Citation16–18] and presence of trihydroxylated sphinganines.[Citation17,Citation19]
Given the vast heterogeneity of ganglioside structures, the development of unique high-performance liquid chromatography techniques that are complementary to existing techniques is important for accurate determination of individual ganglioside molecules in biological samples, as well as purification of homogenous ganglioside standards. There are two categories of HPLC techniques that are currently employed in the separation of gangliosides in HPLC-mass spectrometry: hydrophilic interaction liquid chromatography (HILIC), using amino[Citation20–24] and silica[Citation25,Citation26] columns and reversed-phase HPLC, using C5,[Citation27] C8,[Citation28] C18[Citation29–33] and C30[Citation23,Citation24] columns.
The HILIC technique separates the gangliosides based on the hydrophilic oligosaccharide portion of the ganglioside[Citation21,Citation22] and is advantageous in that the retention time reflects the number of sialic acid groups in the ganglioside. However, there is minimal or no separation of the gangliosides within a particular sialic acid class that differs in their non-polar ceramide composition.[Citation20,Citation21,Citation23] Thus, for example, while the GM1s are separated from the GD1s, GT1s, and GQ1s, there is no separation of the GM1s differing in their ceramide carbon length, resulting in co-elution of all GM1s.
On the other hand, reversed-phase HPLC techniques separate gangliosides based principally on their ceramide difference. For a C30 column, there was overlap or co-elution of gangliosides differing in oligosaccharide composition but having the same ceramide carbon chain length.[Citation23,Citation24] In the case of a C18 column, the gangliosides of a particular ceramide carbon chain length that differ in their sialic acid or oligosaccharide content are separated in a cluster within a particular retention time window.[Citation33] Thus, in this work, all the d34:1 ceramide gangliosides (GQ, GT, GD, GM) will cluster, which will elute prior to the d36:1 ceramide ganglioside cluster (GQ, GT, GD, GM), which will elute prior to the d38:1 ceramide cluster (GQ, GT, GD, GM), and so forth. For this C18 column, the ceramide clusters were separated and the gangliosides within each cluster were partially or fully resolved. For a C5[Citation27] and a C8[Citation28] column, 8–12 GM3 gangliosides that differ in ceramide carbon length were separated. In the same studies, 8–12 GD3 gangliosides were also separated. However, there was considerable overlap in the range of retention times for the GM3 and GD3 gangliosides. Thus, for all reversed-phase columns in the literature cited above, the gangliosides of a particular sialic acid class, differing in ceramide carbon length, were distributed over a wide retention range of the chromatogram, with increasing retention time for increasing ceramide carbon chain length. As a result, unlike the HILIC technique, all the gangliosides of a particular sialic acid or saccharide chain composition do not have a characteristic retention time range (window) in a reversed-phase separation.
In the present work, a phenyl-hexyl column technique shows unique separation capability for gangliosides compared to what is reported in the literature for the reversed-phase HPLC and HILIC techniques, addressing the disadvantages of these latter two chromatographic techniques.
Materials and methods
Chemicals and materials
Monosialoganglioside GM1 (ammonium salt), disialoganglioside GD1a (ammonium salt), trisialoganglioside GT1b (ammonium salt) and tetrasialoganglioside GQ1b (ammonium salt) and the internal standard N-omega-CD3-octadecanoyl monosialoganglioside GM1 (ammonium salt) from the bovine brain were from Matreya LLC (State College, PA, USA). Methanol and acetonitrile, both Optima LC/MS grade, were from Fisher Scientific (Fair Lawn, NJ, USA). Ammonium hydroxide (28% ammonia), formic acid (reagent grade, ≥95%), and anhydrous chloroform (≥99%, containing 0.5–1.0% ethanol as stabilizer) were from Sigma-Aldrich (St. Louis, MO, USA). HPLC grade water was obtained from a Barnstead Nanopure Model 7148 water purification system from Thermo Scientific (West Palm Beach, FL, USA). A 2 mL Potter–Elvehjem glass-teflon homogenizer tube and Sep-Pak C18 3 cc Vac Cartridges, 200 mg, 55–105 µm (Waters Corporation, Milford, MA, USA) were both used in the preparation of the mouse brain sample, with the latter also used in the preparation of ganglioside standard solutions.
Preparation of mouse brain sample
A 5.5 mg of superior colliculus brain tissue from a single C57BL/6J mouse was collected to which 275 µL of 40% methanol in water (ice cold) was added and subsequently homogenized with a 2 mL Potter–Elvehjem glass-teflon homogenizer tube for 10 min in an ice bath. A volume of 100 μL of the homogenized tissue sample was then subjected to a modified Svennerholm and Fredman liquid–liquid extraction procedure used in ganglioside analysis,[Citation22] with an additional solid phase extraction (SPE) procedure, as done in another published work,[Citation29] with modification, as given below.
After completing the liquid–liquid extraction steps, the collected supernatant was applied to a C18 SPE cartridge. The SPE cartridges were conditioned with 3 mL of 100% methanol, twice, followed by 2 mL of 30% methanol in water, twice. The following steps of SPE were done under gravitational force, as vacuum elution resulted in poor recoveries. The extracted sample was loaded and the collected effluent was again passed through the cartridge to bind any non-retained gangliosides of the first pass. Two 3 mL volumes of 30% methanol in water were then passed through the SPE cartridges. Gangliosides were then eluted with two portions of 2 mL 100% methanol, with the eluent being collected. The final eluent was near dried under vacuum and reconstituted with 200 µL of 50% methanol in water. An aliquot of 20 µL of this constituent was additionally diluted 100-fold with 50% methanol in water for LC-MS/MS analysis.
Preparation of ganglioside standard solutions
Standard ganglioside solutions were run as calibrators to quantify the gangliosides in the mouse superior colliculus sample. One stock solution containing all four ganglioside classes (GM1, GD1a, GT1b, and GQ1b, 1 mg/mL of each) in 50% methanol in water was prepared with the bovine brain gangliosides. Standard solutions of 25, 50, 100, 250, 500, and 1000 ng/mL of total ganglioside content for each ganglioside sialic acid class were prepared from serial dilutions of the stock solution with 50% methanol in water. A volume of 100 μL of each standard solution was taken through each step of the liquid–liquid extraction and SPE procedures (except for the final 100x dilution step), as given in the subsection above. The standard solutions were subjected to the sample preparation steps to account for any losses of gangliosides in the sample preparation of the mouse brain sample.
HPLC instrumentation, column, and parameters
HPLC analysis was performed using a high pH Shimadzu Nexera X2 LC-30AD binary pump system (Columbia, MD, USA). A phenyl-hexyl column (XBridge BEH Phenyl, 2.1 × 50 mm, 3.5 μm, 130 Å pore size) with a pre-column (XBridge BEH Phenyl VanGuard Cartridge, 2.1 × 5 mm, 3.5 μm, 130 Å pore size), both from Waters (Milford, MA, USA), was used. A 20 μL volume of the standards and the mouse brain sample was injected with a Shimadzu autosampler (SIL-30AC with a PEEK needle seal for high pH operation) at 4 °C. The column oven (CTO-20A) was maintained at 40 °C. Mobile phase A was 0.028% (v/v%) ammonium hydroxide in water and mobile phase B was 0.028% (v/v%) ammonium hydroxide in pure methanol. The ammonium hydroxide concentration in the mobile phase was the actual concentration, taking into account that ammonium hydroxide is a 28% ammonia solution. The gradient program was 0–2 min 25% B isocratic, followed by a linear gradient of 25–100% B in 20 min, then 100% B for an additional 8 min. The flow rate was 0.2 mL/min. The column was equilibrated prior to the start of the next run for 10 min, pumping 25% B at a flow rate of 0.2 mL/min. The mobile phase solutions were filtered through 0.2 μm PTFE membrane filters from EMD Millipore (Billerica, MA, USA).
The holdup time for the column with pre-column was determined to be 0.88 min by injecting 20 µL of 0.1% formic acid in 100% acetonitrile into a 50:50 acetonitrile:water mobile phase. The time that a gradient change reaches the MS detector from the pumps with the column and pre-column in place and the autosampler loop in the inject position was determined to be 1.43 min by a breakthrough experiment (switching the mobile phase from 5% acetonitrile to 95% acetonitrile in water). The total ion current was monitored for determining both these system times at a flow rate of 0.2 mL/min.
A Zorbax Eclipse Plus C18 (2.1 × 150 mm, 3.5 µm, 95 Å) from Agilent (Santa Clara, CA, USA) was used to compare results with that which was obtained with the phenyl-hexyl column.
Mass spectrometer settings
The chromatographic system was interfaced to a Shimadzu QQQ LCMS-8040 instrument (Columbia, MD, USA) via an electrospray ion source. MS parameters were as follows: 2.5 L/min and 12 L/min for the nebulizer gas flow and drying gas flow, respectively; 250 °C and 400 °C for the desolvation line and heat block temperatures, respectively; and 3.5 kV for the interface voltage. Argon gas was used for the collision ion dissociation at 230 kPa pressure. Collision energy and pre-bias voltages were optimized for individual multiple reaction monitoring (MRM) transitions ().
Table 1. Mass spectrometric parameters for optimal determination of the various gangliosides.
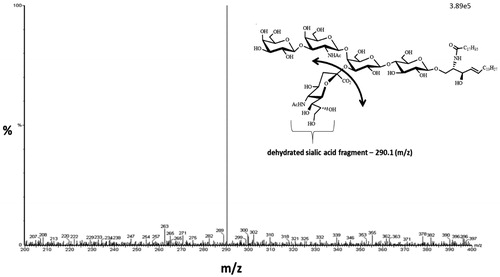
Precursor ganglioside ions from Q1 were fragmented in the collision cell to generate the product ion detected in Q3. The product ion monitored was the dehydrated sialic acid fragment (290.1), per the well-established protocol for MRM determination of gangliosides. The Q3 scan for GM1 is given in showing the 290.1 m/z signal for dehydrated sialic acid product ion. The MS analysis was done in the negative ion mode for both the precursor and product ion.
Figure 2. GM1 gangliosides (d36:1) product ion scan. Product ion scan (Q3 scan) showing the fragmentation of GM1 ganglioside (m/z 1545) resulting in the dehydrated sialic acid product ion (m/z 290.1).
Precision and accuracy
Interday precision was determined for the peak area ratio of ganglioside/internal standard of three standard solutions of low, mid, and high concentrations of ganglioside (all being 100 ng/mL GM1-D3 in internal standard). It was determined for each of the d36:1 ganglioside sialic acid classes for three runs of one run per day for three days (n = 3).
Accuracy was assessed by quantifying matrix effects determined from dilution studies.[Citation22] Gangliosides from the mouse superior colliculus brain sample were extracted by the modified Svennerholm and Fredman ganglioside isolation procedure and were serially diluted (10×, 100×, 400×) with reconstitution solvent, 50% methanol in water. The ganglioside peak areas of the diluted samples of the superior colliculus were compared to that of the standard calibration plot for the d36:1 and d38:1 GM1, GD1, GT1 and GQ1 gangliosides. The concentration of the various gangliosides in the brain sample was determined from the ganglioside peak areas of the 400× diluted sample, estimating the concentration from the standard calibration plot, with the concentration of gangliosides in the 100× and 10× diluted samples determined to be a factor of 4 and 40 times that determined for the 400× diluted sample, respectively. If there is minimal matrix effect, then the peak areas of the 100× and 10× diluted samples will be found to be close to that of the standard calibration plot. The matrix effect was quantified as the percent deviation of the peak area for the 100× diluted sample compared to the theoretical peak area given by the standard calibration plot.
Recovery of gangliosides from samples taken through the sample preparation procedure also impacts accuracy. Any recovery issues were addressed by having both the samples and standards taken through the sample preparation procedure, so that any factor of loss of gangliosides in the sample will also occur in the standard.
Results and discussion
Optimization of the phenyl-hexyl column separation of the gangliosides
Experiments were done to optimize the separation of the GM1, GD1a, GT1b and GQ1b bovine brain gangliosides on the phenyl-hexyl column. Initial experiments were done with no pH adjustment to assess which mobile phase organic modifier to use. As shown in there is a significant difference in the chromatography of gangliosides on a phenyl-hexyl column using a gradient in acetonitrile compared to that using methanol. It is seen that the acetonitrile modifier is a stronger modifier, with the GD1a d36:1 peak having a retention factor (k) that is more than 3-times less in the acetonitrile run (at significantly less organic modifier) than the methanol run. The stronger elution quality of acetonitrile compared to methanol on phenyl columns has been previously reported, which is attributed to acetonitrile’s π–π interaction with the column’s phenyl group that causes suppression of the phenyl group’s π effect on an analyte’s retention.[Citation34–38] This is compared to methanol, which does not suppress the phenyl group’s π effect.[Citation37,Citation38]
Figure 3. Mobile phase optimization studies: methanol versus acetonitrile and addition of ammonium hydroxide. LC-MS/MS chromatograms of GT1b (red), GD1a (green) and GM1 (black) bovine brain gangliosides. (a) Linear gradient of mobile phase A (100% water) and mobile phase B (100% acetonitrile), 45% B to 90% B in 8 min. (b) Linear gradient of mobile phase A (100% water) and mobile phase B (100% methanol), 55% B to 95% B in 8 min. (c) Linear gradient of mobile phase A (100% water adjusted to pH 9 with ammonium hydroxide) and mobile phase B (100% methanol adjusted to pH 9 with ammonium hydroxide), 60% B to 100% B in 8 min.(d) Linear gradient of mobile phase A (100% water adjusted to pH 10 with ammonium hydroxide) and mobile phase B (100% methanol adjusted to pH 10 with ammonium hydroxide), 55% B to 95% B in 8 min.
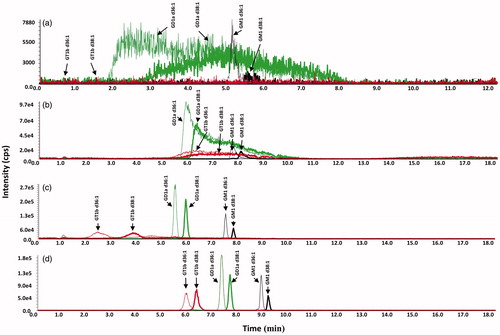
In the chromatography of the GM, GD, and GT gangliosides it is also noted in that there is a substantial broadening of the multi-sialic acid ganglioside peaks for both organic modifiers, with there being more peak broadening noted in the acetonitrile gradient run. The gradient acetonitrile run shows broader GD peaks and no significant GT peaks; while in the methanol gradient run, the GD peaks are more distinct, although there is considerable tailing, and GT peaks are present, although they are wide. Given the relatively better chromatographic performance using a methanol gradient, further optimization studies with respect to pH were done with methanol as the organic modifier.
The narrower peak widths for the GM1 peaks seen in (as well as ) provides evidence to support a hypothesis that the broad peak widths and peak tailing noted for the GD and GT gangliosides results from the presence of multiple sialic acid groups on these gangliosides. It is further thought that this band broadening/tailing of the multi-sialic acid ganglioside peaks seen in results from the heterogeneity in the charge states of these gangliosides. Even though the pKa of a single sialic acid is low (pKa = 2.6), there is expected to be a successive increase in the pKa of the sialic acids because of the close proximity of the multiple acid groups in the GD, GT and GQ gangliosides, as noted in their structures given in . The heterogeneous charge states of the GD, GT and GQ gangliosides results from the pH-dependent variability in the number of de-protonated sialic acids in the ganglioside. Thus, at a particular pH, there could be several charge forms of the GT gangliosides that are chromatographed, leading to broadening/tailing of the peak.
Given this hypothesis, ammonium hydroxide was added to the mobile phase to de-protonate all the sialic acids of the gangliosides. This was done in order that all ganglioside molecules of a particular class would be chromatographed in one ionic state, which is the ganglioside’s maximum negative charge, equaling the number of sialic acid groups. If heterogeneity of the sialic acid de-protonation state was the cause of the band broadening/tailing, chromatographing one ionic form of any individual ganglioside molecule should address this issue. LC-MS/MS experiments were done injecting standard gangliosides onto a phenyl-hexyl column employing a methanol gradient with sufficient ammonium hydroxide added to the mobile phase, such that all gangliosides are in one charge state, equal to the number of sialic acid groups. Given in are chromatograms of gangliosides employing a mobile phase methanol gradient with added ammonium hydroxide in the mobile phase. It is noted that there is tailing of the GT ganglioside peaks in run in which ammonium hydroxide is added to the mobile phase. Further increasing the concentration of ammonium hydroxide in the mobile phase for the run plotted in showed narrow peaks for the GT gangliosides, as well resolving individual molecular species (having a different number of carbons in the ceramide moiety) within each ganglioside class.
For optimized separation of all gangliosides, including GQ gangliosides, the ammonium hydroxide concentration was adjusted to 0.028% in both mobile phases A and B. Also, a shallower methanol gradient was employed.
Precision and accuracy
Interday precision for the d36:1 of each sialic acid class of gangliosides is given in . Considering all the ganglioside sialic acid classes, interday percent coefficient of variation (% CV) was 6–21% CV for low standards (8–12 ng/mL), 2–9% CV for mid standards (34–50 ng/mL), and 0.2–10% CV for high standards (340–500 ng/mL).
Table 2. Interday precision: percent coefficient of variation (% CV) of peak area ratio [standard/internal standard (IS)].
Given that both the standards and the samples were taken through the sample preparation steps to account for any recovery issues, the accuracy will be dependent on matrix effects. The deviation due to matrix effects were assessed by dilution studies of the mouse superior colliculus brain tissue, comparing peak areas of the diluted sample with the theoretical peak area from the standard calibration plot. The percent deviation of the 100x diluted sample in the various gangliosides is given in . The GM1 gangliosides were at the limit of acceptable accuracy with a deviation of 26% and 20% for d 36:1 and d38:1 GM1 ganglioside, respectively, showing a matrix enhancement of the signal. All other ganglioside classes showed substantial negative deviation, from −40% to −79%.
Table 3. Matrix effect studies: comparison of ganglioside peak areas of 100x diluted mouse superior colliculus sample with theoretical peak areas from the standard calibrator.
Ganglioside bovine brain standard results
A phenyl-hexyl HPLC technique was developed and characterized for the separation of pooled bovine brain standards of GQ1b, GT1b, GD1a and GM1 gangliosides. In this work, with the instrumentation used, GM1, GD1a, GT1b, and GQ1b gangliosides of a certain ceramide subclass were quantified from 8 to 500 ng/mL.
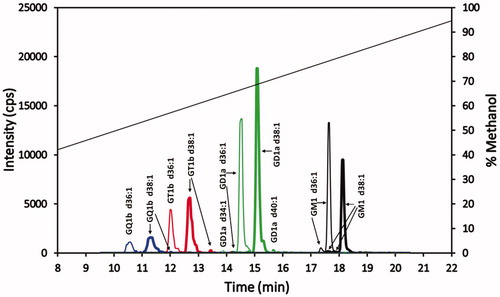
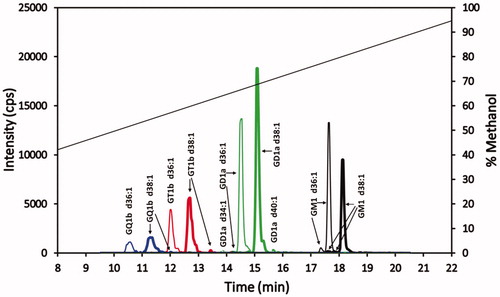
The chromatogram in shows the baseline separation of the most prevalent gangliosides in the standard. Trace isomer peaks or shoulders on peaks are also noted for some of these gangliosides. Of note is the fact that all the gangliosides of a particular sialic acid class differing in ceramide length are well resolved and clustered together in a retention time window specific for that ganglioside class. The retention times of the sialic acid class clusters show an inverse relationship with the number of sialic groups, with the elution order from earliest to latest being GQ1bs, GT1bs, GD1as, and GM1s. Although the GQ1b and GT1b cluster of peaks show the separation of their respective clusters when considering the major gangliosides (that is, the d36:1 and the d38:1 ceramide gangliosides), the GT1b d34:1 ganglioside(s) (a minor component in the standard that is plotted but is not evident in ) elutes within the GQ1b d38:1 peak, while GQ1b d40:1 (a minor component in the standard that is plotted but is not evident in ) elutes within the GT1b d36:1 peak. Hence, there is an overlap of the longer carbon chain ceramide GQ1bs with the shorter carbon chain GT1bs. However, there is a large separation of GD1a and GM1 ganglioside clusters. The expanded chromatographic time region between these two classes is large enough such that peaks for GM1 with ceramides less than 36 carbons and peaks for GD1a with more than 40 carbons are anticipated to be separated from each other. There is also a moderately wide retention time gap between the GT1bs and GD1as, allowing for resolution of the ganglioside clusters from samples having smaller ceramide GD1a and/or larger ceramide GT1b gangliosides. Finally, the wider peaks of GQ1b gangliosides could be due to 20 µL injection volume (11% of column holdup volume), introducing a sample in 50% methanol onto the column. Although this phenyl-hexyl column technique shows good baseline separation of the GQ1b gangliosides, sharper GQ1b peaks might be expected with lower injection volumes (1–5% of the column holdup volume).
Figure 4. Superimposed MRM chromatograms of bovine brain gangliosides. Chromatograms of GQ1b (blue), GT1b (red), GD1a (green) and GM1 (black), each at 25 ng/mL. The 25 ng/mL is the combined concentration for all gangliosides differing in ceramide for each particular class (GQ1b 25 ng/mL, GT1b 25 ng/mL, GD1a 25 ng/mL, and GM1 25 ng/mL). The diagonal line plot is the actual % methanol vs time, corrected for time for a gradient change to reach the mass spectrometer detector (1.43 min). Not evident, because of the scale used, are trace amounts of GT1b d34:1 and GQ1b d40:1 eluting at 11.4 and 12.2 min, respectively.
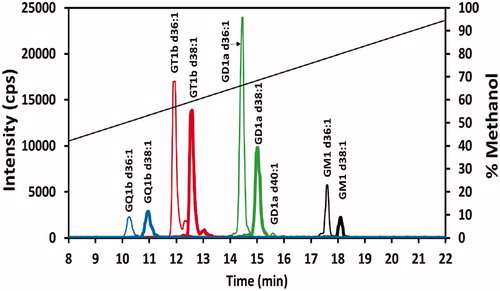
Mouse brain results
A sample of the superior colliculus from a single C57BL/6J mouse brain was prepared, as described in the Materials and methods section. The chromatogram is given in . Given the results of the matrix effect studies, only the GM1 gangliosides could be reasonably estimated (+20% to +26% deviation), determined to be 50 pmol/mg tissue and 7 pmol/mg tissue (not correcting for a matrix effect) for the GM1 d36:1 and GM1 d38:1 gangliosides, respectively. The GD1a, GT1b, and GQ1b could not be reasonably estimated due to significant matrix effect deviation (−40% to −79% deviation). More work on sample preparation needs to be done to remove this matrix interference for accurate quantification of these gangliosides.
Figure 5. Ganglioside profile of mouse superior colliculus sample. Chromatogram of 20 ng/mL of a superior colliculus sample from a single mouse prepared as given in the Materials and methods section. See caption for color identification of ganglioside class and diagonal line specifics.
Separation mechanism of the gangliosides by the phenyl-hexyl column
A plausible mechanism for the noted profile of gangliosides chromatographed on the phenyl-hexyl column draws upon the dual nature of the phenyl-hexyl group, with the phenyl group acting both electrostatically and, in conjunction with the hexyl group, hydrophobically. The electrostatic characteristic of the phenyl group in methanol mobile phases arises from the negative potential resulting from the π electrons of the phenyl group. The π cloud presents a repulsive electrostatic force toward negatively-charged molecules. The relative magnitude of the repulsive force experienced by the chromatographed gangliosides is as follows: GQ14− > GT13− > GD12− > GM11−, reflecting the number of negatively-charged sialic groups. This explains the elution order of the ganglioside classes, with the highest negatively-charged GQ1s eluting first and the lowest negatively-charged GM1s eluting last. A computational study supports this mechanism by documenting that a phenyl group has a repulsive potential toward approaching negatively-charged chloride and fluoride ions.[Citation39]
Studies have reported a π-electron effect in phenyl-column chromatography. However, the effect reported in these studies is attractive (not repulsive) to certain types of analytes. Aromatic compounds[Citation35,Citation37,Citation38,Citation40,Citation41] and electron-poor aromatic compounds (such as aromatic compounds containing nitro substituents)[Citation35,Citation37,Citation41] are selectively retained by a π–π interaction when a methanol organic modifier is used in the mobile phase, leading to specific retention. Concerning possible repulsive effects of the phenyl’s π electrons on negatively-charged analytes, a study was done characterizing the adsorption isotherm of the negatively-charged hexafluorophosphate on a hexyl-phenyl column.[Citation42] However, to the best of our knowledge, the present work is the first reported example of the separation of negatively-charged analytes resulting from a repulsive π-electron mechanism. Other studies involving acidic phenyl analytes chromatographed on phenyl columns have been done, however, these acids were chromatographed as neutral molecules in low pH mobile phases.[Citation36,Citation43]
The second characteristic of the phenyl group, with its hexyl moiety, is its non-polar nature, which imparts a reversed-phase retention and separation mechanism in the chromatography of the gangliosides. The particular commercial phenyl-hexyl column used in this work has been characterized to have similar reversed-phase characteristics as a C8 column, with the retention of acenaphthene being comparable to nineteen C8 columns from different manufacturers.[Citation44] This capability of the phenyl-hexyl column is the presumed mechanism for not only retaining the gangliosides on the column, but it is also important in separating the different gangliosides of a particular sialic acid class that differ in their ceramide content. Thus, the various gangliosides of a particular sialic acid class are separated within a particular retention time window in an elution order of shorter to longer ceramide carbon chain length and separated, for the most part, from the gangliosides of a different sialic acid class.
What distinguishes the phenyl-hexyl ganglioside chromatographic profile from that of a conventional reversed-phase ganglioside profile is how the gangliosides cluster in the chromatogram. While the phenyl-hexyl column clusters the gangliosides according to the ganglioside sialic acid class, the conventional reversed-phase column clusters the ganglioside according to the ceramide subclass. In the conventional reversed-phase column separation, all the gangliosides of a particular ceramide carbon length, but which have a different sialic acid/oligosaccharide content, cluster in a particular retention time window. This is seen in a previously published work employing a C18 column in the chromatography of gangliosides.[Citation33] In this work, the C18 column separates clusters of gangliosides in the order of increasing hydrophobicity with increasing ceramide carbon chain length. Within the cluster of gangliosides of a particular ceramide carbon chain length, there is a fine separation of gangliosides in the cluster differing in the sialic acid content, with the elution order being GT before GD before GM.
Comparing the phenyl-hexyl and conventional reversed-phase HPLC techniques, there is a different primary separation mechanism that separates the clusters, as well as a different secondary mechanism/effect that separates gangliosides within the cluster. For the phenyl-hexyl column, the primary separation effect occurs because of the difference in the sialic acid content of the various sialic acid ganglioside classes, which results in different repulsion effects by the column phenyl group (greater repulsion the more sialic acid groups), resulting in significant separation of the ganglioside sialic class clusters. Contrast this with the conventional reversed-phase separation of gangliosides, in which the sialic acid content has a moderate modulation effect on the ceramide hydrophobicity and thus is only a secondary effect.
Conversely, ceramide hydrophobicity leads to a substantial primary effect in the conventional reversed-phase technique, causing all gangliosides having a particular ceramide carbon chain length to cluster together, with the clusters being well separated in the order of increasing carbon chain length. Within each cluster, the sialic acid content modulates this hydrophobicity causing fine separation of the gangliosides in the conventional reversed-phase technique (gangliosides eluting in the cluster in the order of higher to lower sialic acid content, as seen in the published work, referred to above[Citation33]). Contrast this with the phenyl-hexyl column technique, in which the ceramide hydrophobicity, although being instrumental in retention of gangliosides on the column, this hydrophobicity has a critical secondary effect in separating the different ceramide gangliosides (in the order of increasing ceramide carbon chain length) within each particular sialic acid class cluster.
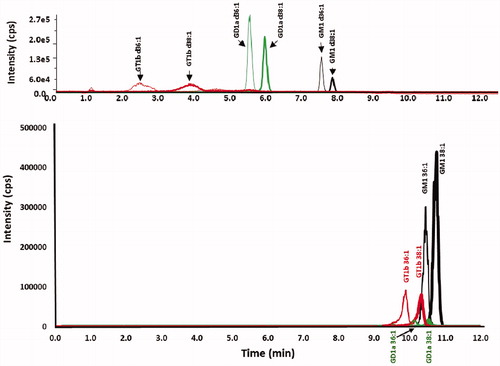
Support for the primacy of the column phenyl group in separating the ganglioside sialic acid classes is seen in experiments in which gangliosides are chromatographed at the same conditions (basic pH, same methanol gradient), comparing the phenyl-hexyl and C18 columns, as given in . These results show that for both columns there is baseline separation of the d36:1 and d38:1 gangliosides of a particular sialic class. However, there is a striking difference in the separation of the sialic acid classes, in which the ganglioside classes are substantially separated on the phenyl hexyl column, while only slightly separated on the C18 reversed-phase column and also showing overlapping peaks of the different ganglioside classes. This lends credence to the conclusion that the phenyl group separates the ganglioside sialic acid classes by a different mechanism than the C18 column, being much more than simply lowering the hydrophobicity of the ganglioside, as is the mechanism in the C18 column. It is proposed here that the primary effect of the phenyl group is a π-repulsion of the negatively-charged gangliosides.
Figure 6. Chromatograms of ganglioside standards chromatographed with a methanol gradient at pH 9 on both a phenyl-hexyl and a C18 column. Chromatograms of GT1b (red), GD1a (green) and GM1 (black) standards for (top) XBridge BEH phenyl-hexyl column and (bottom) Zorbax Eclipse Plus C18 column, employing a linear gradient of mobile phase A (100% water adjusted to pH 9 with ammonium hydroxide) and mobile phase B (100% methanol adjusted to pH 9 with ammonium hydroxide), 60% B to 100% B in 8 min.
Conclusions
The present technique has the advantage over both of the currently-used HPLC techniques, combining the hydrophilic separating capability of the HILIC technique with the non-polar separating capability of the reversed-phase technique. The present technique has the advantage over both of the currently-used HPLC techniques, combining the hydrophilic separating capability of the HILIC technique with the non-polar separating capability of conventional reversed-phase techniques. The phenyl-hexyl technique, like HILIC, separates gangliosides based on the sialic acid number (i.e., separates the classes) and thus is advantageous, in that the retention time reflects the number of sialic acids in the ganglioside. However, unlike HILIC, the present technique separates gangliosides of different ceramide carbon chain lengths within a particular sialic acid class, with the resolved peaks being clustered within a characteristic retention time window for all the gangliosides of that particular sialic acid class. Unlike the conventional reversed-phase technique, in which the gangliosides of each of the sialic acid classes are distributed throughout the chromatogram according to the ceramide carbon chain length, the phenyl-hexyl technique, as just mentioned, clusters all the ganglioside of a particular sialic acid class together in a particular retention time window. Furthermore, within this window, there is a specific retention time for a ganglioside based on its non-polar ceramide, with retention times increasing with increased ceramide chain length.
This phenyl-hexyl HPLC technique adds to the arsenal of existing reversed-phase and HILIC chromatographic techniques, providing a complementary separation capability needed for analysis of gangliosides in biological samples, samples which inevitably contain a large number of different gangliosides. Having more complementary chromatographic techniques for gangliosides can better address the problem of suppression of MS signals that occurs for co-eluting gangliosides, as well as giving alternate separation results to deal with matrix effects that might interfere in the determination of specific gangliosides using a particular type of column.
Acknowledgments
The authors thank Dr. Tariq M. Haqqi for use of the LC-MS/MS instrumentation in his laboratory at Northeast Ohio Medical University, Rootstown, Ohio. This work was conducted with support from the Faculty and Research Development Program and the Dissertation Research Award program at Cleveland State University. This work was also supported by the Neurodegenerative Diseases and Aging Research Focus Area at Northeast Ohio Medical University. The National Science Foundation Major Research Instrumentation Grant (CHE-0923398) supported the requisition of the ABSciex QTrap 5500 mass spectrometer instrument which was used in the optimization studies. The authors also thank Dr. Sameh Helmy for his contribution to finding a pertinent literature article to support the hypothesized chromatographic mechanism.
Disclosure statement
The authors and institutions declare no financial interest or benefit from the work presented in this manuscript
References
- Aureli, M.; Mauri, L.; Ciampa, M. G.; Prinetti, A.; Toffano, G.; Secchieri, C.; Sonnino, S. GM1 Ganglioside: Past Studies and Future Potential. Mol. Neurobiol. 2016, 53, 1824–1842. DOI: 10.1007/s12035-015-9136-z.
- Chester, M. A. Nomenclature of Glycolipids. Pure Appl. Chem. 1997, 69, 2475–2487. DOI: 10.1351/pac199769122475.
- Gobburi, A. L. P.; Zhang, R.; Willard, B.; Inman, D.; Anderson, D. Heterogeneous Ganglioside Standards in LC-MS/MS: Sensitive Method for Quantifying the Major Molecular Components in Mono-Sialo Ganglioside Standards. J. Anal. Bioanal. Tech. 2015, S13, 009. DOI: 10.4172/2155-9872.s13-009.
- Tettamanti, G.; Anastasia, L. Chemistry, Tissue and Cellular Distribution, and Developmental Profiles of Neural Sphingolipids. In Handbook of Neurochemistry and Molecular Neurobiology: Neural Lipids, Vol. 14; Lajtha, A., Ed., Tettamanti, G., Goracci, G., Volume Eds.; Springer Science + Business Media LLC: New York, NY, 2009; pp. 99–169.
- Ando, S.; Yu, R. K. Fatty Acid and Long-Chain Base Composition of Gangliosides Isolated from Adult Human Brain. J. Neurosci. Res. 1984, 12, 205–211. DOI: 10.1002/jnr.490120208.
- Sonnino, S.; Chigorno, V. Ganglioside Molecular Species Containing C18- and C20- Sphingosine in Mammalian Nervous Tissues and Neuronal Cell Cultures, Biochim. Biophys. Acta. 2000, 1469, 63–77. DOI: 10.1016/S0005-2736(00)00210-8.
- Yu, R. K.; Yanagisawa, M.; Ariga, T.; Glycosphingolipid Structures. In Comprehensive Glycoscience: From Chemistry to Systems Biology Volume 1 - Introduction to Glycoscience: Synthesis of Carbohydrates; Kamerling, J. P., Editor- in-Chief, Boons, G. –J., Lee, Y. C., Suzuki, A., Taniguchi, N., Voragen, A. G. J., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2007; pp. 73–122. DOI: 10.1016/B978-044451967-2/00003-9.
- Sarbu, M.; Dehelean, L.; Munteanu, C. V. A.; Vukelić, Ž.; Zamfir, A. D. Assessment of Ganglioside Age-Related and Topographic Specificity in Human Brain by Orbitrap Mass Spectrometry. Anal. Biochem. 2017, 521, 40–54. DOI: 10.1016/j.ab.2017.01.010.
- Flangea, C.; Fabris, D.; Vukelić, Ž.; Zamfir, A. D. Mass Spectrometry of Gangliosides from Human Sensory and Motor Cortex. Aust. J. Chem. 2013, 66, 781–790. DOI: 10.1071/CH13173.
- Zamfir, A. D.; Fabris, D.; Capitan, F.; Munteanu, C.; Vukelić, Ž.; Flangea, C. Profiling and Sequence Analysis of Gangliosides in Human Astrocytoma by High-Resolution Mass Spectrometry. Anal. Bioanal. Chem. 2013, 405, 7321–7335. DOI: 10.1007/s00216-013-7173-x.
- Hama, H. Fatty Acid 2-Hydroxylation in Mammalian Sphingolipid Biology. Biochim. Biophys. Acta. 2010, 1801, 405–414. DOI: 10.1016/j.bbalip.2009.12.004.
- Bouhours, J.-F.; Bouhours, D.; Hansson, G. C. Developmental Changes of Gangliosides of the Rat Stomach. Appearance of a Blood Group B-Active Ganglioside. J. Biol. Chem. 1987, 262, 16370–16375.
- Ostrander, G. K.; Levery, S. B.; Hakomori, S.; Holmes, E. H. Isolation and Characterization of the Major Acidic Glycosphingolipids from the Liver of the English Sole (Parophrys Vetulus). Presence of a Novel Ganglioside with a Forssman Antigen Determinant. J. Biol. Chem. 1988, 263, 3103–3110.
- Keränen, A. Fatty Acids and Long-Chain Bases of Gangliosides of Human Gastrointestinal Mucosa. Chem. Phys. Lipids. 1976, 17, 14–21. DOI: 10.1016/0009-3084(76)90032-3.
- Van Dessel, G. A. F.; Lagrou, A. R.; Hilderson, H. J. J.; Dierick, W. S. H.; Lauwers, W. F. J. Structure of the Major Gangliosides from Bovine Thyroid. J. Biol. Chem. 1979, 254, 9305–9310.
- Ledeen, R.; Salsman, K. Fatty Acid and Long Chain Base Composition of Adrenal Medulla Gangliosides. Lipids. 1970, 5, 751–756. DOI: 10.1007/BF02531387.
- Puro, K.; Keränen, A. Fatty Acids and Sphingosines of Bovine-Kidney Gangliosides. Biochim. Biophys. Acta. 1969, 187, 393–400. DOI: 10.1016/0005-2760(69)90013-7.
- Yu, R. K.; Ledeen, R. W. Gangliosides of Human, Bovine, and Rabbit Plasma. J. Lipid Res. 1972, 13, 680–686.
- Nekrasov, E.; Hubl, U. Gangliosides. In Sialobiology: Structure, Biosynthesis and Function. Sialic Acid Glycoconjugates in Health and Disease; Tiralongo, J., Martinez-Duncker, I., Eds.; Bentham Science Publishers: Oak Park, IL, 2013; pp. 313–380. DOI: 10.2174/97816080538651130101.
- Garcia, A. D.; Chavez, J. L.; Mechref, Y. Rapid and Sensitive LC-ESI-MS of Gangliosides. J Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2014, 947–948, 1–7. DOI: 10.1016/j.jchromb.2013.11.025.
- Ikeda, K.; Taguchi, R. Highly Sensitive Localization Analysis of Gangliosides and Sulfatides Including Structural Isomers in Mouse Cerebellum Sections by Combination of Laser Microdissection and Hydrophilic Interaction Liquid Chromatography/Electrospray Ionization Mass Spectrometry with Theoretically Expanded Multiple Reaction Monitoring. Rapid Commun. Mass Spectrom. 2010, 24, 2957–2965. DOI: 10.1002/rcm.4716.
- Fong, B.; Norris, C.; Lowe, E.; McJarrow, P. Liquid Chromatography-High-Resolution Mass Spectrometry for Quantitative Analysis of Gangliosides. Lipids. 2009, 44, 867–874. DOI: 10.1007/s11745-009-3327-1.
- Oikawa, N.; Matsubara, T.; Fukuda, R.; Yasumori, H.; Hatsuta, H.; Murayama, S.; Sato, T.; Suzuki, A.; Yanagisawa, K. Imbalance in Fatty-Acid-Chain Length of Gangliosides Triggers Alzheimer Amyloid Deposition in the Precuneus. PLoS One. 2015, 10, e0121356. DOI: 10.1371/journal.pone.0121356.
- Nagafuku, M.; Okuyama, K.; Onimaru, Y.; Suzuki, A.; Odagiri, Y.; Yamashita, T.; Iwasaki, K.; Fujiwara, M.; Takayanagi, M.; Ohno, I.; Inokuchi, J.-I. CD4 and CD8 T Cells Require Different Membrane Gangliosides for Activation. Proc. Natl. Acad. Sci. USA. 2012, 109, E336–E342. DOI: 10.1073/pnas.1114965109.
- Khoury, S.; Masson, E.; Sibille, E.; Cabaret, S.; Berdeaux, O. Rapid Sample Preparation for Ganglioside Analysis by Liquid Chromatography Mass Spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2020, 1137, 121956. DOI: 10.1016/j.jchromb.2019.121956.
- Masson, E. A. Y.; Sibille, E.; Martine, L.; Chaux-Picquet, F.; Bretillon, L.; Berdeaux, O. Apprehending Ganglioside Diversity: A Comprehensive Methodological Approach. J. Lipid Res. 2015, 56, 1821–1835. DOI: 10.1194/jlr.D060764.
- Sørensen, L. K. A Liquid Chromatography/Tandem Mass Spectrometric Approach for the Determination of Gangliosides GD3 and GM3 in Bovine Milk and Infant Formulae. Rapid Commun. Mass Spectrom. 2006, 20, 3625–3633. DOI: 10.1002/rcm.2775.
- Zhang, J.; Ren, Y.; Huang, B.; Tao, B.; Pedersen, M. R.; Li, D. Determination of Disialoganglioside GD3 and Monosialoganglioside GM3 in Infant Formulas and Whey Protein Concentrates by Ultra-Performance Liquid Chromatography/Electrospray Ionization Tandem Mass Spectrometry. J. Sep. Sci. 2012, 35, 937–946. DOI: 10.1002/jssc.201101039.
- Giuffrida, F.; Elmelegy, I. M.; Thakkar, S. K.; Marmet, C.; Destaillats, F. Longitudinal Evolution of the Concentration of Gangliosides GM3 and GD3 in Human Milk. Lipids. 2014, 49, 997–1004. DOI: 10.1007/s11745-014-3943-2.
- Lee, J.; Hwang, H.; Kim, S.; Hwang, J.; Yoon, J.; Yin, D.; Choi, S. I.; Kim, Y.-H.; Kim, Y.-S.; An, H. J. Comprehensive Profiling of Surface Gangliosides Extracted from Various Cell Lines by LC-MS/MS. Cells. 2019, 8, 1323. DOI: 10.3390/cells8111323.
- Li, Q.; Sun, M.; Yu, M.; Fu, Q.; Jiang, H.; Yu, G.; Li, G. Gangliosides Profiling in Serum of Breast Cancer Patient: GM3 as a Potential Diagnostic Biomarker. Glycoconj. J. 2019, 36, 419–428. DOI: 10.1007/s10719-019-09885-z.
- Hu, T.; Jia, Z.; Zhang, J.-L. Strategy for Comprehensive Profiling and Identification of Acidic Glycosphingolipids Using Ultra-High-Performance Liquid Chromatography Coupled with Quadrupole Time-of-Flight Mass Spectrometry. Anal. Chem. 2017, 89, 7808–7816. DOI: 10.1021/acs.analchem.7b02023.
- Ikeda, K.; Shimizu, T.; Taguchi, R. Targeted Analysis of Ganglioside and Sulfatide Molecular Species by LC/ESI-MS/MS with Theoretically Expanded Multiple Reaction Monitoring. J. Lipid Res. 2008, 49, 2678–2689. DOI: 10.1194/jlr.D800038-JLR200.
- Bocian, S.; Skoczylas, M.; Goryńska, I.; Matyska, M.; Pesek, J.; Buszewski, B. Solvation Processes on Phenyl-Bonded Stationary Phases-The Influence of Polar Functional Groups. J. Sep. Sci. 2016, 39, 4369–4376. DOI: 10.1002/jssc.201600799.
- Croes, K.; Steffens, A.; Marchand, D. H.; Snyder, L. R. Relevance of π-π and Dipole-Dipole Interactions for Retention on Cyano and Phenyl Columns in Reversed-Phase Liquid Chromatography. J. Chromatogr. A. 2005, 1098, 123–130. DOI: 10.1016/j.chroma.2005.08.090.
- Yang, M.; Fazio, S.; Munch, D.; Drumm, P. Impact of Methanol and Acetonitrile on Separations Based on π-π Interactions with a Reversed-Phase Phenyl Column. J. Chromatogr. A. 2005, 1097, 124–129. DOI: 10.1016/j.chroma.2005.08.028.
- Marchand, D. H.; Croes, K.; Dolan, J. W.; Snyder, L. R.; Henry, R. A.; Kallury, K. M. R.; Waite, S.; Carr, P. W. Column Selectivity in Reversed-Phase Liquid Chromatography VIII. Phenylalkyl and Fluoro-Substituted Columns. J. Chromatogr. A. 2005, 1062, 65–78. DOI: 10.1016/j.chroma.2004.11.014.
- Stevenson, P. G.; Kayillo, S.; Dennis, G. R.; Shalliker, R. A. Effects of π-π Interactions on the Separation of PAHs on Phenyl-Type Stationary Phases. J. Liq. Chromatogr. Relat. Technol. 2008, 31, 324–347. DOI: 10.1080/10826070701780607.
- Wheeler, S. E.; Houk, K. N. Are Anion/π Interactions Actually a Case of Simple Charge-Dipole Interactions? J. Phys. Chem. A. 2010, 114, 8658–8664. DOI: 10.1021/jp1010549.
- Stevenson, P. G.; Soliven, A.; Dennis, G. R.; Gritti, F.; Guiochon, G.; Shalliker, R. A. π-Selective Stationary Phases: (III) Influence of the Propyl Phenyl Ligand Density on the Aromatic and Methylene Selectivity of Aromatic Compounds in Reversed Phase Liquid Chromatography. J. Chromatogr. A. 2010, 1217, 5377–5383. DOI: 10.1016/j.chroma.2010.05.029.
- Euerby, M. R.; Petersson, P.; Campbell, W.; Roe, W. Chromatographic Classification and Comparison of Commercially Available Reversed-Phase Liquid Chromatographic Columns Containing Phenyl Moieties Using Principal Component Analysis. J. Chromatogr. A. 2007, 1154, 138–151. DOI: 10.1016/j.chroma.2007.03.119.
- Kazakevich, I. L.; Snow, N. H. Adsorption Behavior of Hexafluorophosphate on Selected Bonded Phases. J. Chromatogr. A. 2006, 1119, 43–50. DOI: 10.1016/j.chroma.2006.02.094.
- Goss, J. D. Improved Liquid Chromatography of Salicylic Acid and Some Related Compounds on a Phenyl Column. J. Chromatogr. A. 1998, 828, 267–271. DOI: 10.1016/S0021-9673(98)00655-4.
- https://www.waters.com/waters/promotionDetail.htm?id=10048475&alias=Alias_selectivitychart__CHEMISTRY&locale=en_US