Abstract
The present study correlates the mechanism of α-amylase denaturation under acidic condition and its structural stabilization in presence of selected cosolvents (sorbitol, sucrose, trehalose, and glycerol). The objective of the present study was to minimize the enzyme inactivation at lower pH by stabilizing the enzyme structure. The above cosolvents were found to be an effective stabilizer of α-amylase against denaturation at extreme low pH. The optimum activity of α-amylase was found to be in the pH range of 4.5 to 7. Decreasing the pH of enzyme solution below this range results in a decrease in enzyme activity. The intrinsic and ANS fluorescence indicated the gradual unfolding of protein below pH 4 and resulted in exposure of hydrophobic cluster. The pH-induced unfolding also resulted in protein aggregation. The intrinsic and 8-anilinonaphathalene-1-sulphonic acid (ANS) fluorescence, acrylamide quenching, near and far UV-CD spectra of α-amylase at lower pH (pH 2.25) indicated the complete loss of tertiary structure and substantial loss of secondary structure. Cosolvents were shown to prevent the acid induced unfolding of the enzyme to a certain extent and help in retaining the secondary structure of enzyme equivalent to native state. Among all the cosolvents used, sorbitol was proven to be the most efficient stabilizer. At lower pH, in presence of cosolvents the enzyme was shown to retain significant amount of secondary structure and poorly defined tertiary structure. This structure resembles the molten globule of the protein, which has substantial amount of secondary structure but poorly defined tertiary structure.
INTRODUCTION
Although the structural stability of protein largely depends on the primary structure of protein,[Citation1] the pH of the protein solution has a very crucial role in determining the overall stability. At extreme acidic or alkaline pH, away from isoelectric points, the protein tends to unfold due to electrostatic repulsion between like charges formed due to ionization of functional groups in response to a change in pH.[Citation2,Citation3] At extreme pH, proteins gradually undergo denaturation and results in the formation of unfolded structure containing substantial amounts of secondary structure and poorly defined tertiary structure, which resemble the molten globule state of protein in protein folding pathways. Characterization of intermediates of protein unfolding pathways is very crucial for studying the structural stability and designing the protein molecule with novel characteristics. The partially unfolded protein can be refolded in various ways depending upon the conditions provided. Many proteins at low pH undergo partial denaturation, which loses the tertiary structure and retains a certain degree of secondary structure, known as molten globule. Molten globules are likely to form during early stages of protein folding after translation.[Citation4] In order to understand the cascade of folding-unfolding reaction, it is important to characterize the forces, which are required to integrate the protein structure in the native state. In protein folding-unfolding reactions, the intermediates are transiently stable. These intermediates are largely fluctuating structures and their stability is largely determined by a delicate balance of interactions, such as electrostatic repulsion between charged residues and hydrophobic interactions.[Citation5] A large number of proteins are demonstrated to undergo acid induced unfolding and formation of stable molten globule at low pH.[Citation2,Citation6–9]
The solvent perturbation has a great role in providing the structural stability of protein molecule. The interaction of protein with solvent depends on the various physicochemical parameters of solvents. The compositions and conditions determine various types of forces operating in protein molecule responsible for protein stability. The cosolvents are known to stabilize several proteins against thermal denaturation, mainly by alteration of preferential interaction parameters.[Citation10] In this paper the terms cosolvents, additives, excipients, polyols, and sugars have been used interchangeably.[Citation10–12] In addition to stabilizing the native state of protein, cosolvent can also stabilize the intermediate state of protein folding pathway, which may provide valuable information in understanding the nature of these intermediates. Several studies indicated that cosolvents stabilize the protein by enhancing the hydrophobic interaction.[Citation13–17]
In the present investigation α-amylase (α-1,4-D-glucan glucanohydrolase, EC: 3.2.1.1), a well-studied enzyme with complete sequence available, was selected as a model protein. It is a metalloenzyme; requires at least one calcium ion for its structure and function.[Citation18] It contains a characteristic (β/α)8 structure, called a triose-phosphate-isomerase-barrel catalytic domain with highly symmetrical folds of eight inner parallel β-strands surrounded by eight α-helices.[Citation19] The whole structure of enzyme is constituted by three domains named as domain A, B, and C. The domain B is inserted between the 3rd β-strand and 3rd α-helix of (β/α)8 structure of (β/α)8 fold. Each enzyme in this family contains one glutamic acid and two aspartic acids residues in the catalytic active site.[Citation19,Citation20] The domain C is thought to stabilize the catalytic domain by shielding the hydrophobic domain.[Citation21] The stability of α-amylase has been studied extensively using cosolvents and found that stability has significantly improved.[Citation16] In the present investigation, efforts have been made to correlate the structural alteration and stabilization of protein at low pH in presence of selected cosolvents. Study has also been extended to correlate the formation of stable molten globule state of α-amylase at low pH in presence of cosolvents. This may be a new characteristic of cosolvents that not only stabilize the native protein but may also reverse the denaturation reaction.
MATERIAL AND METHODS
Chemicals and Reagents
α-Amylase type II (A6380), trehalose, sorbitol, sucrose, glycerol, starch, dinitrosalisylic acid, 8-anilinonaphathalene-1-sulphonic acid (ANS), bovine serum albumin (BSA), and CaCl2 were procured from Sigma Chemicals Company (St. Louis, MO, USA). The 2-Chloro-p-nitrophenyl-α-D-maltotrioside (CNPG3) was procured from Pointe Scientific, Inc. (Canton, MI, USA). KCl, NaCl, and HCl were obtained from Qualigen Fine Chemicals Pvt. Ltd. (India). Acrylamide, NaOH, glycine, and dibasic sodium phosphate were procured from E-Merck (Mumbai, India). All chemicals were of analytical grade. The crystalline α-amylase was dissolved and dialyzed in 0.02 M citrate buffer (pH 5.9) to remove the excipients, freeze-dried (−80°C) and desiccated at 0°C for future use. Quartz triple distilled water was used for all the experiments. All the cosolvents were analyzed for calcium ion contamination by using atomic absorption spectroscopy. The calcium ion content was found to be approximately 0.1 mM in 50% aqueous solution of each cosolvent, which was low enough to make any appreciable change in activity and structural stability of enzyme.
Protein Estimation
Protein concentration was determined by measuring the absorbance of enzyme sample at 280 nm on Shimadzu UV-spectrophotometer model UV-1601 using an extinction coefficient (E1% 1cm) of α-amylase as 14.46.[Citation16] Alternatively, the protein concentration was determined by the Lowery method using BSA as standard protein.[Citation22]
α-Amylase Activity Measurement
α-Amylase activity was measured by using the Bernfeld method[Citation23] for estimation of reducing sugar. The crystalline amylase was dissolved in 0.02 M citrate buffer, pH 5.9, containing 1 mM of CaCl2 and final concentration of enzyme was adjusted to 1 μg/mL. The reaction mixture containing 1 mL of 1% starch solution and 1 mL of enzyme solution incubated for 5 min on a temperature-controlled circularly rotating water bath at 37°C. The reaction was terminated by addition of 2 mL of 1% alkaline dinitrosalicylic acid solution to the reaction mixture. The whole solution was then subjected to heat in a boiling water bath for 10 min. After cooling, it was diluted five times using triple distilled water, mixed properly, and absorbance was recorded at 540 nm. The activity was calculated by using maltose standard plot. The unit of enzyme activity is defined as the amount of enzyme required for starch hydrolysis to produce 1 μmol of maltose equivalent under given conditions.
Alternatively, the α-amylase activity was estimated by using an artificial substrate CNPG3. α-Amylase hydrolyzes the CNPG3 to release 2-Chloro-nitrophenol, maltotriside, and glucose.[Citation24,Citation25] The amount of 2-Chloro-nitrophenol released reflects the degree of hydrolysis, which could be monitored by spectrophotometry. The 25 μL of enzyme solution was added to 1 mL of CNPG3 solution and incubated at 37°C for 3 min, and absorbance was recorded at 405 nm in Shimadzu UV-1601 UV-Visible spectrophotometer. The enzyme activity was determined using the equation:
where MMA is the milimolar absorptivity of CNPG3, SV is sample volume, and l is the path length of light.
pH Induced Inactivation of α-Amylase
A series of enzyme solutions were prepared in 20 mM solution of different buffers having different pH ranging from 1.5 to 10.00 (KCl–HCl buffer, pH 1.5–2.5, citrate buffer pH 3.0–6.0, citrate-phosphate buffer pH 6.0–7.5, and glycin-NaOH buffer pH 8–10). These enzyme solutions were incubated for 12 h to equilibration at 4°C before the activity measurement. The pH of enzyme solutions was measured before and after the experiment.
Estimation of Protein Aggregation
The pH induced aggregation of α-amylase at different pH was measured by monitoring the absorbance at 360 nm in the Shimadzu UV-1601 UV-Visible spectrophotometer (Japan). The absorbance at 360 nm arise mainly due to scattering of light by particulate matter in the solution, and at this wavelength scattering can be measured free from interference caused by intrinsic fluorescence or light absorption of the enzyme.[Citation26] The α-amylase samples were incubated at different pH at 25°C before taking the absorbance.
Intrinsic and ANS Fluorescence Measurement
Fluorescence measurement of α-amylase in presence of cosolvents at different pH values was performed on a Shimadzu spectroflurophotometer model RF-5000 equipped with temperature-controlled device. Intrinsic fluorescence spectra of α-amylase were measured at 25°C. The pH of protein solution was maintained as 1.5 to 10.00 (KCl–HCl buffer, pH 1.5–2.5, citrate buffer pH 3.0–6.0, citrate-phosphate buffer pH 6.0–7.5, and glycin-NaOH buffer pH 8–10). The enzyme samples were prepared in different concentrations of cosolvents at different pH and incubated for 12 h before recording the fluorescence spectra. For intrinsic fluorescence excitation it was set at 280 nm and emission was recorded in the range of 300–400 nm using slit width of 10 and 5 nm for excitation and emission, respectively. For ANS fluorescence, the molar ratio of ANS and protein was 100:1, excitation was set at 380 nm, and emission spectra were recorded in the range of 400–600 nm, using slit width of 10 and 5 nm for excitation and emission, respectively. Protein concentration for all the fluorescence experiments was 0.05 mg/mL.
Acrylamide Quenching
Aliquots of 10 μL of acryamide stock solution (2M) were added to 2 mL of α-amylase solutions incubated at different pH, mixed by inverting or with magnetic stirrer set with spectrofluorophotometer chamber. Excitation was set at 280 nm and emission maxima were recorded at 340 nm after each addition of acrylamide stock solution. The acrylamide fluorescence quenching of protein samples were analyzed by the Stern-Volmer equation:[Citation27]
where F 0 and F are the fluorescence intensity in absence and presence of quencher, respectively. Ksv is Stern-Volmer constant, which can be obtained from the slope of the curve of F 0/F versus quencher concentrations.
Circular Dichroism (CD) Measurement
CD measurement was carried out on a Jasco spectropolarimeter model J-810 equipped with computer. The instrument was calibrated with D-10 camphorsulphonic acid. α-Amylase samples were equilibrated at pH 7 and 2.25 in presence of different concentrations of each cosolvent and CaCl2 for 12 h at 4°C before CD measurement. Far and near UV-CD was measured using protein concentration of 0.268 and 1 mg/mL, respectively. The path length of far and near UV-CD were 1 mm and 10 mm, respectively, with the scan speed of 50 nm/min. The molar ellipticitiy was expressed as mean residue ellipticity in deg–cm2 dmol−1 using 110 g/mol residual mass of α-amylase. Each spectrum was an average of three independent scans. The secondary structure of α-amylase was analyzed using the program (J-810) of Yang et al.[Citation28] The statistical analysis of the data was carried out using OriginPro 7 software.
RESULTS
pH Induced Inactivation of α-Amylase
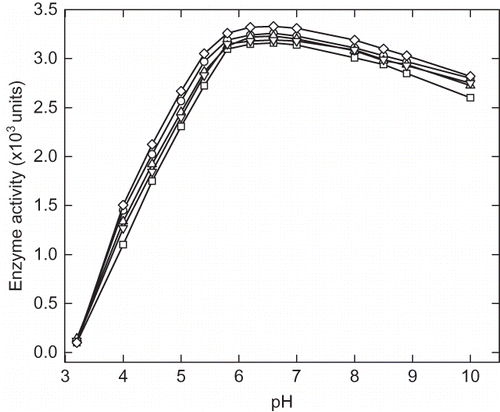
α-Amylase activity was measured in the pH range of 3.2 to 10.00 and it showed optimum activity in the range of pH 5.5–7.5 (). Enzyme samples were incubated at different pH from 3.20 to 10 in appropriate buffer for 12 h followed by activity measurement. indicates the activity of α-amylase in presence of 40% of each cosolvents, viz. trehalose, sorbitol, sucrose, and glycerol. Decreasing the pH below 4.5 resulted in drastic reduction in enzyme activity, and at pH 3.2 there was complete loss of activity. Addition of cosolvents was shown to have protective effect on the enzyme activity under condition of relatively low pH but at pH 3.2 no activity was observed even in presence of cosolvents. The maximum activity at pH 4, 4.5, and 5.0 were found to be 1100, 1740, 2300, and 2720 units, respectively, and in presence of 40% trehalose the activity was reported to be 1500, 2120, 2660, and 3050 units, respectively. The relative activity of enzyme in the presence of cosolvents at low pH was found in the order of trehalose > sucrose > glycerol > sucrose. In alkaline region, the activity enzyme was not affected significantly until pH 10. The pH induced enzyme inactivation was found to be reversible in certain ranges of pH (5–4). The loss of activity at pH below 4.0 was found to be irreversible. At pH close to optimum activity range, enzyme may not undergo any major structural change and the inhibition of enzyme in this range may be because of change in ionization potential of amino acid residues in the catalytic sites.
Figure 1 Effect of cosolvents on α-amylase activity at different pH. The curves represent the α-amylase activity of (∀) control, and in presence 40% (w/v) (8) glycerol, (X) sorbitol, (–) trehalose, and (M) sucrose.
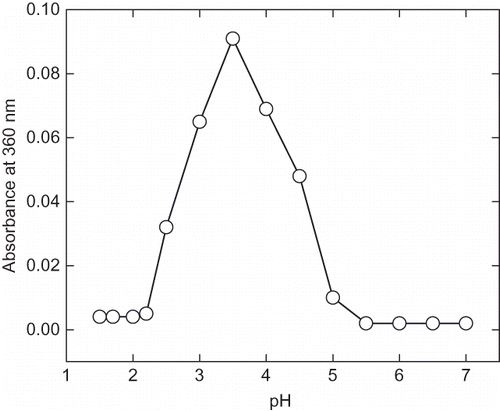
At pH less than 4.5 enzymes show irreversible inactivation and accompanied by pH-induced aggregation (). These protein aggregates were easily soluble in aqueous solution of 0.5% SDS, which is an indication of hydrophobic nature of the aggregates.[Citation29,Citation30] The pH-induced aggregation was prominently found in the range of pH 2.5–4.4. On further, lowering the pH aggregation decreases due to the reburial of hydrophobic residue in the protein interior. The pH-induced change structure indicates that the protein-folding pathway may primarily be governed by the hydrophobic interactions.
Figure 2 pH induced aggregation of α-amylase. The protein aggregation was monitored by recording the absorbance at 360 nm at each pH ranging from 1.5 to 7.
Intrinsic and ANS Fluorescence
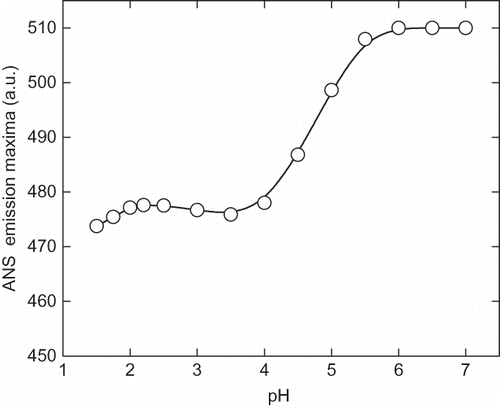
The pH-induced unfolding can be monitored by intrinsic and ANS fluorescence in the ranges of pH 1.5 to 7.0. At pH above 7 (up to 10) there was no appreciable change in the intrinsic and ANS fluorescence spectra. The intrinsic fluorescence intensity gradually decreases the pH until 2.25 (). Although, no significant change was observed in the fluorescence intensity of the enzyme on further lowering of pH, the fluorescence emission maxima (λmax) was found to be altered significantly. The λmax of native enzyme (at pH 7.0) was found to be 340 nm and with decreasing the pH a red shift was observed in the λmax, and at pH 2.25 it was recorded to be 343 nm. Further lowering the pH resulted in a blue shift in λmax, which is an indication of reorganization of unfolded enzyme molecule, although this structure can not be compared with the native enzyme. This reflects the changes in the micro environment of tryptophan and tyrosin residues in the protein. A similar result was reported with glucose/xylose oxidase enzyme where extreme low pH-induced an structuring effect.[Citation31] The ANS fluorescence has been extensively used to probe the conformational changes that occurred during protein denaturation.[Citation32] Generally, the protein denaturation leads to exposed hydrophobic clusters on the surface of enzyme and, hence, it becomes accessible to solvents. ANS is a hydrophobic dye that preferentially binds with the hydrophobic cluster on protein surface and fluoresces in the range of 400–600 nm. As shown in , , and ANS fluorescence intensity increases with the lowering of pH with significant change in the λmax. This indicates the gradual unfolding of α-amylase under acidic condition. The maximum ANS intensity was recorded at pH 2.25, indicating the maximum exposure of hydrophobic clusters on the surface. A decrease in intensity was observed by further lowering the pH. This could be due to the reburial of hydrophobic core of protein molecules away from aqueous medium, which creates variations in the structural compactness and flexibility in enzyme molecules at different pHs.
Figure 3 Intrinsic fluorescence spectra of α-amylase at different pH ranging from pH 1.5 to 7. The enzyme samples were incubated at different pH for 12 h before recording the spectra. The fluorescence spectra are represented as: (a) pH 7, (b) pH 6, (c) pH 5, (d) pH 4, (e) pH 3, (f) pH 2.75, (g) pH 2.25, and (h) pH 1.5.
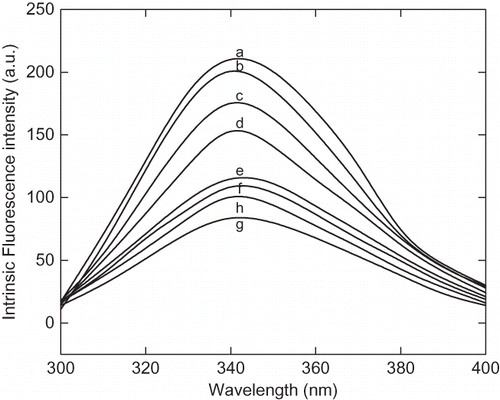
Figure 4a The 8-anilinonaphathalene-1-sulphonic acid (ANS) fluorescence spectra of α-amylase at different pH ranging from pH 1.5 to 7. The curves are represented as: (a) pH 7, 6.5 and 6.00, (b) pH 4.5, (c) pH 4.00, (d) pH 3.5, (e) pH 3.00, (f) pH 2.5, (g) pH 2.25, (h) pH 2.00, (i) pH 1.75, and (j) pH 1.5.
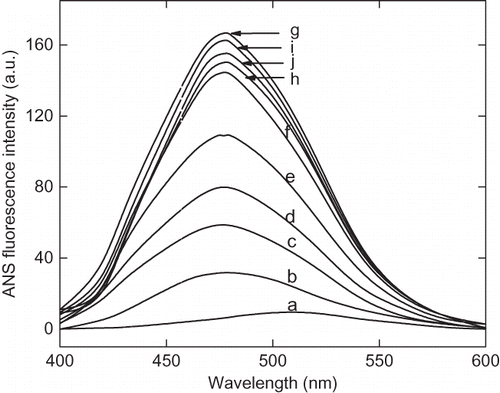
Figure 4b Variation of 8-anilinonaphathalene-1-sulphonic acid (ANS) fluorescence intensity of α-amylase with change in pH at 25°C. After incubating the enzyme solution at different pH the aliquots ANS stock solution was added, mixed and stand for 20 min in dark. The excitation wavelength was fixed at 380 nm and emission spectra were collected between 400–600 nm.
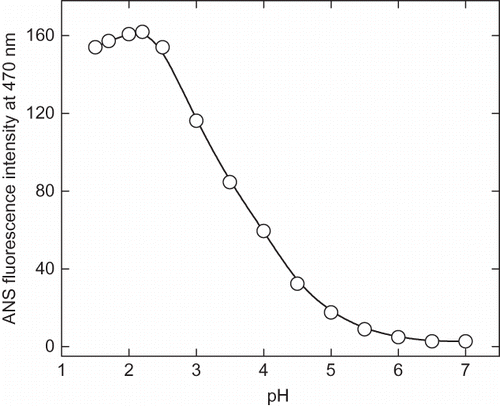
Figure 4c Change in 8-anilinonaphathalene-1-sulphonic acid (ANS) emission maximum as a function of pH. The emission spectra were collected between 400–600 nm and excitation wavelength was fixed at 380 nm.
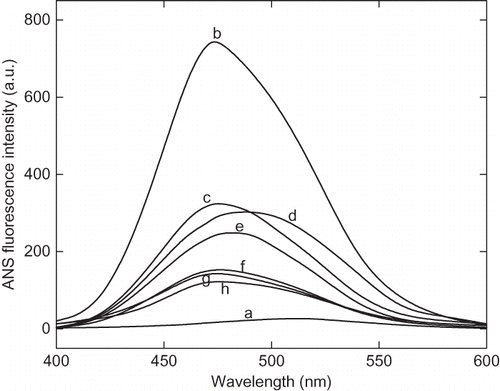
The fluorescence quenching study was carried out in order to confirm the degree of compactness of proteins at pH 7, 2.25, and 1.5. Acrylamide is a neutral molecule, and therefore, efficient quencher of intrinsic fluorescence has been used in this study.[Citation27] shows the Stern-Volmer plot of acrylamide quenching of fluorescence by acrylamide in native and acid denatured state. The protein at pH 2.25 shows very high quenching of intrinsic fluorescence compared to native protein at pH 7.0. The degree to which an amino acid side chains exposed to solvents depends upon the relative compactness of protein molecule. It shows that solvent accessibility of acid-denatured state is comparatively higher than the native enzyme (). The presence of additives, such as polyols-trehalose, sorbitol, sucrose, and glycerol, and salts-NaCl and CaCl2 has significantly reduced the ANS fluorescence in acidic condition, as shown in . The decrease in ANS fluorescence intensity in presence of cosolvents and additive indicates the stabilization of protein at low pH. This result suggests that these additives prevent the acid-induced denaturation of α-amylase.
Figure 5 Stern-Volmer plot of acrylamide quenching of intrinsic fluorescence of enzyme at pH (a) 7.0, (b) 2.25, and (c) 1.5. Aliquot of 2M stock solutions of acrylamide added sequentially and excitation wavelength was fixed at 280 nm and emission maxima were recorded at 340 nm. The F0 and F are the fluorescence intensity of the enzyme in absence and presence of acrylamide, respectively. The ratio of F0 and F indicates the relative quenching of intrinsic fluorescence under given condition.
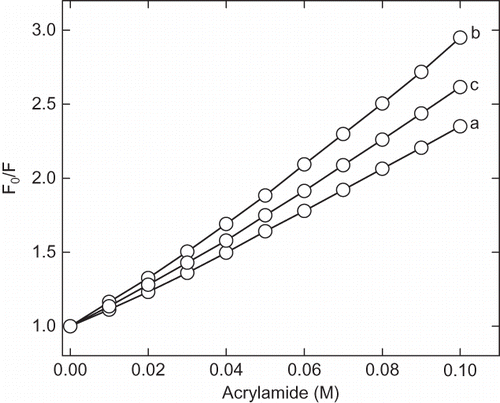
Figure 6 The 8-anilinonaphathalene-1-sulphonic acid (ANS) fluorescence spectra of enzyme at native and acidic pH in presence of cosolvents. Enzyme solutions were incubated at acidic pH in presence of cosolvents for 12 h before recording the spectra. The curves (a) and (b) represent pH 7 and 2.25 in absence of cosolvents, respectively. Curves (c), (d), (e), and (f) represent the spectra in presence of 30% of trehalose, sorbitol, sucrose, and glycerol at pH 2.25, respectively. The curve (g) and represent 100 mM CaCl2 and 250 mM of NaCl, respectively.
Near and Far UV-CD Spectra
The near and far UV-CD spectra of native and acid induced denatured state of α-amylase were recorded after equilibration at each pH. shows the near UV-CD spectra of amylase at pH 7.0 and 2.25. The spectrum in near UV-CD mainly contributed by the conformation of aromatic amino acid residues. The near UV-CD spectra showed the native orientation of aromatic amino acid residues. The appearance of major shoulders at 265 and 295 nm reflect the alteration in the tertiary structure and more flexible environment of aromatic amino acid residues. The far UV-CD spectra of amylase at pH 7.0 and 2.25 are showed in . The curve of native α-amylase has two minima at 222 nm and 208 nm, which are the characteristics of the α-helical structure. The presence of cosolvents did not show any effect on the secondary structure of the native enzyme (pH 7.0). In the acidic pH, cosolvents had significant protective effect on the secondary structure of enzyme. The α-helical content of α-amylase was found to be 16% and 7% at pH 7.0 and 2.25, respectively. Our purpose was to stabilize α-amylase against the acid-induced denaturation using selected cosolvents and salts. Before recording the far UV-CD spectra enzyme samples, they were equilibrated for 12 h in presence of different concentration of each cosolvent and salt at pH 7.0 and 2.25. summarizes the secondary structural element of enzyme in acidic condition in presence of various cosolvents. The presence of cosolvents helped in retaining the α-helical structure and the retention capacity of each cosolvent was found to be depending on the concentration used. Out of all four cosolvents used, sorbitol was proven to be the most efficient stabilizer at all concentrations against acid-induced denaturation. At low concentrations (up to 20% w/v) of other cosolvents, such as glycerol, trehalose, and sucrose, there is no significant change in the ellipticity, but at higher concentrations (30–40% w/v) of trehalose and sucrose, more than 75% the α-helical structure of native enzyme was protected. Glycerol, even at 40% concentrations, was not able to make any significant change. Although it has been shown that glycerol at 80% concentration retained up to 98% α-helical structure of the native human placental cystatin, whereas sorbitol at the same concentration has negligible effect.[Citation33] Since the acid-induced denaturation is a complete function of electrostatic repulsion, the neutralization of charged groups may substantially reduce the electrostatic repulsion by counter ion effect and render the protein stable. Therefore, investigation was also made in the presence of different concentration of salts (i.e., NaCl and CaCl2). Our purpose was also to see the effect of counter ions on the acid-induced denaturation α-amylase. It is well known that calcium is essential for providing structural stability and activity of α-amylase; it may also provide the suitable counter ion which can minimize the electrostatic repulsive forces. The NaCl is neutral salt ionizes completely in the aqueous medium and form the Na+ and Cl−which interact with the counter ion on the protein surface. The secondary structural elements of enzyme in different concentration of each salt are summarized in . The results showed that in the presence of 100 mM of CaCl2 more than 95% of α-helical content was preserved, whereas up to 85% of the native α-helical structure was retained in presence of 25 mM of NaCl.
Figure 7 (a) Near UV-circular dichroic spectra of enzyme at pH (1) 7.0 and (2) 2.25. The enzyme solutions were incubated for 12 h before the circular dichroism measurement. The protein concentrations were maintained to be 1 mg/mL. The [θ]MRW is molar residual elipticity of enzyme. (b) Far UV-circular dichroic spectra of α-amylase at pH (1) 7.0 and (2) 2.25. The enzyme solutions were incubated for 12 h before the CD measurement. The protein concentrations were maintained to be 0.26 mg/mL. The [θ]MRW is molar residual ellipticity of enzyme.
![Figure 7 (a) Near UV-circular dichroic spectra of enzyme at pH (1) 7.0 and (2) 2.25. The enzyme solutions were incubated for 12 h before the circular dichroism measurement. The protein concentrations were maintained to be 1 mg/mL. The [θ]MRW is molar residual elipticity of enzyme. (b) Far UV-circular dichroic spectra of α-amylase at pH (1) 7.0 and (2) 2.25. The enzyme solutions were incubated for 12 h before the CD measurement. The protein concentrations were maintained to be 0.26 mg/mL. The [θ]MRW is molar residual ellipticity of enzyme.](/cms/asset/2c19350c-69de-447d-ac96-9e9d641db17d/ljfp_a_459788_o_f0009g.gif)
Table 1 Comparison of secondary structural elements of α-amylase at pH 7.0 and 2.25 in presence of cosolvents
Table 2 Comparison of secondary structural element of α-amylase at pH 2.25 in the presence of CaCl2 and NaCl
DISCUSSION
Under mild denaturation conditions, such as low pH, temperature, and low concentration of denaturants, a number of protein exist in a stable conformation which neither resemble with native state nor fully denatured state, and can be classified as molten globule state: the intermediate between native and unfolded state.[Citation4,Citation8,Citation15,Citation29] It has been shown that in the presence of low concentration of urea, papain, and human serum albumin exist as molten globule and fully denatured at higher denaturant's concentrations.[Citation34,Citation35] A number of proteins are shown to undergo denaturation under the heavy acidic condition. It is reported that the α-amylase exist as molten globule-like structure at pH 3.0 with secondary structure equivalent to native enzyme but altered tertiary structure.[Citation36] Both, the cosolvents and salts (CaCl2 and NaCl), have a protecting effect of α-amylase against acid-induced denaturation. ANS binds preferentially to the hydrophobic clusters on the protein surface.[Citation32] Acid-induced (pH 2.25) denatured state of α-amylase has a maximum ANS fluorescence intensity, suggesting the exposure of hydrophobic patches on the protein surface. Although the addition of cosolvent decreases the ANS fluorescence intensity significantly, yet it was found to be more than the native enzyme. The decrease in intrinsic fluorescence might be due to the relocation of tryptophan and tyrosin residues to polar environment after acid denaturation, which reflects the alteration in the tertiary structure.[Citation37,Citation38] This suggests that presence of cosolvents helped in retention of secondary structure equivalent to native enzyme, but the acid denatured protein have not regained the native structure. The reduction in fluorescence intensity in the presence of cosolvents varies to different extent in presence of different cosolvents indicating the difference in the mechanism of stabilization. In the presence of CaCl2 and NaCl, the ANS fluorescence was close to native enzyme. Addition of additives did not affect the intrinsic fluorescence intensity and it was similar to the acid denatured state. The secondary structure of enzyme in presence of 50% of cosolvents found to be similar or close to the native enzyme. The mechanism of salt-induced refolding can be explained on the basis of neutralization of protonated side chains of amino acids by negative ions of salts, which decreases the internal repulsive force, which favor unfolding.[Citation9,Citation15]
Cosolvents generally stabilized the protein by altering the solvent distribution in the solution. In a previous report, it is shown that the presence of cosolvents in solution results in preferential hydration of α-amylase.[Citation39] It is also shown that molten globule state of papain is more hydrated compared to the native one.[Citation33] The preferential exclusion of cosolvents is directly proportional to surface area of the proteins and favors the protein to occupy minimum surface area.[Citation40] The preferential exclusion of cosolvents leads to increase compactness of the protein molecule compared to acid denatured state of enzyme in absence of cosolvents, which might provide the more rigid and stable structure compared to a native one.[Citation41–43] In the presence of cosolvents and salts, although the secondary structure was comparable to native state, the enzyme remains inactive, and the ANS fluorescence remains nearer to the denatured state. These results are indicative of the formation of molten globule state of enzyme, which has substantial amount of secondary structure and poor in tertiary structure.
CONCLUSION
Cosolvents were shown to have stabilizing effect on α-amylase at low pH. The stabilizing effects of cosolvents were found to be proportional to their concentration used. Although presence of cosolvent in the enzyme solution indicated the structural stabilizing effect, still the enzyme did not achieve the native functional state. This might be probably due to the loss of stereochemistry of tertiary structure. In this situation, the structures of the enzyme seems similar to the molten globule state of protein, which has significant secondary structure and poorly defined tertiary structure. Addition of salts (NaCl and CaCl2) also has significant effect on structural stabilization of α-amylase, although salts and cosolvents follow different mechanism of structural stabilization of the enzyme.
ACKNOWLEDGMENTS
Jay Kant Yadav gratefully acknowledges the Council of Scientific and Industrial Research (CSIR), New Delhi, India, for providing financial support in the form of Junior and Senior Research Fellowship during the course of work.
Related Research Data
REFERENCES
- Dill , K.A. 1990 . Dominant forces in protein folding . Biochemistry , 29 ( 30 ) : 7133 – 7155 .
- Goto , A. and Fink , A.L. 1989 . Conformational states of beta-lactamase: Molten globule state at acidic and alkaline pH with high salt . Biochemistry , 28 ( 3 ) : 945 – 952 .
- Volkin , D.B. and Klibanov , A.M. 1989 . “ Minimizing protein interactions ” . In Protein Function: A practical approach , Edited by: Creighton , T.E. 1 – 24 . Oxford, , UK : Information Press .
- Christina , R. , Smith , R.A.G. and Dobson , C.M. 1994 . Structural characterization of highly- ordered ‘molten globule’ at low pH . Nature Structural Biology , 1 : 23 – 29 .
- Kamiyama , T. , Sadahide , Y. , Nogusa , Y. and Gekko , K. 1999 . Polyols-induced molten globule of cytochrome c: An evidence for stabilization by hydrophobic interaction . Biochimica et Biophysica Acta (BBA)-Protein Structure and Molecular Enzymology , 1434 : 44 – 57 .
- Kataoka , M. , Hagihara , Y. , Mihara , K. and Goto , Y. 1993 . Molten globule of cytochrome c studied by small angle X-ray scattering . Journal of Molecular Biology , 229 : 591 – 596 .
- Nishii , I. , Kataoka , M. , Tokunaga , F. and Goto , Y. 1994 . Cold denaturation of molten globule states of apomyoglobin and a profile for protein folding . Biochemistry , 33 ( 16 ) : 4903 – 4909 .
- Christensen , H. and Pain , R.H. 1991 . Molten globule intermediates and protein folding . European Biophysics Journal , 19 : 221 – 229 .
- Potekhin , S. and Pfeil , W. 1989 . Microcalorimetric studies of conformational transitions of ferrycytochrome c in acidic solution . Biophysical Chemistry , 34 ( 1 ) : 55 – 62 .
- McClements , D.J. 2002 . Modulation of globular protein functionality by weakly interacting cosolvents . Critical Reviewes in Food Sciences and Nutrition , 42 : 417 – 471 .
- Gekko , K. and Timasheff , S.N. 1981 . Thermodynamic and kinetic examination of protein stabilization by glycerol . Biochemistry , 20 ( 16 ) : 4677 – 4686 .
- Gekko , K. and Ito , H. 1990 . Competing solvent effects of polyols and guanidine hydrochloride on protein stability . The Journal of Biochemistry , 107 : 572 – 577 .
- Gekko , K. 1981 . Mechanism of polyol-induced protein stabilization: Solubility of amino acids and diglycine in aqueous polyol solutions . The Journal of Biochemistry , 90 ( 6 ) : 1633 – 1641 .
- Xie , G. and Timasheff , S.N. 1997 . Mechanism of stabilization of ribonuclease A by sorbitol: Preferential hydration is greater for the denatured than for the native protein . Protein Science , 6 ( 1 ) : 211 – 222 .
- Goto , Y. , Calciano , L.J. and Fink , A.L. 1990 . Acid induced unfolding of proteins . Proceedings of National Academy of Sciences, USA , 87 : 573 – 577 .
- Rajendran , S. , Radha , C. and Prakash , V. 1995 . Mechanism of solvents induced thermal stabilization of α-amylase from Bacillus amyloliquefaciens . International Journal of Peptide and Protein Research , 45 : 122 – 128 .
- Fink , A.L. , Calciano , L.J. , Kurotsu , T. and Palleros , D.R. 1994 . Classification of acid denaturation of proteins: Intermediates and unfolded states . Biochemistry , 33 : 12,504 – 12,511 .
- Marco , J.L. , Bataus , L.A. , Valencia , F.F. , Ulhoa , C.J. , Astolfi-Filho , S. and Felix , C.R. 1996 . Purification and characterization of a truncated Bacillus subtillis α-amylase produced by Escherichia coli . Applied Microbiology and Biotechnology , 44 : 746 – 752 .
- Svensson , B. 1994 . Protein engineering in α-amylase family: Catalytic mechanism, substrate specificity, and stability . Plant Molecular Biology , 25 : 141 – 157 .
- MacGregor , E.A. , Janecek , S. and Svensson , B. 2001 . Relation of sequence and structure to specificity in the alpha amylase family of enzymes . Biochimica et Biophysica Acta , 1546 : 1 – 20 .
- Dauter , Z. , Dauter , M. , Brzozowski , A.M. , Christensen , S. , Borchert , T.V. , Beier , L. , Wilson , K.S. and Devies , G.J. 1999 . X-ray structure of Novamyl, the five domain “maltogenic” alpha-amylase from Bacillus stearothermophilus: Maltose and ascarbose complexes at 1.7 A° resolution . Biochemistry , 38 : 8385 – 8392 .
- Lowry , O.H. , Rosenborough , N.J. , Farr , A.L. and Randall , R.J. 1951 . Protein measurement with the Folin phenol reagent . Journal of Biological Chemistry , 193 : 265 – 275 .
- Bernfeld , P. 1955 . α- and β- Amylases . Methods in Enzymology , 1 : 149 – 158 .
- Foo , A.Y. and Bais , R. 1998 . Amylase measurement with 2-chloro-4-nitrophenyl maltotrioside as substrate . Clinica Chimica Acta , 272 : 137 – 147 .
- Dupuy , G. , Hilaire , G. and Aubry , C. 1987 . Rapid determination of α-amylase activity by use of a new chromogenic substrate . Clinical Chemistry , 33/4 : 524 – 528 .
- Dragani , B. , Cocco , R. , Principe , D.R. , Cicconetti , M. and Aceto , A. 2000 . Structural characterization of acid induced intermediates of human glutathione transferase P1 . 1. The International Journal of Biochemistry and Cell Biology , 32 : 725 – 736 .
- Eftink , M.R. and Ghiron , C.A. 1982 . Fluorescence quenching studies with proteins . Analytical Biochemistry , 114 : 199 – 227 .
- Yang , J.T. , Wu , C.S.C. and Martinez , H.M. 1986 . Calculation of protein conformation from circular dichroism . Methods in Enzymology , 130 : 208 – 269 .
- Edwin , F. , Sharma , Y.V. and Jagannadham , M.V. 2002 . Stabilization of molten globule state of papain by urea . Biochemical and Biophysical Research Communication , 290 : 1441 – 1446 .
- Wang , W. 1999 . Instability, stabilization, and formulation of liquid protein pharmaceuticals . International Journal of Pharmacology , 185 : 129 – 188 .
- Pawar , S.A. and Deshpande , V.V. 2000 . Characterization of acid induced unfolding intermediates of glucose/xylose isomerase . European Journal of Biochemistry , 267 : 6331 – 6338 .
- Engelhard , M. and Evans , P.A. 1995 . Kinetics of interaction of partially folded proteins with a hydrophobic dye: Evidence that molten globule character is maximal in early folding intermediates . Protein Science , 4 : 1553 – 1562 .
- Rashid , F. , Sharma , S. , Baig , M.A. and Bano , A. 2006 . Effect of polyols and salts on the acid- induced state of human placental cystatin . Biochemistry (Moscow) , 71 : 619 – 626 .
- Sathish , H.A. , Kumar , P.R. and Prakash , V. 2002 . Effect of urea at lower concentration on the structure of papain: Formation of a stable molten globule and its characterization . Indian Journal of Biochemistry and Biophysics , 39 : 155 – 162 .
- Muralidhara , B.K. and Prakash , V. 2002 . Molten globule intermediates of human serum albumin in low concentration of urea . Indian Journal of Biochemistry and Biophysics , 39 : 318 – 324 .
- Asghari , S.M. , Khajeh , K. , Moradian , F. , Ranjbar , B. and Naderi-Manesh , H. 2004 . Acid-induced conformational changes in Bacillus amyloliquefaciens α-amylase: Appearance of molten globule likes state . Enzyme Microbial Technology , 35 : 51 – 57 .
- Machius , M. , Wiegand , G. and Huber , R. 1995 . Crystal structure of calcium depleted Bacillus licheniformis alpha-amylase at 2 . 2A resolution. Journal of Molecular Biology , 246 : 545 – 559 .
- Jiang , J.X. and Landon , E. 1990 . Involvement of denaturation-like changes in Pseudomonas exotoxin A, hydrophobicity, and membrane penetration determined by characterization of pH and thermal transitions: Roles of two distinct conformationally altered states . Journal of Biological Chemistry , 265 : 8636 – 8641 .
- Yadav , J.K. and Prakash , V. 2009 . Thermal stability of amylase in aqueous cosolvent system . Journal of Biosciences , 43 ( 3 )
- Kendrick , B.S. , Chang , B.S. , Arakawa , T. , Peterson , B. , Randolph , T.W. , Manning , M.C. and Carpenter , J.F. 1997 . Preferential exclusion of sucrose from recombinant interleukin-1 receptor antagonist: Role in restricted conformational mobility and compaction of native state . Proceedings of National Academy of Sciences, USA , 94 : 1917 – 1922 .
- Liu , Y. and Bolen , D.W. 1995 . The peptide backbone plays a dominant role in protein stabilization by naturally occurring osmolytes . Biochemistry , 34 : 12884 – 12891 .
- Devaraj , K.B. , Kumar , P.R. and Prakash , V. 2009 . Characterization of acid-induced molten globule like state of ficin . International Journal of Biological Macromolecules , 45 ( 3 ) : 248 – 254 .
- Kulkarni , A. , Gaikwad , S. and Rao , M. 2008 . pH induced structural alterations in an aspartic protease from Vigna radiate indicating an alkali induced molten globule state . International Journal of Biological Macromolecules , 43 ( 4 ) : 375 – 376 .