
Abstract
Chemical interactions and protein conformations changes during the formation of silver carp surimi gel were studied by textural analysis, chemical methods, laser Raman spectroscopy, and circular dichrosim. The optimum setting time at 40°C was 60 min. During surimi gel formation, ionic bonds and hydrogen bonds decreased significantly (P ≤ 0.05), while hydrophobic interactions, disulfide bonds, and non-disulfide covalent bonds increased significantly (P ≤ 0.05). Hydrophobic interactions, disulfide bonds, and non-disulfide covalent bonds were the main chemical interactions maintaining the stable structure of surimi gel. Secondary structural analysis of surimi protein showed that 94.11% α-helix existed in native myosin and it partly changed into β-turn and random coil during heating. Myosin gel was made up of 33.70% α-helix, 12.40% β-turn and 53.90% random coil. These three kinds of secondary structures were the main protein conformations in surimi gel.
INTRODUCTION
Myofibrillar proteins were the main fractions participating in surimi gel formation, and mainly included myosin and actin. Surimi gels were induced by heat treatment. Generally, Silver carp surimi gel was obtained by two steps of heating: high temperature setting at 40°C and cooking at 90°C. Protein gelation involved changes in chemical interactions and protein conformation with inter-, intra-protein and water-protein interactions,[Citation1,Citation2] which took very important roles in maintaining stable surimi gel network. Hydrophobic interactions, the sulfhydryl-disulfide interactions and the nondisulfide covalent bond catalyzed by transglutaminase (TGase) in the fish muscle, varied in the gel formation.[Citation3–Citation6] However, there was still some uncertainty about the mechanism responsible for silver carp gel formation. This was, in part, due to less work dealing with the analysis in situ of the structural changes occurring in the surimi proteins.[Citation2,Citation7–Citation9] There was little information on chemical interactions and conformation changes of silver carp surimi protein in gel formation.[Citation6] In the present investigation, chemical interaction changes during gel formation were assayed by soluble protein in some chemicals. Circular dichroism (CD) and Raman spectroscopy were used to further study the effect of heat treatments on the conformation of surimi proteins.
CD spectroscopy was a valuable optical technique for studying protein secondary structures in diluted solutions and provides quantitative analysis of protein secondary structures.[Citation10,Citation11] Raman spectroscopy had the advantage of being able to investigate in situ protein structural changes during denaturation, aggregation, and gelation of solid, liquid or aggregated food samples.[Citation12] It was usually more suitable for aqueous protein samples since water had weak Raman scattering and thus less interference in Raman spectroscopy.[Citation12] Hence, Raman spectroscopy had a distinct advantage over FTIR spectroscopy which could not be used in aqueous systems due to strong interference of the water band.[Citation11] This technique was successfully employed to investigate the protein structure in fish muscle and its gel.[Citation2,Citation7,Citation8]
MATERIALS AND METHODS
Suwari Gel and Surimi Gel Preparation
Surimi was prepared from fresh silver carp which was purchased in the Wuhan, Hubei province. The weight and length of purchased silver carp was 1500 ± 100 g, 56 ± 5 cm. The fish was killed 1 h after purchase. The fish was beheaded, degutted, deboned, and washed with running water. The white muscle was minced. The minced meat was washed three times with running water at the ratio of fish meat to water 1:5 (w/v). Each washing cycle was 10 min. The meat was dewatered by wrapping it in gauze and squeezing by hand. The dewatered meat, without any cryoprotectant, was chopped in a food maker (Braun electric apparatus K600, Shenzhen, China) for 1 min with 2.5% salt. This was followed by 1 min additional chopping with water for the moisture content of 78%. The quantities of salt and water were based on a dewatered meat weight basis. The surimi paste was stuffed into polyvinylidine casing (2.5 cm diameter) and both ends were sealed tightly. Then fish sausages were heated at 40°C for 30, 60, 90, and 120 min, respectively. After heating for the designated time, they were cooled immediately in running water, and stored at 4°C. The gels obtained were referred to as “suwari gels”. After the suwari gels were cooked at 90°C for 30 min, surimi gels were obtained. Finally, surimi gels were also immediately cooled and stored at 4°C.
Instrumental Texture Analysis
This test was carried out using a TA-XTPlus Texture Analyser (Stable Micro Systems, Surrey, England) at room temperature. A ball probe with 1/2 inch diameter was used to penetrate the sample 15 mm and the first peak of the curve was used as a penetration force. The results were expressed in terms of gel strength (g × mm), which is the multiplication of penetration force (g) by deformation (mm). To maintain stability during the test, gels were cut into cylinders of 25 mm diameter × 25 mm length.
Preparation of Myosin
Myosin was prepared immediately from antemortem silver carp (Hypophthalmichthys molitrix) by the method of Park and Lanier[Citation13] with some modifications. The composition of solutions used in the preparation were as follows: Solution A: 0.1 mol/L KCl, 0.02% NaN3 and 20 mmol/L Tris-HCl buffer, pH7.5; Solution B: 0.45 mol/L KCl, 5 mmol/L β-mercaptoethanol, 0.2 mol/L Mg(CH3COO)2, 1 mmol/L ethylene glycolbis N, N, N′, N′-terraacctic (EGTA), and 20 mmol/L Tris-maleate buffer, pH 6.8; Solution C: 0.5 mol/L KCl, 5 mmol/L β-mercaptoethanol, and 20 mmol/L Tris-HCl buffer, pH 7.5; Solution D: 0.8 mol/L KCl, 5 mmol/L β-mercaptoethanol, and 0.2 mol/L Mg(CH3COO)2. All solutions used for myosin preparation were kept at 4°C to minimize proteolysis and protein denaturation. Minced muscle was washed with 10 volume solution A (w/v) and held at 4°C for 15 min, and centrifuged at 1000 g for 10 min. The supernatant was discarded and pellet was suspended in 5 volume solution B (w/v), added ATP to final concentration 5 mmol/L, and held at 4°C for 1 h and centrifuged at 10,000 g for 15 min. The supernatant was diluted with 5 volume of 1 mmol/L KHCO3 (w/v), and held at 4°C for 15 min and centrifuged at 12,000 g for 10 min. Pellet was re-suspended with 2.5 volume solution C, homogenized in an inline dispersing homogenizer (Model FJ-200, Shanghai specimen and models factory, China), held at 4°C for 10 min, diluted with 2.5 volume of 1 mmol/L KHCO3 (w/v) and added MgCl2 to final concentration 10 mmol/L, then held at 4°C overnight and centrifuged at 20,000 g for 15 min. Pellet was suspended in 1 volume solution C (w/v) and homogenized. Myosin purity was checked using gel electrophoresis (PAGE) according to Laemmli[Citation14] without sodium dodecyl sulfate. illustrated the electrophoretic profile of the extracted myosin. The purity of the myosin samples was more than 95% as determined by the densitometry (Gel Logic 200 Imaging System, Kodak Co., USA).
Figure 1 PAGE of extracted native myosin.
Preparation of Actomyosin (AM)
White muscle was used to extract AM. AM was extracted using a modified method according to Ogawa, Nakamura, Horimoto, An, Tsuchiya, and Nakai.[Citation15] Minced fish meat (100 g) was added to 400 mL of 3.38 mmol/L Na2HPO4-15.5 mmol/L NaH2PO4 buffer (pH 7.5) and homogenized for 2 min at 15°C. The homogenate was centrifuged at 8000 g for 5 min. The supernatant containing sarcoplasmic proteins was discarded. The precipitates were washed twice using the same buffer. The pellet was homogenized with 300 mL of 0.45 mol/L NaCl in 3.38 mmol/L Na2HPO4-15.5 mmol/L NaH2PO4 buffer (pH 7.5) and placed overnight at 4°C. Then the suspension was centrifuged at 15,000 g for 10 min. The supernatant containing myofibrillar protein was filtered through three-layers of gauze to remove the connective tissue and lipid. The filtrate was stirred in 3 L of deionized water to precipitate myofibrillar protein for 30 min. The supernatant was removed from the pellet. The precipitate (AM) was collected at 12,000 g for 5 min. AM was dissolved in 0.6 mol/L NaCl.
Determination of Soluble Protein
According to Gómez-Guillén, Borderías, and Montero,[Citation16] gels were treated with chemicals selected for their capacity to cleave certain kinds of bond: 0.05 mol/L NaCl (SA), 0.6 mol/L NaCl (SB), 0.6 mol/L NaCl + 1.5 mol/L urea (SC), and 0.6 mol/L NaCl + 8 mol/L urea (SD). Proteins were partially solubilized with these solutions in order to determine the existence of nonspecific associations (soluble protein in SA), ionic bonds (difference between soluble protein in SB and in SA), hydrogen bonds (difference between soluble protein in SC and in SB), hydrophobic interactions (difference between soluble protein in SD and in SC). Two grams of chopped gel were homogenized with 10 mL of each solution in an inline dispersing Model FJ-200 homogenizer (Shanghai specimen and models factory, China) for 2 min. The resulting homogenates were stirred at 4°C for 1 h, then centrifuged for 15 min at 20,000g in a CR21 centrifuge (Hitachi, Japan). Protein concentration in supernatants was determined using the Lowry method.[Citation17] The soluble protein content was expressed as g soluble protein/L of homogenate.
Determination of Disulfide Bonds Content
Disulphide bond content was determined using 2-nitro-5-thiosulphobenzoate (NTSB) assay according to the method of Balange and Benjakul.[Citation18] 3 mL of freshly prepared NTSB assay solution was added to 0.5 mL of natural AM (1.0 mg/mL). The mixture was incubated in the dark at 26°C for 25 min. Absorbance at 412 nm was measured. Disulphide bond content was calculated using the extinction coefficient of 13,900 M−Citation1 cm−Citation1.
Solubility Studies
The solubility of samples in 20 mmol/L Tris–HCl, pH 8.0, containing 1% (w/v) SDS, 8 mol/L urea and 2% (v/v) β-mercaptoethanol (β-ME), was determined as described by Benjakul, Visessanguan, and Chantarasuwan.[Citation19] The sample (1 g) was homogenized in 20 mL of solution for 1 min using a Model FJ-200 homogenizer. The homogenate was heated in boiling water (100°C) for 2 min and stirred at room temperature for 4 h. The resulting homogenate was centrifuged at 10,000 g for 30 min, using a CR21 centrifuge. Protein in the supernatant (10 mL) was precipitated by the addition of 50% (w/v) cold TCA (4°C) to a final concentration of 10%. The mixture was kept at 4°C for 18 h and then centrifuged at 10,000 g for 30 min. The precipitate was washed with 10% TCA and solubilized in 0.5 mol/L NaOH. To obtain the total amount of protein, surimi directly solubilized in 0.5 mol/L NaOH. The protein content was measured using the Lowry method.[Citation17] The solubility was expressed as percent of the total protein.
Laser Raman Spectroscopy
Raman spectra were recorded on a Renishaw inVia Raman Imaging Microscope (system 1000) equipped with a 514 nm model 70 Argon ion laser excitation (Spectra Physics, Mountain View, CA, USA). Raman frequency was calibrated by a silicon slide at 520 nm. The laser was focused on the solid samples which were dispersed on microscope slides. Each spectrum was collected at room temperature under the following conditions: 100 mW of laser power, 1 cm−Citation1 resolution, 30 s exposure time, and 40 scans. The experiments were performed in a dark environment to avoid interference from light and cosmic radiation. Spectral data from the scans of samples were baseline-corrected and normalized against the phenylalanine band at 1003 ± 1 cm−Citation1 using the GRAMS/32 AI Software (Galactic Industries Corporation, Salem, NH, USA). Raman spectra (300–3800 cm−Citation1) were plotted as relative intensity (arbitrary units) against Raman shift in wave number (cm−Citation1).
Circular Dichrosim Spectroscopy
CD spectra were recorded on a Jasco J-810 spectropolarimeter (Jasco International Co., Ltd., Tokyo, Japan) equipped with a Peltier thermostat supplied by the manufacturer. The myosin samples were placed in a 1 mm light-path length rectangular quartz cell. Myosin concentration was 0.1190 mg/mL for the ellipticity (θ) measurements in the far UV region (240–190 nm). Temperature scans were performed between 4 and 90°C at a rate of 1°C per minute. Spectra were averaged over three scans and background corrected. A mean residue weight of 110 g/mol has been assumed. The percentages of α-helix, β-sheet, β-turn, and random coil structures were determined using the protein secondary structure estimation program (Yang’s method) provided with a Jasco J-810 spectropolarimeter.
Statistical Analysis
Each sample was determined at least three times. The least significant difference (LSD) at 5% was applied to define significant difference between mean values. All analysis was performed using SAS software v 8.1 (SAS Institute Inc., Cary, NC, USA, 2000).
RESULTS AND DISCUSSION
Effect of Heating Treatment on Gel Strength
As shown in , breaking force, deformation, and gel strength of surimi gels increased with extended setting time and reached maximum values, 533.15 g, 11.32 mm, and 5331.53 g × mm, after setting 60 min at 40°C. Subsequently, it decreased with the increase of setting time. In the process of surimi setting, myosin heavy chain (MHC) underwent polymerization via the formation of non-sulfide covalent cross-links, which was catalyzed by an endogenous TGase.[Citation5,Citation20] Moreover, protein conformation changed and the reactive groups (glutamine and lysine) which were involved in the cross-linking reaction via TGase, were exposed.[Citation19] Furthermore, endogenous protease hydrolyzed protein molecule chains to disrupt surimi gel network.[Citation19,Citation20] When setting time was more than 60 min, endogenous protease led to gel structure disintegration or softening. For surimi gel, heating temperature passed through the active temperature of protease (50–60°C). So gel softening occurred and gel strength decreased.
Table 1 Effect of setting time on surimi gel properties
Changes of Chemical Interactions During Surimi Gel Formation
Soluble protein in a number of solutions selected for their capacity to disrupt certain kinds of bonds was shown in . As the setting time increased from 30 to 120 min, the contribution of ionic bonds to suwari gels gradually decreased and was more than that of the surimi gels significantly (P ≤ 0.05). The difference of soluble protein in SB and SA was very low (less than 1 g/L) in all surimi gels, thus indicating very minor participation of ionic bonds in gel formation. The content of soluble protein of sardine surimi gels in SB was also less than 1 g/L.[Citation16]
Figure 2 Variations of chemical interactions during the formation of suwari gels and surimi gels. In horizontal axis, the numbers 1, 2, 3, 4, 5, 6, 7, 8, and 9 represent surimi, suwari gel setting at 40°C for 30 min, suwari gel setting at 40°C for 60 min, suwari gel setting at 40°C for 90 min, suwari gel setting at 40°C for 120 min, surimi gel setting at 40°C for 30 min, surimi gel setting at 40°C for 60 min, surimi gel setting at 40°C for 90 min, and surimi gel setting at 40°C for 120 min, respectively.
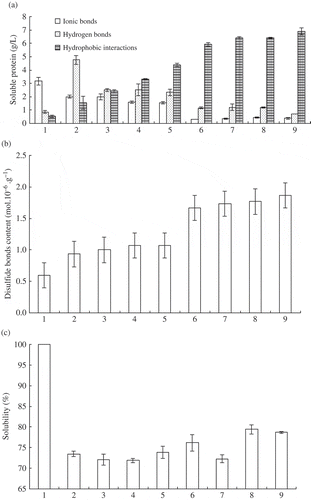
With regard to hydrogen bonds, the difference of soluble protein of surimi gels in SC and SB was lower than that of the suwari gels significantly (P ≤ 0.05) (). In other words, the contribution of hydrogen bonds to suwari gels was more than that to surimi gels. For instance, the content of soluble protein in suwari gel and surimi gel setting for 30 min were 4.77 g/L and 1.16 g/L, respectively. With the increase of setting time, the content of soluble protein for these two kinds of gels decreased. After setting for 120 min, the content of soluble protein in suwari gel was only 2.33 g/L. Further cooking for 30 min at 90°C, the content of soluble protein reduced significantly (P ≤ 0.05) and was only 0.71 g/L. Hydrogen bond energy was low, 2–40 kJ/mol. During heat treatment, hydrogen bonds between water and protein molecules were easily interrupted. Although hydrogen bonds might form through cooling, they were less. Ionic bonds and hydrogen bonds decreased significantly (P ≤ 0.05) during surimi gel formation. However, gel strength of surimi gels was higher than that of the corresponding suwari gels and increased remarkably (P ≤ 0.05) (). As a result, the contribution of ionic bonds and hydrogen bonds to surimi gel network was small.
The highest values of soluble protein were obtained with SD (0.6 mol/L NaCl + 8 mol/L urea) as a measure of hydrophobic interactions. With the increase of setting time, the content of soluble protein increased significantly from 1.55 g/L (30 min) to 4.36 g/L (120 min) in suwari gels (P ≤ 0.05) (). After cooking 30 min at 90°C, the increasing trend was also significant (P ≤ 0.05). For surimi gel setting 120 min, the value increased to 6.9 g/L. Yongsawatdigul and Park[Citation21] also reported Threadfin bream AM surface hydrophobicity increased from 30 to 70°C. The change of disulfide bond content was similar to hydrophobic interactions. The content of disulfide bonds increased from 0.93 mol/106 g (suwari gel, setting 30 min) to 1.87 mol/106 g (surimi gel, setting 120 min) (). During protein heating denaturation, more hydrophobic groups and sulphydryl groups were exposed. Exposed hydrophobic groups aggregated with each other by surface hydrophobicity, and sulphydryl groups linked with each other and formed disulfide bonds. These interactions contributed to the formation of gel network. The changes of hydrophobic interactions and disulfide bonds were almost in accordance with gel strength when setting time was in 60 min ( and ). When setting time was more than 60 min, hydrophobic bonds and disulfide bonds increased, but gel strength did not ( and ).
Solution containing SDS, urea, and β-mercaptoethanol was used to solubilise protein by destroying all bonds, except non-disulfide covalent bonds, particularly the ϵ-(γ-glutamyl) lysine linkage.[Citation22] Therefore, the decrease in solubility indicated that non-disulfide covalent bond formed. Endogenous TGase catalyzed MHC cross-linking, which led to the decrease of solubility.[Citation23] Compared with surimi, the solubility of suwari gels and surimi gels were lower significantly (P ≤ 0.05; ). This illustrated that non-disulfide covalent bonds formed, which was catalyzed by an endogenous TGase. For suwari gels, solubility decreased with the setting time increase to 60 min. But it increased with further setting at 40°C. Hydrophobic bonds and disulfide bonds had no significant difference among all surimi gels (P > 0.05). Both of them had no contribution to the difference of gel strength, but solubility did. The decrease and increase of solubility corresponded with the increase and decrease of gel strength ( and ). The lowest solubility corresponded to the highest gel strength. Solubility was relative to non-disulfide covalent bond formation and proteins hydrolysis. Therefore, both of MHC cross-linking and surimi proteins hydrolysis affected gel strength. As a result, gel strength increased when effect of MHC cross-linking interactions was more than that of surimi protein hydrolysis.
Raman Spectroscopy
Protein secondary structure
The Raman spectra in surimi, setting surimi (setting 60 min at 40°C) and surimi gel (setting 60 min at 40°C, then cooking 30 min at 90°C) were shown in . The -CO-NH- amide or peptide bond had several distinct vibrational modes, with the amide I band near 1650 cm–Citation1 and the amide III band near 1250 cm–Citation1 being the most easily indentified in the Raman spectra of proteins. The exact location of these bands depended on the secondary structure of the polypeptide chain, and these features were therefore useful for estimating secondary structure fractions of proteins. Due to possible overlap of bands from the solvent water in the amide I region and miscellaneous C–H bending and aromatic ring vibration in the amide III region, it was recommended that both regions should be analyzed in order to obtain a more reliable interpretation of the changes in the secondary structure of proteins.[Citation8]
Figure 3 Laser Raman spectroscopy. A: surimi; B: suwari gel setting for 60 min at 40°C; C: surimi gel setting for 60 min at 40°C and cooking for 30 min at 90°C.
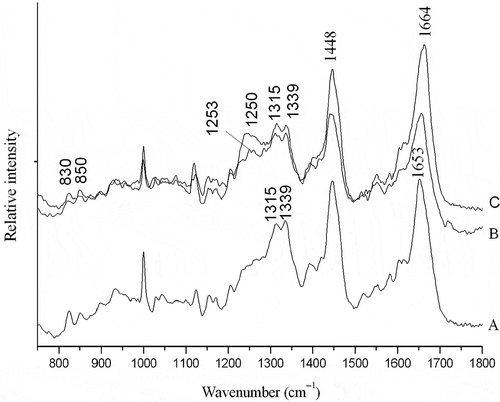
The amide I band resulted primarily from in-plane peptide C=O stretching vibrations and also had contributions from C–N vibrations and C–C–N vibrations.[Citation23] The band correlated with the amount of the types of protein backbone conformation.[Citation24] Generally, proteins with high α-helical content showed that an amide I band centered around 1658–1650 cm–Citation1, while those with predominantly β-sheet structures had a strong band at 1680–1665 cm–Citation1. Proteins with a high proportion of undefined or random coil structure had the amide I band centered 1665–1660 cm–Citation1.[Citation25] Comparison of the amide I band of surimi and gel samples revealed a shifting of the maximum band towards higher frequencies from surimi to the gel (). For surimi, the major feature of the amide I band was centered at 1653 cm–Citation1 (), indicating a predominance of α-helical structure, while suwari gel and surimi gel had a strong band at 1664 cm–Citation1(), suggesting a shift of α-helical structure to undefined or random coil.
The amide III band provided additional information of polypeptide backbone,[Citation26,Citation27] and was complementary of the amide I. The amide III band arose primarily from C-N stretching and N-H in-plane bending vibrations as well as contributions from C-C stretching and C=O in plane bending of the peptide bond. Proteins with high content of α-helical structures had a weak band, located in a broad region above 1275 cm–Citation1. Random coil structures usually appeared near 1250 cm–Citation1, while β-sheet structures led to a tense band 1238–1245 cm–Citation1.[Citation8] Surimi appeared α-helical structure at 1315 cm–Citation1 and 1339 cm–Citation1 (). Besides these two bands, the bands of 1250 cm–Citation1 and 1253 cm–Citation1 occurred in suwari gel and surimi gel, respectively (). This also showed that α-helical structure partly converted into random coil structure. α-helical structures of myosin mainly existed in myosin tail and its unfolding initiated setting.[Citation7] The results were in agreement with the results shown in the amide I region. Ogawa et al.[Citation7] and Sánchez-González et al.[Citation2] reported that α-helix content decreased and converted into other secondary structure during surimi gelation.
Tyrosine doublet
The doublet bands located near 830 and 850 cm–Citation1 could be used to monitor the microenvironment around tyrosyl residues.[Citation25] This doublet band was assigned to vibrations of the para-substituted benzene ring of tyrosine residues which were affected by the environment and the involvement of the phenolic hydroxyl group in hydrogen bonding. In the case of tyrosine residues which were exposed to the aqueous or polar environment or which acted as simultaneous acceptor and donor of moderate to weak hydrogen bonds, the intensity ratio of the doublet bands (I850/830) usually ranged from 0.90–l.45, but could be as high as 2.5. On the other hand, I850/830 values for tyrosine residues which were buried in a hydrophobic environment and which tended to act as hydrogen donors usually ranged between 0.7 and 1.0, and could be as low as 0.3 in the case of extremely strong hydrogen bonding to a negative acceptor.[Citation24,Citation25] The intensity ratios I850/830 of surimi was 0.98 (), suggesting that some tyrosine residues was mainly buried in a more hydrophobic environment and was involved as strong hydrogen bond donors. For suwari gel and surimi gel, the I850/830 ratios increased to 1.01 and 1.05, respectively (). This indicated that tyrosine residues were mainly exposed and able to participate in moderate or weak hydrogen bonding. This shift showed that hydrophobicity of surimi proteins increased as a result of tyrosine exposing. The result was consistent with hydrophobic interactions determined by soluble protein ().
CD Spectroscopy
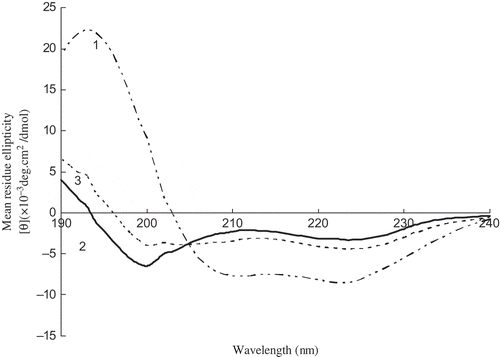
The change in the CD spectral characteristics of myosin was shown in . A positive band at 193 nm and two negative bands at 210 and 222 nm were observed, and also the characteristic CD features of α-helical structure in native myosin. The native myosin spectrum was consistent with the general features of α-helices. The CD spectra of α-helices had an intense positive band at 193 nm and two negative bands at about 208–210 nm and 222 nm, while that of β-sheets had a fairly intense positive band at about 198 nm and a negative band at about 215–220 nm.[Citation10,Citation28] As shown in , quantitative estimation of the relative amounts of α-helical and random coil structure in native myosin was 94.11 and 5.89%, respectively. The calculated percentage of α-helix in native myosin differed somewhat from those obtained from a previous CD study[Citation2,Citation8,Citation9,Citation21,Citation29] which reported a range between 35 and 67%. The differences might be due to different protein composites or fish species differences. Since high α-helical content was found, the native myosin could be considered as a protein with highly ordered and stable protein conformation. After heating at 40°C, α-helical structure converted partly into β-turn and random coil (). Further heating at 90°C, the relative amounts of secondary structures were α-helix 33.7%, β-turn 12.4% and random coil 53.9%, which were different from Alaska pollock surimi gel[Citation2] consisting of α-helix, β-sheet, β-turn, and random coil structure.
Table 2 Percent of secondary structure units of myosin
Figure 4 CD spectra of myosin during heating. The numbers 1, 2, and 3 represent native myosin (pH 7.5), myosin heating for 60 min at 40°C, myosin heating for 60 min at 40°C, and then heating for 30 min at 90°C, respectively.
CONCLUSION
Endougenous TGase and protease might exist in silver carp surimi. They caused MHC cross-linking and protein hydrolysis, and affected surimi gel strength together. Hydrogen bonds, ionic bonds, hydrophobic interactions, disulfide bonds, and non-disulfide covalent bonds participated in surimi gel network. The main chemical interactions in stabilizing surimi gel network were hydrophobic interactions, disulfide bonds, and non-disulfide covalent bonds. The present CD and Raman data illustrated that surimi, especially myosin, contained a high level of α-helical structure compared with other fish species. The conformation of surimi protein was influenced by heating treatment. α-helix converted into other secondary structures, β-turn, and random coil. α-helix, β-turn, and random coil were the main secondary structure in surimi gel.
ACKNOWLEDGEMENT
The research project was supported by College Students Creative Fund of Ludong University (10y009 and 11g005) and A Project of Shandong Province Higher Educational Science and Technology Program (J12LD03).
REFERENCES
- Lanier, T.C.; Carvajal, P.; Yongsawatdigul, J. Surimi gelation chemistry. In: Surimi and Surimi Seafood; Park, J.W.; Eds.; Marcel Dekker: New York, 2004; 451–470.
- Sánchez-González, I.; Carmona, P.; Moreno, P.; Bordeías, J.; Sánchez-Alonso, I.; Rodríguez-Casado, A.; Careche, M. Protein and water structural changes in fish surimi during gelation as revealed by isotopic H/D exchange and Raman spectroscopy. Food Chemistry 2008, 106, 56–64.
- Niwa, E. Role of hydrophobic bonding in gelation of fish fresh paste. Nippon Suisa Gakkaishi 1975, 53, 2011–2020.
- Seki, N.; Uno, H.; Lee, N.; Kimura, I.; Toyoda, K.; Fujita, T.; Arai, K. Transglutaminase activity in Alaska pollack muscle and surimi, and its reaction with myosin B. Nippon Suisa Gakkaishi 1990, 56, 125–132.
- Kimura, I.M.; Sugimoto, M.; Toyoda, K.; Seki, N.; Arai, K.; Fujita, I. A study on the cross-links reaction of myosin in kamaboko ‘suwari’gels. Nippon Suisan Gakkaishi 1991, 57, 1386–1396.
- Liu, R.; Zhao, S.; Liu, Y.; Yang, H.; Xiong, B.; Xie, S.; Qin, L. Effect of pH on the gel properties and secondary structure of fish myosin. Food Chemistry 2010, 121, 196–202.
- Ogawa, M.; Kanamaru, J.; Miyashita, H.; Tamiya, T.; Tsuchiya, . Alpha-helical structure of fish actomyosin: Changes during setting. Journal of Food Science 1995, 60, 297–299.
- Bouraoui, M.; Nakai, S.; Li-Chan, E. In situ investigation of protein structure in Pacific whiting surimi and gels using Raman spectroscopy. Food Research International 1997, 30, 65–72.
- Thawornchinsombut, S.; Park, J.W.; Meng, G.T.; Li-Chan, E.C.Y. Raman spectroscopy determines structural changes associated with gelation properties of fish proteins recovered at alkaline pH. Journal of Agricultural and Food Chemistry 2006, 54, 2178–2187.
- Greenfield, N.J. Applications of circular dichroism in protein and peptide analysis. Trends in Analytical Chemistry 1999, 18, 236–244.
- Choi, S.M.; Ma, C.Y. Structural characterization of globulin from common buckwheat (Fagopyrum esculentum Moench) using circular dichroism and Raman spectroscopy. Food Chemistry 2007, 102, 150–160.
- Li-Chan, E.C.Y. The applications of Raman spectroscopy in food science. Trends in Food Science and Technology 1996, 7, 361–370.
- Park, J.W.; Lanier, T.C. Scanning calorimetric behavior of tilapia myosin and actin due to processing of muscle and protein purification. Journal of Food Science 1989, 54, 49–51.
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685.
- Ogawa, M.; Nakamura, S.; Horimoto, Y.; An, H.; Tsuchiya, T.; Nakai, S. Raman spectroscopic study of changes in fish actomyosin during setting. Journal of Agricultural and Food Chemistry 1999, 47, 3309–3318.
- Gómez-Guillén, M.C.; Borderías, A.J.; Montero, P. Chemical interactions of nonmuscle proteins in the network of Sardine (Sardina pilchardus) muscle gels. Lebensmittel-Wissenschaft und-Technologie 1997, 29, 602–608.
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with Folin phenol reagent. Journal of Biological Chemistry 1951, 193, 256–275.
- Balange, A.; Benjakul, S. Enhancement of gel strength of bigeye snapper (Priacanthus tayenus) surimi using oxidised phenolic compounds. Food Chemistry 2009, 113 (1), 61–70.
- Benjakul, S.; Visessanguan, W.; Chantarasuwan, C. Effect of high-temperature setting on gelling characteristic of surimi from some tropical fish. International Journal of Food Science and Technology 2004, 39, 671–680.
- Takeda, H.; Seki, N. Enzyme-catalyzed cross-linking and degradation of myosin heavy chain in walleye Pollack surimi paste during setting. Fisheries Science 1996, 62, 462–467.
- Yongsawatdigul, J.; Park, J.W. Thermal denaturation and aggregation of threadfin bream actomyosin. Food Chemistry 2003, 83, 409–416.
- Benjakul, S.; Visessanguan, W.; Srivilai, C. Porcine plasma proteins as gel enhancer in bigeye snapper (Priacanthus tayenus) surimi. Journal of Food Biochemistry 2001, 25, 285–305.
- Krimm, S.; Bandekar, J. Vibrational spectroscopy of peptides and proteins. Advances in Protein Chemistry 1986, 38, 181–360.
- Tu, A.T. Proteins [M]. In: Raman Spectroscopy in Biology: Principles and Applications; Tu, A.T.; Eds.; Wiley Interscience: New York, 1982; 65–116.
- Li-Chan, E.C.Y.; Nakai, S.; Hirotsuka, M. Raman spectroscopy as a probe of protein structure in food systems. In: Protein Structure Function Relationships in Foods; Yada, R.Y.; Jackman, R.L.; Smith, J.L.; Eds.; Chapman and Hall Inc: New York, 1994; 163–197.
- Lippert, J.L.; Tyminski, D.; Desmules, P.J. Determination of the secondary structure of proteins by laser Raman spectroscopy. Journal of the American Chemical Society 1976, 98, 7075–7080.
- Yu, T.J.; Lippert, J.L.; Peticolas, W.L. Laser Raman studies of conformational variations of poly-L-lysine. Polymers 1973, 12 (9), 2161–2176.
- Woody, R.W. Theory of circular dichroism of proteins. In: Circular Dichroism and the Conformational Analysis of Biomolecules; Fasman, G.D.; Eds.; Plenum Press: New York, 1996; 25–67.
- Moosavi-Nasab, M.; All, I.; Ismail, A.; Ngadi, M. Protein structural changes during preparation and storage of surimi. Food and Chemical Toxicology 2005, 70, C448–C453.