
Abstract
Fructose, glucose, and sucrose in sugarcane molasses are determined simultaneously by high performance liquid chromatography using maltose as internal standard with refractive index detector. The mixture of the diluted samples and internal standard was purified with Sep-Pak C18 solid-phase extraction and filtered through a 0.22-μm membrane before injection. The results showed that the linear ranges for fructose, glucose, and sucrose were 3.30–16.48, 1.80–9.02, and 5.94–29.70 g/L with the squared correlation coefficients (R2) being 0.9986, 0.9987, and 0.9955, respectively. The method is simple, quantified, and time-saving for determination of sugars in sugarcane molasses.
INTRODUCTION
Sugarcane molasses, a by-product from sugarcane refining, is a thick and dark syrup, resulting from the crystallization and removal of the majority of sucrose from the original juice. The current total supply of molasses is 300–400 million tons per year in China. In general, molasses contains about 50% sugar by dry weight, predominantly sucrose, fructose, and glucose. Furthermore, unlike refined sugars, molasses contains trace amounts of vitamins and several minerals. Because of its unusual properties, molasses is widely used in baking, as an animal feed additive, or as a fermentation feedstock. Therefore, determining accurately the content of fructose, sucrose, and glucose in molasses is important for the development and effective utilization of molasses.
The conventional approaches used to determine sugars are either polarimetry or chemical assay methods, such as Lane-Eynon method for determining total sugar[Citation1] and DNS method for reducing sugar.[Citation2] Both the classical and official methods for determining sugars in molasses require considerable time, contain inherent errors, and are based on empirically derived constants.[Citation3] The analysis methods of the oligosaccharides include the near-infrared spectroscopy,[Citation4] the thin layer chromatography method, gas chromatography (GC),[Citation5] and high performance liquid chromatography (HPLC).[Citation6,Citation7] GC has been used to separate carbohydrates employing several types of derivatization modes. Since the nonvolatility of carbohydrates is caused by polar hydroxyl, amino, and carboxyl groups, the derivatization of these groups can greatly increase the volatility of carbohydrates, but the usual challenge to GC is the appearance of multiple peaks in the chromatogram due to the presence of tautomeric forms of reduced sugars. The GC method is particularly accurate in determining low sugar concentrations, but sample preparation is time consuming. The HPLC method can directly determine oligosaccharide with a simple sample preparation. Thus, HPLC is one of the most promising methods for sugar analysis, due to its universality, time efficiency, accuracy, and selectivity for the quantification of carbohydrates.[Citation8] Authors, such as Nejib et al.[Citation9] and Sims,[Citation10] have reported the HPLC method as an established and preferred method for the determination of individual sugars in carbohydrate mixtures, for its accuracy and simplicity.
Generally, carbohydrates do not absorb ultraviolet (UV) and visible radiation or emit fluorescence without suitable prior derivatization, because they possess neither chromophore nor fluophore. Some carbohydrates absorb near UV radiation in the region of 180–220 nm. Based on measurement of the absorption in this range, however, the detection is nonselective. But carbohydrates can be measured by high performance liquid chromatography-evaporative light scattering detector (HPLC-ELSD).[Citation11] While HPLC-ELSD has high sensitivity and can use gradient elution, it has good reproducibility only at low sugar concentrations. High-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD) is one of the most useful techniques for oligosaccharide determination,[Citation12] but amperometric detection possesses poor stability, and new electrode materials have been constantly explored. High performance liquid chromatography-refractive index detector (HPLC-RID) today has achieved considerable improvement in constant velocity and optical element temperature control for the detection stability. Thus, the HPLC-RID method is widely used in carbohydrate analysis,[Citation13] due to the reliability of the detection results of glucose, fructose, and sucrose. For the above reasons, the HPLC-RID method was used in this study, together with an Agilent Zorbax carbohydrate column.
Although numerous methods have been published concerning the HPLC determination of carbohydrates in natural products,[Citation14,Citation15] the internal standard (IS) method has seldom been used.[Citation16] An IS is preferred over the external standard (ES) because the IS can eliminate the problems concerning the injection of exact volumes into the liquid chromatography, variations in mobile phase composition, and potential changes in the aging of the column. In this study, maltose was selected as the IS for the HPLC determination of the sugars because its physical and chemical characteristics were similar to those of the three sugars and the used molasses contained no maltose.[Citation3,Citation17]
For the molasses detection by HPLC, different sample pretreatment procedures of decolorization and purification were tested. This was necessary because of the presence of pigment and nitrogen compound mixtures in molasses, which could interfere with the detection and could shorten the lifespan of the chromatographic column.[Citation18] In this study, solid-phase extraction (SPE) was selected to clean samples, and results were calculated from peak areas automatically generated by a computing integrator. This work aimed to establish and to apply simple extraction and analytical conditions to identify and quantify carbohydrates in molasses by HPLC using RID and an amino-bonded silica column.
MATERIALS AND METHODS
Instrumentation and Reagents
The HPLC system consists of a model 1525 Binary pump, a model 7725 manual sampler, a model 2414 RID (Waters Assoc., Milford, MA, USA), and Agilent Zorbax carbohydrate column (250 × 4.6 mm I.D., 5 μm) protected with a guard column (12.5 × 4.6 mm I.D.). UtimateTM XB-NH2 column (250 × 4.6 mm I.D., 5 μm) was from Welch Material Technology (Shanghai) Co., Ltd. UV2802SH-type ultraviolet-visible spectrophotometer was purchased from UNICO (Shanghai) Instruments Co., Ltd. (USA). TGL-19G centrifuge (5 mL) was supplied by Shanghai Anke company, Ltd. (China).
HPLC-grade acetonitrile (A998-4) was purchased from Thermo Fisher Scientific (USA), and HPLC-grade methanol was acquired from Kermel Chemical Reagent Co., Ltd. (Tianjin, China). Water used in the study was double-deionized water (Milli-Q, Millipore Corp., Milford, MA, USA) of 18.2 MΩ/cm resistivity. Molasses was obtained from Zhanjiang Sugarcane Research Center of Guangzhou Sugarcane Industry Research Institute (Guangdong Province, China). HPLC grade sugar standards (sucrose, fructose, glucose, and maltose) were supplied by Aladdin Chemistry Co. Ltd. (China).
Membrane filters (0.22 μm) were purchased from MEMBRANA Company (Germany). Macroporous resins (Seplite LX-38, LXA-8, LX-17, XDA-8) were acquired from Xi’an LanXiao Technology Co., Ltd. (China) and resin D101 was from Tianjin Bochum Resin Technology Co., Ltd. (China). Waters Sep-Pak C18 SPE cartridge (WAT020805) was purchased from Waters Company (Milford, MA, USA). The SPE column was preconditioned by passing it through 2.0 mL of methanol, followed by 4.0 mL of double-deonized water.
Solutions
A mixed standard stock solution of sucrose, fructose, and glucose was prepared by dissolving 2.970 g of sucrose, 1.648 g of fructose, and 0.902 g of glucose in 100 mL of water, which was stable for ≥6 months at −20°C.The working solutions of sugars were prepared by pipetting 0.4, 0.8, 1.2, 1.6, and 2.0 mL of the above mixed standard stock solution into five 2-mL amber glass volumetric flasks, and then making up to the mark with water, respectively. Maltose (IS) solution (10.095 g/L) was prepared by dissolving 2.019 g of maltose in 200 mL deionized water, which was stable for ≥6 months at −20°C.
Calibration standard solutions were prepared by pipetting 1.0 mL of the IS stock solutions into five 2.0-mL amber glass volumetric flasks and making up to the mark with 1.0 mL of the above sugars working solutions separately. Mobile phase (75% acetonitrile in water) was obtained by adding 250 mL deionized water to 750 mL acetonitrile and mixing well by sonication.
Sample Preparation
The use of the linear calibration equations was mandatory in the indicated range, and thus the molasses was required to be diluted by mass ratio as follows: 50 g of molasses was weighed and poured into a 250-mL glass-stoppered Erlenmeyer flask before being diluted to 150 g with water, and then mixed uniformly. A 100-mL aliquot of solution was used for determination of the Brix (Bx). The above diluted molasses (26.7 °Bx) was accurately measured (1.0 mL aliquot), poured into a 10-mL volumetric flask, and diluted to the scale mark with water, which was used as the working molasses solution. Next, a 4.0-mL aliquot of the working molasses solution and a 4.0-mL aliquot of IS solution were added to 10-mL centrifuge tubes and mixed thoroughly, followed by the procedures:
Direct injection—The sample-IS solution was filtered through 0.22-μm membranes before injection.
SPE cleanup—The sample-IS solutions were passed through the activated Sep-Pak C18 SPE column like this: 8 mL sample-IS solutions were first poured into the activated C18 SPE cartridge, followed by inserting the plunger, discarding the initial 3-mL eluate, and collecting the follow-up 3-mL eluate, which was passed through 0.22-μm membrane prior to HPLC at the optimum chromatographic conditions.
Macroporous resin decolorization method—3 mL of the above working molasses solution was pipetted into 5 mL of centrifugal tubes and mixed with 50 and 100 mg of five different Macroporous resins, respectively (Seplite LX-38, Seplite LXA-8, Seplite LX-17, Seplite XDA-8, and D101). Next, the centrifugal tubes were sealed with PARAFILM and incubated at 30°C, 150 rpm for 3 h in the shaking table. After adsorption, 3 mL of liquid supernatant was harvested by centrifugation for 10 min at 12000 rpm using a TGL-19G centrifuge. Finally, 1 mL of the harvested supernatant was used for the analysis of sugars using HPLC, and 2 mL of the supernatant was used for the absorbance measurements using the UV2802SH-type ultraviolet-visible spectrophotometer at a wavelength of 560 nm. The decoloration rate (Dec.) on the samples was calculated as follows:
where A0 is the original absorbance of the working molasses solution, and A is the absorbance of the working molasses solution pretreated as described above.
HPLC
The chromatographic separation of sugars (sucrose, fructose, and glucose) and the IS method was achieved with a Waters HPLC system in an isocratic mode. The samples were analyzed on a carbohydrate column (250 × 4.6 mm I.D., 5 μm,) equipped with a guard column (12.5 × 4.6 mm I.D.). The column and RID temperatures were set at 30 and 35°C, respectively. The mobile phase was composed of acetonitrile and water (75:25, v/v), and the flow rate was 1.0 mL/min. The injection volume was 10 μL. Peak detection and integration were done using a Breeze Chromatographic System (Waters Company, Milford, MA, USA). Maltose was used as an IS and the internal method was applied for quantification. After the column was equilibrated with a mobile phase, the flow rate was kept at 1.0 mL/min and 10 μL of standard solution was injected to the chromatographic column. The sample-IS solutions were determined by standards, and the detection of each sample-IS solution was repeated in triplicate. The column was washed at the end of each experiment for more than 30 min with the mobile phase.
Method Validation
The calibration curves were prepared for each sugar by injecting the five concentrations of calibration standard solutions into the HPLC system (10 μL). Linear regression calibration curves (y = ax + b) based on five points, which included 0, represented by the plots of the peak-area ratios of each sugar to IS multiplied by the IS concentration (y) versus the concentration of each sugar calibration standards (x) were constructed using the weighted (1/x2) linear least-squares regression as the mathematical model.[Citation19] The limits of detection (LOD) was determined by the signal to noise (S/N) ratio method. It was estimated as the minimum concentration of analyte providing a S/N ratio of 3:1[Citation20,Citation21] by injecting a series of diluted solutions with known concentration. The precision of the HPLC method was assessed for each sugar (sucrose, fructose, and glucose) by repeating five times the analysis of the sample-IS solution prepared as described in “sample preparation-method (b).” The precision, reported as RSD, was calculated by the following formula:
Formulas
The capacity factor (k) of the columns was calculated using the following equation:
RESULTS AND DISCUSSION
Comparison of Two Sample Pretreatment Methods
Besides saccharides, sugarcane molasses contains pigments, nitrogen compounds, and inorganic ions, such as potassium, calcium, sodium, and magnesium. Pigments in sugarcane molasses include polyphenols plant pigments and pigments generated in the course of production, such as pseudo-melanin caused by reducing sugar decomposition, the reaction between reducing sugar and amino acid, and caramel pigments resulting from charred sucrose in the process of heating. Nitrogenous compounds encompass proteins, amino acids, and amide. The presence of the substances influences the separation of sugars, and particularly the pigment substances can even pollute chromatographic column easily, reducing separation degree and lifespans of the column.[Citation14,Citation18] Despite little difference in saccharide peak shapes between pretreated and unpretreated samples, the long-term accumulation of the pigments in the chromatographic column can reduce the column lifespan. So it is necessary to pretreat the sample before injection.
The macroporous resin decolorization method and the Sep-Pak C18 SPE column purification method were used to pretreat sugarcane molasses samples in this study. The decolorizing effect of XDA-8 resin on molasses samples was more significant than that of the other resins (data not shown). However, the sugar loss of the macroporous resin method was more significant than that of the SPE column purification method, which affected the accuracy of the subsequent measurement (see ).
TABLE 1 Effect of sample pretreatment on sugars in molasses
TABLE 2 Comparison of two columns on the sugar separationa
As shown in , Dec. of these two methods were both about 87%, but the total sugar loss for the macroporous resin method (11.32%) was about 17-fold higher than that of the SPE method (0.69%). The sample-IS solutions were passed through the Sep-Pak C18 cartridges that had been primed with 4.0 mL of methanol and 8.0 mL of water. The substances, including pigments and polyphenols, in the sample were loaded onto the Sep-Pak C18 cartridge, but the sugars were not absorbed due to their stronger hydrophilicity, indicating that substances, including pigments, polyphenol, and lipid, can be removed. In summary, the SPE cleanup method is of higher discoloration efficiency, lower sugar loss, and better chromatographic peak separation. Therefore, SPE was used to remove the interferents of molasses samples in this study.
Selection of Chromatography Column
Chromatographic separation column is the core of the HPLC method, which directly influences the component resolution and analysis results. An Agilent Zorbax carbohydrate column and UtimateTM XB-NH2 column were compared on the separation and determination of carbohydrate material. As shown in , the retention time of the three sugars on the carbohydrate column increased gradually, suggesting the moderate interval and good separation effect, while the retention time of fructose and glucose on the XB-NH2 column was too close to be well separated. As to capacity factor (k) of the columns, the larger the capacity factor, the greater capacity of the component that stationary phase fixes, which indicates that the components flow more slowly from the column and has the longer retention time. If the difference between the capacity factors of two components on the same column is greater, the difference between retention times of the two components on the same column may be significant, leading to better separation of the two components. As shown in , capacity factors (k) of the three sugars on the carbohydrate column are all larger than that of the three sugars on the XB-NH2 column. Moreover, when performed with common amino based columns (XB-NH2 column), it often results in the formation of a Schiff base (reaction between the sugar and the amine group of the stationary phase), which might have a significant influence on the stationary phase of the chromatography (loss of amine group and cannot be regenerated). The sugar that is bound to the surface of the stationary phase cannot be eluted from the column, thereby affecting quantification and shortening the lifespan of the column. In addition, the pressure of the XB-NH2 column was relatively higher than that of the carbohydrate column, which may decrease the lifespan of the XB-NH2 column with a long-term use. For the above reasons, the carbohydrate column was selected to separate and detect sugars in the present study.
Optimization of Mobile Phase
Due to polar groups contained in the sugar molecules, solvents with strong polarity (such as water, methanol, and acetonitrile) should be chosen as the mobile phase. It has been shown that a single solvent cannot achieve ideal separation of the sugars, and thus a mixed solvent is necessary for the mobile phase. Wei and Ding[Citation22] reported that the content of water in the mobile phase had a great effect on the retention capacity of the carbohydrates, and the carbohydrates would be eluted more quickly with increasing content of water in the mobile phase. In the present work, mobile phase proportion was optimized by choosing acetonitrile and water at the ratios of 60:40, 65:35, 70:30, 75:25, and 80:20 (v/v), respectively. As a result, the resolution (the degree of separation of two adjacent peaks in chromatogram) was improved and the retention time was extended with increasing acetonitrile concentration. The acetonitrile-water ratio of 80:20 exhibited the optimum separation with the longest retention time, but the ratio of 70:30 was unable to separate fructose and glucose well. In contrast, the ratio of 75:25 compromised on the analysis time and separation degree between fructose and glucose. Hence, the acetonitrile-water ratio of 75:25 (v/v) was chosen for further experiments.
Chromatographic Separation
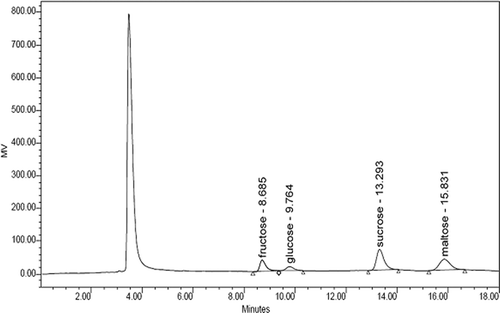
Under the chromatographic conditions, a good separation could be achieved among sucrose, fructose, and glucose (see ). The retention time of fructose, glucose, sucrose, and maltose in mixed standard solutions were 8.62, 9.70, 13.22, and 15.74 min, respectively. The retention time of fructose, glucose, sucrose, and maltose in the molasses-IS samples were 8.69, 9.76, 13.29, and 15.83 min, respectively. Comparing the HPLC sugar profiles of samples with the commercial standards available, the fructose, glucose, and sucrose have been identified. The chromatograms of standard mixture and molasses samples are shown in and .
FIGURE 1 HPLC-RID chromatogram of a standard mixture of three carbohydrates with a maltose internal standard. Determination conditions: mobile phase of acetonitrile and water (75: 25, v/v), flow rate 1.0 mL/min, column temperature 30°C, and RID temperature 35°C.
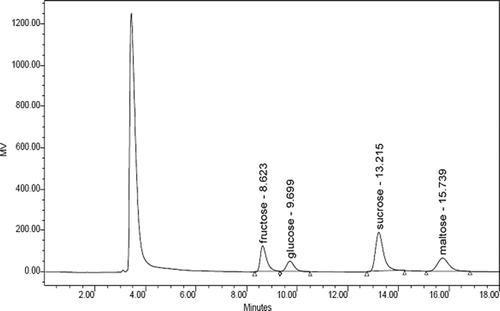
FIGURE 2 HPLC-RID chromatogram of three carbohydrates in molasses samples with a maltose internal standard. Determination conditions: mobile phase of acetonitrile and water (75: 25, v/v), flow rate 1.0 mL/min, column temperature 30°C, and RID temperature 35°C.
Linear Range and Limit of Detection
The calibration curves were consistently linear from 5.94–29.70 g/L for sucrose, 3.30–16.48 g/L for fructose, and 1.80–9.02 g/L for glucose. The mean squared correlation coefficients (R2) were all generally ≥0.995. LODs of three carbohydrates were investigated and the results were shown in . Three carbohydrates in the range exhibit an excellent linear relationship. The studies on LODs were also reported by other workers using HPLC. Kakita et al.[Citation8] analyzed monosaccharides and oligosaccharides by HPLC with postcolumn fluorescence derivatization, and the LODs of glucose and maltohexaose were 1.78 and 2.59 pmol, respectively. Scotter et al.[Citation18] developed a selective and rapid procedure by HPLC and PDA for the determination of coumarin in foodstuffs with LOD ranging from 0.05–2.5 mg/kg. Wei and Ding[Citation22] reported that LODs were between 0.2 and 1.2 μg for different carbohydrates in drinks by HPLC with ELSD.
TABLE 3 Parameters of quantitative analysis for three carbohydrates
Precision and Recovery
and summarize the precision and recovery validation of molasses samples as described above. The RSDs of precision experiments of the three carbohydrates were 4.81, 4.42, and 3.43%, respectively, each of which was less than 5.0%, indicating that the method was precise. Suitable amounts of the carbohydrate standards were added to the molasses samples of known carbohydrate content, and the mixtures were analyzed as described in the “sample preparation-method (b).” The standard addition recoveries were 100.33, 100.30, and 96.40%, respectively. RSDs were less than 5.0%, indicating that this method was accurate. Wei and Ding[Citation22] analyzed carbohydrates in drinks by HPLC with a dynamically modified amino column and the recoveries of carbohydrates were between 95.8 and 103.0%. Sanchez-Mata et al.[Citation23] determined monosaccharide, disaccharide, and oligosaccharide in legumes by HPLC using an amino-bonded silica column with recovery percentages between 93.33 and 101.45%.
TABLE 4 The precision for fructose, glucose, and sucrose (n = 5)a
TABLE 5 Results of spiked recovery test (n = 3)a
Reproducibility and Analysis of the Samples
The reproducibility of our HPLC method was tested by repeating the analysis of one sugarcane molasses sample four times. The sample pretreatment method followed SPE cleanup of sample preparation procedures. The results are presented in . At the same time, we also adopted “sample preparation-method (a),” and the results are shown in . and show that the RSDs range from 0.57 to 2.28% and 0.97 to 4.13%, respectively, which are less than 5.0%, indicate that the methods possess good reproducibility. The difference detected between the two methods indicates that the SPE absorbs less sugar. Therefore, the SPE-HPLC method is appropriate for the quantification of sugars in sugarcane molasses.
TABLE 6 Results of sample determination by SPE (n = 4)a
TABLE 7 Results of sample determination by direct injection (n = 3)a
CONCLUSION
SPE-HPLC with IS method was successfully developed for the separation and determination of fructose, glucose, and sucrose in sugarcane molasses. The sample preparation of the established method was simple and easy to perform, and the results were accurate and quantified. The present investigation suggests that SPE-HPLC with IS method is of high practical value for the determination of sugars in molasses.
FUNDING
This work was supported by the National Natural Science Foundation of China (Grant Nos. 51078147 and 51278200); Guangdong Natural Science Foundation (Grant No. S2012010010380); the Opening Project of State Key Laboratory of Pulp and Paper Engineering (South China University of Technology) [Grant No. 201484); and the Guangdong Provincial Science and Technology Program (Grant Nos. 2012B091100163, 2011B090400033, and 2010A010500005).
REFERENCES
- Ranganna, S. Manual of Analysis of Fruit and Vegetable Products. Tata McGraw-Hill Publishing Company: New Delhi, 1977; 9–13.
- Miller, G.L. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Analytical Chemistry 1959, 31, 426–428.
- Heikkila, H. Separating sugars and amino acids with chromatography. Chemical Engineering 1983, 24, 50–52.
- He, Y.; Wu, D.; Feng, S.J. Fast measurement of sugar content of yogurt using Vis/NIR-spectroscopy. International Journal of Food Properties 2007, 10, 1–7.
- Ruiz-Matute, A.I.; Montilla, A.; Castillo, M.D.D.; Martínez-Castro, I.; Sanz, M.L. A GC method for simultaneous analysis of bornesitol, other polyalcohols and sugars in coffee and its substitutes. Journal of Separation Science 2007, 30, 557–562.
- Ouchemoukh, S.; Schweitzer, P.; Bey, M.B.; Djoudad-Kadji, H.; Louaileche, H. HPLC sugar profiles of Algerian honeys. Food Chemistry 2010, 121, 561–568.
- Legua, P.; Melgarejo, P.; Martinez, J.J. Evaluation of Spanish pomegranate juices: Organic acids, sugars, and anthocyanins. International Journal of Food Properties 2012, 15, 481–494.
- Kakita, H.; Kamishima, H.; Komiya, K.; Kato, Y. Simultaneous analysis of monosaccharides and oligosaccharides by high-performance liquid chromatography with postcolumn fluorescence derivatization. Journal of Chromatograph A 2002, 961, 77–82.
- Nejib, H.; Rania, J.; Messaoud, M. Organic acids, sugars, and anthocyanins contents in juices of Tunisian Pomegranate fruits. International Journal of Food Properties 2011, 14, 741–757.
- Sims, A. HPLC analysis of sugars in foods containing salt. Journal of Agricultural and Food Chemistry 1995, 43, 377–380.
- Bhandari, P.; Kumar, N.; Singh, B.; Kaul, V.K. Simultaneous determination of sugars and picrosides in Picrorhiza species using ultrasonic extraction and high-performance liquid chromatography with evaporative light scattering detection. Journal of Chromatograph A 2008, 1194, 257–261.
- Morales, V.; Corzo, N.; Sanz, M.L. HPAEC-PAD oligosaccharide analysis to detect adulterations of honey with sugar syrups. Food Chemistry 2008, 107, 922–928.
- Barreira, J.C.M.; Pereira, J.A.; Oliveira, M.B.P.P. Sugars profiles of different chestnut (Castanea sativa Mill.) and almond (Prunus dulcis) cultivars by HPLC-RI. Plant Food for Human Nutrition 2010, 65, 38–43.
- Sajwani, A.M.; Eltayeb, E.A.; Farook, S.A. Sugar and protein profiles of Omani honey from Muscat and Batinah regions of Oman. International Journal of Food Properties 2007, 10, 675–690.
- Vaccar, G.; Lodi, G.; Tanburin, E.; Bernardi, T.; Tosi, S. Detection of oligosaccharides in sugar products using planar chromatography. Food Chemistry 2001, 74, 99–110.
- Monticelli, E.; Aman, C.S.; Costa, M.L.; Rota, P.; Bogdan, D.; Allevi, P.; Cighetti, G. Simultaneous free and glycosylated pyridinium crosslink determination in urine: Validation of an HPLC-fluorescence method using a deoxypyridinoline homologue as internal standard. Journal of Chromatograph B 2011, 879, 2764–2711.
- Hsu, F.; Nurok, D.; Zlatkis, A. The determination of sucrose in molasses by high-performance thin-layer chromatography. Journal of Chromatograph A 1978, 158, 411–415.
- Scotter, M.J.; Roberts, D.P.T.; Gareth, O.R. Development and single-laboratory validation of an HPLC method for the determination of coumarin in foodstuffs using internal standardization and solid-phase extraction cleanup. Analytical Methods 2011, 3, 414–419.
- Mei, Y.H.; Xu, J.; Zhao, J.H.; Feng, N.P.; Liu, Y.; Wei, L. An HPLC method for determination of oridonin in rabbits using isopsoralen as an internal standard and its application to pharmacokinetic studies for oridonin-loaded nanoparticles. Journal of Chromatograph B 2008, 869, 138–141.
- Shabir, G.A. Validation of high-performance liquid chromatography methods for pharmaceutical analysis: Understanding the differences and similarities between validation requirements of the US food and drug administration, the US pharmacopeia and the international conference on harmonization. Journal of Chromatograph A 2003, 987, 57–66.
- Ribani, M.; Bottoli, C.B.G.; Collins, C.H.; Jardim, I.S.F.; Melo, L.F.C. Validation for chromatographic and electrophoretic methods. Química Nova 2004, 27, 771–780.
- Wei, Y.; Ding, M.Y. Analysis of carbohydrates in drinks by high-performance liquid chromatography with a dynamically modified amino column and evaporative light scattering detection. Journal of Chromatograph A 2000, 904, 113–117.
- Sanchez-Mata, M.C.; Penuela-Teruel, M.J.; Canmara-Hurtado, M.C.; Diez-Marques, C.; Torija-Isasa, M.E. Determination of mono-, di-, and oligosaccharides in legumes by high-performance liquid chromatography using an amino-bonded silica column. Journal of Agricultural and Food Chemistry 1998, 46, 3648–3652.