
ABSTRACT
A new protease was isolated from the intestine of small red scorpionfish. After ammonium sulfate precipitation, the enzyme was purified to homogeneity by a two-step chromatography with 6.69% recovery and fivefold increase in specific activity. The molecular weight of the protease was about 24 kDa as estimated by size exclusion chromatography and sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The enzyme optimum pH and temperature were pH 10.0 and 40°C, respectively. The protease was stable at temperatures below 40°C and over a broad pH range (8.0–11.0). The Km and Kcat values were 0.36 mmol L−1 and 1.61 s−1, respectively. Interestingly, the enzyme presented specificity for casein, egg albumin, bovine serum albumin, and gelatin with obtained degrees of hydrolysis ranging from 3.14% to 6.58%, after 2 h of hydrolysis. Moreover, gluten hydrolysate analysis confirmed protein hydrolysis and solubilization. These results suggested the potential use of the protease in the preparation of food protein hydrolysates with interesting properties.
Introduction
Proteases constitute the most important group of industrial enzymes accounting for at least 60% of all global industrial enzyme sales and have several industrial applications such as in the detergent, food, pharmaceutical, leather and silk, waste treatment, and bioactive peptide synthesis.[Citation1,Citation2] Marine animals have adapted to different environmental conditions, and these adaptations have resulted in digestive proteases with certain unique properties. These characteristics include high activity over a wide range of pH (2.0–11.0), low-temperature conditions (30–65°C), and more flexible enzyme structure associated with higher catalytic activity and lower thermostability (e.g. thermal denaturation temperature of pepsins from cold water animals is about 20°C lower than mammalian pepsins and thus are more readily inactivated by milder heat treatment).[Citation3,Citation4]
Fish viscera are rich potential sources of various enzymes that can be valorized for an optimal utilization. In fact, according to the last FAO reports, 21.7 million tones of fish processing wastes has been discarded or destined to nonfood uses (e.g. fishmeal and fish oil) in 2012.[Citation5] These by-products include frame bones, heads, tails, skin, and viscera.[Citation6] Trypsin (EC 3.4.21.4), one of the main digestive proteases detected in fish viscera, has specificity for the peptide bonds involving the carbonyl group of arginine or lysine residues. Trypsins play major roles in biological processes, including digestion and activation of zymogens of the pancreatic enzymes, including itself. Today, there is an increasing demand for marine trypsins in food processing because they are most active at alkaline pH range (from 7.5 to 10.0) and relatively low temperatures (from 35 to 65°C) which is advantageous in some food processing operations.[Citation7,Citation8]
A number of studies on trypsins from fish viscera have been carried out. Recently, we isolated and characterized fish trypsins from the following species: true sardine (Sardinops melanostictus) and arabesque greenling (Pleuroprammus azonus), atlantic bonito (Sarda sarda), gray triggerfish (Balistes capriscus), bogue (Boops boops), zebra blenny (Salaria basilisca), and Tunisian barbel (Barbus callensis).[Citation9–Citation14]
Wheat gluten protein, a by-product of the wheat starch industry, are very complex proteins containing hundreds of components present as monomers, oligomers, and polymers that by definition are not soluble in water. Enzymatic hydrolysis of gluten proteins is generally carried out to improve the functional properties of proteins, changing solubility or producing low molecular peptides with low allergenic effects.[Citation15,Citation16]
In this study, we describe the purification of a novel intestinal protease from small red scorpionfish (Scorpaena notata) and investigate its main biochemical and kinetic characteristics. The potential use of the protease in the production of industrial protein hydrolysates with new characteristics was then investigated.
Materials and methods
Fish sample
Small red scorpionfishes (S. notata) were purchased from the fish market of Tunis. Fishes were placed in ice with a fish/ice ratio of 1:2 (w/w) and transported to the laboratory. Upon arrival, fishes were washed and filleted and the intestines were collected, rinsed with distilled water, and used immediately for protease preparation and purification.
Preparation of crude extract
Preparation of crude extract from Small red scorpionfishes was performed following the method of Jellouli et al.[Citation11] The intestines (200 g) from S. notata were homogenized three times for 60 s with 400 mL of cold (4°C) extraction buffer (10 mmol L−1 Tris–HCl, pH 8.0). The homogenate was then centrifuged at 7000g for 30 min at 4°C to remove the pellet and the supernatant, referred to as crude protease extract, was collected.
Proteolytic activity assay
Proteolytic activity was evaluated as described by Segers et al. with modifications.[Citation17] The reaction mixture contained 50 µL of crude protease extract, 100 µL of reaction glycine–NaOH buffer (0.1 mol L−1, pH 10.0) (Buffer A), and 50 µL of 5% azocasein (w/v) dissolved in Buffer A. The assays were carried out at 40°C and stopped after 30 min by adding 600 µL of 10% (w/v) trichloroacetic acid. After 15 min incubation on ice, the reaction mixture was centrifuged at 7000g for 10 min to remove the precipitated protein. Supernatant (600 µL) was neutralized by adding 700 µL of 1.0 mol L−1 NaOH and absorbance at 440 nm was recorded with an UV/visible spectrophotometer (Shimadzu model 1240, Tokyo, Japan). One unit of enzyme activity was defined as the amount, which yielded an increase A440 of 0.01 in 30 min at 40°C under the assay conditions, as mentioned above.
Chromatographic procedures for protease purification
The crude extract containing the enzyme was first subjected to ammonium sulfate precipitation at 80% saturation. The protein precipitate was obtained after centrifugation at 5000g for 30 min. It was suspended in a minimal volume of glycine–NaOH buffer (0.02 mol L−1, pH 10.0) (Buffer B) and dialyzed overnight against the same buffer. The sample was then loaded onto a Toyopearl HW-50 column (1.6 × 40 cm) using an ÄKTA FPLC fast-performance liquid chromatography system from GE Healthcare (Supplanta, Sweden) and pre-equilibrated with Buffer B. Elution was carried out at a flow rate of 0.5 mL min−1 with Buffer B and enzyme fractions of 2.0 mL which were assayed individually for protein at 280 nm and for protease activity as described above. Fractions with protease activity were pooled and further purified by anion exchanger chromatography.
Mono Q-Sepharose anion-exchange chromatography column (2.0 cm × 10 cm) was equilibrated with Buffer B and elution was carried out with a linear gradient from 0 to 1 M NaCl in the equilibrating buffer at a flow rate of 1 mL min−1. The fractions of 2.0 mL with high protease activity were collected and stored at 4°C for further analysis. Fractions showing maximum activity were further analyzed for purity by electrophoresis.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis analysis
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) was performed using 5% stacking gel and 12% separating gel following the method of Laemmli.[Citation18] Samples were mixed with sample buffer containing SDS and β-mercaptoethanol and heated at 100°C before electrophoresis. Electrophoresis was performed at 100 V and protein bands were visualized after staining the gel with 0.25% Coomassie Brilliant Blue R250 in 30% ethanol–10% acetic acid and destained with 30% ethanol–10% acetic acid. The apparent molecular weight of protein bands was determined by comparing with the bands of standard molecular mass marker.
Detection of protease activity by zymography
Zymography was performed on SDS–PAGE according to the method of Schmidt et al. with slight modification.[Citation19] The resolving gel was copolymerized with 0.05% gelatin. After migration at 100 V and 4°C, the gel was submerged in Buffer A containing 2.5% Triton X-100, with shaking for 30 min to remove SDS. Triton X-100 was then removed by washing the gel three times with Buffer A and the gel was incubated in Buffer A for 60 min at 40°C. Finally, the gel was stained with 0.25% Coomassie Brilliant Blue R250 in 30% ethanol–10% acetic acid and destained with 30% ethanol–10% acetic acid. A clear zone against a dark blue background of the gel indicated protease activity.
Determination of native molecular weight
The molecular weight of the purified enzyme was determined using gel filtration chromatography on a Toyopearl HW-40 column (1.0 × 36 cm) using an ÄKTA FPLC system from GE Healthcare (Supplanta, Sweden). Purified protease (15 µg) was loaded to the column and elution with Buffer B was performed at a flow rate of 0.5 mL min−1. The column was previously calibrated with bovine serum albumin (BSA) (Mr 66,000 Da), ovalbumin (Mr 44,000 Da), and lysozyme (Mr 14,000 Da) (Sigma Chemical Co., St. Louis, MO, USA). The elution volume (Ve) was measured for each protein standard and for the purified enzyme. Protease activity was tested in the chromatographic fractions by azocasein assay.
Effect of temperature and pH on protease activity and stability
In order to determine optimal temperature, the enzymatic assay was carried out at different temperatures (25–70°C), at pH 10.0. Thermal stability was determined by preincubating the enzyme at temperatures ranging from 20 to 70°C for 60 min and the residual activity was measured at 40°C in buffer A and expressed as percentage of initial activity taken as 100% (non-heated enzyme). Optimum pH was determined by assaying in buffers (0.1 mol L−1) of varying pH at 40°C (phosphate buffer, pH 7.0; Tris–HCl buffer, pH 8.0–9.0; glycine–NaOH buffer, pH 10.0–11.0–12.0–13.0). For pH stability, the enzyme was kept at 4°C for 12 h in different buffers at 0.1 mol L−1, at pH values ranging from 4.0 to 11.0. Residual proteolytic activity was estimated as described earlier and expressed as percentage of the initial activity taken as 100%.
Effect of enzyme inhibitors and surfactants on enzyme activity
The effect of enzyme inhibitors (2.0 and 5.0 mmol L−1) on the purified protease was studied using phenylmethylsulfonyl fluoride (PMSF), β-mercaptoethanol, and ethylenediaminetetraacetic acid (EDTA). The purified enzyme was preincubated with each additive for 30 min at 4°C, and the remaining enzyme activity was determined using azocasein as substrate. The activity of the enzyme assayed in the absence of inhibitors was taken as 100%. Enzyme stability in surfactants Tween 20, Triton X-100, and SDS was studied by preincubating the purified protease for 30 min at 4°C with each additive at different concentrations (1% and 5%). The residual activity was measured at pH 10.0 and 40°C. The activity of the enzyme in the absence of surfactant was taken as 100%.
Effect of metal ions on enzyme activity
The effect of various metal ions (5.0 and 10.0 mmol L−1) on protease activity was determined by adding the monovalent (Na+ and K+) and divalent (Ca2+, Mn2+, Zn2+, Cu2+, Mg2+) metal ions to the reaction mixture. After incubation for 30 min at 4°C, substrate was added and the residual activity of enzyme was measured. Protease activity without metal ions was taken as 100%.
Effect of salt on enzyme activity and stability
The effect of salt on enzyme activity was studied by adding NaCl into reaction mixture to obtain final concentrations ranging from 0% to 30% (w/v). The enzymatic assay was then carried out at optimum conditions (40°C, pH 10.0). For stability, the purified enzyme was preincubated for 60 min at 4°C with NaCl concentrations ranging from 0% to 30% (w/v). The enzyme activity assayed in absence of salt was taken as 100%.
Kinetic studies
The kinetic parameters of the enzyme were determined by assaying the protease under varying azocasein concentrations (0.211–1.055 mmol L−1) (in Buffer A and under optimized assay conditions). Kinetic constants, including the apparent Michaelis–Menten constant (Km) and the maximal velocity (Vmax), were calculated from Lineweaver–Burk plot. The value of the turnover number (Kcat) was calculated from the following equation: Kcat = Vmax/[E], where [E] is the active enzyme concentration.
Wheat gluten hydrolysis
Two different concentrations (1% and 3%, w/v) were used when testing the hydrolysis of wheat gluten (Sigma Chemical Co., St. Louis, MO, USA) using the intestinal protease of S. notata. Wheat gluten was first dissolved in distilled water, then the suspension was adjusted to appropriate temperature and pH of the enzyme (i.e., T = 40°C, pH = 10.0). The hydrolysis reaction was started by the addition of the enzyme preparation at an enzyme–substrate ratio of 5:1 (U mg−1) and incubated for 24 h. During hydrolysis, the pH of the mixture was maintained at the desired value using 0.1 mol L−1 NaOH. After hydrolysis, the solution was heated at 90°C for 20 min to inactivate the enzyme and hydrolysate was centrifuged at 5500g for 30 min to remove non-hydrolyzed residues. The supernatant was stored at −20°C and used for further analyses as described below.
The degree of hydrolysis (DH), defined as the percent ratio of the number of peptide bonds broken (h) to the total number of peptide bonds in the substrate studied (htot), was calculated from the amount of base (NaOH) added to keep the pH constant during the hydrolysis as given below:
with B is the amount of alkali consumed (mL) to keep the pH constant during hydrolysis, Nb is the normality of the base, Mp is the mass of the substrate (g) (N × 6.25), htot is the content of peptide bonds, and α is the average degree of dissociation of the α-NH2 groups released during hydrolysis expressed as
where pH and pK are the values at which the proteolysis was conducted. The total number of peptide bonds (htot) in the protein substrate was assumed to be 8.38 meq g−1 gluten proteins.[Citation20]
Enzymatic hydrolysis of natural industrial proteins
The substrate specificity of S. not-protease toward different natural substrates including casein, egg albumin, BSA, and gelatin was examined. The protein substrate was first dissolved in distilled water at a concentration of 3% and the hydrolysis reaction was conducted as described above for 2 h. The DH was calculated from the amount of base (NaOH) added to keep the pH constant during the hydrolysis and the total number of peptide bonds (htot) in the protein substrates was assumed to be 8.2, 7.67, 8.8, and 11.1 meq g−1 of casein, egg albumin, BSA, and gelatin, respectively.[Citation20]
Protein assay
Protein was estimated according to the method of Bradford with BSA as the standard.[Citation21] The protein content was calculated from a standard curve (data not shown). During chromatographic purification steps, protein determination of each fraction was estimated by measuring its absorbance at 280 nm.
Statistical analysis
All the tests were performed in triplicate. Data were presented as means ± standard deviations (SD). Statistical analysis was done using the Statistical Analysis System. Means were compared using Duncan’s multiple range test. Differences were considered significant at p < 0.05.
Results and discussion
Protease purification from S. notata intestine
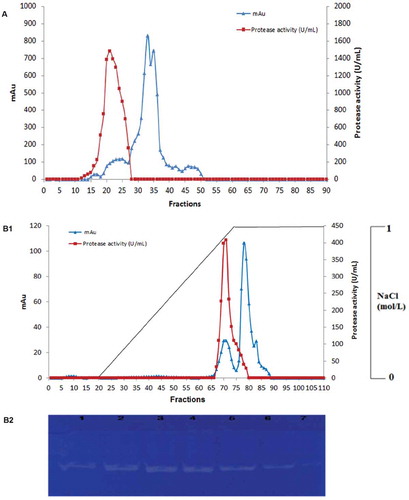
S. not-protease was purified by ammonium sulfate precipitation, gel filtration chromatography on Toyopearl HW-50 column, and anion exchange chromatography. After gel filtration chromatography, several protein fractions were obtained with only a single peak presenting a proteolytic activity (fractions 13–25) (). Fractions were pooled and submitted to anion exchange chromatography on Mono Q-Sepharose anion-exchange chromatography column. Binding proteins were eluted with a linear gradient of NaCl (0–1 mol L−1). Protease was eluted on a single peak at a NaCl concentration of 0.9 M (). Analysis by zymogram activity staining of different obtained fractions revealed the presence of a single clear band of proteolytic activity against the blue background indicating the presence of only one isoform (). summarizes the results of the purification process. About fivefold purification and 6.69% recovery were achieved. Specific activity of the protease increased from 138,060 U mg−1 for the crude protease extract to 675,949 U mg−1 after the final purification step.
Table 1. Purification of the intestinal protease of Scorpaena notata.
Figure 1. (A) Elution profile of S. not-protease after gel filtration chromatography on Toyopearl HW-50 column (0.5 mg of the protein extract was loaded to the column). (B1) Elution profile of S. not-protease on Mono Q-Sepharose anion-exchange chromatography column (0.2 mg of the protein extract was loaded to the column). Elution was carried out with a linear gradient of 0–1 mol L−1 NaCl. (B2) Zymogram detection of proteolytic activity of different fractions (F68, F69, F70, F71, F72, F73, F74) of the purified S. not-protease.
Molecular weight analysis of S. not-protease
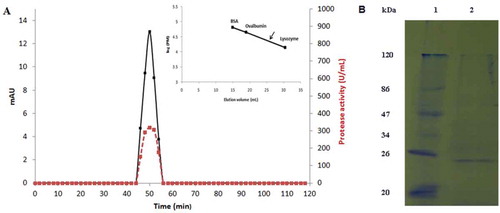
The apparent molecular mass of the purified S. not-protease was estimated to be 24 kDa after FPLC gel filtration chromatography analysis on Toyopearl HW-40 column (). The purified protease was also analyzed by SDS–PAGE and migrated as a single band corresponding to a molecular weight estimated to be 24 kDa (). SDS polyacrylamide gel under reduced conditions confirmed the homogeneity and the monomeric state of the intestinal protease. The molecular weight of S. not-protease was similar to digestive proteases purified from other fish species such as tongol tuna (Thunnus tonggol), skipjack tuna (Katsuwonus pelamis), and Tunisian barbel (Barbus callensis).[Citation14,Citation22,Citation23]
Figure 2. (A) Elution profile of S. not-protease after gel filtration chromatography on Toyopearl HW-40 column using an FPLC system (15 µg of the purified protease was loaded to the column). Elution volume of S. not-protease is showed by an arrow in the calibration curve. Protein content is shown in continuous line and activity in discontinuous line. (B) SDS–PAGE analysis of the purified S. not-protease. Lane 1, standard molecular weight markers; Lane 2, purified S. not-protease (2 µg).
Effect of temperature and pH on protease activity and stability
Temperature profile of S. not-protease is shown in . The low molecular weight protease was active in the temperature range of 30 and 50°C with an optimum at 40°C. The relative activities at 30 and 50°C were about 87.7% and 88.31%, respectively. These findings are in accordance with several reports showing high activity of digestive proteases at low temperatures such those reported by Jellouli et al. concerning the purification of a trypsin from the intestine of gray triggerfish (Balistes capriscus) and Espósito et al. for the characterization of a trypsin from the lane snapper (Lutjanus synagris).[Citation11,Citation24] However, the optimal temperature of this enzyme is lower than that of other low molecular weight proteases.[Citation25,Citation26]
Figure 3. Effect of temperature on enzyme activity (A) and stability (B). Remaining protease activity was measured under standard assay conditions. Effect of pH on purified S. not-protease activity (C) and stability (D). The stability was estimated as residual enzyme activity after pre-incubation in different buffers for 12 h at 4°C. The results represent mean values of three independent experiments each performed in triplicates and error bars indicate standard deviation.
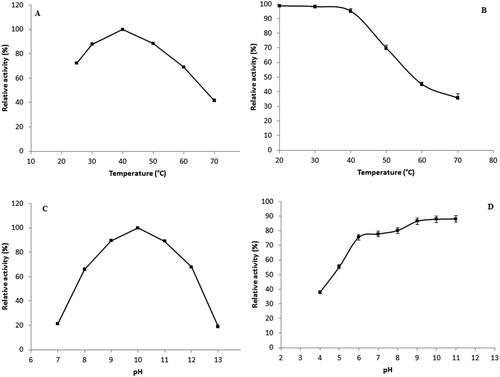
The thermostability of the protease was carried out by preincubating the enzyme for 60 min at a temperature range from 20 to 70°C as shown in . The intestinal protease was stable at low temperatures since it retains 98.77%, 98.26%, and 95.16% of its initial activity after 60 min of preincubation at 20, 30, and 40°C, respectively. This property is of great interest for enzyme utilization in main industrial applications such as production of peptides and amino acids, detergent formulations, and meat tenderization since the enzyme can be active and stable in the solution for a long period of time throughout a wide range of temperatures. However, preincubation for 60 min at higher temperature (70°C) resulted in a loss of activity and the protease retains not more than 35.78% of its initial activity. The observed thermal stability was similar to the ones previously reported for digestive proteases (95% and 100% of residual activity after 60 min of preincubation at 40°C for proteases of Tunisian barbel and sardinelle, respectively).[Citation14,Citation26] However, the stability of the protease at 70°C is better than that of trypsins isolated from the intestine of gray triggerfish and from Tunisian barbel viscera.[Citation11,Citation14]
The pH activity profile of S. not-protease showed that the enzyme is active in the pH range of 8.0–12.0 with an optimum at pH 10.0 suggesting that it is an alkaline protease. This is an important characteristic since the protease can be used in several applications such as protein hydrolysis and in laundry detergents. The relative activities at pH 8.0 and 12.0 were about 65.9% and 67.88%, respectively (). Several proteases purified from fish viscera showed similar results for optimum pH such as a trypsin purified from the viscera of zebra blenny (Salaria basilisca) and a trypsin purified from Tunisian barbel (Barbus callensis).[Citation13,Citation14] Nevertheless, this pH value was higher than that of other low molecular weight proteases such as the collagenase purified from the mackerel (S. japonicas) (optimum activity at pH 7.5) and the protease purified from sardinelle (Sardinella aurita) (optimum activity at pH 8.5).[Citation26,Citation27] The pH stability profile showed that the purified protease was highly stable over a wide pH range (8.0–11.0) maintaining approximately 80% of its original activity at pH 8.0 and 88.09% of its activity at pH 11.0 ().
Effect of enzyme inhibitors and surfactants on S. not-protease activity
In order to determine the nature of the intestinal protease, different enzyme inhibitors such as specific group reagents and a chelating agent were preincubated with the purified enzyme and their effect on enzyme activity was then investigated (). The intestinal protease was strongly inhibited by PMSF. It retained only 9.09% of its initial activity after 30 min incubation in the presence of 5.0 mmol L−1 PMSF. High inhibition of protease activity by PMSF indicates that the enzyme is a serine protease. In fact, PMSF is known to sulfonate the essential serine residue on the active site of the enzyme resulting in the complete loss of enzyme activity.[Citation28] This result is in accordance with several studies reporting the purification and characterization of serine proteases from fish species.[Citation29–Citation32] The chelating agent EDTA showed minimal inhibitory effect on protease activity (14.4% inhibition in the presence of 5.0 mmol L−1 EDTA) suggesting that the enzyme most likely required metal ion(s) for its activity. The decrease of fish proteases activity in the presence of EDTA was also reported for trypsins from the spleen of tongol tuna,[Citation22] the spleen of yellowfin tuna,[Citation33] and the intestine of hybrid tilapia.[Citation34] Nevertheless, β-mercaptoethanol was practically without influence on the activity of the purified enzyme, indicating its stability in the presence of reducing agents. Similar results were obtained for trypsins purified from the intestine of gray triggerfish[Citation11] and the viscera of sardinelle.[Citation26]
Table 2. Effect of various enzyme inhibitors on the activity of S. not-protease.†
The effect of surfactants on the protease activity is shown in . The enzyme was relatively stable in the presence of the nonionic surfactants Tween 20 and Triton X-100 at the two tested concentrations 1% and 5% and retained about 99.22% and 89.14% of its activity in the presence of 5% Tween 20 and Triton X-100, respectively. However, the presence of a strong anionic surfactant like SDS in the mixture caused a moderate inhibition of 65.89% and 59.68% at a concentration of 1% and 5%, respectively. The relative stability of the enzyme in the presence of nonionic surfactants is a very important characteristic for its eventual use in detergent formulations.
Table 3. Effect of surfactants on enzyme activity of S. not-protease.†
Effect of metal ions on S. not-protease activity
The effects of several metal ions (at two different concentrations, 5.0 and 10.0 mmol L−1) on the protease activity were investigated (). K+, Na+, Mg2+, and Mn2+ did not affect the enzyme activity at concentrations of 5.0 and 10.0 mmol L−1. Ca2+ increased enzyme activity to 115% at a concentration of 10.0 mmol L−1. However, we have observed that the protease activity was strongly affected by the addition of the metal ions Cu2+ and Zn2+. In fact, the enzyme retains no more than 28.36% and 42.35% of its initial activity after preincubation with 10.0 mmol L−1 Cu2+ and Zn2+, respectively. These findings are similar to those of Espósito et al. and Silva et al.[Citation24,Citation35] The mechanism of the inhibitory action of certain ions is not clear. Metal ions may influence binding of substrate with enzyme by various mechanisms.[Citation36] In fact, the metal ion Cu2+ can react with cysteines promoting their oxidation and formation of cystine (Cys–Cys), probably affecting enzyme structure and/or the access to the active site.[Citation37]
Table 4. Effect of metal ions on enzyme activity.†
Effect of salt on enzyme activity and stability
The effect of NaCl concentration (0–30%) on the activity and stability of the intestinal protease is shown in . The activity of the enzyme was not affected in the presence of 5% salt. However, its activity decreased gradually with increasing NaCl concentration. The decrease in activity might be explained by the denaturation of the enzyme. In fact, the presence of high salt concentrations alters electrostatic interactions between charged amino acids, leading to a stronger protein–protein interaction probably associated with enzyme denaturation and precipitation.[Citation38] The intestinal protease showed high activity even in the presence of 30% NaCl with a relative activity of approximately 53.32%.
Figure 4. Effect of NaCl concentrations on activity and stability of purified S. not-protease.
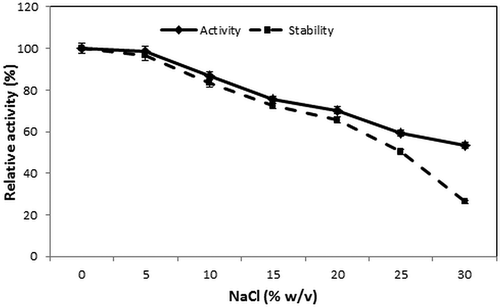
For stability, the purified enzyme was preincubated for 60 min at 4°C with NaCl concentrations ranging from 0% to 30% (w/v). Results revealed that the enzyme retains 50.23% of its activity at salt concentration of 25% (w/v). Nevertheless, its relative activity decreased to reach 26.24% after preincubation in the presence of 30% (w/v) NaCl. These results confirmed that the enzyme can have a potential application in the protein hydrolysis of high-salt products, such as fish sauce.
Kinetic properties
The kinetic parameters of the purified intestinal protease were studied by varying azocasein concentrations at pH 10.0. The Km and Kcat values determined through a Lineweaver–Burk plot were 0.36 mmol L−1 and 1.61 s−1, respectively. The Km value of the intestinal protease was lower than those of other proteases purified from zebra blenny (Salaria basilisca) viscera (0.6 mmol L−1) and sardinelle (Sardinella aurita) viscera (1.67 mmol L−1).[Citation13,Citation26] This suggests that it has higher substrate-binding capacities that can be explained by the differences in their substrate-binding region. The determined turnover number (Kcat) value was higher than that of other reported fish visceral proteases indicating a more efficiency and a greater amount of substrate molecules that are converted into product (Kcat values of 1.32, 0.93, and 0.024 for proteases purified from zebra blenny, silver mojarra, and flatfish, respectively).[Citation13,Citation35,Citation39] The catalytic efficiency (Kcat/Km) of the intestinal protease (4.47 s−1 mmol L) was higher than other previously cited fish proteases ().[Citation13,Citation14,Citation26,Citation39] These results suggest that S. not-protease is an efficient catalyst at low temperatures.
Table 5. Biochemical and kinetic properties of S. not-protease and different fish proteases.
summarizes the characteristics of S. not-protease, along with the corresponding values for other fish proteases obtained from the literature. In fact, S. not-protease is the first protease that was isolated from the intestines of S. notata. S. not-protease is an alkaline enzyme that can be used mainly in detergent formulations and peptide synthesis. It is active at a low temperature (40°C) compared with other fish trypsins (35–65°C). This catalytic property is of great interest since it could present a positive economic impact by reducing energy requirements in industrial processes and destruction of essential food components. This novel protease showed higher kinetic properties than many other fish proteases. S. not-protease presents higher substrate-binding capacities and is more efficient (higher Kcat/Km) which could be an important biotechnological tool for peptide synthesis and food industries.
Enzymatic hydrolysis of natural industrial proteins
Protein hydrolysates have been widely used for animal feed and in the recent years, their potential applications in the food industry have been thoroughly studied. Enzymatic hydrolysis is a widely used method for improving the solubility of proteins, preparing hydrolysates with unique functional and biological properties as well as dietetic and hypoallergenic products. Furthermore, protein hydrolysis can be employed in the release of peptones, source of nutrient for growth of certain microorganisms. This method could have a considerable economic impact in comparison with commercial peptones.
S. not-protease was assayed for its ability to hydrolyze natural substrates. As shown in , the purified enzyme presented broad substrate specificity and was able to degrade various industrial proteins such as casein, egg albumin, BSA, and gelatin with high DH ranging from 3.14% to 6.58%, after 2 h of hydrolysis. The protease showed a high level of hydrolytic activity against gelatin (DH of 6.58%) and moderate hydrolysis of casein, egg albumin, and BSA. Thus, S. not-protease could have various applications especially for the production of functional foods (human and animal feed), peptones, and bioactive peptides.
Table 6. Substrate specificity of S. not-protease toward natural substrates.
Enzymatic hydrolysis of wheat gluten
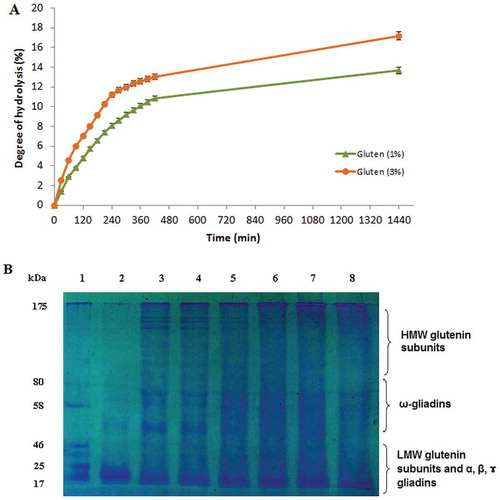
The hydrolysis reaction using S. not-protease was performed at two different concentrations of wheat gluten (1% and 3%, w/v). After 24 h of incubation, the extent of protein degradation reached 13.69% and 17.17% at gluten concentrations of 1% and 3%, respectively (). A concentration of 3% gave the highest DH value at all reaction times confirming that the reaction rate increased as the amount of substrate increased. The obtained DH value was higher than that previously obtained DH using different proteases (Protamex, Neutrase, Pancreatin) with DH values varying from 6.2% to 11.5% after 360 min of incubation.[Citation40] The shape of the hydrolysis curves obtained in this study was similar to those previously reported for gluten hydrolysis.[Citation40–Citation42] The hydrolysis curves proceeded at a rapid rate during the first 4 h of hydrolysis indicating that the maximum cleavage of proteins occurred during this period of time. Thereafter, a slower rate of proteolytic reaction was observed. The rate of hydrolysis decreases with time could be due to three possible reasons: decrease in the amount of peptide bonds that are prone to be cleaved, enzyme inhibition by products, or denaturation of enzyme.
Figure 5. (A) Enzymatic hydrolysis of wheat gluten at two different concentrations (1% and 3%, w/v) with S. not-protease at an enzyme–substrate ratio of 5:1 (U mg−1). (B) SDS–PAGE analysis of gluten hydrolysate at a concentration of 3% at different reaction times. Lane 1, standard molecular weight markers; Lane 2, gluten supernatant at t = 0; Lanes 3–8, gluten hydrolysate supernatant at t = 1, 2, 4, 6, 7, and 24 h, respectively.
Solubility and SDS–PAGE analysis of wheat gluten hydrolysate
The hydrolysate with the highest DH was analyzed by SDS–PAGE as shown in . During the first 4 h of hydrolysis, a larger amount of high molecular weight peptides (MW higher than 40 kDa) and lower amount of peptides of low molecular weights are observed which could correspond to the HMW subunits of glutenin and some components of gliadins.[Citation43] This can be explained by the low DH that would imply that less peptide bonds were cleaved during hydrolysis. Thus, larger peptides are found in the hydrolysates of 1, 2, and 4 h of hydrolysis.
However, in lanes 6, 7, and 8 corresponding to a higher time of hydrolysis, small molecular sized peptides (MW below 20 kDa) are obtained with a remarkable extinction of HMW proteins, which confirmed the obtained DH and the efficiency of the enzyme.
Moreover, the hydrolysis reaction can be used for gluten solubilization. In fact, in lane 2, corresponding to gluten supernatant at t = 0, we can observe that gluten is insoluble in the reaction buffer and that solubility increased with reaction time. The low solubility of gluten can be explained by the strong intermolecular interaction induced by hydrogen bonds and hydrophobic interactions, and the large size of the proteins. This property can limit its application in many food processes. Several studies demonstrated that gluten hydrolysis improved its solubility.[Citation40,Citation44] Such behavior could be explained by the fact that smaller, more hydrophilic, and more solvated polypeptide units were produced as a consequence of enzymatic hydrolysis. Furthermore, enzymatic reaction could have an impact in dissociation of possible formed aggregates. Thus, wheat gluten hydrolysates can be used in the food industry essentially as foaming and emulsifying improvers.
Conclusion
In the present study, a new protease designated S. not-protease was purified to homogeneity by a three step procedure from the intestine of small red scorpionfish. The enzyme was active at 40°C and pH 10.0 and its molecular weight was estimated to be 24 kDa. It was stable over a pH range of 8.0–11.0 and in the presence of different metal ions. Then, we have demonstrated that S. not-protease can be used in enzymatic hydrolysis of low-cost industrial proteins. Thus, this enzyme can be considered as promising tool in food processing and production of peptones. Moreover, proteolytic degradation of wheat gluten allows its solubilization and the production of a hydrolysate that can be a beneficial ingredient for functional foods against gluten intolerance.
Acknowledgments
This work was supported by the financial project of LIP-MB Laboratory, INSAT, Carthage University, Ministry of Higher Education and Scientific Research of Tunisia. The authors declare that they have no competing interests.
References
- Gupta, R.; Beg, Q. K.; Khan, S.; Chauhan, B. Bacterial Alkaline Proteases: Molecular Approaches and Industrial Applications. Applied Microbiology and Biotechnology 2002, 59, 15–32.
- Aissaoui, N.; Abidi, F.; Hardouin, J.; Abdelkafi, Z.; Marrakchi, N.; Jouenne, T.; Marzouki, M. N. Two Novel Peptides with Angiotensin I Converting Enzyme Inhibitory and Antioxidative Activities from Scorpaena Notata Muscle Protein Hydrolysate. Biotechnology and Applied Biochemistry 2016 10.1002/bab.1478.
- Simpson, B. K.;. Digestive Proteinases from Marine Animals. In Seafood Enzymes: Utilization and Influence on Postharvest Seafood Quality; Haard, N. F., Simpson, B. K. Eds.; Marcel Dekker: New York, 2000; 531–540.
- Shahidi, F.; Kamil, J. Y. V. A. Enzymes from Fish and Aquatic Invertebrates and Their Application in the Food Industry. Trends in Food Science and Technology 2001, 12, 435–464.
- FAO. The State of World Fisheries and Aquaculture 2012: Opportunities and Challenges; Food and Agricultural Organization of the United Nation: Rome, 2014; 4–7.
- Guerard, F.; Decourcelle, N.; Sabourin, C.; Floch-Laizet, C.; Le Grel, L.; Le Floch, P.; Gourlay, F.; Le Delezir, R.; Jaouen, P.; Bourseau, P. Recent Developments of Marine Ingredients for Food and Nutraceutical Applications. Journal Des Sciences Halieutique Et Aquatique 2010, 2, 21–27.
- Kishimura, H.; Klomklao, S.; Benjakul, S.; Chun, B. Characteristics of Trypsin from the Pyloric Ceca of Walleye Pollock (Theragra Chalcogramma). Food Chemistry 2008, 106, 194–199.
- De Vecchi, S.; Coppe, Z. L. Marine Fish Digestive Proteases in Food Industry and Its Relevance to the South-West Atlantic Region. Journal of Food Biochemistry 1996, 10, 193–214.
- Kishimura, H.; Hayashi, K.; Miyashita, Y.; Nonami, Y. Characteristics of Trypsins from the Viscera of True Sardine (Sardinops Melanostictus) and the Pyloric Caeca of Arabesque Greenling (Pleuroprammus Azonus). Food Chemistry 2006, 97, 65–70.
- Klomklao, S.; Benjakul, S.; Visessanguan, W.; Kishimura, H.; Simpson, B. K. A 29 kDa Protease from the Digestive Glands of Atlantic Bonito (Sarda Sarda): Recovery and Characterization. Journal of Agricultural and Food Chemistry 2007, 55, 4548–4553.
- Jellouli, K.; Bougatef, A.; Daassi, D.; Balti, R.; Barkia, A.; Nasri, M. New Alkaline Trypsin from the Intestine of Grey Triggerfish (Balistes Capriscus) with High Activity at Low Temperature: Purification and Characterization. Food Chemistry 2009, 116, 644–650.
- Barkia, A.; Bougatef, A.; Nasri, R.; Fetoui, E.; Balti, R.; Nasri, M. Trypsin from the Viscera of Bogue (Boops Boops): Isolation and Characterization. Fish Physiology and Biochemistry 2010, 36, 893–902.
- Ktari, N.; Ben Khaled, H.; Nasri, R.; Jellouli, K.; Ghorbel, S.; Nasri, M. Trypsin from Zebra Blenny (Salaria Basilisca) Viscera: Purification, Characterization and Potential Application as a Detergent Additive. Food Chemistry 2012, 130, 467–474.
- Sila, A.; Nasri, R.; Jridi, M.; Balti, R.; Nasri, M.; Bougatef, A. Characterization of Trypsin Purified from the Viscera of Tunisian Barbel (Barbus Callensis) and Its Application for Recovery of Carotenoproteins from Shrimp Wastes. Food Chemistry 2012, 132, 1287–1295.
- Lahl, W. J.; Braun, S. D. Enzymatic Production of Protein Hydrolysates for Food Use. Food Technology 1994, 48, 68–71.
- Gonzalez-Tello, P.; Camacho, F.; Jurado, E.; Paez, M. P.; Guadex, E. M. Enzymatic Hydrolysis of Whey Protein: II. Molecular- Weight Range. Biotechnology and Bioengineering. 1994, 44, 529–534.
- Segers, R.; Butt, T. M.; Kerry, B. R.; Peberdy, J. F. The Nematophagous Fungus Verticillium Chlamydosporium Produces a Chymoelastase-Like Protease Which Hydrolyses Host Nematode Proteins in Situ. Microbiology 1994, 140, 2715–2723.
- Laemmli, U. K.;. Cleavage of Structural Proteins during Assembly of Head of Bacteriophage T4. Nature 1970, 257, 680–685.
- Schmidt, T. M.; Bleakley, B.; Nealson, K. H. Characterization of an Extracellular Protease from the Insect Pathogen Xenorhabdus Luminescens. Applied and Environmental Microbiology 1988, 54, 2793–2797.
- Adler-Nissen, J.;. A Review of Food Hydrolysis Specific Areas. In Enzymic Hydrolysis of Food Proteins; Adler-Nissen, J. Ed.; Elsevier Applied Science Publishers: Copenhagen, 1986; 57–109.
- Bradford, M.;. A Rapid and Sensitive Method for the Quantification of Microgram Quantities of Protein Utilizing the Principle of Protein Dye Binding. Analytical Biochemistry 1976, 72, 248–254.
- Klomklao, S.; Benjakul, S.; Visessanguan, W.; Kishimura, H.; Simpson, B. K. Purification and Characterization of Trypsin from the Spleen of Tongol Tuna (Thunnus Tonggol). Journal of Agricultural and Food Chemistry 2006, 54, 5617–5622.
- Klomklao, S.; Kishimura, H.; Nonami, Y.; Benjakul, S. Biochemical Properties of Two Isoforms of Trypsin Purified from the Intestine of Skipjack Tuna (Katsuwonus Pelamis). Food Chemistry 2009, 115, 155–162.
- Espósito, T. S.; Marcuschi, M.; Amaral, I. P.; Carvalho, L. B.; Bezerra, R. S. Trypsin from the Processing Waste of the Lane Snapper (Lutjanus Synagris) and Its Compatibility with Oxidants, Surfactants and Commercial Detergents. Journal of Agricultural and Food Chemistry 2010, 58, 6433–6439.
- Bougatef, A.; Jellouli, K.; Balti, R.; Haddar, A.; Ellouz-Triki, Y.; Barkia, A.; Nasri, M. Low Molecular Weight Oxidant-Stable Protease from the Intestine of Smooth Hound (Mustelus Mustelus). Purification and Characterization. In Progress in Food Chemistry; Koeffer, E. N. Ed.; Nova Science Publishers: New York, 2008; 183–199.
- Ben Khaled, H.; Nasri, R.; Bougatef, A.; Ghorbel, S.; Nasri, M. Low Molecular Weight Serine Protease from the Viscera of Sardinelle (Sardinella Aurita) with Collagenolytic Activity: Purification and Characterization. Food Chemistry 2011, 124, 788–794.
- Park, P. J.; Lee, S. H.; Byun, H. G.; Kim, S. H.; Kim, S. K. Purification and Characterization of a Collagenase from the Mackerel, Scomber Japonicas. Journal of Biochemistry and Molecular Biology 2002, 35, 576–582.
- Adinarayana, K.; Ellaiah, P.; Prasad, D. S. Purification and Partial Characterization of Thermostable Serine Alkaline Protease from a Newly Isolated Bacillus Subtilis PE-11. AAPS PharmSciTech 2003, 4, 440–448.
- Chong, A. S. C.; Hashim, R.; Chow-Yang, L.; Ali, A. B. Partial Characterization and Activities of Proteases from the Digestive Tract of Discus Fish (Symphysodon Aequifasciata). Aquaculture 2002, 203, 321–333.
- Kishimura, H.; Tokuda, Y.; Yabe, M.; Klomklao, S.; Benjakul, S.; Ando, S. Trypsins from the Pyloric Ceca of Jacopever (Sebastes Schlegeli) and Elkhorn Sculpin (Alcichthys Alcicornis): Isolation and Characterization. Food Chemistry 2007, 100, 1490–1495.
- Elhadj-Ali, N.; Hmidet, N.; Bougatef, A.; Nasri, R.; Nasri, M. A Laundry Detergent Stable Alkaline Trypsin from Striped Seabream (Lithognathus Mormyrus) Viscera: Purification and Characterization. Journal of Agricultural and Food Chemistry 2009, 57, 10943–10950.
- Kanno, G.; Yamaguchi, T.; Kishimura, H.; Yamaha, E.; Saeki, H. Purification and Characteristics of Trypsin from Masu Salmon (Oncorhynchus Masou) Cultured in Fresh-Water. Fish Physiology and Biochemistry 2010, 36, 637–645.
- Klomklao, S.; Benjakul, S.; Visessanguan, W.; Kishimura, H.; Simpson, B. K.; Saeki, H. Trypsins from Yellowfin Tuna (Thunnus Albacores) Spleen: Purification and Characterization. Comparative Biochemistry and Physiology 2006, 144, 47–56.
- Wang, Q.; Gao, Z. X.; Zhang, N.; Shi, Y.; Xie, X. L.; Chen, Q. X. Purification and Characterization of Trypsin from the Intestine of Hybrid Tilapia (Oreochromis Niloticus × O. Aureus). Journal of Agricultural and Food Chemistry. 2010, 58, 655–659.
- Silva, J. F.; Esposito, S. T.; Marcuschi, M.; Ribeiro, K.; Cavalli, R. O.; Oliveira, V.; Bezerra, R. S. Purification and Partial Characterisation of a Trypsin from the Processing Waste of the Silver Mojarra (Diapterus Rhombeus). Food Chemistry 2011, 129, 777–782.
- Bougatef, A.;. Trypsins from Fish Processing Waste: Characteristics and Biotechnological Applications- Comprehensive Review. Journal of Cleaner Production 2013, 57, 257–265.
- Rigo, A.; Corazza, A.; Di Paolo, M. L.; Rossetto, M.; Ugolini, R.; Scarpa, M. Interaction of Copper with Cysteine: Stability of Cuprous Complexes and Catalytic Role of Cupric Ions in Anaerobic Thiol Oxidation. Journal of Inorganic Biochemistry 2004, 98, 1495–1501.
- Bougatef, A.; Balti, R.; Nasri, R.; Jellouli, K.; Souissi, N.; Nasri, M. Biochemical Properties of Anionic Trypsin Acting at High Concentration of NaCl Purified from the Intestine of a Carnivorous Fish: Smooth Hound (Mustelus Mustelus). Journal of Agricultural and Food Chemistry 2010, 58, 5763–5769.
- Misook, K.; Yoonhwa, J. Purification and Characterization of a Trypsin-Like Protease from Flatfish (Paralichthys Olivaceus) Intestine. Journal of Food Biochemistry 2013, 37, 732–741.
- Kong, X.; Zhou, H.; Qian, H. Enzymatic Hydrolysis of Wheat Gluten by Proteases and Properties of the Resulting Hydrolysates. Food Chemistry 2007, 102, 759–763.
- Berends, P.; Appel, D.; Eisele, T.; Rabe, S.; Fischer, L. Performance of Enzymatic Wheat Gluten Hydrolysis in Batch and Continuous Processes Using Flavourzyme. LWT - Food Science and Technology 2014, 58, 534–540.
- Hardt, N. A.; Boom, R. M.; van der Goot, A. J. Starch Facilitates Enzymatic Wheat Gluten Hydrolysis. LWT - Food Science and Technology 2015, 61, 557–563.
- Shewry, P. R.; Tatham, A. S. Biotechnology of Wheat Quality. Journal of the Science of Food and Agriculture 1997, 73, 397–406.
- Linarès, E.; Larré, C.; Lemeste, M.; Popineau, Y. Emulsifying and Foaming Properties of Gluten Hydrolysates with an Increasing Degree of Hydrolysis: Role of Soluble and Insoluble Fractions. Cereal Chemistry 2000, 77, 414–420.