
ABSTRACT
We identified physicochemical, volatile compound, and microbial community differences between three grades of roasted-sesame-like flavored Daqu. The Daqu grades had different physicochemical characteristics. The concentrations and proportions of various esters reflected sensory evaluations of the Daqu classes but no rule linking the flavor compound types and concentrations to Daqu grade was identified. The prokaryotic microorganism communities were similar in premium- and first-grade Daqu but very different in general-grade Daqu. The eukaryotic microorganism communities in premium- and general-grade Daqu were similar, but the genus abundances were more similar in the premium- and first-grade Daqu samples than in premium- and general-grade Daqu.
Introduction
Baijiu, one of the six traditional distilled beverages around the world, is produced through spontaneous mixed-culture solid-state fermentation by a saccharifying and fermenting agent called Daqu.[Citation1] Daqu, which is a natural fermentation starter made from cereals, plays a key role in determining the final flavor characteristics of the Baijiu.[Citation2] Daqu provides some of the brewing material, as well as being a source of microorganisms and crude enzymes, so, Baijiu produced without Daqu will not have the characteristic aroma and flavor.[Citation3] The contribution of the Daqu used to the Baijiu produced is well known and sometimes described using the phrases: “Daqu is the bone or heart of Baijiu” and “better Daqu, better Baijiu”.[Citation4] Daqu contributes 61%–80% of the fungi present during Baijiu fermentation and is closely related to the yield and flavor of fresh Baijiu.[Citation5] Most manufacturers of high-quality Baijiu therefore pay close attention to the quality of the Daqu used.
Daqu is made from wheat, barley, and/or peas using a traditional solid-state fermentation (SSF) process that involves ingredient formulation, grinding, mixing, shaping, incubation, fermentation, and aging (drying and ripening).[Citation6] Daqu is produced in an open environment and involves natural inoculation and spontaneous fermentation, with no sterilization procedures.[Citation7,Citation8] Most of the microbes are supplied from the environment, i.e., the workplace, the air, and the feedstock.[Citation9] The composition of the microbial community in the Daqu will change during fermentation, and many metabolites, including enzymes able to degrade substrates and produce flavor compounds, will be produced.[Citation2,Citation9] Some metabolites formed during Daqu production may be further converted into flavor components that remain in the finished Baijiu.[Citation2] Daqu therefore not only starts the brewing process but also provides important crude enzymes, complex microbial flora, and aroma precursors that determine the flavor of the final product.[Citation10]
Daqu is prepared under semi-controlled conditions, so the Daqu quality will vary.[Citation7] Daqu is graded by the producers to decrease variability in the Baijiu production process caused by variability in Daqu quality.[Citation4] To some degree this makes the Baijiu brewing process stable. However, no scientific grading standards for Daqu have been developed, so the grading method is very subjective and does not fully achieve the intended goal of a stable Baijiu brewing process. Daqu grading is currently based mainly on sensory evaluations (appearance, fractured surface, and aroma) by experienced producers.[Citation4] Some companies also grade Daqu using physicochemical indicators such as acidity, esterification activity, fermentation activity, liquefication activity, pH, and saccharification activity.[Citation11] However, most of the Daqu grading process is subjective, so some uncertainty in the grading of Daqu is inevitable. This causes different Baijiu batches to have different qualities. Many companies use mixtures of Daqu of different grades when producing Baijiu to suit the actual fermentation conditions.[Citation12] The root problem is a lack of a scientific basis for grading Daqu because our understanding of the overall characteristics of Daqu is limited.
In this study, we comprehensively assessed the differences between roasted-sesame-like flavored (RSF) Daqu samples that had been graded by performing sensory evaluations. The traits that were analyzed were the flavor compound concentrations, microbial community structure, physicochemical properties, and specific effects during the simulated SSF of Baijiu. The results will be useful for establishing a scientific method for grading Daqu.
Materials and methods
Sampling
Daqu of three grades, premium grade (sample A), first grade (sample B), and general grade (sample C), were produced from RSF liquor at Shandong Bandaojing (Shandong, China). Experienced workers determined the Daqu grades from the acidity, appearance, aroma, and fractured surface color. Daqu bricks were produced through SSF of wheat grains in fall 2017. Each sample was a mixture of randomly selected aliquots from the upper, middle, lower, and interior of the relevant Daqu pile. Each sample was ground to a powder, mixed well, and then subsampled using the quartering method (Supplementary Figure S1).[Citation13] Each sample was placed in a sterile bag, sealed, and stored at −80°C for analysis.
Simulated SSF
Simulated SSF was performed following a method described by Fan et al., and the aroma-producing characteristics of the different grades of Daqu were assessed.[Citation14] Oven-dried sorghum and rice husks, were used to ferment the Baijiu. The dried sorghum grains were ground to break each grain into four or five pieces. A 20 g aliquot of rice husks and 80 g of cracked sorghum were mixed, soaked in water at 80°C for 24 h, and then steamed for 1.5 h to completely destroy the crystalline starch structures in the sorghum. The mixture was cooled and placed in a 1.5 L ceramic jar with 10 g of crushed Daqu, and water was added to bring the moisture content to 50%. The jar was covered and sealed with water, and the mixture in the jar was allowed to ferment at room temperature (Supplementary Figure S2). Three jars were prepared using each Daqu grade. The mixtures were allowed to ferment under stable conditions for 30 d. Three control jars were also prepared using crushed wheat (the raw material used to make the Daqu). Flavor compounds were detected by headspace solid-phase microextraction gas chromatography mass spectrometry (HS-SPME-GC-MS).
Physicochemical properties
The physicochemical properties of each Daqu sample were determined in triplicate using standard methods.[Citation15] The physicochemical properties that were determined were the moisture, protein, and starch contents, the acidity, the pH, and the esterification, fermentation, liquefication, and saccharification activities. The data were analyzed by performing principal component analysis (PCA) using SPSS 16.0 software (SPSS, Chicago, IL, USA).
Volatile compound analysis
Volatile compounds in the Daqu and SSF samples were detected by HS-SPME-GC-MS using a Trace GC TSQ 8000 Evo instrument (Thermo Fisher Scientific, Waltham, MA, USA), using a previously published method.[Citation16] Individual volatile compounds were identified by comparing the acquired spectrum with mass spectral data in the NIST 05a library (Thermo Fisher Scientific) and quantified using the response for the internal standard 2-octanol, (Sigma-Aldrich, St. Louis, MO, USA). Each volatile compound concentration is expressed in milligrams per kilogram of Daqu and is the mean concentration found for three analyses.
DNA extraction and quantification
Genomic DNA from the Daqu samples was extracted using PowerSoil DNA Isolation kits (Mo-Bio, Carlsbad, CA, USA) and prepared using a method described by Zhang et al.[Citation17] The DNA was quantified by spectrophotometry (using the ratio of the optical densities at 260 and 280 nm), then the concentration was adjusted to the required concentration before the DNA was used as a template for polymerase chain reaction (PCR) amplification.
PCR amplification and illumina MiSeq sequencing
The bacterial communities were analyzed using an Illumina HiSeq system to sequence 16S rRNA gene V3–V4 region amplicons with the forward primers 340F (5ʹ-barcode-CCTACGGGNBGCASCAG-3ʹ) and reverse primers 805R (5ʹ-GACTACNVGGGTATCTAATCC-3ʹ). The 18S ribosomal RNA fungi gene was amplified using the primer set 512F (5ʹ-TATTCCAGCTCCAATAGCG-3ʹ)/978R (5ʹ-barcode-GACTACGATGGTATCTAATC-3ʹ). The primers contained an error-correcting barcode to distinguish between samples. The PCR reactions and amplicon extraction were performed following a previously published method.[Citation16] The amplicons were quantified and purified, then they were pooled in equimolar quantities and subjected to paired-end sequencing (2 × 250 bp) using the Illumina HiSeq2500 platform (Illumina, San Diego, CA, USA) following instructions provided by the supplier. The raw sequencing data were submitted to the GenBank Sequence Read Archive database (PRJNA494030).
Processing sequencing data
The raw sequences were processed in accordance with the QIIME (version 1.8) quality control process. Reads that could not be assembled were removed, and chimeric sequences were discarded using UCHIME. Operational taxonomic units (OTUs) were clustered using UPARSE version 7.1 (http://drive5.com/uparse/) using a 97% identity threshold. The taxonomic classification of each 16S rRNA/18S rRNA gene sequence at a confidence level of 70% was determined using the Ribosomal Database Project Classifier (http://rdp.cme.msu.edu/) and the Silva (SSU115) 16S rRNA and 18S rRNA database.
Alpha diversity indices, including Good’s coverage, Chao 1, ACE, and Shannon and Simpson indices, were calculated using the QIIME suite of programs. Beta diversity was assessed using the UniFrac method. The similarities of different Daqu samples were evaluated by performing UniFrac analyses. Principal coordinate analysis was achieved by performing weighted and unweighted calculations. Correlations between community structures and environmental variables were assessed by performing redundancy analysis (RDA) and canonical correspondence analysis (CCA) using Vegan software, and a heatmap was constructed using the pheatmap package in R software (http://vegan.r-forge.r-project.org/).
Statistical analysis
Each treatment was performed in triplicate. All the statistical analyses were performed using SPSS 16.0 software.
Results and discussion
Differences in the sensory characteristics of different Daqu grades must be caused by differences in the production process parameters. The most important role of Daqu when brewing Baijiu is to enrich the microbial community. Differences in the Daqu microbial community structures in Daqu of different grades will cause different physicochemical indices and flavors, and be manifest as differences in sensory characteristics. The differences between Daqu of different grades were studied here to provide a scientific basis for classifying Daqu.
Physicochemical properties
As shown in , most of the physicochemical properties were different for the different Daqu samples. The moisture contents of the different Daqu grades were different, but they all met the moisture requirement of <13% for Daqu.[Citation18] PCA indicated that the moisture content played an important role in the classification of Daqu (Supplementary Figure S3). Acidity is one of the most important quality indices for Daqu and is also an important index for determining the Daqu grade.[Citation4] According to the acidity specification in the Daqu quality indicator system, samples A and B were superior-grade Daqu and sample C was general-grade Daqu.[Citation4] The Daqu classification system used by Shandong Bandaojing is mainly based on sensory characteristics and acidity. Samples A, B, and C met the Daqu standard in terms of acidity, and this was confirmed by the PCA results (Supplementary Figure S3). The starch content of Daqu will reflect the amount of starch consumed by the microorganisms when the Daqu is prepared. Higher starch consumption rates indicate more microorganism reproduction and metabolism has occurred, meaning the metabolite content of the Daqu will be higher.[Citation19] It can be seen from that the starch content of sample C was lower than the starch contents of samples A and B, indicating that the microorganisms in sample C were better able than the microorganisms in samples A and B to decompose starch. This was consistent with the liquefication and saccharification activities, and reflected the ability of the microorganisms or enzymes in the Daqu to hydrolyze starch to give sugar.[Citation20] This may explain the presence of “fire circles” on a cross-section and the rough surface and uneven coloration of sample C. These characteristics are generally regarded as indicating low-grade Daqu. Sample C also had a higher esterification activity than samples A and B. The esterification activity is closely related to the synthesis of esters, which are important flavors of Baijiu. Fermentation activity, which reflects the wine production capacity of Daqu, was higher for sample A than for samples B and C. In summary, there was no effective relationship between Daqu grade determined from sensory and acidity indices and the biochemical activity (premium- and first-grade Daqu do not necessarily have higher biochemical activities than ordinary-grade Daqu), but different biochemical activities probably caused the different appearances of the Daqu samples. The PCA results indicated that the physicochemical indices played important roles in Daqu classification (Supplementary Figure S3). Variations in the biochemical activities of Daqu cause a poor appearance. For example, Daqu with high liquefication and saccharification activities easily undergoes the Maillard reaction at a high temperature, and this causes “fire circles” to form.[Citation21] We concluded that classification using the method described above will not reflect the composition and viability of the microorganisms and enzymes in Daqu but that the classification can be seen as an evaluation of the Daqu appearance (i.e., the uniformity of the Daqu and whether fire circles are present).
Table 1. Changes in physicochemical properties of RSF Daqu during aging
Volatile compounds
A total of 38 volatile compounds (five alcohols, six aldehydes, six alkanes, eight benzodiazepines, seven esters, four ketones, one nitrogen-containing compound, and one phenol) in the Daqu samples were identified by HS-SPME-GC-MS (Supplementary Table S1). There were no significant differences in the types of flavor compounds present in the three samples. Samples A, B, and C contained 33, 29, and 33 volatile flavor compounds, respectively, and 27 of these flavor compounds were found in all three samples. The main differences in the flavor compound contents of the three samples were that the ester content was higher for the premium-grade Daqu than the other samples; the aldehyde, alkane, ketone, and nitrogen-containing compound contents were higher for the first-grade Daqu than the other samples; and the alcohol, benzodiazepine, and phenol contents were higher for the ordinary-grade Daqu than for the other samples. Esters (which have fruity aromas) are the most important compounds in Baijiu and are important indicators of Baijiu quality.[Citation22] Esters also strongly affect the Daqu grade. The grades of the Daqu samples we analyzed were strongly related to the ester contents. Phenethyl alcohol contributes a rose-like scent, and the phenethyl alcohol content is an essential contributor to the aroma of RSF Baijiu. Tetramethylpyrazine has a nutty aroma with roast and barbecue elements, and was detected in all the samples.[Citation23]
A total of 75 flavor compounds (one acid, seven alcohols, two aldehydes, six alkanes, five benzodiazepines, 45 esters, one furan, three ketones, and five phenols) were detected in the SSF samples (Supplementary Table S2). SSF produced from the premium-grade, first-grade, and ordinary-grade Daqu contained 61, 55, and 62 flavor compounds, respectively, and 44 of these flavor compounds were found in all the SSF samples. The flavor compound contents were higher for the SSF samples produced from first-grade Daqu than for the SSF samples produced from the other Daqu grades. However, the acid and ketone contents were higher for the SSF samples produced from ordinary-grade Daqu than for the SSF samples produced from the other Daqu grades and the benzodiazepine contents were higher for the SSF samples produced from premium-grade Daqu than for the SSF samples produced from the other Daqu grades. The flavor compound contents of the SSF samples produced from Daqu with high biochemical activities were not high. This may have been related to the compositions and proportions of microorganisms and enzymes in the Daqu.[Citation24] Baijiu production is a long and dynamic process, so it is not possible to predict the Baijiu quality from the biochemical activities of the Daqu used. Indeed, the microbial structure of the Daqu and dynamic changes in the microorganisms during brewing are probably more important indicators of Baijiu quality.[Citation7] There were almost twice as many flavor compounds in the SSF samples as in the Daqu samples. The difference was mainly in the number of esters and, to a lesser extent, alcohols. The contents, of most of the main flavor compounds were higher in the SSF samples than the Daqu samples. Many flavor compounds in the SSF samples originated in the Daqu, but others were derivatives of the flavor compounds in the Daqu. This confirms that Daqu is not only a source of microbes, enzymes, and material during the brewing of Baijiu but is also an important source of flavors or flavor precursors to Baijiu.[Citation3] The flavor components in the control samples were also found in the SSF samples produced from Daqu, and the contents of these components were higher in the SSF samples than the control samples, indicating that the raw material also provided flavors to the Baijiu.
Prokaryote community structure and diversity
Prokaryote microbe diversity in the Daqu samples was determined by performing sequencing using the Illumina HiSeq system. Raw tag counts of 207257, 185973, and 84091 were found for samples A, B, and C, respectively. After filtering out low-quality reads and detecting chimera, samples A, B, and C had 141321, 126747, and 57154 effective tags, respectively, with average read lengths of 453, 452, and 455 bp, respectively (). The sample A, B, and C OTU numbers (determined using a 97% identity 16S rRNA sequence cutoff) were 2626, 2485, and 1338, respectively. As shown in , Chao1 and the Shannon diversity index indicated that the prokaryotic microorganism diversity decreased in the order premium-grade Daqu > first-grade Daqu > ordinary-grade Daqu. Rarefaction analysis indicated that adequate sampling and sequencing efforts had been performed, and rarefaction curve analysis indicated a clear-cut asymptotic plateau (Supplementary Figure S4a). The relative abundances of the OTUs and taxonomic ranks from the genus to OTU level indicated that the prokaryote communities in the Daqu samples formed two clusters, (I) samples A and B and (II) sample C (). UniFrac clustered analysis and principal coordinate analysis also indicated that the prokaryote communities formed two groups (Supplementary Figures S5 and S6). These results indicated that the Daqu samples became more different as the Daqu grade decreased.
Table 2. Summary of sequencing results and the alpha diversity statistical analysis of RSF Daqu samples
Figure 1. Taxonomic classification of sequences from prokaryotic communities of three samples (A, premium-grade Daqu; B, first-grade Daqu; C, general-grade Daqu) at genus level. The relative abundance was calculated by dividing the number of classified tags by the total tags number of each sample. The top 50 genera detected in RSF Daqu are shown. The relative abundance of each genus was indicated by color intensity in a heat map
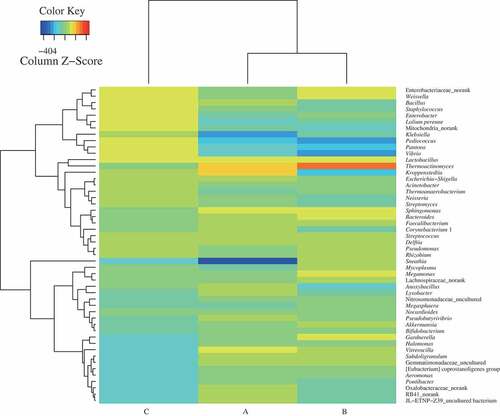
The taxonomic distributions at the phylum level for the samples are shown in . A total of 39 prokaryotic phyla were found in the three RSF Daqu samples. There were 33, 33, and 22 prokaryotic phyla in samples A, B, and C, respectively. These Daqu samples were richer in bacterial species than Daqu samples previously analyzed.[Citation25] As shown in , the compositions and proportions of prokaryotic microbes were similar at the phylum level in samples A and B, but prokaryotic microbes were much less diverse in sample C. Firmicutes and Proteobacteria were the main prokaryotic phyla in all three samples, and contributed 73.1%, 69.8%, and 87.1% of the total abundances of bacteria in samples A, B, and C, respectively. Similar results were found in previous studies.[Citation2] Firmicutes was the most abundant phylum in all the Daqu samples. The Firmicutes abundance was similar in each sample, contributing 47.7%, 46.5%, and 44.1% of the total abundances of bacteria in samples A, B, and C, respectively. Proteobacteria was the next most abundant phylum. Proteobacteria contributed 25.3%, 23.3%, and 42.9% of the prokaryotic sequences in samples A, B, and C, respectively. Proteobacteria were more abundant in sample C than in samples A and B. The Acidobacteria, Actinobacteria, Bacteroidetes, Chloroflexi, Gemmatimonadetes, Nitrospirae, Planctomycetes, and Verrucomicrobia contributions to the prokaryotic sequences were similar for the premium- and first-grade Daqu samples and markedly higher than the contributions for the ordinary-grade Daqu. Cyanobacteria contributed 10.7% of the prokaryotic sequences for the ordinary-grade Daqu, markedly more than the contributions for the other samples. The Deinococcus-Thermus and Parcubacteria contributions to the prokaryotic sequences were markedly higher but the Fusobacteria and Tenericutes contributions to the prokaryotic sequences were lower for the premium-grade Daqu than for the first-grade Daqu. Overall, the prokaryotic microbes were more diverse in the premium- and first-grade Daqu samples than the ordinary-grade Daqu, and the contributions of many species to the total prokaryotic sequences in the premium- and first-grade Daqu samples were similar.
Figure 2. Analysis of prokaryotic community composition in three samples (A, premium-grade Daqu; B, first-grade Daqu; C, general-grade Daqu) using high-throughput sequencing. Results of taxonomy at the phylum level and genus level are shown in (a) and (b), respectively. The relative abundance defines sequence percentages in samples as depicted by the colors in the bar chart. The top 20 phyla and genera detected in RSF Daqu are shown in (a) and (b), respectively. The rest of phyla and genera detected in all samples were classified as others
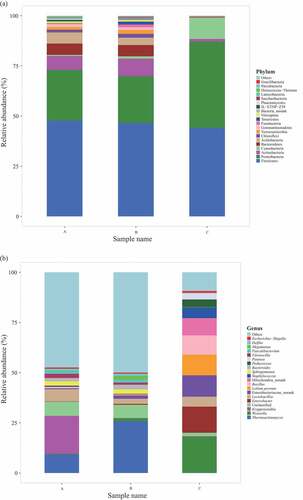
A total of 743 abundant genera were found in the three Daqu samples, 543 in sample A, 508 in sample B, and 246 in sample C. Venn analysis indicated that 322 genera were found in both samples A and B, 173 genera were found in both samples A and C, 180 genera were found in both samples B and C, and 163 genera were found in all three samples. In general, the bacterial structures in the different Daqu grades were quite different at the genus level (). The dominant genera with relative abundances >1% in sample A were Kroppenstedtia (which contributed 18.9% of the genera), Thermoactinomyces (9.3%), unclassified (7.1%), Lactobacillus (6.1%), Sphingomonas (2.2%), Vitreoscilla (2.1%), Bacteroides (1.6%), Faecalibacterium (1.3%), and Oxalobacteraceae_norank (1.1%). The dominant genera in sample B were Thermoactinomyces (25.8%), unclassified (6.6%), Lactobacillus (2.5%), Bacteroides (2.3%), Sphingomonas (2.2%), Megamonas (1.9%), Enterobacteriaceae_norank (1.6%), Gardnerella (1.6%), Weissella (1.6%), Faecalibacterium (1.5%), Delftia (1.1%), Mycoplasma (1.1%), Vitreoscilla (1.0%), and Lachnospiraceae_norank (1.0%). The dominant genera in sample C were Weissella (18.0%), Enterobacter (13.0%), Enterobacteriaceae_norank (10.7%), Lolium perenne (10.3%), Bacillus (9.6%), Mitochondria_norank (8.6%), Staphylococcus (5.4%), Lactobacillus (4.9%), Pediococcus (3.8%), Pantoea (3.1%), unclassified (1.7%), and Vibrio (1.5%) (). The predominant genera in samples A and B were relatively similar, and the predominant genera in sample C were somewhat different from the predominant genera in samples A and B. Samples A and B contained large numbers of bacterial species and large proportions of low-abundance (<1.0%) bacterial flora, which contributed 50.4% and 48.3% of the bacteria in samples A and B, respectively. Low-abundance bacterial flora contributed only 10.2% of the bacteria in sample C. These results indicated that better quality Daqu contains more diverse microorganisms. The low acidities of samples A and B were probably caused by the prevalence of Kroppenstedtia in sample A and Thermoactinomyces in sample B. In previous studies of correlations between Thermoactinomyces distributions in Daqu and the physicochemical properties of Daqu it was found that acidity negatively correlated with the amount of Thermoactinomyces in the Daqu, which agreed with our conclusion.[Citation26] Kroppenstedtia and Thermoactinomyces were probably the dominant prokaryotic microorganisms in the premium- and first-grade Daqu, respectively. In contrast, the high acidity of sample C was probably caused by the dominance of Enterobacter and Weissella, which were typical prokaryotic microorganisms in general-grade Daqu. The highly acidic environment of sample C may also explain the lower microbial diversity in that sample.[Citation6,Citation27]
Eukaryotic community structure and diversity
A total of 314542 high-quality reads with a mean read length of 440 bp corresponding to the 18S rDNA gene sequences were found after low quality and chimera reads had been removed and the PCR primers had been trimmed (). The rarefaction curves for all the samples leveled off strongly, indicating that eukaryotic communities were captured well by the analysis (Supplementary Figure S4b). The eukaryotic microbes were less diverse than the prokaryotic communities. The valid sequences (determined using a 97% similarity value) in samples A, B, and C had 97, 98, and 104 OTUs, respectively. The microbial richness (Chao1 and ACE) of the eukaryotic community was higher for sample C than sample B and lowest for sample A. The microbial diversity (Shannon and Simpson indices) was significantly higher for sample A than for samples B and C (). UniFrac cluster analysis and principal coordinate analysis indicated that sample B formed one cluster, and samples A and C formed another cluster (Supplementary Figures S7 and S8).
Seven eukaryotic phyla (Alveolata, Ascomycota, Basidiomycota, Euglenozoa Ochrophyta, unclassified, and Zygomycota) were found in the Daqu samples. Ascomycota and Zygomycota were the dominant eukaryotic communities in all the Daqu samples, which agreed with the results of previous studies.[Citation8,Citation25,Citation28] These two phyla contributed >90.0% of the total abundances in the Daqu samples (), but the separate contributions of Ascomycota and Zygomycota were related to the Daqu grade. Zygomycota contributed more in the lower-grade Daqu than in the higher-grade Daqu (85.5% in sample A, 92.5% in sample B, and 97.6% in sample C), and Ascomycota contributed more in the higher-grade Daqu than in the lower-grade Daqu (8.0% in sample A, 4.9% in sample B, and 1.9% in sample C). The main components of Zygomycota are Absidia, Lichtheimia, Rhizomucor, Rhizopus, Thermomucor, and other genera. These are the main producers of amylase and are likely to be responsible for the high saccharification and liquefication activities of Daqu.[Citation5,Citation6,Citation20,Citation29] Ascomycota, which includes Hanseniaspora, Metschnikowia, Saccharomyces cerevisiae, and Saccharomycopsidaceae, was the most abundant group of yeasts and the most efficient producer of ethanol during Baijiu fermentation. Baijiu produced by these yeasts typically has higher “harmony” scores, and higher floral aroma scores than Baijiu produced by other yeasts.[Citation30,Citation31] This explained to some extent the high liquefication and saccharification activities of sample C, and the high fermentation activity of sample A. A total of 26 genera were detected in the Daqu samples. There were 21 genera in sample A, 23 in sample B, and 22 in sample C (). Samples A and B had 20 genera in common, samples A and C 19, and samples B and C 20. A total of 19 genera were found in all three samples (determined by Venn analysis). Seven abundant genera (>1% of the total genera) were detected in sample A. These were Absidia (43.1%), unclassified (39.2%), Trichocomaceae_norank (5.7%), Gregarina (5.1%), Thermomucor (2.4%), Saccharomycetaceae_norank (1.3%), and Rhizomucor (1.1%). Six abundant genera were found in sample B. These were Absidia (52.3%), unclassified (36.5%), Trichocomaceae_norank (2.8%), Thermomucor (2.6%), Gregarina (1.5%), and Saccharomycopsidaceae (1.2%). Four abundant genera in two phyla were found in sample C. These were Absidia (50.8%), Rhizopus (2.9%), and Thermomucor (2.7%) in the Zygomycota phylum and one unclassified genus (39.8%). The dominant eukaryotic microorganism genera in the premium- and first-grade Daqu samples were similar, as was the case for prokaryotic microorganisms.
Figure 3. Analysis of eukaryotic community composition in three samples (A, premium-grade Daqu; B, first-grade Daqu; C, general-grade Daqu) using high-throughput sequencing. Results of taxonomy at the phylum level and genus level were shown in (a) and (b), respectively. The relative abundance defines sequence percentages in samples as depicted by colors in the bar chart. The top 20 phyla and genera detected in RSF Daqu are shown in (a) and (b), respectively. The rest of phyla and genera detected in all samples were classified as others
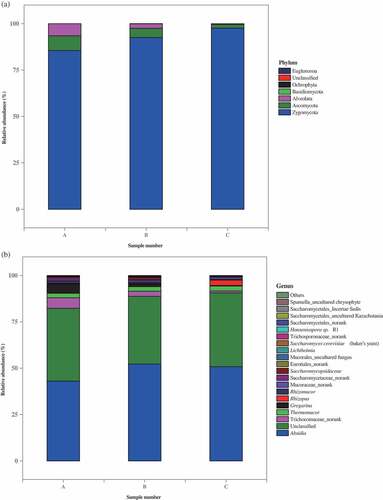
Absidia, Thermomucor, and an unclassified genus were the dominant genera in all the Daqu samples. Absidia, the most frequently found and important fungi in the Daqu, will produce large amounts of saccharifying hydrolases that transform starch into sugars. Absidia are important contributors to Baijiu fermentation.[Citation20,Citation30,Citation32] Interestingly, Thermomucor, which has been found in Daqu in only a few studies, can secrete large amounts of various hydrolyzing enzymes, including protease, glucosidase, glucoamylase, lipase, and pectinase, which can decompose macromolecules.[Citation28,Citation33–Citation39] Thermomucor is therefore expected to be an important contributor to the saccharification and liquefication activities and to cause small metabolites and volatile compounds to be produced during Baijiu production.
Relationships between physicochemical properties and microbial communities in the Daqu
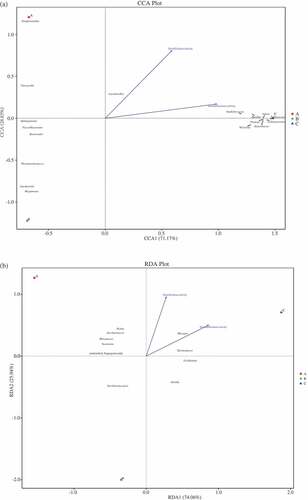
Correlations between microbial communities were identified by performing CCA and RDA (CCA for prokaryotic communities, Detrended correspondence analysis (DCA) = 3.74; RDA for eukaryotic communities, DCA = 0.42) and physicochemical properties (). It can be seen from that Kroppenstedtia, Lactobacillus, and Vitreoscilla in sample A positively correlated, which agreed with the results presented above (). In these genera, Lactobacillus positively correlated with the esterification and saccharification activities (). Lactobacillus has previously been found to produce a rich enzyme system including amylase, glucosidase, and proteolytic activity and to produce lactic acid from glucose or starch to provide a substrate for esterification by yeasts.[Citation6,Citation27] Correlation analysis using the two-way orthogonal partial-least-squares method indicated that Lactobacillus positively correlated with the ester content, particularly the ethyl acetate content.[Citation7] Positive correlations were also found between Bacteroides, Faecalibacterium, Gardnerella, Megamonas, Sphingomonas, and Thermoactinomyces in sample B. The CCA results also indicated that Bacillus, Enterobacter, Lolium perenne Pantoea, Pediococcus, Staphylococcus, Vibrio, and Weissella positively correlated in sample C. These genera contributed to the high esterification activity and particularly the high saccharification activity in sample C, possibly because they could produce amylase, lipase, and other enzymes ().[Citation20] For example, Bacillus, Enterobacter, and Staphylococcus produce lipase.[Citation40] Pediococcus and Weissella are lactic acid bacteria and produce not only lactic acid as a major end-product of carbohydrate fermentation but also provide substrates for esterification by yeasts.[Citation27] The RDA results for the eukaryotes () indicated that there were significant positive correlations between Pichia, Rasamsonia, Rhizomucor, Saccharomyces, and unidentified Eugregarinorida in sample A and between Absidia, Lichtheimia, Rhizopus, and Thermomucor in sample C. The RDA results indicated that Lichtheimia, Rhizopus, and Thermomucor, positively correlated with the saccharification activity and Pichia, Rhizopus, Saccharomyces, and Thermomucor positively correlated with the esterification activity. This confirmed that these organisms were the main producers of amylase, esterase, or glucosidase. Rhizopus is a functional microbe that produces amylase, esterase, glucoamylase, lipase, and protease.[Citation41–Citation44] As mentioned earlier, Thermomucor would have been one of the main contributors of the saccharification and esterification activities because it can produce glucoamylase, glucosidase, lipase, pectinase, protease, and other enzymes.[Citation34,Citation35,Citation38,Citation39] It has been found in some studies that Lichtheimia is strongly saccharogenic and causes starch hydrolysis.[Citation45] It has also been found that these biota can produce esterase or lipase, which can give high esterification activities.[Citation46–Citation49] Pichia are aroma-producing yeasts (also called ester-producing yeasts), and mainly cause esterification and enhance the ester fragrances of liquors.[Citation50] Saccharomyces are the main producers of ethanol in Baijiu, but also produce important flavor compounds such as esters and higher alcohols.[Citation31] This may explain the high fermentation activity of sample A.
Figure 4. Canonical correspondence analysis (CCA) of prokaryotic communities at genus level and physicochemical properties (a) (DCA = 4.10), and redundancy discriminate analysis (RDA) of eukaryotic communities at genus level and physicochemical properties (b) (DCA = 1.15) found in Daqu. Arrows represent different physicochemical properties; the symbols ■, ●, and▲, represent samples A (premium-grade Daqu), B (first-grade Daqu), and C (general-grade Daqu), respectively
Correlations between microbial communities and flavor compound concentrations
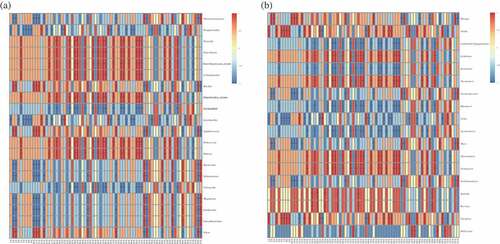
Correlations between the microbial communities and flavor compound concentrations in the SSF samples (Supplementary Table S2) were identified from the heatmap shown in . Significant correlations were found between the microbial communities and flavor compound concentrations through clustering (). Correlation analysis, indicated that the flavor compounds could be divided into eight clusters (Supplementary Table S3). As shown in , Absidia and Clavispora positively correlated with cluster I and negatively correlated with cluster IV. In contrast, Kroppenstedtia, Lactobacillus, and Pichia negatively correlated with cluster I and positively correlated with cluster IV. The heatmap also indicated strong positive associations between Bacillus, Rhizopus, Staphylococcus, Thermoactinomyces, Vibrio, and cluster II and negative associations between Bacillus, Rhizopus, Staphylococcus, Thermoactinomyces, Vibrio, and cluster VII. Strong positive associations were found between Bacteroides, Gardnerella, Faecalibacterium, Megamonas, Mucor, Saccharomycopsis, Sphingomonas, Wickerhamomyces, and cluster II, and negative associations were found between Bacteroides, Gardnerella, Faecalibacterium, Megamonas, Mucor, Saccharomycopsis, Sphingomonas, Wickerhamomyces, and cluster VII. More genera were associated with clusters III and VIII. Enterobacter, Enterobacteriaceae_norank, Hanseniaspora, Lichtheimia, Lolium perenne, Mitochondria_norank, Pantoea, Pediococcus, Thermomucor, Trichosporon and Weissella positively correlated with cluster III and were negatively associated with cluster VIII. Rasamsonia, Rhizomucor, Saccharomyces, unclassified, unidentified Eugregarinorida, and Vitreoscilla, were negatively associated with cluster III and positively correlated with cluster VIII. Boeremia and Spumella were associated with cluster V, and Millerozyma positively correlated with cluster VI. The abundances of these genera in the Daqu of different grades and the relative concentrations of the flavor compounds in the SSFs indicated that these genera were all mutual inhibited during fermentation. An alternative explanation is that the products were substrates of the other genera, so may have been converted into other flavor compounds or partly degraded.[Citation51] Relationships between some genera and flavors (e.g., Bacillus with 1-octen-3-ol, Pichia with phenylethyl alcohol, and Saccharomycopsis with 2-methoxy-4-vinylphenol) have been found in previous studies.[Citation10,Citation52,Citation53] However, some relationships (e.g., between Lichtheimia and octanoic acid ethyl ester) have not previously been described, and further work will be required to verify these relationships. Some relationships may have been caused by interactions between microorganisms rather than by the actions of an individual genus, so multiple genera may have been associated with a single flavor compound. The genera mentioned above may therefore be key microorganisms in different Daqu grades, and may play important roles in the production of distinctive Baijiu. Differences in the compositions and proportions of flavor compounds during SSF by Daqu of different grades will be caused by the different structures of these key microbial flora in the Daqu. This may also explain why mixing Daqu of different grades (typically mostly premium- or first-grade Daqu) in appropriate proportions is necessary to produce high-quality Baijiu.[Citation54,Citation55]
Figure 5. Correlation between volatile compounds (V1-V75: the codes of different flavor compounds in Supplementary Table S2) and prokaryotic communities (a) and eukaryotic communities (b) found in Daqu. Scale bar colors denote the nature of the correlation, with 1 indicating a perfectly positive correlation (read) and −1 indicating a perfectly negative correlation (blue) between them. “*” shows significant correlations (P <0.05), and “NA” shows no correlations
Conclusions
Differences between Daqu of different grades in terms of physicochemical properties, flavor components, and microbial communities were comprehensively compared. The results for the three Daqu grades matched the traditional Daqu classification criteria, and the sensory evaluation classification grades correlated to some extent with the physicochemical properties. The ability to assess Daqu quality is essential to the production of high-quality Baijiu. However, assessing quality subjectively rather than using quantifiable indicators has drawbacks. This means that some uncertainty related to Baijiu production remains (particularly in terms of the use of mixed Daqu), which could lead to poor uniformity in Baijiu quality. We speculate, from the results, that a comprehensive scientific standard for evaluating the grade of Daqu could be established. The microbial flora community and dominant genera would be the most important classification indicators, and physicochemical properties (e.g., acidity and enzyme activities) and flavor compounds (types and contents) would be used as supplementary indicators. Establishing such a standard would make classifying Daqu more scientifically based than is currently the case and would promote development of the Baijiu industry.
Supplemental Material
Download Zip (22.4 MB)Acknowledgments
We thank Gareth Thomas and Austin Schultz, PhD, for providing language help. And, this research was supported by the National Natural Science Foundation of China [No. 31830069, 31701592 and 31671798], and the Foundation of Beijing Technology and Business University [No. LKJJ2017-10]. The data used to support the findings of this study are available from the corresponding author upon request. The authors declare that they have no competing interests.
Supplementary Material
Supplementary data for this article can be accessed here.
Additional information
Funding
References
- Liu, J.-J.; Chen, J.-Y.; Fan, Y.; Huang, X.-N.; Han, B.-Z. Biochemical Characterisation and Dominance of Different Hydrolases in Different Types of Daqu - a Chinese Industrial Fermentation Starter. J. Sci. Food Agric. 2018, 98(1), 113–121. DOI: 10.1002/jsfa.8445.
- Xiao, C.; Lu, Z.-M.; Zhang, X.-J.; Wang, S.-T.; Ao, L.; Shen, C.-H.; Shi, J.-S.; Xu, Z.-H. Bio-Heat Is a Key Environmental Driver Shaping the Microbial Community of Medium-Temperature Daqu. Appl. Environ. Microbiol. 2017, 83 (23) . DOI: 10.1128/AEM.01550-17.
- Zhang, C.-L.; Ao, Z.-H.; Chui, W.-Q.; Shen, C.-H.; Tao, W.-Y.; Zhang, S.-Y. Characterization of the Aroma-Active Compounds in Daqu: A Tradition Chinese Liquor Starter. Eur. Food Res. Technol. 2012, 234(1), 69–76. DOI: 10.1007/s00217-011-1616-4.
- Ming, H.-M.; Zhou, J.; Zhu,L.-L.; Xu, D.-F.; Liu, Y.-H.; Yao, X. A Comparative Study of Traditional/New Daqu Quality Indicators System. Liquor-Making Sci. Technol. 2015, (4), 32–36. DOI:10.13746/j.njkj.2014453.
- Wang, X.-S.; Du, H.; Zhang, Y.; Xu, Y. Environmental Microbiota Drives Microbial Succession and Metabolic Profiles during Chinese Liquor Fermentation. Appl. Environ. Microbiol. 2018, 84(4). DOI: 10.1128/AEM.02369-17.
- Li, P.; Aflakpui, F.-W.-K.; Yu, H.; Luo, L.-X.; Lin, W.-T. Characterization of Activity and Microbial Diversity of Typical Types of Daqu for Traditional Chinese Vinegar. Ann. Microbiol. 2015, 65(4), 2019–2027. DOI: 10.1007/s13213-015-1040-2.
- Pang, X.-N.; Han, B.-Z.; Huang, X.-N.; Zhang, X.; Hou, L.-F.; Cao, M.; Gao, L.-J.; Hu, G.-H.; Chen, J.-Y. Effect of the Environment Microbiota on the Flavour of Light-Flavour Baijiu during Spontaneous Fermentation. Sci. Rep. 2018, 8. DOI: 10.1038/s41598-018-21814-y.
- Zhang, Y.-Y.; Zhu, X.-Y.; Li, X.-Z.; Tao, Y.; Jia, J.; He, X.-H. The Process-Related Dynamics of Microbial Community during a Simulated Fermentation of Chinese Strong-Flavored Liquor. BMC Microbiol. 2017, 17. DOI: 10.1186/s12866-017-1106-3.
- Y.-H.;, Yi, Z.-L.; Jin, Y.-L.; Huang, M.-J.; He, K.-Z; Liu, D.-Y.; Luo, H.-B.; Zhao. D.; He, H.; Fang, Y.; Zhao, H. Metatranscriptomics Reveals the Functions and Enzyme Profiles of the Microbial Community in Chinese Nong-Flavor Liquor Starter. Front. Microbiol. 2017, 8. DOI: 10.3389/FMICB.2017.01747.
- Wang, P.; Wu, Q.; Jiang, X.-J.; Wang, Z.-Q.; Tang, J.-L.; Xu, Y. Bacillus Licheniformis Affects the Microbial Community and Metabolic Profile in the Spontaneous Fermentation of Daqu Starter for Chinese Liquor Making. Int. J. Food Microbiol. 2017, 250, 59–67. DOI: 10.1016/j.ijfoodmicro.2017.03.010.
- W.-Q.;Ao, Z.-H.; Zhang, C.-L.Shen, C.-P.; Tao, W.-Y.; Lu, Z.-M.; Wang, X.-J . Study on the Correlations between Sensory Characteristics and Microbe, Physiochemical Indexes and Biochemical Property of Luzhou Laojiao Daqu. J. Food Sci. Biotechnol. 2011, 30(5), 761–766.
- Chen, Z.-X.; Lin, L.; Han, Y.; Yang, F.; Wang, D.-Q.; Wang, L. Application of Functional High-Temperature Daqu Based on the Composition of Flavor Compounds. China Brewing. 2017, 36(1), 98–101. DOI:10.11882/j.issn.0254-5071.2017.01.020.
- Wang, X.-S.; Du, H.; Xu, Y. Source Tracking of Prokaryotic Communities in Fermented Grain of Chinese Strong-Flavor Liquor. Int. J. Food Microbiol. 2017, 244, 27–35. DOI: 10.1016/j.ijfoodmicro.2016.12.018.
- G.-S.;Du, Y.-H.;Fu, Z.-L.; Chen, M.; Wang, Z.; Liu, P.-X.; Li, X.-T. Characterization of Physicochemical Properties, Flavor Components and Microbial Community in Roasted Sesame-Like Flavor Daqu. J. Inst. Brew. 2019.DOI:10.1002/jib.583.
- QB. People’s Republic of China Professional Standard. General Methods of Analysis for Daqu (QB/T 4257-2011), In China, Ministry of Industry and Information Technology of the People’s Republic of China, QB/T 4257-2011, China Light Industry Press, Beijing, pp. 1–15, 2011.
- Fan, G.-S.; Sun, B.-G.; Fu, Z.-L.; Xia, Y.-Q.; Huang, M.-Q.; Xu, C.-Y.; Li, X.-T. Analysis of Physicochemical Indices, Volatile Flavor Components and Microbial Community of a Light-Flavor Daqu. J. Am. Soc. Brew. Chem. 2018, 76(3), 209–218. DOI: 10.1080/03610470.2018.1424402.
- Zhang, L.-Q.; Wu, C.-D.; Ding, X.-F.; Zheng, J.; Zhou, R.-Q. Characterisation of Microbial Communities in Chinese Liquor Fermentation Starters Daqu Using Nested PCR-DGGE. World J. Microbiol. Biotechnol. 2014, 30(12), 3055–3063. DOI: 10.1007/s11274-014-1732-y.
- Z.-K.;Feng, Y.-F.;Meng, Q.-Y.; Wang, X.-Z.; Guo, J.-Y.; Fang, H.-Z.; Du, J.; Duan, K.-L. Research on the Perspective of Microbial Diversity and Physicochemical Properties of Xifeng-Daqu’s Culturable Microorganisms. Liquor Making. 2015, 42(3), 36–41. DOI:10.3969/j.issn.1002-8110.2015.03.013.
- Wang, Q.; Guo, D.-Y.; Wang, X.-L.; Wu, S.; Wang, D.-L. Comparative Analysis of Physiochemical Properties of Baijiu (Liquor)Starter of Different Flavor Types. Liquor-Making Sci. Technol. 2015, (6), 6–10. DOI: 10.13746/j.njkj.2014450.
- Zheng, X.-W.; Yan, Z.; Han, B.-Z.; Zwietering, M.-H.; Samson, R.-A.; Boekhout, T.; Nout, M.-J.-R. Complex Microbiota of a Chinese “Fen” Liquor Fermentation Starter (Fen-Daqu), Revealed by Culture-Dependent and Culture-Independent Methods. Food Microbiol. 2012, 31(2), 293–300. DOI: 10.1016/j.fm.2012.03.008.
- Zhang, L.-X. Water Cycle and Fire Cycle of Xifeng Daqu. Liquor-Making Sci. Technol. 2005, (11), 38–39. DOI: 10.13746/j.njkj.2005.11.007.
- Fan, G.-S.; Sun, B.-G.; Xu, D.; Teng, C.; Fu, Z.-L.; Du, Y.-H.; Li, X.-T. Isolation and Identification of High-Yield Ethyl Acetate-Producing Yeast from Gujinggong Daqu and Its Fermentation Characteristics. J. Am. Soc. Brew. Chem. 2018, 76(2), 117–124. DOI: 10.1080/03610470.2017.1396849.
- Wang, X.-X.; Fan, W.-L.; Xu, Y. Comparison on Aroma Compounds in Chinese Soy Sauce and Strong Aroma Type Liquors by Gas Chromatography-Olfactometry, Chemical Quantitative and Odor Activity Values Analysis. Eur. Food Res. Technol. 2014, 239(5), 813–825. DOI: 10.1007/s00217-014-2275-z.
- Liu, H.-L.; Sun, B.-G. Effect of Fermentation Processing on the Flavor of Baijiu. J. Agric. Food Chem. 2018, 66(22), 5425–5432. DOI: 10.1021/acs.jafc.8b00692.
- Fan, G.-S.; Fu, Z.-L.; Sun, B.-G.; Zhang, Y.-H.; Wang, X.-L.; Xia, Y.-Q.; Huang, M.-Q.; Li, X.-T. Roles of Aging in the Production of Light-Flavored Daqu. J. Biosci. Bioeng. 2019, 127(3), 309–317. DOI: 10.1016/j.jbiosc.2018.08.005.
- X.-J.;Feng, H.-J.;Zhai, L.; Bai, X.-B.; Xu, L.; Yu, P.-P.; Cheng, C.; Yao, S. . The Dynamics of Thermoactinomyces and Their Functional Genes in High Temperature Daqu of Sesame-Flavored Baijiu. Food Ferment. Ind. 2019. DOI: 10.13995/j.cnki.11-1802/ts.019959.
- J.; Zhong, Q.-P.;Yang, Y.-Y.; Li, H.-R.; Wang, L.; Tong, Y.-G.; Fang, X.; Liao, Z.-L.. Comparison of Bacterial Diversity between Two Traditional Starters and the Round-Koji-Maker Starter for Traditional Cantonese Chi-Flavor Liquor Brewing. Front. Microbiol.2018, 9. DOI: 10.3389/fmicb.2018.01053.
- Hu, Y.-L.; Dun, Y.-H.; Li, S.-A.; Fu, B.-A.; Xiong, X.-M.; Peng, N.; Liang, Y.-X.; Zhao, S.-M. Changes in Microbial Community during Fermentation of High-Temperature Daqu Used in the Production of Chinese “Baiyunbian” Liquor. J. Inst. Brew. 2017, 123(4), 594–599. DOI: 10.1002/jib.v123.4.
- Zheng, X.-W.; Tabrizi, M.-R.; Nout, M.-J.-R.; Han, B.-Z. Daqu - A Traditional Chinese Liquor Fermentation Starter. J. Inst. Brew. 2011, 117(1), 82–90. DOI: 10.1002/j.2050-0416.2011.tb00447.x.
- Cai, H.-Y.; Zhang, T.; Zhang, Q.; Luo, J.; Cai, C.-G.; Mao, J.-W. Microbial Diversity and Chemical Analysis of the Starters Used in Traditional Chinese Sweet Rice Wine. Food Microbiol. 2018, 73, 319–326. DOI: 10.1016/j.fm.2018.02.002.
- Liu, P.-L.; Xiong, X.-M.; Wang, S.; Miao, L.-H. Population Dynamics and Metabolite Analysis of Yeasts Involved in a Chinese Miscellaneous-Flavor Liquor Fermentation. Ann. Microbiol. 2017, 67(8), 553–565. DOI: 10.1007/s13213-017-1286-y.
- Gou, M.; Wang, H.-Z.; Yuan, H.-W.; Zhang, W.-X.; Tang, Y.-Q.; Kida, K. Characterization of the Microbial Community in Three Types of Fermentation Starters Used for Chinese Liquor Production. J. Inst. Brew. 2015, 121(4), 620–627. DOI: 10.1002/jib.272.
- Wang, X.-D.; Ban, S.-D.; Qiu, S.-Y. Analysis of the Mould Microbiome and Exogenous Enzyme Production in Moutai-Flavor Daqu. J. Inst. Brew. 2018, 124(1), 91–99. DOI: 10.1002/jib.v124.1.
- Kumar, P.; Islam, A.; Ahmad, F.; Satyanarayana, T. Characterization of a Neutral and Thermostable Glucoamylase from the Thermophilic Mold Thermomucor Indicae-Seudaticae: Activity, Stability, and Structural Correlation. Appl. Biochem. Biotechnol. 2010, 160(3), 879–890. DOI: 10.1007/s12010-009-8666-0.
- Merheb-Dini, C.; Chaves, K.-S.; Gomes, E.; da Silva, R.; Gigante, M.-L. Coalho Cheese Made with Protease from Thermomucor Indicae-Seudaticae N31: Technological Potential of the New Coagulant for the Production of High-Cooked Cheese. J. Food Sci. 2016, 81(3), C563–C568. DOI: 10.1111/1750-3841.13217.
- Li, Z.-M.; Chen, L.; Bai, Z.-H.; Wang, D.-L.; Gao, L.-P.; Hui, B.-D. Cultivable Bacterial Diversity and Amylase Production in Two Typical Light-Flavor Daqus of Chinese Spirits. Front. Life Sci. 2015, 8(3), 264–270. DOI: 10.1080/21553769.2015.1041188.
- Martin, N.; Guez, M.-A.-U.; Sette, L.-D.; Da Silva, R.; Gomes, E. Pectinase Production by a Brazilian Thermophilic Fungus Thermomucor Indicae-Seudaticae N31 in Solid-State and Submerged Fermentation. Microbiology. 2010, 79(3), 306–313. DOI: 10.1134/S0026261710030057.
- Pereira, J.-D.; Leite, R.-S.-R.; do Prado, H.-F.-A.; Martins, D.-A.-B.; Gomes, E.; da Silva, R. Production and Characterization of Beta-Glucosidase Obtained by the Solid-State Cultivation of the Thermophilic Fungus Thermomucor Indicae-Seudaticae N31. Appl. Biochem. Biotechnol. 2015, 175(2), 723–732. DOI: 10.1007/s12010-014-1332-1.
- Ferrarezi, A.-L.; Ohe, T.-H.-K.; Borges, J.-P.; Brito, R.-R.; Siqueira, M.-R.; Vendramini, P.-H.; Quilles, J.-C.; Nunes, C.-D.-C.; Bonilla-Rodriguez, G.-O.; Boscolo, M.; Da-Silva, R.; Gomes, E. Production and Characterization of Lipases and Immobilization of Whole Cell of the Thermophilic Thermomucor Indicae Seudaticae N31 for Transesterification Reaction. J. Mol. Catal. B Enzym. 2014, 107, 106–113. DOI: 10.1016/j.molcatb.2014.05.012.
- Qiao, Y.-X.; Zha, X.-Y.; Zhu, J.; Tu, R.; Dong, L.-B.; Wang, L.; Dong, Z.-Y.; Wang, Q.-H.; Du, W.-B. Fluorescence-Activated Droplet Sorting of Lipolytic Microorganisms Using a Compact Optical System. Lab Chip. 2018, 18(1), 190–196. DOI: 10.1039/C7LC00993C.
- Yu, X.-W.; Zhu, S.-S.; Xiao, R.; Xu, Y. Conversion of a Rhizopus Chinensis Lipase into an Esterase by Lid Swapping. J. Lipid Res. 2014, 55(6), 1044–1051. DOI: 10.1194/jlr.M043950.
- Li, S.; Yang, Q.; Tang, B.; Chen, A. Improvement of Enzymatic Properties of Rhizopus Oryzae Alpha-Amylase by Site-Saturation Mutagenesis of Histidine 286. Enzyme Microb. Technol. 2018, 117, 96–102. DOI: 10.1016/j.enzmictec.2018.06.012.
- Hiol, A.; Jonzo, M.-D.; Rugani, N.; Druet, D.; Sarda, L.; Comeau, L.-C. Purification and Characterization of an Extracellular Lipase from a Thermophilic Rhizopus Oryzae Strain Isolated from Palm Fruit. Enzyme Microb. Technol. 2000, 26(5–6), 421–430. DOI: 10.1016/S0141-0229(99)00173-8.
- Muthukumaran, N.; Dhar, S.-C. Purification and Properties of a Glucoamylase Fraction from the Culture Filtrate of Rhizopus Nodosus. Ital. J. Biochem. 1983, 32(4), 239–253.
- Carroll, E.; Tran, N.-T.; Son, H.; Lee, Y.-W.; Seo, J.-A. Comprehensive Analysis of Fungal Diversity and Enzyme Activity in Nuruk, a Korean Fermenting Starter, for Acquiring Useful Fungi. J. Microbiol. 2017, 55(5), 357–365. DOI: 10.1007/s12275-017-7114-z.
- Chopra, A.-K.; Chander, H. Factors Affecting Lipase Production in Syncephalastrum Racemosum. J. Appl. Bacteriol. 1983, 54(2), 163–169. DOI: 10.1111/j.1365-2672.1983.tb02602.x.
- Lv, M.; Chen, M.-B.; Zhen, D. Isolation of Esterase-Producing Strain HSM from Nong-Flavor Daqu and Study on Its Esterase-Producing Nutritional Conditions. Liquor-Making Sci. Technol. 2014, (1), 12–15. DOI:10.13746/j.njkj.2014.01.006.
- Wu, X.-W.; Zheng, R.-C.; Zheng, Y.-G. Overexpression of Talaromyces Thermophilus Lipase (TTL) and Its Application in Biosynthesis of Pregabalin Intermediate. Chin. J. Bioprocess Eng. 2016, 14(5), 37–44. DOI: 10. 3969 /j. issn. 1672–3678. 2016. 05. 008.
- Zhou, W.; Liu, X.-H.; Ye, L.; Feng, M.-Q.; Zhou, P.; Shi, X.-L. The Biotransformation of Astragalosides by a Novel Acetyl Esterase from Absidia Corymbifera AS2. Process Biochem. 2014, 49(9), 1464–1471. DOI: 10.1016/j.procbio.2014.05.026.
- Kong, Y.; Wu, Q.; Zhang, Y.; Xu, Y. In Situ Analysis of Metabolic Characteristics Reveals the Key Yeast in the Spontaneous and Solid-State Fermentation Process of Chinese Light-Style Liquor. Appl. Environ. Microbiol. 2014, 80(12), 3667–3676. DOI: 10.1128/AEM.04219-13.
- Zou, W.; Ye, G.-B.; Zhang, K.-Z. Diversity, Function, and Application of Clostridium in Chinese Strong Flavor Baijiu Ecosystem: A Review. J. Food Sci. 2018, 83(5), 1193–1199. DOI: 10.1111/1750-3841.14134.
- Owens, J.-D.; Allagheny, N.; Kipping, G.; Ames, J.-M. Formation of Volatile Compounds during Bacillus Subtilis Fermentation of Soya Beans. J. Sci. Food Agric. 1997, 74(1), 132–140. DOI: 10.1002/(SICI)1097-0010(199705)74:1<132::AID-JSFA779>3.0.CO;2-8.
- Huang, C.-J.; Lee, S.-L.; Chou, C.-C. Production and Molar Yield of 2-Phenylethanol by Pichia Fermentans L-5 as Affected by Some Medium Components. J. Biosci. Bioeng. 2000, 90(2), 142–147. DOI: 10.1263/jbb.90.142.
- Li, Q.; Xiang Li, J.-X. Constrast of Fermenting Performance of Three Kinds of Fenjiu Daqu. Liquor-Making Sci. Technol. 2011, (4), 63–64. DOI: 10.13746/j.njkj.2011.04.026.
- Tang, Q.-W.; Huang, J.-K.; Tang, Z.-J. High Temperature Koji Mixed Use in the Production of Yellow Rice Wine in the Applied Research. Liquor Making. 2011, 38(5), 75. DOI:10.3969/j.issn.1002-8110.2011.05.029.