
Abstract
In 2012, the WHO classified diesel emissions as carcinogenic, and its European branch suggested creating a public health standard for airborne black carbon (BC). In 2011, EU researchers found that life expectancy could be extended four to nine times by reducing a unit of BC, vs reducing a unit of PM2.5. Only recently could such determinations be made. Steady improvements in research methodologies now enable such judgments.
In this Critical Review, we survey epidemiological and toxicological literature regarding carbonaceous combustion emissions, as research methodologies improved over time. Initially, we focus on studies of BC, diesel, and traffic emissions in the Western countries (where daily urban BC emissions are mainly from diesels). We examine effects of other carbonaceous emissions, e.g., residential burning of biomass and coal without controls, mainly in developing countries.
Throughout the 1990s, air pollution epidemiology studies rarely included species not routinely monitored. As additional PM2.5. chemical species, including carbonaceous species, became more widely available after 1999, they were gradually included in epidemiological studies. Pollutant species concentrations which more accurately reflected subject exposure also improved models.
Natural “interventions” - reductions in emissions concurrent with fuel changes or increased combustion efficiency; introduction of ventilation in highway tunnels; implementation of electronic toll payment systems – demonstrated health benefits of reducing specific carbon emissions. Toxicology studies provided plausible biological mechanisms by which different PM species, e.g., carbonaceous species, may cause harm, aiding interpretation of epidemiological studies.
Our review finds that BC from various sources appears to be causally involved in all-cause, lung cancer, and cardiovascular mortality, morbidity, and perhaps adverse birth and nervous system effects. We recommend that the U.S. EPA rubric for judging possible causality of PM2.5. mass concentrations, be used to assess which PM2.5. species are most harmful to public health.
Implications: Black carbon (BC) and correlated co-emissions appear causally related with all-cause, cardiovascular, and lung cancer mortality, and perhaps with adverse birth outcomes and central nervous system effects. Such findings are recent, since widespread monitoring for BC is also recent. Helpful epidemiological advances (using many health relevant PM2.5 species in models; using better measurements of subject exposure) have also occurred. “Natural intervention” studies also demonstrate harm from partly combusted carbonaceous emissions. Toxicology studies consistently find biological mechanisms explaining how such emissions can cause these adverse outcomes. A consistent mechanism for judging causality for different PM2.5 species is suggested.
A list of acronyms will be found at the end of the article.
Aims of Critical Review
This critical review (CR) consists of three main sections. First, we review recent major regulatory and scientific assessments of black carbon/elemental carbon (BC/EC) diesel emission. Second, changes in the understanding of health effects of BC in the United States, Canada, and Western Europe, including usage of improved methodologies for assessing health effects of different PM2.5 species, are discussed. Third, we survey emerging literature concerning health effects of incompletely combusted carbonaceous species in the developing world. Specifically, the CR will:
Highlight recent major current regulatory and scientific assessments of BC/EC/diesel emissions.
Explain how the knowledge base for the health effects of BC has changed over the years in the United States, Canada, and Western Europe highlighting these topics:
Early understanding of PM effects—Great London Fog, marked by black smoke among other emissions; Six Cities (1993) and American Cancer Society (1995) studies, which did not consider black smoke or BC; other early reviews.
Evolution of enhanced assessment methodologies, and of monitoring networks measuring major PM2.5 components, advancing understanding of potentially harmful PM2.5 sources and species.
Population-based epidemiology studies using newer assessment methodologies for comparison with older studies or studies with less accurate methodologies.
Human panel studies that included BC/EC as a monitored PM2.5 species.
Relevant toxicology of different PM2.5 species, especially BC.
Health outcomes from BC exposure with regard to all-cause, cardiovascular, and cardiopulmonary mortality and morbidity, as well as birth outcomes, cognition, and lung cancer.
Ambiguities with other combustion emissions, mainly co-emissions of BC such as organic carbon, and polycyclic aromatic hydrocarbons, but also including PM2.5 metals, road wear particles, and noise.
Survey the emerging literature concerning health effects of incompletely combusted carbonaceous species (e.g., biomass and residential coal use) in the developing world.
In order to provide a regulatory context to this CR, we suggest use of five U.S. Environmental Protection Agency (EPA) assessment criteria (U.S. EPA, 2009) to assess whether a given PM2.5 species is or is not likely to be causally related to mortality or cardiovascular disease (CVD) morbidity endpoints. These criteria are as follows:
“Consistency of the observed association”—Do many studies, using different designs, in multiple locations, consistently show elevated risks for a given mortality or morbidity endpoint?
“Coherence”—Are consistent associations in population based epidemiological studies supported by other findings from human panel and animal toxicology studies?
“Biological plausibility”—Do experimental studies provide evidence for biological mechanisms supporting links between exposure to an agent and adverse effects in humans?
“Biological gradient”—Is there a dose-response function, such that increasing exposure is linked with increasing adverse effects?
“Experimental evidence”—Does a change in exposure bring about a change in occurrence of health effects?
Rebecca Klemm

Richard B. Schlesinger

Thomas J. Grahame

Introduction
Epidemiological studies linking vehicular emissions, particularly diesel (primary particulate diesel emissions, agglomerated spherical graphitic particles 20 to 30 nm in size when freshly emitted, usually monitored as BC or EC; Watson et al., Citation2005), to morbidity and mortality are now common (full discussion and definitions of BC and EC in Supplemental Material; shorter definitions in the following). Fifteen years ago, such studies were relatively nonexistent in North America. PM2.5, and PM2.5 species other than BC/EC, were seen as culpable, partly because they were monitored frequently and thus could be included specifically in epidemiological studies, whereas BC/EC was not. Prior to the rest of this review, we provide short technical definitions of BC and EC.
Definitions
EC is nonvolatile carbon in graphite-like form, found in mainly nanometer-size fractions (<56 nm; 56–100 nm; and 100–180 nm; Mauderly and Chow, Citation2008); however, EC is never found in pure form in the atmosphere, because various organic carbon (OC) and other compounds are adsorbed onto, and/or mixed with the EC core (Li and Nel, Citation2006). EC containing a coating of mixed OC and inorganics, with adsorbed carbonaceous material, is referred to as BC. Atmospheric EC and organic carbon (OC), commonly determined on PM2.5 (particulate matter with aerodynamic diameters <2.5 μm) filters, are operationally defined based on thermal oxidation (Chow et al., Citation2001). The U.S. long-term urban Chemical Speciation Network (CSN) and non-urban Interagency Monitoring of PROtected Visual Environments (IMPROVE) network determine EC by thermal/optical method (Chow et al., Citation2007). BC is generally determined by light absorption by filter or by photoacoustic system measurement. These devices measure light absorption, which is then converted to BC concentration in micrograms per cubic meter (μg/m3) based on specific absorption efficiency (Chow et al., Citation2001). BC and EC are often used interchangeably by epidemiologists, as they are proxies for the same emissions; for simplicity, we use BC to refer to carbonaceous PM assessed using either method of measuring amounts of solid, light-absorbing, refractory graphitic carbonaceous material in ambient air, material that is insoluble in water and common organic solvents (Lack et al., Citation2014). As there are several different methods for measuring both BC and EC, there exists measurement ambiguity, depending on measurement method used (Watson et al., Citation2005; Supplemental Material). BC is highly correlated with other carbonaceous compounds, many gases or semivolatile organic compounds (SVOCs), and with trace metals in lubricating oil. Finally, BC should not be confused with brown carbon, a product of inefficient combustion of biomass, which also is light absorbing, but more toward the ultraviolet (UV) part of the spectrum (Andreae and Gelencser, Citation2006). In places where substantial amounts of biomass are burned inefficiently, brown carbon can thus bias measurements of BC (Andreae and Gelencser, Citation2006).
OC refers to all carbonaceous species that are not BC/EC (other than carbonates). Many carbonaceous gases from anthropogenic as well as natural sources are chemically changed in the atmosphere into low-volatility particulate forms, called secondary organic carbon (SOC), also a subset of OC.
Total carbon (TC) is the sum of OC and BC. The health effects observed with exposure to sources of BC are likely due in some part not just to BC, but also to specific types of highly correlated co-emissions (e.g., SVOC, volatile organic carbon [VOC], and OC, including PAHs). (Later, we discuss the roles of size and of chemistry in causing adverse effects.)
Sources of BC
provides estimated 2010 BC emissions by source (power, industrial, residential, transport, and biomass) for major geographical regions of the world (Organization for Economic Cooperation and Development [OECD] Europe, Eastern Europe, South America, and East Asia, for example), as well as for specific countries. These estimates are derived from known amounts of fuels used and various appropriate emission factors.
Table 1. Global BC emission estimates by region and by sector in 2010 based on RCP 4.5 (unit: Gg/year)
The transportation source category is the main source of BC/EC emissions in the United States and Western Europe (). Within that category, BC emissions from present-day diesels are more than 8 times as high as those from gasoline powered mobile sources in the United States (U.S. EPA, Citation2012). However, industrial and residential coal burning and residential biomass burning, each with little or no particulate controls, are major daily BC/EC sources in much of the developing world. Irregular biomass burning (e.g., forest fires, agricultural burning) are also important sources of BC and OC, especially in the developing world (). A more detailed description and definition of BC/EC and OC in the atmosphere is found in the Supplementary Material.
Total estimated worldwide 2010 ambient BC emissions are 8,111 Gg/yr. East Asia is the largest contributing region (1,876 Gg/yr). Southeast Asia contributes 877 Gg/yr, the United States 321 Gg/yr, OECD Europe 301 Gg/yr, and Africa 1,788 Gg/yr. The largest single source in the United States and OECD Europe is estimated to be transport (mostly diesel, 216 and 212 Gg/yr, respectively), while the leading sources in Asia are industry and residential (East Asia, 996 and 660 Gg/yr) and residential and biomass (Southeast Asia, 282 and 339 Gg/yr). Domestic and industrial (excluding electrical generation) coal burning without particulate controls is the main source of industrial and residential emissions in East Asia. In Africa and in Central and South America, open biomass burning is the largest source of BC. Irregular burning of biomass for clearing land and naturally occurring fires are included in under open burning, and are also large sources of TC. Such estimates, of course, cannot take into account all “real-world” factors, so it is not surprising that there is variation in such estimates. For example, Chow et al. (Citation2011) review differing BC and OC source profiles in the United States from two different compilations. Worldwide BC estimates of Bond et al. (Citation2004) differ somewhat from the estimates in . We use the estimates in because they are developed with a consistent methodology and include not just regions but also important countries. All estimates are uncertain and subject to change, as emissions factors are updated, and as newer technologies (such as on road diesels with catalyzing filters which oxidize almost all BC and much OC) replace older technologies. (The remaining tables referred to in the text are found in the Supplemental Material, section C.)
Because ambient sources of BC in the United States and Western Europe are primarily diesel related, while the major sources and mix of BC elsewhere in the world are different, we initially discuss the United States and Western Europe.
Evolution of Our Understanding of Health Effects From BC in North America and Europe
Within the last 2 years, there have been three major reports that solidify the assessment of harm from BC and from diesels. WHO (Citation2012) called for consideration of an air pollution health standard for BC, separate from that for PM2.5, suggesting that such a standard “may be useful in evaluating local action aimed at reducing the population's exposure to combustion PM (for example, from motorized traffic).” This appears to be the initial occasion when a major governmental body has suggested that BC per se be regulated. In 2012, the IARC upgraded its 1988 finding that diesel emissions (DEE) were “probably carcinogenic to humans,” to a finding that diesel emissions are “carcinogenic to humans” (IARC, Citation2012). Janssen et al. (Citation2011) found that reducing a unit of BC will lengthen life four to nine times more than reducing a comparable amount of PM2.5 mass (e.g., if 1 μg/m3 of each were reduced). We have not identified other studies directly comparing life-extending effects from BC with those from PM2.5. It should be noted that these three assessments relate to current on- and-off road older diesels, not to emissions from post-2006 heavy-duty on-road diesels in the United States, which are subject to more stringent PM emission regulations that greatly reduce BC and associated emissions (McDonald et al., Citation2004). It should also be noted that sulfur in fuels needed to be further reduced to avoid poisoning catalysts for meeting post-2006 emission standards. The U.S. EPA (2012) suggested that health effects of BC are similar to those of PM2.5 mass simply because BC is a component of PM2.5 mass. It notes, “BC mitigation strategies, which lead to reductions in PM2.5, can provide substantial public health and environmental benefits.”
Since 2007, new on-road heavy-duty diesels in the United States have a filter that oxidizes or captures virtually all BC and much of other carbonaceous emissions when in working order (McDonald et al., Citation2004). The emission reductions reduce oxidative stress and inflammation in vivo that were found with diesel exhaust not treated by the new oxidizing filter (McDonald et al., Citation2004).
Slightly more than a decade ago, BC was rarely evaluated as part of epidemiological models, and diesel emissions were rarely examined in relation to the toxicology of particulate emissions. What changed, to allow the three conclusions just described about the harm caused by BC to be drawn?
Early evaluations of PM effects
We review in detail in the Supplemental Material, Part B, seminal studies and events in early evaluation of PM effects. Initial studies include those of the Great London Fog of 1952 and a study of a similar inversion in Dublin, Ireland, in 1982. Both events involved pollution that was mainly derived from domestic burning of coal for heat, without pollution controls. These European studies demonstrated that partly burned hydrocarbons and other pollutants that result from burning poor-quality coals without pollution controls, when concentrated during multiday stagnant episodes, can cause sharply increased numbers of excess deaths. Black smoke, a measure of the darkness of material in a filter as measured by optical reflectance, was the measure of PM used in these European studies. Quincey (Citation2007) reported that these methods are similar to aethalometry, one of the principal methods of measuring BC.
We then discuss several early U.S. reports and studies, the first one of which is from a 1975 U.S. EPA advisory board (U.S. EPA Scientific Advisory Board [SAB], 1975). This is followed by two pivotal studies of the 1980s, namely, Evans et al. (1984) and Ozkaynak and Thurston (Citation1987). Perhaps understandably, due to the emphasis on acid rain as a major pollution research topic at the time, these two studies emphasized sulfate rather than other PM species. BC, EC, diesel, polycyclic aromatic hydrocarbons (PAHs), and vehicular emissions were not examined.
Evans et al. (1984) specifically highlighted methodological issues that remain important today in assessing possible causality. These include:
Exposure misclassification, due to use of a single monitor, or monitor averages, to represent exposure of individuals citywide to a pollutant with substantial local spatial variability.
Confounding variables, where a potentially causal agent is not in the model, but is correlated with agents that are included. Given the dearth of information about other PM2.5 species at the time, and about biological mechanisms generally, Evans et al. (1984) were mostly concerned about confounding due to sociological factors, for example, lower income people living in more polluted areas. The potential importance of confounding by other PM2.5 species came later.
The variability of chemical species in aerosols classified by size (in this case, TSP).
Plausibility of sulfate as a cause of adverse health outcomes, given that ammonium sulfate was seen as “a relatively innocuous species.”
Health relevance of pollutants not included (specifically, cancer researchers would want information on organic chemical compounds, including those from vehicular emissions).
Ambiguities in the “accuracy and precision of pollution measurements,” specifically in the context of individual Hi-Vol measurements.
Stern et al. (Citation1988)
Stern et al. (Citation1988), a study of arteriosclerotic heart disease (ASHD) mortality risks for bridge and tunnel officers (BTOs) in New York City from 1952 through 1982, was a rare exception to the trend in the 1980s of not evaluating the potential importance of vehicular emissions for mortality and morbidity risks. Officers mainly collected tolls, but also directed and observed traffic. Bridge officers had less exposure to vehicular emissions, as wind provided natural ventilation, whereas there was no ventilation in tunnels until ventilation systems were installed around 1970. The ventilation systems allowed for an “intervention” study, where an event in the physical world causes pollution levels or types to change, enabling an evaluation of the effect of the change on public health with change in exposure at the same venue. Heart disease and arteriosclerotic heart disease (ASHD) mortality risks were each calculated in comparison to the mortality record for the New York City population as a whole. Amount of time worked as a BTO was categorized as less than 10 years; 10 years or more; and combined (total). Significantly elevated risks of mortality, both for all heart disease and for ASHD, were found for tunnel officers who had worked for more than 10 years, and for all tunnel officers combined. The study design also allowed for examination of mortality risk in tunnel officers compared to bridge officers for the period after ventilation was installed within the tunnels. The ASHD mortality risk for each of three age cohorts of tunnel officers declined in a constant manner, relative to the average mortality risk for all bridge workers, after ventilation was introduced (). Thus, Stern et al. (Citation1988) provides evidence that elevated vehicular emissions are associated with elevated risks of dying of ASHD, both in the higher mortality of tunnel officers compared to bridge officers prior to ventilation of tunnels, and with the reduction of mortality risks for tunnel officers compared to bridge officers after ventilation was introduced.
Figure 1. Risk of arteriosclerotic hearth disease mortality in tunnel officers relative to bridge officers, as a function of age and calendar year, after introduction of ventilation in 1970. Adapted from Stern et al. (Citation1988). © Oxford University Press. Reproduced by permission of Oxford University Press. Permission to reuse must be obtained from the rightsholder.
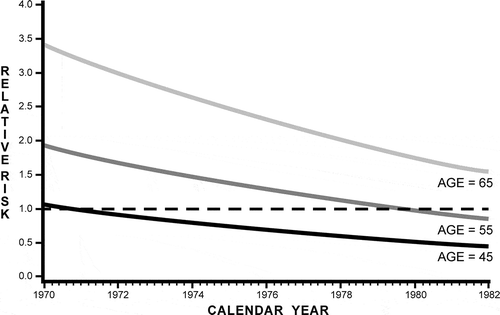
Stern et al. (Citation1988), however, do not present any associations with specific pollutants. CO concentrations for years before and after ventilation, and concentrations of NOx, PAHs, lead, and asbestos in 1981 (a decade after ventilation) were, however, provided. CO levels in 1970, monitored over 38 consecutive days, were 63 ppm in tunnel and 13 ppm in bridge toll booths. In 1981, daily sampling for 2 weeks found mean CO levels of 38.3 ppm in tunnels and 23.0 ppm on bridges. Carboxyhemoglobin levels were higher in tunnel officers than in bridge officers (nonsmokers 2.12% to 2.93%; smokers 3.90% to 5.01%) prior to ventilation. A decade after ventilation, the National Institute for Occupational Safety and Health (NIOSH) found similar carboxyhemoglobin levels in bridge (4.9%) and tunnel (4.5%) officers. However, the findings of this study appear not to have been considered by those concerned about adverse effects of PM, perhaps because the authors attributed the elevated mortality to carbon monoxide (CO), calling concerns about effects of “nitric oxides, hydrocarbons, particulates, lead, sulfur, or aldehydes … highly speculative.”
Six cities and American Cancer Society studies
We next review the two pivotal studies of the early 1990s, the Six Cities study (Dockery et al., Citation1993) and the American Cancer Society (ACS) study (Pope et al., Citation1995). Both studies examined sulfate, and Dockery et al. (Citation1993) examined aerosol acidity, but no other PM species were included in these studies. The possible role of specific industrial emissions, perhaps of importance in each study, is not discussed. Associations between lung cancer and PM are discussed, but studies that found extremely elevated lung cancer rates among coke oven workers are not cited (e.g., Cohen and Pope, Citation1995). The International Agency for Research on Cancer (IARC) 1988 (IARC, Citation2012) finding that diesel emissions were a probable cause of lung cancer were not noted, nor were BC, EC, diesel, and PAHs mentioned. The cursory summary in this paragraph does not do justice to either study, nor to our fuller review of them; readers are urged to read the more complete summary in the Supplemental Material. The main point here is the lack of emphasis in these studies on vehicular or carbonaceous emissions.
CASAC and NRC considerations
In 1996, the U.S. EPA advisor, the Clean Air Scientific Advisory Committee (CASAC), sent a letter to the U.S. EPA Administrator (CASAC, Citation1996) as part of the National Ambient Air Quality Standards (NAAQS) setting process for PM2.5. The letter noted that the CASAC members were divided as to whether there should be new PM2.5 standards (in particular, a new annual PM2.5 standard), and how stringent the regulations should be, because of a variety of “unanswered questions and uncertainties” detailed in the letter. The reasons included exposure misclassification, measurement error, influence of confounders, lack of understanding of biological mechanisms, and possible alternative explanations. These uncertainties echoed those of Evans et al. (1984) and foreshadowed studies performed post 2000.
In 1998, the National Research Council (NRC) released a report (“Research Priorities for Airborne Particulate Matter”) that reviewed and reported on aspects of the U.S. EPA research plan for particulate matter (PM) (NRC, Citation1998). The report recognized the research need to pinpoint which types of particles are harmful, and their mechanism of causing harm. This report states:
The biological basis of most of the [PM] associations is essentially unknown … There is … limited scientific information about the specific types of particles that might cause adverse health effects, … [and] the toxicological mechanisms by which the particles might cause adverse health effects … [Important research priorities include] (NRC 1998, 2)
Assess through toxicological and epidemiological studies the most biologically important physical and chemical characteristics and constituents of particulate matter that produce adverse health effects…
Investigate the toxicological mechanisms by which particulate matter produces mortality and acute or chronic morbidity, using laboratory-animal models, human clinical studies, and in vitro test systems … (NRC 1998, 6)
The Committee recommends that EPA consider more fully the possibility that future research results might indicate that the expensive monitoring program is not measuring the most biologically important aspects of particulate matter.
As such, this report in conjunction with the CASAC letter might be viewed as the end of an initial era of PM research, in which researchers became satisfied that statistical associations between PM and mortality and morbidity reflected causation but did not yet understand how biological effects occurred and, thus, which types of particles might be causally involved.
2006 AWMA Critical Review
Pope and Dockery (Citation2006), the 2006 Air & Waste Management Association (AWMA) Critical Review (CR), can be seen both as the summary of our knowledge about PM2.5 effects at the time and as the beginning of the transition to fully examining effects of specific PM2.5 species, rather than of only total PM2.5 mass. The great majority of studies in the tables in Pope and Dockery (Citation2006) used PM2.5 or PM10 as the measure of pollution associated with mortality or morbidity. However, several European studies associating black smoke with mortality and two studies associating mortality with proximity to major roads are cited. Diesel is mentioned several times, in reference to toxicology studies. Twenty studies of HRV changes are summarized, but none include BC as the pollutant of interest. Birth outcomes are discussed in relation to PM, not in relation to vehicular emissions, but it is noted that much remains to be understood. Carbon is mentioned briefly, noting that it is part of the PM mixture. The issue of co-pollutants is introduced; co-pollutants to PM from traffic were said to include secondary nitrates and coexposure to NO2 and CO, but neither PAHs nor vehicular hazardous air pollutants (HAPs) are mentioned. Exposure misclassification is briefly mentioned.
Pope and Dockery (Citation2006) concluded that the evidence for PM2.5 harming human health is substantial, partly because of consistent results among different researchers, using different study designs (the first of the U.S. EPA criteria described earlier in this review). Progress in understanding potential biological mechanisms (inflammation, oxidative stress, HRV, pro-thrombotic effects, modulated immunity, etc.) is discussed, with a finding that none of these pathways have been adequately explored. One of the major research gaps noted is the “role of various characteristics and constituents of PM, and … the relative importance of various sources and related co-pollutants,” and that “there remains a need to further elucidate the biological mechanisms.”
At the time Pope and Dockery (Citation2006) appeared, the great majority of studies linking diesel emissions, or BC, to various health effects had not yet been published. These studies include: population-based epidemiology in which BC and several other PM2.5 species are specifically included in the model (Tables S3, S4, and S5); human panel studies in which BC and other emissions are in the model (Tables S6 and S7); controlled human exposure studies of diesel, biomass smoke, and BC (Table S8); studies associating BC and traffic emissions with adverse birth outcomes (Table S9); and studies associating BC and traffic emissions with effects on the brain, cognition and behavior (Table S10). Again, all tables except are found in the Supplementary Material.
In the upcoming sections and in the Tables in the Supplementary Material, we discuss 139 of these newer studies, all of which include BC, traffic density, diesel emissions per se, or other measures of vehicular emissions, and virtually all of which find associations with BC or other measures of vehicular emissions. A large majority of the findings are with BC. Many studies are “highway proximity” studies, associating adverse health effects for those living in close proximity to major roads (Table S2), versus those living further away.
Studies examining associations between health endpoints and specific components of PM2.5 and/or PM10 continue to be published. Billionet et al. (Citation2012) and Domenici et al. (Citation2010), for example, discuss PM species in the context of multipollutant approaches. Billionet et al. (Citation2012) state that a major challenge in examining multipollutant mixtures is to distinguish harmful pollutants from surrogates without direct causal effects. Domenici et al. (Citation2010) also addressed this theme, finding that “PM chemical composition varies greatly across the U.S … and chemical composition may affect toxicity.” Domenici et al. (Citation2010) continue: “High-priority questions … have shifted from health effects of total mass to … size fractions [and] chemical components … primarily responsible for various health outcomes.” Thus, these newer PM studies continue the themes noted already, namely, that different PM2.5 species have different toxicities, and that researchers need to distinguish harmful species from those that are merely acting as surrogates for harmful PM2.5 species. As our interest in this current CR lies specifically with BC and related PM2.5 species, we do not review further studies that do not include BC (and in almost all cases, many other PM2.5 species), with very limited exceptions.
The next era (roughly, post 2001) gradually expanded the research community's understanding of those methodologies important in determining which PM species are most, or least, harmful. This understanding remains incomplete. But as the next section shows, the research community has learned a great deal more in recent years.
Importance of exposure determination for epidemiologic studies and toxicology
In this section, we focus on all-cause and cardiovascular mortality, and cardiovascular morbidity effects, leaving out the substantial literature regarding respiratory effects due to length limitations. The Health Effects Institute (HEI) Research Special Report 17 (HEI, Citation2010) is an excellent discussion of respiratory issues related to diesel and vehicular emissions
There are several specific indoor pollutants in North America and Europe (e.g., cooking, wood smoke from fireplaces and poorly ventilated wood stoves, smoking tobacco), but indoor sources in these locations are not discussed for reasons of space. Indoor sources elsewhere, such as heating with coal and biomass and cooking with biomass, that produce very high PM levels are of major public health interest, and are discussed in a later section.
With regard to exposure to outdoor pollutants, until fairly recently, an outdoor monitor reading (or average of readings from several monitors) was used as the surrogate for actual personal exposure. The PM concentrations were usually annual averages, daily averages, or multiday averages.
Improvements in methodologies, and empirical studies in the United States, have now demonstrated that central monitors can present grossly inaccurate exposure information for epidemiological studies for emissions, such as vehicular, having substantial local spatial variability. BC and other vehicular emissions can vary by a factor of 10 over just one section of a metro area (Maciejczyk et al., Citation2004), but central monitor measurements historically assigned the same concentration as a proxy for exposure to everyone in the entire area. Thus, central monitor concentrations may greatly overestimate, or underestimate, actual subject exposures.
Two recent studies found significant health effects with locally variable emissions when exposure was measured on specific study subjects, or inside and outside a subject's residence, but not when a central monitor exposure proxy was considered in the analysis (Suh and Zanobetti, Citation2010, for BC; Hsu et al., Citation2011, for Ni). Thus, reasonably accurate exposure estimation is crucial for improving accuracy of observed statistical associations, for both long-term population-based epidemiology and studies of short-term acute effects. For example, concentrations from a monitor placed in the outside or inside of a home, or from a model that can portray yearly average pollutants at a residence, will for most people in an urban area be a superior proxy for actual exposure than would concentrations from a central monitor for the same time period, because these will express actual subject exposure more accurately. However, activity patterns will differ for people in the same house, which can mean that different residents of the home may have somewhat different exposure to spatially variable emissions such as BC (Dons et al., Citation2011).
If a pollutant's “measured” exposure poorly represents actual exposure, statistical associations might “migrate” from poorly measured variables to variables that better express subject exposure (less measurement error) (Goldberg and Burnett, Citation2003; Grahame, Citation2009). Grahame (Citation2009) examined whether associations of BC/EC with changes in measures of heart-rate variability (HRV; variation in the interval between consecutive beats) would vary among studies, depending on whether exposure to BC/EC concentrations were estimated from a central monitor or from a more accurate method of estimating subject exposure. In studies where BC/EC exposure was reasonably accurate (see Grahame [Citation2009] for different definitions of “reasonably accurate”), either BC/EC or urban air was significantly associated with adverse HRV changes, but sulfate or regional air masses with high PM2.5 mass were not. However, in studies with the poorest BC/EC exposure, BC/EC was not, but sulfate was, associated with HRV outcomes. This suggests the possibility of associations “migrating” from poorly monitored PM2.5 species to better monitored ones. These findings were later supported by the results of Suh and Zanobetti (Citation2010), in that BC associations with HRV were found only when subject exposure was accurate, but sulfate was not so associated.
Among the first post-2000 epidemiological findings showing the potential importance of vehicular emissions to health were “highway proximity” studies. Highway emissions, such as BC and ultrafine PM, have been shown to be most elevated within 100 m of major roads (Zhu et al., Citation2002 [Los Angeles]; Roorda-Knape et al., Citation1998 [Europeans cities, black smoke]). Therefore, comparing effects for those living in close proximity to major roads with effects for those living further away might provide a better understanding of health consequences of highway emissions than a single BC concentration, as if one concentration accurately represented everyone's exposure to BC (or other primary vehicular emissions) across a metropolitan area.
Early studies of health effects for those living within 100 meters of highways or within 50 meters of a major urban road showed elevated rates of mortality compared to those living further away (e.g., Hoek et al. [Citation2002] for cardiopulmonary mortality). Finkelstein et al. (Citation2004a) found elevated risks of all-cause mortality related to the same proximity to roadways, with a mortality rate advancement period of 2.5 years for those living closer relative to those living farther away. Finkelstein et al. (Citation2005) found sharply elevated risks of circulatory disease mortality for those living in proximity to roadways (relative risks = 1.40, CI = 1.08 to 1.81). Furthermore, some studies showed that health effects were more elevated if nearby roads were travelled by diesel emitters, such as buses and trucks (Medina-Ramon, Citation2008; Lin et al., Citation2002), suggesting that perhaps a marker for diesels, such as BC, might be as good a marker or a better marker for exposure than proximity to a major road, assuming good subject exposure assessment.
Other methods for determining more accurate BC exposure were developed that used:
Monitors inside and outside homes of retirees (CitationDelfino et al., 2008, Citation2009).
Mobile monitors that travelled with retirees (Adar et al., 2007).
Monitors within 100 m of a major urban road, when the subjects also lived in close proximity to the road, not far from the monitor (Gold et al., Citation2005; Schwartz et al., Citation2005a).
Concurrently, new methods for assessing effects of diesel emissions, and identifying biological mechanisms of harm, were being used. For example, Sauvain et al. (Citation2011) found that oxidative stress levels in workers at a bus depot were elevated after a workday, compared to before work, and were more highly elevated at the end of a second consecutive workday than after the first workday. Relative to the general U.S. population, workers in the trucking industry most consistently exposed to higher levels of diesel exhaust (drivers and dock workers) had significantly elevated rates of both lung cancer and ischemic heart disease (Laden et al., Citation2007), while those with less exposure (hostlers and clerks) did not. Finkelstein et al. (Citation2004b) found that ischemic heart disease mortality was significantly elevated among heavy equipment operators, relative to other, less exposed union members in Ontario, Canada.
In addition to the issue of better exposure assessment, there is the issue of components (PM2.5 species) used within any model. If a health-relevant PM2.5 species is missing from a model, two consequences follow: (1) Effects cannot be found for a pollutant not in the model; and (2) the model might suggest associations with pollutants included in the model that might not have been so dentified, had a more health-relevant pollutant been included (e.g., associations might “migrate” from a pollutant not in a model, to a pollutant included in a model). Furthermore, data on percentage of residences with central air conditioning (AC) are useful, in multicity and multicounty studies, because on hot days in warmer cities, with much central AC (with little infiltration of outdoor air), people tend to be inside in homes. In parts of the United States such as the Northeast, with less central AC, opening windows is more prevalent on hot summer days (Bell et al., Citation2009b). Thus, the percentage of residences with central AC modifies exposure to outdoor pollutants (Janssen et al., Citation2002; Bell et al., Citation2009b). Studies that utilize this knowledge, in conjunction with inclusion of many relevant PM2.5 species, may produce more dependable results due to improved exposure assessment.
Source apportionment studies
Receptor modeling studies since 2000 include source apportionment methods such as chemical mass balance and factor analyses (Watson and Chow, Citation2005b). Factor analysis examines the correlations between various PM and gaseous pollutant components so as to differentiate pollution information into weighted “factors.” Each factor is characterized by one or more PM2.5 species.
More than 100 of these studies, relating “factors” (which can sometimes be associated with source types) with adverse health outcomes, have been published. Although these analyses vary both in the numbers of factors identified (even for the same locations) and in the factors they potentially associate with health outcomes, factor analyses as a whole appear to share at least four commonalities:
The number of factors identified, and the percentage of PM in each factor, usually vary as additional relevant emissions are included in the model. Therefore, the meaningful number of factors, and the mass of pollutants in each factor, cannot easily be quantitatively determined.
Those factors associated with health outcomes often change as additional pollutants are included in a model, or as different model methodologies are employed.
Markers or compounds that are taken to represent one source may be emitted by several sources, complicating attribution and possible health interpretations.
Because individual exposure information, or reasonable proxies for such exposure, may not be available, particularly for emissions which are more spatially variable, factor analyses can have the same exposure misclassification issues as do population-based epidemiology studies using central monitors (e.g., Suh and Zanobetti, Citation2010).
These issues are discussed briefly in HEI (Citation2010) and at length in Grahame and Hidy (Citation2007a). The implication is that factor analyses and other forms of source apportionment may face challenges in adequately interpreting any adverse health associations that may appear to be revealed in such a study. Liu et al. (Citation2005, Citation2006) illustrate how factors change in number and magnitude as additional relevant pollutants are added to the model. (The following four paragraphs are extracted from Grahame and Hidy [Citation2007a], with limited alteration [no italics in original].)
Liu et al. (Citation2005) [… identified …] 10 factors, including a secondary sulfate factor, a primary coal-combustion emissions factor, and a vehicular emissions factor [note: no separation was made into diesel vs. gasoline emissions]. Data were taken from four localities, two urban (Atlanta, GA, and Birmingham, AL) and two rural (Yorkville, GA, and Centreville, AL).
The coal combustion emissions factor had three features suggesting possible co-mingling of coal emissions with urban emissions in the two urban areas. First, the EC/Se ratios in this “coal” factor in the urban locations were 5 to 10 times higher than in the two rural locations … Secondly, the coal emissions factor was enriched with Ca only in the two urban locations. Third, the coal combustion factor exhibited a weekday–weekend emissions variation that [… was …] unlikely to reflect variability in power plant operations, but that could reflect differences in weekday versus weekend use of diesel vehicle transportation.
… Liu et al. (Citation2006) … expanded their analysis of data from the same sites to include a total of eight thermally-differentiated carbon fractions and four gaseous components (CO, SO2, NOy, and HNO3). The authors were now able to estimate factors associated with diesel and gasoline emissions, separately, in three of the four localities (including both urban locations). Liu et al. (Citation2005), however, had been unable to resolve vehicular emissions at either rural location. The factor attributed to primary coal combustion reported by Liu et al. (Citation2006) decreased in magnitude between the first and second studies, dropping from 3% of PM2.5 mass to 1.9% in Atlanta, from 6.0% to 0.7% in Yorkville, and from 5.0% to 1.5% in Centreville. In absolute terms, the coal combustion factor in Liu et al. (Citation2006) represented 100 ng/m3 of PM2.5 in Yorkville, and 190 ng/m3 in Centreville. In the eastern United States, PM2.5 concentrations of this magnitude are reasonable estimates of coal fly ash from coal fired power plants (William Aljoe, NETL, personal communication).
The contrast in findings between the two studies suggests the importance of employing as many specific PM2.5 and gaseous components as possible to enhance the ability to identify factors, to minimize co-mingling of PM2.5 species into different factors, and to more accurately determine the size of factors. We are not aware of any other factor analysis other than Liu et al. (Citation2006) to utilize this number of added emissions.
Laden et al. (Citation2000) further illustrates potential issues with source apportionment models. The authors used PM2.5 species in each of the Six Cities to construct several factors, including ones marked by Pb (traffic factor), Se (coal combustion factor), and Si (soil and crustal material). (Pb was used, apparently, because BC/EC was not routinely monitored in the time frame and locations of the study.) For the Pb (traffic) factor, significant associations were found for risks of daily mortality in two of the cities, including one of the two large ones (St. Louis, MO) but not the other (Boston, MA), and in combined data for all six locations. For the Si (soil and crustal material) factor, there were no significant positive associations. For the Se (coal combustion) factor, associations were found only in Boston and in the combined data. In an assessment with increase in mass of additional elements, sulfur (as sulfate) showed the same pattern as that for Se, that is, significant associations only in Boston among the cities, and in the combined data. The findings for the Se factor showed the opposite of a dose-response function (World Health Organization [WHO], Citation2009), in that Se levels in Boston were the lowest among all locations (7 times lower than in Steubenville, OH, 3 times lower than in St. Louis, MO). A similar pattern was found for sulfate, with higher sulfate levels in St. Louis, Steubenville, and Harriman, TN, than in Boston.
Using U.S. EPA data, Grahame and Hidy (Citation2004) found that in Boston, about three-fourths of the Se appeared to result from burning of residual oil for electricity generation (there were 1700 MW of residual oil power plants within 8 km of the monitor), and about half the sulfate was also from local residual oil power production. Rahn and Lowenthal (Citation1985) illustrated similar results; that is, half the pollution found at monitors in two New England locations resulted from local combustion marked by high levels of vanadium (V), a marker for residual oil use (widespread in New England at the time), even though Midwestern SO2 emissions were 10 times higher than were those in New England.
Thus, Grahame and Hidy (Citation2004) suggested that Se was not a marker for one source (nor was sulfate), but rather two sources, with substantial amounts of Se and sulfate coming from residual oil burning, but only in Boston. The known toxicity of residual oil components caused Se to be associated with daily mortality in Boston. The apparent finding is that co-mingled emissions from one of these sources (residual oil burning) might more likely be causally related to daily mortality. Finally, if much of the Se in Boston is from a different source than elsewhere, it would be inappropriate to combine data to look for an association if Se represented differing sources of emissions having disparate toxicities in different localities. The finding that associations might be the result of such confounding is one of the concerns highlighted by Evans et al. (1984).
A source apportionment study with accurate exposure should, in theory, provide more reliable health associations with potential source types (factors). The Riediker et al. (Citation2004) study of troopers in their cars, working mainly on freeways and busy streets, may meet this criterion. The authors looked only for factors derived from in-vehicle exposure, reflecting on-road and near-road emissions or dust particles. Such a study improves the ability to create factors accurately reflecting composition of near-roadway and roadway sources.
The preceding discussion suggests that most source apportionment studies may, in fact, add uncertainty, instead of reducing uncertainty, in attempting to determine which PM species are particularly harmful—or less so—to humans. This is because we cannot know whether a given study has identified the correct number of factors, and whether the factors are appropriately identified or whether there has been co-mingling of common species among factors, and because subject exposure to spatially variable “factors” will be poor. In our view, these uncertainties are why factor analyses so often give contradictory results, even for similar locations. With a goal of reducing uncertainty, we refrain from discussing further source apportionment analyses that do not meet the exposure criteria in the preceding paragraph.
Modeling pollution levels to residences for use in epidemiologic studies
By the late 2000s, several research groups modeled pollutants to residences of subjects admitted to hospitals or deceased, as a way of improving the ability to accurately link exposures to spatially variable emissions with adverse health effects. Maynard et al. (Citation2007) used BC, while Jerrett et al. (Citation2009) used NO2 as a proxy for exposure to vehicular emissions. Furthermore, attempts have been made to make better use of available data in studies where accurate exposure information is not available. These new studies attempt to use different assessment methodologies than those previously employed (Mostofsky et al., 2012).
Epidemiology studies using improved methodologies to assess all-cause and cardiovascular/cardiopulmonary mortality
Highway proximity studies and studies of traffic volume near residences
Table S2 presents results of 15 available studies that associate mortality risks with close proximity to highways (100 m unless noted otherwise) and/or major urban roads (50 m unless noted otherwise) in North America and Western Europe. These are distances in which roadway emissions are substantially elevated (Zhu et al., Citation2002; Roorda-Knape et al., Citation1998), relative to further distance from roads. Table S2 also includes studies that examine morbidity (CVD or stroke) associations with high levels of traffic density near residences.
These studies include cohorts ranging from 1389 to more than 121,000 people, and found that close proximity to roadways and major urban roads, and/or higher traffic density, are associated with early mortality, all-cause mortality, and/or cardiovascular or cardiopulmonary mortality. The reduction of both vehicular pollution and mortality, with greater distance from major roadways, is similar to the reduction of mortality in , where pollution was reduced not by increasing distance from roads, but by ventilation of roadway tunnels in which tunnel officers worked. In both cases, a reduction in vehicular pollution was associated with significant reduction in mortality.
A number of additional studies associate close residential distance to highways with increased symptoms related to cardiovascular and other disease states. Examples of such studies include Hoffman et al. (Citation2007) with incidence of coronary heart disease; Jacobs et al. (Citation2011) with increased carbon load in airway macrophages and increased oxidized low-density lipoprotein (LDL); Hoffman et al. (Citation2006) with increased coronary artery calcification; CitationHoffmann et al. (2009) with increased risks of peripheral artery disease for those living within 50 m of a major road, versus 200 m or more distant; Lue et al. (Citation2013) with reduced renal function (glomerular filtration rate) equivalent to a 4-year increase in biological age; Van Hee et al. (Citation2009) with higher left ventricular mass index (LVMI), equivalent to 5.6 mm Hg greater systolic blood pressure; and Kuenzli et al. (Citation2010) with progression of atherosclerosis (like Van Hee et al. [Citation2009], Kuenzli et al. [Citation2010] found that total PM2.5 mass associations failed to reach significance).
While Allen et al. (Citation2009) did not find any associations between proximity to traffic and development of aortic atherosclerosis, almost half the subjects in Allen et al. (Citation2009) were from New York and Chicago. Since Restrepo et al. (Citation2004) showed that highway pollutants rapidly become less concentrated with vertical distance from a highway, Allen et al. (Citation2009) consequently speculated that associations with vehicular emissions might have been reduced for subjects living in high-rise buildings in these localities.
Effects found in the studies just noted parallel those of Peters et al. (Citation2004), who found a strong association between having an initial myocardial infarction (MI) and being in traffic an hour prior to the heart attack—whether in a car, on mass transit, or on a bicycle.
Studies beginning in 2002 shown in Table S2 were groundbreaking in that they used improved exposure information regarding highway emissions to suggest that the elevated pollution near such roads may be causally related to various forms of mortality, especially from cardiovascular and cardiopulmonary causes. Although this breakthrough in exposure assessment to locally variable emissions clarified that living near major roads is likely to accelerate CVD disease states and reduce life expectancy, other exposure assessment methods might offer further improvements in understanding how pollution from roadways cause these adverse health effects.
Highway proximity studies appear to fulfill the first criteria used by the U.S. EPA to determine causality, which is consistency in making similar findings among many studies, with different authors, in different locations.
In theory, assessing health effects associated with specific pollutants, with accurate subject exposure information for BC and other vehicular emissions (including those from tire and brake wear), could be at least as useful as highway proximity studies, for two reasons: (1) Roadways with similar traffic counts might substantially differ in numbers of diesel trucks and buses; and (2) traffic counts on major roads can substantially differ. In order to directly compare effects of different PM species, we now discuss studies associating measures of multiple pollutants with adverse health effects. Investigations using toxicology and human panel studies should aid identification of which specific pollutants might be causally related to disease.
Studies using BC/EC or other vehicular emissions, including several different centrally monitored PM2.5 species
Table S3 shows mortality associations from eight studies that use multiple PM species, including BC/EC, but that do not model pollutants to the home of subjects or decedents. As reasonably expected from studies that derive subject exposure for a large area from a central monitor, the results are not consistent for spatially variable emissions, because subject exposure among the studies can reflect different levels of exposure misclassification.
Lipfert et al. (Citation2006b, Citation2009) examined all-cause mortality associations with many different pollutants in 206 urban and rural counties across the United States. Lipfert et al. (Citation2006b) used 33 pollution measures, 15 of which were elements (mostly metals), plus EC, OC, sulfate, nitrate, various gases, and traffic density. The most consistent associations among single and multiple pollutant models are with traffic density; both single- and multipollutant models show strong associations with EC, nitrate, Ni, and V. Lipfert et al. (Citation2009) used 12 HAPs (hazardous air pollutants, classified by the U.S. EPA, three of which are diesel exhaust, characterized by diesel particulate matter [DPM], benzene, and formaldehyde) plus EC, sulfate, oxides of nitrogen, SO2, and traffic density. Lipfert et al. (Citation2009) found strong associations with traffic density, but found that associations were strongest with traffic-related air pollutants, that is, EC, DPM, oxides of nitrogen, benzene, and formaldehyde; mortality risks were significant for Ni but not for Si. Neither Lipfert et al. study found any associations with sulfate. However, Lipfert et al. (Citation2009) found significant mortality risk associations with SO2, but only in high-traffic-density counties, “where SO2 appears to be a surrogate variable,” presumably related to vehicular emissions. Once again, an association was identified that might represent causality by a co-emission, not by the “surrogate variable.”
Lipfert et al. (Citation2006a) included traffic density with a smaller set of pollutants: PM2.5, sulfate, nonsulfate PM2.5, coarse PM, peak CO, NO2, and peak O3. Increased traffic density was significantly associated with increased mortality risks, in one-, two-, three-, and four-pollutant models. Only peak O3 was associated with increased mortality risks in multipollutants models, but in single-pollutant models, PM2.5, nonsulfate PM2.5, coarse PM, and peak O3 were associated. BC was not monitored, but roughly implied in traffic density measures, similar to the way proximity to major roads roughly implies BC.
In another cohort study, Ostro et al. (Citation2007) examined associations of 14 elements plus PM2.5, EC, OC, nitrate, and sulfate, with all-cause, CVD, respiratory, and daily mortality for people >65 years of age, for each of two time periods (annual, winter months), thus allowing for up to eight associations with daily mortality for a given pollutant. The authors found EC associations for risks of CVD mortality annually. A number of associations were also found for PM2.5 and several PM2.5 metal species; the authors noted that many of the PM species appeared to be associated with either vehicular emissions or with tire or brake wear (e.g., EC, Cu, Zn). Si was associated with all-cause mortality risks and risks in winter months for those >65 years of age, while Ni was not associated with any adverse health outcomes. Ostro et al. (Citation2007, Citation2010) noted that exposure misclassification might have attenuated associations with spatially variable emissions, due to use of central monitor data as a proxy for exposure for people across a wide area, and also noted that associations with a given emission might reflect either toxicity of that emission or of a substance highly correlated with that emission.
Ostro et al. (Citation2010, including Ostro et al. [Citation2011] erratum) examined associations for eight PM2.5 species at two different geographic scales: for those living within 8 km of a monitor, and for those living within 30 km of a monitor. For those living within 8 km, daily mortality associations were found with all-cause, cardiopulmonary, and ischemic heart disease (IHD) mortality, for all eight pollutants in the model, including EC. When restricted to only study subjects within 30 km of a monitor (data from 2011 erratum), no associations with all-cause or pulmonary mortality were identified. All pollutants were associated with IHD mortality, and several pollutants (excluding EC) were associated with cardiopulmonary mortality. Si was among the pollutants examined, but Ni was not.
Klemm et al. (2011) analyzed daily mortality for a period of 9 years and 5 months in four counties in Atlanta, GA, examining associations with 24-hr average PM2.5, EC, OC, NO3, and SO4; maximum 3-hr O3; and maximum 1-hr NO2, CO, and SO2. All-cause mortality associations were found for same day PM2.5, EC, OC, and CO, suggesting the importance of traffic emissions.
Franklin et al. (2008) used a two-stage procedure. First, they examined the association of PM2.5 with daily mortality in each of 25 different communities (2000–2005 mortality data from same database as in Krall et al., Citation2013, described next). Then, using meta-analysis, they examined how the pooled seasonal associations might be modified by any of 18 emissions, including BC, OC, sulfate, nitrate, the ammonium ion, and various metals. They concluded that the magnitude of PM2.5 mortality associations increased when Al, Si, As, S, and Ni were a larger proportion of PM2.5. This study thus suggested that fugitive dusts containing Al and Si were more harmful than “average” PM2.5, but that BC was not. The authors discussed the importance of exposure error within a city, but also pointed out that the coefficients of variation for BC in their study were small because the city-based ratios of BC to PM showed low variability across the 25 communities examined.
Krall et al. (Citation2013) used death certificate data from the National Center for Health Statistics database to examine short-term mortality associations (2000–2005) in 72 U.S cities. Pollution data included PM2.5 and the seven most abundant PM2.5 species in ambient U.S. air: ammonium ion, EC, nitrate, organic carbon matter (OCM), Si, sodium ion, and sulfate. Krall et al. employed a two-level Bayesian hierarchical model to estimate mortality risk. Significant mortality risk associations were found for PM2.5, OCM, EC, Si, and the sodium ion. The authors found no seasonal or regional variation between PM2.5 and mortality risks; nor did mortality risk vary by region or season for the four PM species with significant associations. Associations with the sodium ion remain perplexing, but a potential hypothesis could be that since a main source of Ni in most parts of the United States is residual oil in shipping, perhaps the sodium ion (most likely from sea salt) might be correlated with Ni, with the effects of Ni perhaps being to some extent attributed to sodium.
The findings of Krall et al. (Citation2013) regarding EC suggest that the significant and positive results with EC in the Lipfert et al. studies relative to Franklin et al. (2008) may be due to greater differences in BC concentrations among 206 counties, or to greater ratios of BC to PM. Differences in findings might result partially from the use of a Bayesian hierarchical approach in Krall et al. (Citation2013), and/or to the larger number of localities used in Krall et al. (Citation2013).
The results of the studies in Table S3 may be summarized as follows with regard to vehicular emissions: Using different methodologies, studying either daily or long-term mortality, traffic density was always robustly associated with mortality risks, and BC/EC was also associated with mortality endpoints, with the exception of one study. Other PM2.5 species were occasionally associated with mortality risks, but less frequently than traffic density or BC. In the Ostro et al. studies, PM species other than EC were found to be significantly associated with a mortality endpoint more frequently than EC.
Janssen et al. (Citation2002) used PM data representing several different sources to examine morbidity effects, that is, whether associations with hospital admissions for heart and lung diseases increased as the proportion of PM10 from different sources increased. The authors obtained U.S. EPA data on PM10 emissions by emissions source, vehicle miles traveled (VMT), and population density, for 14 localities across the United States. Janssen et al. (Citation2002) was the first study to examine whether prevalence of central air conditioning modified pollutant associations. For CVD admissions, in single-pollutant models, significant associations were found for increasing proportion of PM10 from highway vehicles, highway diesels, oil combustion, and metal processing, as well as with population density and VMT/mile. No associations were found for increasing PM10 from coal or wood combustion; a negative significant association was found for fugitive dust PM. In multipollutant models, associations were found only with PM10 from highway vehicles/diesels and oil combustion. Cities with higher percentages of air conditioning had lower CVD hospital admission rates. The authors concluded that both percentage of central air conditioning and proportion of PM from highway vehicles/diesels modified the effect on CVD related hospital admissions.
Table S4 describes nine studies that examined associations of daily hospital admissions with many pollutants, including EC/BC. EC (and other measures of traffic pollution such as CO) was significantly associated with hospital admissions in each study. This summary finding was not observed for any other PM2.5 species. In the two studies that included Ni and V among the PM species, both were significantly associated with hospital admissions (Ni and V are indicators of residual oil combustion). Si was not found to be significantly associated with daily hospital admissions in any of the four studies that included Si among the PM species assessed. Again, despite the BC/EC associations, exposure misclassification might have caused BC risk to be underestimated.
Comparing the studies in Tables S3 and S4, studies that use central monitor data and that include many pollutants, it appears that EC and other measures of traffic emissions were statistically associated with the health endpoint considered in all but one study. No other PM2.5 species was associated as frequently with the health effect analyzed. However, Ni was significantly associated with adverse health risks in five of the six studies, whereas Si was associated with adverse health endpoints in only four of the nine studies.
Thus, Tables S3 and S4 again show consistent, significant health associations with regard to BC/EC and/or traffic density, in different geographic areas, in studies using different study designs, again fulfilling the first U.S. EPA criterion for assessing causality, consistency of associations by multiple research designs in multiple settings.
Studies modeling vehicular emissions to residences
The findings of the multipollutant studies already discussed may be compromised by exposure misclassification, since the studies used centrally monitored emissions data for estimated exposure. In this section we examine studies that modeled traffic emissions of various types to residences of subjects, thus reducing exposure misclassification, and then compare results to those in the previous section. The studies in Table S5 represent substantial geographic diversity among countries in North America and Western Europe.
Gan et al. (Citation2011) modeled four pollutants (BC, NO2, NO, PM2.5) at >450,000 residential homes in Vancouver, Canada, in which the residents were not known to have coronary heart disease (CHD) at baseline. The risks of CHD hospitalization or mortality were examined using BC, NO2, and NO as markers for vehicular emissions. BC was associated with risks of both CHD endpoints with “clear linear exposure-response relationships between BC and coronary events.” PM2.5 was associated with increased risk of hospitalization only, without evidence of a linear relationship. Linear relationships were found for NO2 and NO with CHD mortality; the relationships were generally attenuated after adjustment for BC, suggesting the greater importance of BC.
Jerrett et al. (Citation2009) modeled NO2, O3, and PM2.5 to the homes of 2360 subjects served by a clinic in Toronto, Canada. NO2 was seen as a marker for traffic emissions, not as a causal factor. Associations were found for increased all-nonaccidental and circulatory mortality for an IQR increase in NO2 concentrations. NO2 associations were lower in magnitude but more significant than associations for distance from highways. The authors suggest that modeled vehicular emissions may have less exposure error than “distance to highway,” which does not account for traffic types and amounts. A companion study by Beckerman et al. (Citation2012) found that an increase from the 10th to the 90th percentile of NO2 concentrations was associated with significant risks of IHD.
Chen et al. (Citation2013b) used a random cohort of over 205,000 residents, aged 35 to 85 years, and modeled NO2 levels in three cities in Ontario, Canada, to examine associations with CVD, IHD, and cerebrovascular mortality. They found that each increase of NO2 levels by 5 ppb was significantly associated with increases in CVD and IHD mortality.
In a case-control study, Neupane et al. (Citation2010) modeled average annual NO2, SO2, and PM2.5 concentrations to homes of residents of the industrial (steelmaking) city of Hamilton, Ontario, Canada. The authors found that long term exposure to NO2 and PM2.5, but not to SO2, were associated with increased risks of hospital admissions for pneumonia for people over 65 years of age.
In a case-crossover study, Maynard et al. (Citation2007) examined approximately 100,000 deaths from 1995–2002 in the Boston, MA, area. The authors modeled BC to the homes of residents, and acquired sulfate data from a monitor adjacent to a major urban road with bus and truck traffic. Interquartile range (IQR) increases in previous day's BC and sulfate were associated with increases of 2.3% and 1.1% in all-cause daily mortality, respectively, in single-pollutant models. In a two-pollutant model, BC risks remained significantly associated and nearly identical (2.2% increase), but sulfate risks were no longer significant. Reponen et al. (Citation2003) found sulfate gradients away from major roads, reflecting levels of sulfur in fuels in the period studied by Maynard et al. (Citation2007). Therefore, the location of the monitor adjacent to a major road might suggest commingling of sulfate from long distance transport with vehicular sulfate, possibly explaining why the sulfate association disappeared in a two pollutant model incorporating BC (Grahame and Hidy, Citation2007b).
In another case-control study, Tonne et al. (Citation2007) examined 4565 survivors of acute MIs in the greater Worcester, MA, area. A pollution variable for traffic emissions was created using PM2.5 filter light absorbance and NO2. This variable was then modeled to the homes of subjects. An IQR increase in concentrations of traffic particles was associated with a 10% increase in odds of an acute MI (Tonne et al., Citation2007).
Wellenius et al. (Citation2012) modeled BC to the homes of 1705 subjects who were hospitalized with acute ischemic stroke in the Boston, MA, area. Data on other pollutants (NO2, CO, O3, PM2.5, and SO4) were centrally monitored. Comparing 75th percentile to 25th percentile of pollutant in 24 hr preceding onset of stroke, significant associations for risk of ischemic stroke were found for PM2.5, estimated BC, and NO2. The authors viewed these results as suggesting that traffic emissions may be particularly harmful.
Both Brunekreef et al. (Citation2009) and Beelen et al. (Citation2009) utilized data from a cohort of about 120,000 subjects in the Netherlands. Brunekreef et al. (Citation2009) found mortality risks associated with a 10-μg/m3 increase in black smoke (BS), while similar-sized risks for PM2.5 were not found to be significant. When only the three largest Dutch cities were assessed, the magnitude of BS associations roughly tripled. One possible reason may be is that fresh vehicular emissions in the large urban areas, prior to atmospheric aging and agglomeration, might be more harmful, as suggested by Li et al. (Citation2003). The HEI review committee for this report suggested possible, not mutually exclusive explanations for the higher BS associations when only the three cities were considered: (1) Emissions in the three cities might be more toxic than in the country as a whole; and (2) greater exposure misclassification for the entire cohort, than when only subjects in the three largest cities were included. Beelen et al. (Citation2009) found large and significant associations for cerebrovascular mortality and for heart failure with BS, background (regional) NO2, and background PM2.5.
Beverland et al. (Citation2012) studied short- and long-term associations of modeled BS concentrations with all-cause mortality. Two cohorts were used: One cohort was of elderly subjects (Renfrew/Paisley cohort), living in counties adjacent to Glasgow, Scotland; the second was of younger people across central Scotland recruited in workplaces (collaborative cohort). Using a nested case-control modeling approach, the authors found a 1.8% increase in mortality for an increase of 10 μg/m3 (3-day mean exposure) in modeled BS in the Renfrew/Paisley cohort. This differed from a time-series study using central monitor BS data, where smaller associations (0.2% increase in mortality) were found (Beverland et al., Citation2007). Noting that a time-series study using central monitor data cannot illuminate an individual's specific characteristics, nor specific exposure, the authors suggested that the case-control approach using modeled BS may be superior on methodological grounds.
Newer methodologies when accurate exposure data are not available
Mostofsky et al. (2013) recognized that a deficit of “standard” models that simply associate PM2.5 and PM2.5 constituents in the model with health endpoints is that “constituents that are more strongly correlated with PM2.5 may appear more closely related to adverse health outcomes than other constituents even if they are not inherently more toxic.” Using 18 PM2.5 species (including BC plus elements such as Ni, V, Si, Se, and S), and examining associations with ischemic stroke, the authors created five new methods for examining associations of pollutants with adverse health outcomes, each with possible theoretical advantages relative to standard models, in order to gauge the toxicity of PM2.5 constituents. Two of the newer models were selected as being superior to the other three newer models. shows the comparison from these two new methods with the “standard” method. In all three methods, BC and Ni showed the largest incidence rates for ischemic stroke onset in the 24 hr following exposure among admittees to a Boston medical center. BC was always significantly associated, while Ni was marginally associated. No other emissions showed consistent positive associations that were close to significance, and several showed positive associations in the “standard” model but negative associations in one or both of the newer models (e.g., S, Na, Al, Br, Ca, Zn).
Figure 2. Incidence rate ratios and 95% confidence intervals for the association between an interquartile range increase in particle constituents and ischemic stroke onset in the following 24 hr among 1060 patients hospitalized for acute ischemic stroke who resided in the Boston, MA, metropolitan area, 2003–2008. The results are presented for models with parameters for constituent concentration (a), constituent concentration adjusted for total particulate matter (b), and constituent residuals (c). © Oxford University Press. Reproduced by permission of Oxford University Press. Permission to reuse must be obtained from the rightsholder.
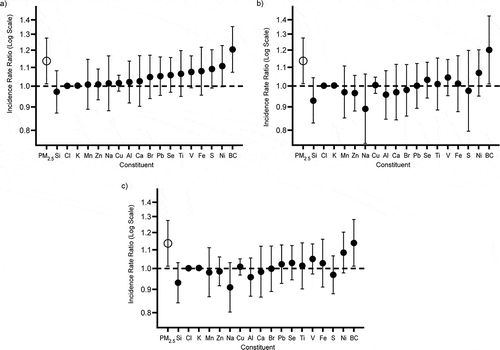
While other studies reviewed herein showed consistent associations with BC in different geographic areas, in different model formulations, and with different mixes of pollutants, Mostofsky et al. (2012) also showed consistent associations between BC and ischemic stroke, but in the context of different model formulations using the same data in each case.
Cardiovascular morbidity—human panel studies
This section examines studies regarding which pollutants may affect the development and/or exacerbation of cardiovascular and cardiopulmonary diseases. Particular advantages of human panel studies with a small number of patients are that researchers can acquire information about a subject's underlying health and can monitor emissions in close proximity to subjects in some cases. Knowing things like genetic capability to resist oxidative stress, whether subjects are taking statins and other drugs, and whether they have diabetes or are overweight, in addition to age, sex, health, and smoking status, can help researchers better understand interactions between pollution and health status, isolate effects of pollution, and identify subgroups that might be more susceptible to effects of pollutants. Among the health parameters studied were blood pressure, inflammation, oxidative stress, heart rate variability, platelet and lipoperoxidation parameters, arrhythmias, tachycardia, and telomere shortening. These studies can help link studies of all-cause and CVD mortality and CVD hospitalization, with biological mechanisms that may help explain associations between pollution and these health outcomes. Each of these studies examines BC and/or EC, and in the great majority of studies, other PM2.5 species as well. None of them include PM2.5 metals, making it impossible to examine health associations with Ni, for example.
We examine two sets of studies. One set is from the Harvard School of Public Health (HSPH) (Table S6), of which many studies used subjects from the Normative Aging Study, a long-studied cohort of veterans in the greater Boston area. These studies allow substantial comparison across health endpoints, and among studies with different degrees of exposure accuracy. The second set of studies is from researchers in southern California, led by Professor Ralph Delfino (Table S7), in which all the studies feature excellent subject exposure measurements. Although each group examines many of the same endpoints, they often do so with different biological methods, and include different sets of pollutants.
Harvard School of Public Health studies
The first 10 studies in Table S6 have good-to-excellent subject exposure. In each study, BC was strongly associated with health endpoints, including HRV (several different measures), ST-segment reduction, carotid intima-media thickness, cognitive function in elderly men, leukocyte telomere length (LTL), systolic and diastolic blood pressure, circulating biomarkers of inflammation, and endothelial response (soluble adhesion molecules). Suh and Zanobetti (Citation2010) and Schwartz et al. (Citation2005a) included BC with other pollutants (PM2.5 and either sulfate or “secondary PM,” lacking BC); either BC alone was associated with changes in the HRV endpoints, or PM2.5 associations were found to be significant only when highly correlated with BC, but not otherwise. Gold et al. (Citation2005) found BC but not PM2.5 to be associated with decreased ST-segment reductions (no other pollutants were included). In Adar et al. (2007), PM2.5 and BC associations were very similar, perhaps because when the subjects were in a diesel bus, both PM2.5 and BC concentrations were sharply higher than at other times. Six of the studies did not measure other pollutants, and thus do not allow for comparison of BC associations with those of other pollutants, nor do they allow evaluation as to whether BC associations might change if other pollutants were included in the model.
The next 27 studies in Table S6 use pollution concentrations from centrally located monitors (generally 10 to 50 ft above street level) as proxies for subject exposure. Associations with BC thus would be more likely to be attenuated than those for pollutants with less local spatial variability—for example, PM2.5 and sulfate, as stated in several of these studies. In most cases, subjects came from the greater Boston area, usually within 25 km of the clinic they visited. Health endpoints include most of those in the first 10 studies, plus several others. A short summary of the findings is as follows:
BC/EC: Examined in 27 studies, significantly associated with endpoint in 20.
PM2.5: Examined in 25 studies, significantly associated with endpoint in 20.
Sulfate: Examined in 14 studies, significantly associated with endpoint in 8.
The fact that BC is as likely to be associated with adverse endpoints as PM2.5, and more likely to be associated than sulfate, is unexpected, since with greater local spatial variability for BC, there should be more measurement error when central monitor concentrations are used as a proxy for subject exposure for BC, than for PM2.5 or sulfate. Greater measurement error for BC should bias BC associations toward the null. Therefore, these findings suggest that BC/EC (and vehicular emissions with which they are correlated) are more likely to be harmful, since they are associated with adverse endpoints more frequently than are emissions for which there is less spatial variation. Several of the studies specifically note that their results suggest effects from traffic. As with studies with good to excellent exposure in Table S6, which examined multiple PM species, BC and traffic emissions were more frequently associated with any given health endpoint than were either PM2.5 per se or other PM species.
The last 3 studies in Table S6 were conducted in Steubenville, OH, using a monitor on a hill over 100 m above, and ˜1.5 km from, subjects’ residences. Just as pollutant concentrations are largely dispersed within a hundred meters or so of horizontal distance from major roadways (Zhu et al., Citation2002), they are also dispersed with vertical distance from roadways, dropping by half or more within 15 vertical meters (Restrepo et al., Citation2004). Therefore, dispersion of such pollutants will be considerably greater more than 100 m higher than the roadway at the top of a hill than at a typical standard monitor, which is usually about 3 to 15 m above ground. Exposure misclassification would, therefore, be potentially worse than in the previous studies; the BC/EC or other traffic emission concentrations measured at this site are quite unlikely to reflect actual exposure of people living ˜1.5 km away and 100 m or more lower in elevation.
These last three studies do not show BC associations. Associations are only with PM2.5 and sulfate. Grahame (Citation2009) suggested that when exposure error for BC was greatest, not only was there less chance of finding BC health associations, but associations might be transferred from the least well measured pollutant to those with less measurement error. The three Steubenville, OH, studies are consistent with but do not prove this possibility. Further analysis is certainly required, including analysis of local emissions (e.g., remaining metals manufacturing) at the time of the study.
Although the HSPH studies add to our understanding of how PM2.5 species affect public health, few HSPH or any other human panel studies included metals, especially Ni. (Ni may be particularly important because of the number of population-based epidemiology studies finding Ni associations [e.g., Bell et al., Citation2009a, 2012; Mostofsky et al., 2012; Lipfert et al., Citation2006b, Citation2009; Zanobetti et al., 2009].) The value of human panel studies will be increased if researchers can jointly assess effects of additional PM2.5 species, including metals such as Ni, Cu, and Fe, with PM2.5 species more commonly assessed today, such as BC and sulfate.
Delfino et al. studies
Three particular features set the seven Delfino et al. studies (Table S7) apart from other human panel studies. One is that the studies used a very specific, large group of urban pollutant metrics, including BC, EC, OC, POC, SOC (see Delfino et al. [Citation2008] for specifics on measurements of these species), particle number, PAHs, hopanes, several different PM size fractions, particle number, and gases such as CO, NO2, NOx, and O3, although not all these pollutants were assessed in every study. A second feature was that pollution monitors were set just outside and inside several retirement communities in the Los Angeles area, and pollutants were measured for up to 9 consecutive days; this ensured that subject exposure was quite accurate, and that any effects examined in the studies (e.g., blood biomarkers) would not be missed should they occur only over a multi-day period. The third feature was that subjects were nonsmokers, aged 65 years or older, who had a history of coronary artery disease, ensuring commonality when comparing across different health endpoints. The combination of these features enabled very precise associations of either primary or secondary emissions with a large number of different biomarkers in blood, such as those for oxidative stress, inflammation, and platelet activation, as well as for cardiovascular endpoints, such as ST-segment depression, HRV, arrhythmia, and blood pressure.
The Delfino et al. studies showed that primary vehicular emissions—mostly BC/EC and POC (and PAHs)—were associated with almost all endpoints examined. SOC was not associated with any of the blood biomarkers or with ST-segment depression, but was associated with blood pressure increases; however, the increased blood pressure associated with an IQR increase in POC was roughly twice as much as for an IQR increase in SOC. For most of the large numbers of effects examined, all of which are relevant for cardiovascular issues, it was primary vehicular emissions that are of most importance. The usually negative SOC findings are of interest, since most studies, if they examine OC at all, use just OC, impeding the ability to know if POC or SOC is driving an association. The SOC findings should be viewed as preliminary, since few studies to date have examined SOC associations with various health endpoints, especially with human subjects.
Toxicology of Relevant PM2.5 species: Cardiopulmonary effects of diesel exhaust
Causality between exposure to specific types of PM and health effects observed in epidemiological studies may be supported if biological mechanistic evidence is consistent with the etiology/pathophysiology of these health outcomes. Toxicological studies can provide indications as to whether the effects of concern are biologically plausible and may reasonably be attributed to the pollutant of interest. This section provides an overview of toxicological evidence for cardiopulmonary effects of diesel engine exhaust (DEE). Details of specific studies are provided in the Supplementary Material.
DEE is a mixture of fine PM (diesel PM, DPM) and gases. It consists of a central carbon core with various adsorbed and absorbed combustion products (e.g., organic chemicals, such as PAHs, and transition metals). The mass concentration of freshly emitted DPM is dominated by particles that are typically 50–100 nm in diameter, but the number concentration is dominated by smaller particles having diameters below 50 nm (Barath et al., Citation2010). The exact composition of DEE varies by engine type and age, operating conditions, fuel composition, and exhaust treatment.
There is much toxicological support for adverse cardiopulmonary effects of DEE that are mediated via inflammation resulting from oxidative stress (Ristovski et al., Citation2011). Lung inflammation is a complex set of responses that involves a sequence of events involving various biological mediators (cytokines) derived from pulmonary macrophages and bronchial epithelial cells. Oxidative stress results when there is an imbalance between the production of reactive oxygen species (ROS) and the availability of endogenous antioxidant defenses. The induction of oxidative stress is a hierarchial event, in which protective responses at lower oxidative stress levels yield to proinflammatory effects at higher levels, with the ultimate biological effect in the respiratory tract being a function of the dynamic equilibrium between these protective and proinflammatory responses (Li et al., Citation2003; Xiao et al., Citation2003).
Exposure to DPM has been demonstrated to result in activation of the dual pathway just noted, and DPM does induce the aforementioned hierarchy of oxidative stress effects ranging from protective to injurious. For example, some organic chemicals derived from DPM induce expression of enzymes in macrophages and bronchial epithelial cells that initially protect against the proinflammatory effects of exposure (Li et al., Citation2004). While ROS are normal products of aerobic metabolism in cells, under conditions where ROS production increases beyond the ability to detoxify these chemicals, molecular damage may occur. Thus, other studies have shown production of proinflammatory cytokines following DEE exposure (Diaz-Sanchez et al., Citation1997; Diaz-Sanchez et al., Citation2000; Wang et al., Citation2005). DEE may also impact the immune response. One result could be increased prevalence of infectious pulmonary disease. For example, DPM was found to suppress cell-mediated cytotoxicity by affecting the activity of natural killer (NK) cells (Muller et al., Citation2013), which may result in a reduced ability to kill virus-infected cells, thus increasing susceptibility to viral infection. Increased viral susceptibility in DEE-exposed animals has been reported (McDonald et al., Citation2004, Citation2011).
Alteration in response to pathogens may also be due to the immune system's role in mediating the inflammatory response following exposure to DEE (Ristovski et al., Citation2011). In the innate immune system, pathogens are recognized by receptors expressed on antigen-presenting cells, which include dendritic cells, macrophages, and B cells. DPM has been shown to increase expression of some of these receptors in human bronchial epithelial cells, which may result in suppression of ROS release from alveolar macrophages (Sawyer et al., Citation2010), affecting the ability to kill microorganisms. Ambient PM in general has been implicated in cardiovascular morbidity and mortality, and DEE appears to play a role in this regard. A number of potential pathophysiological mechanisms may underlie this apparent relationship. DPM has been shown to induce ROS related lipid peroxidation to produce oxidized low-density lipoproteins, which can serve as a stimulus for monocyte migration into subendothelial spaces, representing a potential step in the development of atherogenesis (Ross, Citation1999; Ikeda et al., Citation1995). Active, unstable atherosclerotic plaques are characterized by increased accumulation of lipid and thin fibrous caps (Ni et al., Citation2009). Plaque disruption and atherothrombosis are underlying causes of a large percentage of sudden cardiac deaths (Burke et al., Citation1997). Exposure to DEE has been shown to result in increased numbers of alveolar macrophages and increased plaque lipid content in certain strains of mice (Bai et al., Citation2011). In addition, expression of oxidative markers was increased in these plaques. Thus, it appears that DEE promotes changes in plaques characteristic of unstable plaques, suggesting that DEE-induced oxidative stress affects plaques as well as pulmonary tissue (Bai et al., Citation2011).
Endothelial dysfunction may be another risk factor for future CV disease, and for deaths in patients with existing coronary disease (Newby et al., Citation1999; Newby et al., Citation2001; Heitzer et al., Citation2001). Poss et al. (Citation2013) examined effects of DPM on endothelial progenitor cells, which are circulating cells derived from bone marrow that modulate cardiovascular function and regeneration. Atherogenesis, a specific form of arteriosclerosis in which there is thickening of the arterial wall due to the accumulation of calcium and fatty materials, was increased and production of ROS was elevated. Aortas from certain strains of DEE-exposed mice showed increased markers of vascular remodeling as well as overall changes in plaque composition (Campen et al., Citation2010); filtration of PM from the exposure atmosphere did not alter the types of responses seen with emissions, although the intensity of some of these responses was reduced.
The effects in the toxicological studies just described are consistent with those from controlled human exposures, in that DEE has been implicated in altering parameters that may increase risk for CV disease. Table S8 outlines controlled human exposure studies noted herein. Normal subjects exposed to DEE showed evidence of increased thrombus formation and indication of platelet activation (Lucking et al., Citation2008). Thrombosis plays a central role in the pathogenesis of arteriosclerosis, and thrombosis at the site of a disrupted coronary arterial plaque may result in occlusion of the vessel, resulting in myocardial infarction (Lucking et al., Citation2008). DEE has also been shown to promote thrombosis formation by inhibiting endogenous fibrinolytic capacity, via reduction in release of endothelial tissue plasminogen activator (Mills et al. (Citation2007).
Controlled human exposure studies have also provided evidence for DEE effects on other risk factors for CV disease, such as altered hemodynamics. Normal subjects exposed to DEE showed an immediate, but transient, increase in arterial stiffness (Lundback et al., 2009). Enhanced arterial stiffness may be a pathological process in hypertension (Laurent et al., Citation2001), and may also result in greater central aortic systolic pressure, increased left ventricular afterload, and reduced coronary blood flow (Oliver et al., Citation2003). While the effect in this study was relatively small, it may be more clinically significant in more susceptible individuals, such as those with concurrent CV disease.
Elevation of blood pressure is another risk factor for CV disease, and epidemiological studies have noted a relatively consistent association between short term exposure to PM2.5 (Dvonch et al., Citation2009: three areas of Detroit, MI) and BC and POC (Delfino et al., Citation2010: elderly retirees in Los Angeles, CA, with coronary heart disease) with increased systolic blood pressure. However, controlled exposure studies with normal humans have shown equivocal results, with some studies showing a DEE-induced increase in systolic blood pressure and others showing no effect (Cosselman et al., Citation2012; Mills et al. Citation2005)
The endothelium plays a critical role in controlling vasomotor tone via release of vasoactive substances. Altered endothelial and vasomotor function is involved in the pathogenesis of atherosclerosis and ischemic coronary disease (Zeiher et al., Citation1991; Maseri et al., Citation1989). Normal human subjects exposed to DEE showed attenuated vasodilatory response to various potent vasodilators (Barath et al., Citation2010; Mills et al., Citation2005), indicating a reduction in vasomotor homeostasis that may persist for up to 24 hr post exposure (Tornqvist et al., 2007). The effect was suggested as due to reduced availability of nitric oxide (NO) in the vasculature, postulated, in turn, as due to oxidative stress induced by DEE (Mills et al., Citation2005; Wauters et al., Citation2013). Finally, DEE exposure was found to significantly upregulate vascular endothelial growth factor, which stimulates the migration of monocytes/macrophages, may increase the permeability of the vasculature, and may affect vascular tone (Peretz et al., Citation2007; Park et al., Citation2002; Svedas et al., Citation2003).
It is not clear whether DEE-induced alterations of vascular homeostasis are a direct consequence of oxidative stress to endothelial cells, or the result of systemic inflammation that affects endothelium via interaction with leukocytes (Peretz et al., Citation2007); the vascular endothelium plays a key role in the regulation of arterial tone (Lundback et al., 2009). A proinflammatory response was noted following exposure to DEE, and there was also upregulation of genes coding for end products involved both in the inflammatory response and in oxidative stress in peripheral blood mononuclear cells of healthy subjects (Lundback et al., 2009). However, as in the preceding, it was not possible to determine whether the proinflammatory response was induced by oxidative stress.
Another aspect of vascular function that could be related to CV disease is clotting. Healthy subjects exposed to DEE showed impairment of tissue plasminogen activator (t-PA) release from vascular endothelium, indicating a potential effect on vascular homeostasis (Barath et al., Citation2010). Impairment of t-PA following exposure to DEE has also been observed by Mills et al. (Citation2005).
There is a relationship between autonomic regulation of the cardiac cycle and CV mortality, and some observational studies have noted effects of ambient air pollution on such regulation. One parameter used to assess this regulation is HRV; reduction in HRV is associated with increased risk of CV morbidity and mortality in both healthy individuals and patients following MI (Tsuji et al., 1996; Kleiger et al., 1987). Studies finding associations between BC and reduced HRV, often in older or chronically ill subjects, include Suh and Zanobetti (Citation2010), Schwartz et al. (Citation2005a), Adar et al. (2007), Wheeler et al. (2006), and Park et al. (Citation2007). However, studies examining HRV in normal subjects, and even in an at-risk population with stable coronary heart disease, exposed to DEE showed no reproducible or consistent changes in HRV related to exposure (Peretz et al., Citation2008; Mills et al., Citation2011). CitationTong et al. (2014) found significant HRV associations in a study involving only six healthy, middle aged subjects exposed to 300 μg/m3 of diesel exhaust from an idling engine. Because all subjects had the GSTM1 null genotype, and thus lacked a full protective defense against oxidative stress, HRV changes after exposure to diesel exhaust and BC may be most likely to occur to those without such full defenses (CitationChahine et al., 2007; CitationSchwartz et al., 2005b).
The Relative contribution of particle size and chemistry to biological response
The extent to which effects of BC are due to size and/or to chemistry has been the subject of considerable research. As noted earlier, DEE varies in physicochemical properties depending upon a number of factors, for example, age and repair status of the engine, characteristics of the fuel, operating cycle, and so on. Biological effects will likely depend upon the specific properties of the exposure atmosphere, and therefore results in a controlled setting may differ from those found, for example, when relating emissions from nearby roadways to health outcomes in epidemiological studies. Smaller sized DPM or some combination of various chemical components in DEE may trigger different cell receptors and, therefore, different signaling pathways than would larger particles in the fine mode (Ristovski et al., Citation2011); this may account for some of the differences among various toxicological studies.
One approach to attempt to address the issue of size versus chemistry in eliciting adverse health outcomes is to consider carbon black (CB), an industrial product not found in ambient air but that is almost entirely EC, with less than 1% extractable organic compounds (Long et al., 2013). Using CB thus allows for a reasonable determination of effects of an EC core before any organic or other chemicals are adsorbed and the particle effectively becomes BC. Responses to different sizes of CB have been examined, with smaller sizes generally being more effective in altering various biological endpoints. For example, after intranasal instillation of 125 μg of two different sizes of CB (14 nm and 95 nm), Tin-Tin et al. (2006) found that only the smaller size fraction caused proinflammatory cytokine and chemokine activity in the mouse olfactory bulb. Gilmour et al. (2004) exposed Wistar rats to “uf” CB (114 nm) and to “fine” CB (268 nm), finding that only the smaller CB particles induced significant increases in total bronchoalveolar lavage (BAL) leukocytes and in blood leukocytes. These studies have shown that nearly pure EC particles can induce deleterious effects, at least at highly elevated levels of exposure, and that the smallest such particles are more effective in this regard. This is not surprising, in that ultrafine particles have been found to penetrate the walls of human lung cells and damage organelles far more easily than do larger, fine PM (Li et al., Citation2003). Ambient EC particles are mostly in uf (ultrafine; usually particles 100 nm or smaller in diameter) or near uf size ranges (<0.056 μm; 0.056–0.10 μm; and 0.10–0.18 μm; with another grouping between 0.56 and 1.0 μm; Mauderly and Chow, Citation2008). Since BC is approximately 75% EC (Long et al., 2013), BC occurs within the ultrafine and near ultrafine sizes, which appear to be the most potentially damaging.
While size clearly is a potential determinate of adverse biological responses, chemical makeup also determines the type and extent of response. It is, however, not fully clear which chemical components of the DEE mix (including organic gases) may be responsible for the observed effects, although there appears to be an important role for those associated with ROS production, for example, transition metals and organic chemicals (Risom et al., Citation2005). Organic extracts of DPM have produced proinflammatory responses in lung tissue cells, while DPM that had these organic components extracted did not show this response (Bonvallot et al., Citation2001; Hiura et al., Citation1999; Li et al., Citation2000; Li et al., Citation2002). Furthermore, the aromatic and polar fractions were noted to produce a proinflammatory response (Li et al., Citation2000), while the aliphatic fraction had no such effect (Ristovski et al., Citation2011). The aromatic portion was enriched with PAHs, while the polar fraction was enriched with quinones. Also, transition metals in DPM are potential mediators of inflammation (e.g., Ghio et al., Citation1999; See et al., Citation2007), although this has not been shown by all studies (e.g., Becker et al., Citation1996; Shukla et al., Citation2000). One study in which human bronchial epithelial cells were exposed to DPM found that carbon black particles mimicked the effect of the inorganic portion of diesel exhaust and did not increase ROS production (Baulig et al., Citation2003). Totlandsdal et al. (Citation2012) examined cytotoxicity and expression and release of proinflammatory mediators in human bronchial epithelial cells exposed to whole DPM, methanol DPM extract, or residual DPM left after the extraction. It was found that both whole DPM and DPM extract, but not residual DPM, induced marked expression of an enzyme involved in the inflammatory response, as well as cytotoxicity and release of proinflammatory mediators. A specific gene involved in Phase I metabolism and induced by aromatic compounds was mainly induced by native and residual DPM. The investigators noted that while the majority of PAHs and PAH derivatives were removed in the methanol-extracted samples, certain PAH derivatives, probably their carboxylic isomers, were retained on the residual DPM. Thus, different components of the DPM are likely involved in different aspects of the DPM -induced proinflammatory response.
In a randomized, crossover study, Brauner et al. (2007) exposed 29 healthy volunteers to ambient urban ultrafine PM of different sizes. DNA strand breaks in peripheral blood mononuclear cells, caused by oxidative stress, and FPG repair sites, which repair damaged DNA, were examined. Ultrafine PM in the 12 nm and 212 nm size fractions did not affect DNA strand breaks, but the authors found that the 57-nm “soot” fraction caused systemic oxidative stress with damage to DNA, and both the 57- and the 23-nm size fractions were associated with increased FPG repair sites. The authors reported that the 23-nm size fraction represents condensed semivolatile organic compounds from diesel vehicles. Here, chemistry appears to determine adverse effects, with the smallest uf particles having no effect.
Helfenstein et al. (2008) examined effects of diesel exhaust particles and engineered nanoparticles (TiO2 and single-walled carbon nanotubes [SWCNT]) on neonatal rat heart cells in vitro. A dose-dependent increase in oxidative stress and change in heart function (impulse conduction velocity) was observed with exposure to diesel PM and TiO2, but not with SWCNT. These results are consistent with those reported by Verma et al. (Citation2011), who noted that the oxidative potential of atmospheric semivolatile compounds in ambient PM was correlated strongly with organic carbon and PAH content of the particles.
The role of chemistry in eliciting response is also evident from Li et al. (2010), who examined effects of diesel emissions in idling mode versus in an urban driving schedule. Particles from the urban driving schedule contained higher levels of redox organic compounds and metals on a per PM mass basis. Particles from either mode upregulated stress-response genes and induced superoxide production in human aortic endothelial cells. However, those from the urban driving schedule also induced expression of proinflammatory genes, and induced nuclear factor (NF)-kB activity.
Recognizing that carbon nanotubes can cross the cell wall of rat macrophages, Pulskamp et al. (2007) examined effects of single- and multiwalled carbon nanotubes (CNTs), CB, and quartz on acute toxicity and release of reactive oxygen species in rat macrophages and human lung cells. While no acute toxicity was observed, a dose- and time-dependent increase in ROS and decrease in mitochondrial membrane potential occurred in both cell types with all particles, except for CNT (SWCNT) that were acid treated to remove manufacturing impurities. The authors concluded that traces of metal in the manufacturing process were likely to have caused the effects noted with CNTs not pretreated with acid. This also suggests that chemistry is critical in ultimate response, regardless of size.
Diabate et al. (2011) tested effects of water-soluble and water-insoluble municipal solid waste incinerator fly ash and CB in human lung epithelial cells. Both fly ash and CB induced production of ROS. Fly ash (insoluble fraction only) induced ROS and was correlated with induction of heme oxygenase-1 (HO-1) and with increase of Nrf2, but CB did not induce HO-1. ROS generation and HO-1 inducement were inhibited by preincubation of cells by the antioxidant N-acetylcysteine (NAC), and were reduced by the metal chelator deferoxamine, indicating the role of bioavailable transition metals in causing the effects.
Mills et al. (Citation2011) examined vascular effects of dilute diesel exhaust (median diameter of DPM = 67 nm), filtered diesel exhaust, and pure carbon nanoparticles (median diameter = 37 nm) versus filtered air in healthy young volunteers. Diesel exhaust attenuated vasodilation, but exposure to pure carbon nanoparticles, or to filtered diesel exhaust, had no effect on vasodilation. These effects of DPM were due to both soluble and insoluble fractions on the surface of DPM. Compared to filtered air, both diesel exhaust and filtered diesel exhaust were associated with increased systolic blood pressure (SBP). These results demonstrate the importance of chemical makeup versus the size of particle, and also show that gaseous components of DEE affect some biological endpoints.
Thus, while the effect of size and the effect of chemical composition have been examined in terms of biological response, both seem to have significant influence. Regarding size, ultrafine particles, everything else being equal, are likely more harmful than larger particles of similar composition, because they can penetrate furthest into the lung, can penetrate the cell wall, and have larger surface area per given mass. Yet the chemical makeup of the particle, including any surface-adsorbed materials, often is of greater importance, as shown in some of the examples just discussed.
In summary, specific pathways by which DEE/DEP exposure results in adverse cardiovascular health outcomes have been examined. Toxicological studies strongly suggest that the many adverse health outcomes are likely to occur via production of an inflammatory response mediated by the induction of oxidative stress and the release of proinflammatory cytokines after activation of transduction pathways including mitogen-activated protein kinase (MAPK) and NF-κB. There is also indication that some of the adverse health outcomes may be due to change in the bioavailability of NO within the vascular system. Furthermore, studies in human subjects show that DEE exposure can result in migration of leukocytes into bronchial tissue, another index of inflammation (Salvi et al., 1999, Citation2000). In addition, there appears to be a pathway of innate immunity that is affected by exposure to DEE, and effects on the immune system may also play a role in mediating any inflammatory response to DEE. Parts of the organic portion of DPM appear to be consistently responsible for many of these adverse biological effects, with the extent of response depending upon engine load and other operating variables. The responses observed in the animal toxicology and human controlled clinical studies are consistent with CVD effects noted in epidemiological/observational studies, thus providing a degree of support for causality, based upon the coherence and biological plausibility criteria of the U.S. EPA rubric.
Outcomes other than all-cause, cardiovascular, cardiopulmonary mortality and morbidity: natal health, cognition, and lung cancer
Prenatal health
A “natural intervention” took place in New Jersey and Pennsylvania in the late 1990s, when E-ZPass was introduced in several heavily traveled freeways and parkways. E-ZPass is an electronic device put in a vehicle that allows a driver to drive through a special lane that automatically charges the verhicle's toll. When many drivers use this time-saving device, congestion, vehicle acceleration, and pollution are greatly reduced.
Currie et al. (Citation2009) found that the incidence of prematurity and low birth weight for children of mothers living within 2 km of a toll plaza was reduced by 10.8% and 11.8%, respectively, after introduction of E-ZPass, but that no such improvement occurred for mothers living within 2 km of a freeway but further than 2 km from toll plazas.
Table S9 reviews 19 studies that examine associations between one or more vehicular emissions and various birth outcomes. Most of the studies are from the United States, with the majority from California, with German, Canadian, and Australian studies also included. Most of the outcomes examined have to do with “traditional” adverse effects on both the newborn (preterm birth, small for gestational age, low birth weight, and in one case, spontaneous abortion) and the mother (preeclampsia), but one recent article assesses links of vehicular emissions with childhood cancers.
Studies in Table S9 show that when traffic exposures are reasonably well measured, whether by traffic density or distance to major roadway near residence or by modeling emissions to the residence of the mother, significant associations with adverse birth outcomes or with preeclampsia are usually found. In the California studies, if an air pollution monitor is <1.5 km from the residence of the mother, associations are often found with traffic emissions such as CO, but if the monitor is farther from the home, such associations are rare. Exposure misclassification is discussed in several studies. Thus, exposure to an environmental factor(s) near roadways, highly correlated with vehicular emissions, seems likely to be causally related to these various outcomes.
Several studies suggest the involvement of PAHs may result in adverse birth outcomes. In a sample of 263 African-American and Dominican women living in parts of Harlem and South Bronx in New York City using personal monitors for pollutants, Perera et al. (Citation2003) found high prenatal exposure to PAHs to be significantly associated in African-Americans but not Dominicans with 10% lower birth weight and 2% smaller head circumference after adjusting for potential confounders. Smaller head circumference at birth “correlates with lower IQ as well as poorer cognitive function” (Perera et al., Citation2003). Smokers were excluded from the study. A similar study from the same group found that increased PAH exposures were significantly associated in African-American mothers with greater risks of infants being born small for gestational age (SGA), for having a fetal growth ratio< 85%, or for a fivefold increase in risk or preterm delivery (Choi et al., Citation2008).
Perera et al. (Citation2005) found direct evidence of DNA damage in both mothers and offspring exposed to PAHs. Maternal PAH exposure increased among four locations as follows: PAH exposure in northern Manhattan < near the World Trade Center in the year after 9/11 < Krakow, Poland < Tong Liang, China. There was residential coal use without controls in both Krakow and Tong Liang. DNA adducts are fragments of DNA that are bound to a potential cancer causing chemical, and are used as markers of exposure. Although prenatal exposure of the fetus to PAHs is about one-tenth that of the mother, similar levels of PAH–DNA adducts were found in mothers and children, with the numbers of adducts in each following the order of exposure in the four locations. The similarity of levels of PAH–DNA adducts in mothers and children therefore suggests that the fetus may be 10 times more susceptible to DNA damage from exposure to PAHs than the mother.
Thus, several adverse birth associations found in the 19 studies in Table S9 with various markers of vehicular emissions were associated with PAHs and PAH-DNA adducts, suggesting the possibility that biologically active PAHs might be causally related, or a marker for a highly correlated vehicular emission that might be causally related, to adverse prenatal endpoints.
Lung cancer
In 2012, IARC declared that diesel emissions are a known cause of lung cancer. Rather than review studies that led to that decision, here we briefly review three important U.S. studies for context.
Garshick et al. (Citation2008) obtained work records for 31,135 male workers who were in the trucking industry in 1985. Lung cancer mortality records were obtained through 2000. Jobs in the trucking industry were classified with regard to regularity of exposure to diesel exhausts. The authors found that for more heavily exposed workers (long-haul and pick-up/delivery drivers, dockworkers, combination workers), risks of dying from lung cancer rose with cumulative years of exposure, in most cases monotonically. The authors suggested that these effects were not necessarily due solely to diesel exposure, since different types of workers were exposed to different mixes of vehicular emissions.
Garshick et al. (Citation2004) examined lung cancer prevalence among 54,973 U.S. railroad workers from 1959 (when most locomotives had converted to from coal to diesel fuel) to 1996. Lung cancer mortality was elevated among workers in jobs on locomotives powered by diesel. After adjusting for the healthy worker effect, relative risk of lung cancer mortality among workers involved in operating trains was 1.40 (CI = 1.30, 1.51). The effects appeared to be greater in those with longer exposure to diesel locomotives and shorter exposure to ones powered by steam.
Morello-Frosch and Jesdale (Citation2006) estimated cancer risks associated with ambient air toxics by census tract, after controlling for SES measures. The authors modeled estimated concentrations of 33 air toxics (including DPM) from the U.S. EPA National Air Toxics Assessment, combining this information with cancer potency information. Estimated cancer risk from all sources combined was 632 per million, which after removal of DPM declined to 116 per million (DPM thus representing 82% of cancer risk from air toxics). Mobile sources contributed 88.3% of total cancer risk when diesels were included.
Neurological effects: Cognition and behavior
Table S10 summarizes 12 studies associating traffic emissions with either poorer cognition in elderly, or reduced scores on tests of intelligence, anxiety, attention, nonverbal reasoning, and psychomotor development in children. Four of the studies link cognitive impairment in the elderly to either higher BC concentrations, or higher levels of pollution in Mexico City versus in relatively clean cities, or residence in close proximity to major roads. The other eight studies are of young children, either those whose mothers were exposed to higher levels of PAHs during pregnancy (the Perera et al. studies; Edwards et al., Citation2010) or those who were exposed to higher levels of BC modeled to residences in the Boston area (Suglia et al., Citation2008; Chiu et al., Citation2013). Various deficits are associated with higher levels of the vehicular emissions tested, whether in learning (IQ, nonverbal reasoning, cognitive development), emotion (anxiety, depression, attention), or behavior. The findings are provocative, but further research is needed.
Possible biological mechanisms for neurological effects are suggested by toxicology. Epidemiological and observational studies have suggested a relationship between generalized air pollution and adverse effects on the central nervous system (CNS). PM may cross the blood brain barrier (Peters et al., Citation2006), the tightly regulated capillary endothelium that separates the peripheral blood circulation from the CNS, and this may be a factor in air pollutant-related neuropathology. PM may also enter the brain directly via the cribriform plate of the ethmoid bone (Brenneman et al., Citation2000); direct translocation to the CNS may be especially important for ultrafine PM (Genc et al., Citation2012).
Pollutant-related neuropathology may be due to the development of neuroinflammation (Calderon-Garciduenas et al., Citation2002) or direct translocation of material. On the other hand, PM may not directly enter the CNS, but could produce effects by triggering the release of inflammatory mediators from primary deposition sites, such as the respiratory tract, resulting in altered susceptibility for neuroinflammation in the CNS (Genc et al., Citation2012). The brain may be especially vulnerable to oxidative stress due to its high metabolic demands, low levels of free radical scavengers, and high cellular content of lipids and proteins (Mattson, Citation2001). As noted previously, induction of a respiratory-tract inflammatory response may result in production of systemic effects.
Toxicological studies have shown that exposure to diverse forms of air pollution, including urban PM and some metals (Levesque et al., Citation2011), can result in similar proinflammatory responses and oxidative stress in the brain. For example, exposure to ambient PM from urban air has been found to enhance neuroinflammatory markers and proinflammatory mediators in the brain of mice (Kleinman et al., Citation2008; Campbell et al., Citation2005; Veronesi et al., Citation2005; Campbell et al., Citation2009) and to alter select neuronal and glial cell activities (Morgan et al., Citation2011).
DEE has been shown to elevate proinflammatory factors in select brain regions, as well as to produce generalized neuroinflammation (Cruts et al., Citation2008; Gerlofs-Nijland et al., Citation2010; Mogi et al., Citation1994; van Berlo et al., 2010). While the mechanism of any PM induced neuroinflammation is not certain, DPM has been shown, for example, to activate microglia (Block et al., Citation2004); microglial cells are resident immunocompetent cells that provide tissue maintenance and immune surveillance in the brain. Upon activation, they can produce proinflammatory and neurotoxic mediators. A pathway for microglial-mediated neurotoxicity is via ROS, especially superoxide, and activation of NADPH oxidase, which produces ROS (Block et al., Citation2007). Activation of microglia has been recognized to contribute to the pathogenesis of Parkinson's disease, and there is accumulating evidence that microglial-derived oxidative stress may be involved in selective toxicity to dopamine secreting neurons (Block et al., Citation2004).
One of the key components of the blood–brain barrier is P-glycoprotein, a transporter molecule that limits penetration to the brain of xenobiotics, including various drugs (Kim, Citation2002; Taylor, Citation2002). Impairment of the blood–brain barrier in response to toxic agents may be a factor in enhanced penetration of chemicals into the CNS. DPM has been found to alter the blood–brain barrier function via oxidative stress and proinflammatory cytokine production (Desai et al., Citation2007; Dazert et al., Citation2006). Circulatory production of oxidative stress and inflammatory mediators may contribute to CNS pathology, in addition to direct effects of DEE on microglial cells or neurons.
One study examined effects related to chemistry of the exposure atmosphere. Tin-Tin et al. (2012) exposed female BALB/c mice to clean air, medium-dose nanoparticle-rich diesel exhaust (NRDE), high-dose NRDE, or filtered diesel exhaust for 3 months. The NRDE particles mean size was 23 nm for the medium dose (35.5 mg/m3) and 26 nm for the high dose (122 mg/m3). In a test of memory (Morris water maze), mice in the high-dose group took a significantly longer time to reach a hidden platform, versus the medium-dose and filtered exhaust groups. High-dose mice also were found, in the hippocampus, to have significantly higher levels of expression of an inflammatory cytokine (CCL3), of mRNA expression of NMDA receptor subunit NR2A, and of brain-derived neurotrophic factor (BDNF), compared to the control group. Filtered diesel affected only BDNF. Thus, 3 months of exposure to NRDE affected spatial learning and memory function-related gene expression in the female mouse hippocampus, with higher doses of NRDE more harmful than lower doses or filtered exhaust.
In summary, some percentage of the incidence of neurodegenerative diseases in the population may be due to environmental exposures, perhaps coupled with some inherent susceptibility factors (MohanKumar et al., Citation2008), and there is mounting toxicological evidence that air pollution and at least certain types of PM can produce proinflammatory and inflammatory effects in the brain, contributing to the overall oxidative stress burden. Neuroinflammation appears to be involved in the pathogenesis and/or progression of neurodegenerative diseases, such as Parkinson's and Alzheimer's (Block et al., Citation2007; Glass et al., Citation2010; Frank-Cannon et al., Citation2009). Current thought is that proinflammatory events in the brain likely occur across the entire life span, culminating in neuropathology (Frank-Cannon et al., Citation2009; Carvey et al., Citation2006; Peters et al. Citation2006). Furthermore, respiratory-tract inflammation may also contribute to brain inflammation (Calderon-Garciduenas et al., Citation2003), perhaps by increasing systemic levels of specific cytokines, which then cross the blood–brain barrier and evoke an inflammatory response in the brain (Rivest, Citation2001). Specific characteristics and sources of PM responsible for the oxidative stress burden are not fully identified, although DEE seems a likely source. As noted, environmental agents may damage CNS neurons by direct cytotoxicity, or indirectly via microglial activation (Block et al., Citation2007). Dopaminergic neurons seem to be particularly vulnerable to the effects of microglial activation. Pulmonary inflammation can result in systemic effects, and microglia can be activated in response to peripherally circulating cytokines (MohanKumar et al., Citation2008). Direct physical entry of PM into the CNS is therefore not a requisite for CNS neurotoxicity. Thus, toxicology provides support to the epidemiological and observational study findings that DEE can adversely impact the CNS by suggesting potential mechanisms for biological plausibility, namely, inflammatory and oxidative stress pathways.
Ambiguities: Confounding of BC associations with PM2.5 from brake, tire, engine wear (including metals), and noise
Researchers have suggested that environmental stressors other than engine exhaust, but derived from traffic and, thus, correlated with such emissions, may be related to adverse health effects that have been associated with distance to roadways and/or with BC/EC or other vehicular emissions. PM species from tire, brake, and engine wear (primarily metals) and road noise (possibly linked to health effects via hypertension) have been primarily mentioned.
A study in Stockholm found that Cu and Zn were the primary metal components of brake wear, whereas Zn was the primary such component of tire wear (Hjortenkrans et al., Citation2007). Lough et al. (Citation2005) found that Fe (primarily from engine wear), Cu, and Zn are primary metal components associated with PM from roadways. Among the few studies that allow some insight into multiple effects of wear particles is Riediker et al. (Citation2004). This study examined 10 different health endpoints (supraventricular ectopic beats, two measures of HRV, a number of biomarkers) in healthy young patrol officers after their work shifts on North Carolina highways. The researchers found that neither road dusts including Fe (including from engine wear) nor gasoline combustion components were associated with any of the endpoints, but that that components representing changing speeds of diesels (such as alkanes and sulfate, from acceleration, and Cu from braking) were associated with all 11 endpoints. The extent to which Cu may have causal influence thus requires additional investigation.
A recent review regarding components of road emissions and their health effects was performed by van der Gon et al. (Citation2013). The authors noted that “insufficient evidence is available” to understand health risks of these components, and that “more toxicological and epidemiological” analysis and a “standardized approach” are needed, in particular, to “properly disentangle” effects of PM from wear sources and other PM fractions.
Some epidemiological studies of near-roadway health effects have attempted to take effects of noise into account. The study that perhaps has most thoroughly assessed effects of traffic noise in relation to effects of traffic emissions is Beelen et al. (Citation2009). This study found all-cause and CVD mortality associations for pollution indicators (levels of BS, traffic intensity on nearest roadway) that were “insensitive to adjustment for traffic noise.” The study also found separate CVD associations for exposure to the highest levels of traffic noise (>65 dB). Kan et al. (Citation2008) discussed possible effects of noise, noting that associations of noise with cardiovascular risks are “far less consistent than those between air pollution and cardiovascular disease” and that noise-related cardiovascular effects (hypertension) would follow different physiological pathway than the CVD effects found in Kan et al. (Citation2008). Kan et al. (Citation2008) found CHD associations in non-hypertensive subjects. Hoffmann et al. (Citation2006) noted that men exposed to more than 70 dB during the day had a 1.3-fold increase in risk for an MI, also noting that effects of noise would work via hypertension. The authors’ CVD associations were made after adjusting for hypertension, and they concluded than any residual confounding by noise would have been small, but they also concluded that additional work to parse out effects of noise from effects of pollution is appropriate. Stern et al. (Citation1988) found that CVD mortality was significantly reduced in workers in roadway tunnels, relative to those working on bridges, when ventilation was introduced to the tunnels. Since there was likely no decrease in noise accompanying this intervention, the reduction in mortality can be attributed only to reduced vehicular pollution, not reduced noise.
Syntheses of findings from studies in North America and Western Europe
BC and co-emissions (mainly from diesels) were not the subject of much apparent research prior to about 15 years ago. In this section we seek to explain how it is that BC and co-emissions have in the short time since then come to be viewed as perhaps playing a major role in regard to all-cause and CVD mortality, and CVD morbidity.
Part of the story has to do with BC or EC measurements: Epidemiology cannot assess what isn't measured. Once measurement became available, and a few years of data had been accumulated, epidemiology by the mid-2000s in the United States could begin to assess BC associations in relation to other PM species in the same models. BS measurements had been available in Western Europe for several decades, but in early periods, the source for which BS was primarily a marker was residential burning of coal. Katsouyanni et al. (Citation1997) analyzed mortality in 12 European cities, finding mortality associations with PM10, SO2, and BS. The authors suggested that mortality associations with BS might be either because BS was in the fine fraction (smaller than PM10) or because BS came from diesels, which in Western Europe became the major source of BS, as residential coal use declined. Thus, availability of BS monitoring information in Europe may have allowed slightly earlier associations of carbonaceous vehicular emissions with mortality than in the United States, where widespread BC monitoring had yet to take place.
Poor subject exposure assessment also was an issue. Initial studies, now just over a decade old, found large mortality and morbidity risks for those living close to major roads versus those at more than 100 or 150 m distance, dramatically demonstrating how important it was to know the amounts and identities of pollutants to which subjects were actually exposed. Previously unknown risks identified in these “highway proximity” studies spurred interest in understanding effects from vehicular emissions.
Regarding exposure to different PM species, recent studies have demonstrated that using measurements from a central monitor, instead of a personal monitor or a monitor just inside or outside of a home, can cause associations for pollutants with substantial local spatial variability to switch from large and significant, to small and nonsignificant. Thus, recent studies often either model estimated BC concentrations or other vehicular emissions to a subject's home, use personal monitors, or use monitors inside or just outside subject residences.
Virtually all the epidemiology studies reviewed herein (both human panel and population based) showed associations with BC (and thus for correlated vehicular co-emissions) for the great majority of health endpoints assessed, even when subject exposure to BC was poorly represented by concentrations measured at a central monitor. These endpoints include: CVD and all-cause mortality; CVD hospital admissions; biomarkers of oxidative stress, inflammation, and vascular function; and increased blood pressure, lipoperoxidation, HRV changes, greater ST-segment depression, and reduced production of t-PA. It is surprising that poor exposure assessment did not cause BC associations to become insignificant in more studies than it did. Many of these studies commented that there was greater exposure misclassification for BC than for other pollutants (Chuang et al., 2008) and that poor subject exposure to BC should have attenuated BC associations or biased them toward the null, but did not (Bind et al., 2012; Baja et al., 2010; Baccarelli et al., 2009; O'Neill et al., 2007). Goldman et al. (2010) estimated error in risk estimates due to spatial variability not accounted for when central monitor data were used as a proxy for subject exposure. The authors found that risk ratio reductions were less than 16% for secondary pollutants (ozone, PM2.5 sulfate, nitrate, and ammonium) but between 43% and 68% for primary pollutants (NOx, NO2, SO2, CO, PM2.5, EC), further illustrating how exposure error attenuates associations with pollutants, such as BC, with significant spatial variability.
Associations of BC and other vehicular emissions with adverse health effects are made more convincing when toxicology studies suggest biological mechanisms underlying the effects observed in epidemiology. These biological mechanisms are then more convincing when they are also seen in humans, whether in controlled exposure studies (mainly diesel and wood smoke) or in panel studies.
That said, it is important to understand to what extent the effects of BC are due to its EC core (usually uf or quasi-uf in size) per se; to gases co-emitted with BC; and/or to other particles, such as SVOCs and PAHs, which are diesel co-emissions, many of which absorb onto EC cores and thus operationally become BC.
Cassee et al. (2013, Citation803), in a review of toxicology studies, noted that:
black carbon (measured as [EC]) may not be a major directly toxic component of fine PM … but it may operate as a universal carrier of a wide variety of combustion-derived chemical constituents of varying toxicity to sensitive targets in the human body.
Cassee et al. (Citation2013) thus add to the results of many studies reviewed herein, which found that organic compounds adsorbed or coating graphite-like EC particles were often the causative agent for adverse effects.
Several CVD effects appear to be caused by PM, as opposed to gaseous co-emissions, based upon experiments in which human subjects wore highly efficient facemasks while walking a predetermined route, and then walked the same route without the facemask, in Beijing, China. Langrish et al. (Citation2009) found the use of the facemask by healthy young volunteers significantly reduced SBP, and increased HRV (e.g., toward normal levels). The second study involved patients with coronary heart disease (Langrish et al., Citation2012). In this study, maximal ST-segment depression was reduced, mean arterial pressure was lower, and again HRV measures increased. There were, however, no significant changes in SBP or diastolic blood pressure (DBP). These are among the first studies which use face masks to determine whether ambient particles cause specific health effects, and the studies took place in a highly polluted location. Replication of these studies in polluted parts of the United States or the European Union (EU) would thus be recommended (recognizing the considerably lower level of PM in the United States and EU). Several other studies found adverse effects using only DPM (e.g., Kim et al., Citation2012: increased QT-interval, and ventricular premature contractions in vivo).
However, several studies discussed in the toxicology section (including Supplemental Material) found that both PM and gases from diesels were associated with different health endpoints.
Kleinman (Citation2013) used “quasi-ultrafine” PM (<180 nm) from concentrated ambient air taken from downtown Los Angeles to study CVD effects in apoE‐/‐ mice. The particles were denuded of particle-associated semivolatile material at 120ºC, with the remainder of the PM consisting of elemental carbon and inorganic materials, plus larger organic molecules that did not have sufficient vapor pressure to be stripped from the particles. Kleinman found that both undenuded “quasi-ultrafine” PM and denuded SVOCs accelerated development of atherosclerotic plaque in arteries, but these effects were not seen when mice were exposed only to the denuded concentrated air particles (CAPs) (which were primarily EC). Lipid peroxidation was increased by undenuded particles and by denuded SVOCs, but again, not by the denuded particles. These results suggest that the organic compounds, whether adsorbed onto the EC core or not, caused the adverse CVD effects. At ambient temperatures, BC would thus appear to be quite potent, as they would not be denuded of much of the SVOCs. Kleinman (Citation2013) thus shows a biological mechanism, involving biologically active SVOCs but not EC, for the findings of Jacobs et al. (Citation2011), for example, that increased plasma oxidized LDL (a marker of atherosclerosis) in human subjects was associated with increased loading of carbon in airway macrophages.
Similarly, Verma et al. (Citation2011) examined oxidative potential, as measured by the dithiothreitol (DTT) assay, in “quasi-ultrafine” PM (<180 nm) from concentrated ambient air taken from downtown Los Angeles, CA. Particles were thermally denuded at temperatures of 50, 100, and 200ºC. As particles were progressively denuded of SVOC, low-molecular-weight PAHs, high-molecular-weight PAHs, and OC with increasing temperatures, the oxidative potential of the remaining core particle was diminished, with the oxidative potential being increasingly attributed to the various SVOCs. Denuding EC per se changed oxidative potential of EC very little. Again, biologically active SVOCs, whether on the BC particle or not, caused oxidative stress.
A recent review concluded that chronic oxidative stress likely causes shortened telomeres, which in turn likely shorten life by accelerating biological aging (Grahame and Schlesinger, Citation2012). Telomeres are strands at the end of chromosomes that prevent loss of DNA when cells divide, for example, that act like protective caps and prevent deterioration of the chromosome. With each cell division, they shorten. When they are too short, cells no longer divide and become senescent, with gradual loss of function, for example, accelerated biological aging. Grahame and Schlesinger (Citation2012) linked studies finding that exposure to diesel exhaust or to BC is associated with increased oxidative stress, with studies linking oxidative stress with shortened telomeres and accelerated CVD. They hypothesized that one mechanism by which people who are exposed to higher levels of BC (Krall et al., Citation2013; Lipfert et al., 2000b, Citation2009; Klemm et al., 2011; Maynard et al., Citation2007; Gan et al., Citation2011) or who live near major roads (Finkelstein et al., Citation2004; Gehring et al., Citation2006) might have lower life expectancy and increased CVD risks (Hoffmann et al., Citation2006; Hoffmann et al., Citation2007; Hoffmann et al., Citation2009) is via accelerated shortening of telomeres. Effects of oxidative stress may therefore have greater importance for human health than previously thought, should the hypothesis that oxidative stress caused by BC and co-emissions shortens telomeres be validated by further research.
We conclude that much of the adverse effects caused by biologically active carbonaceous species is caused at ambient temperatures by BC, in large part because of carbonaceous materials such as SVOCs and PAHs that are adsorbed onto BC’s EC core. Lucking et al. (Citation2011) found that a regenerating particle trap greatly reduced particle numbers and mass, and (in a study of healthy young human subjects) increased vasodilation, reduced thrombus formation, and increased release of tPA when the trap was used to filter diesel exhaust (vs. whole diesel exhaust; concentrations of gaseous hydrocarbon did not change). Small size also matters, because uf particles can penetrate the cell wall, and because uf and quasi-uf PM penetrate further into the lung than do larger PM. But not all such carbonaceous uf PM species in DEE will be adsorbed onto EC, and DEE also included carbonaceous gases; it is likely that some of these will also be harmful. Thus, from a regulatory viewpoint, measures that can reduce the great majority of carbonaceous diesel emissions (e.g., the oxidizing catalyst trap required for on-road diesels in the United States since 2007) as well as filter metallic co-emissions would be crucial for public health. McDonald et al., (Citation2004) suggest that BC is virtually eliminated and most other carbonaceous and metal species are largely reduced by the oxidizing trap, and that toxicological effects in mice (e.g., oxidative stress, inflammation, inability to resist a virus) appear to be abolished when the trap is retrofitted. Follow-up research would be useful to confirm that these effects remain after several years of use of the new oxidizing traps, and that other adverse effects are also abrogated by them.
Another relevant question is: To what extent might effects observed in studies of PM2.5 be largely driven by BC and co-emissions? Janssen et al., (Citation2011) suggests that reducing a unit of BC might prolong life by 4 to 9 times more than reducing a unit of PM2.5 (e.g., 1 μg/m3 of BC, vs. 1 μg/m3 of PM2.5). Finkelstein et al. (Citation2004a) found that those living near major roads were likely to live 2.5 years less than those further away, everything else equal. Several studies of truckers show elevated CVD and lung cancer morbidity and mortality for those who work closely with diesels, versus those in the same industry who do not do so (Laden et al., Citation2007; Finkelstein et al., Citation2004b; Garshick et al., Citation2008). Morello-Frosch and Jesdale (Citation2006) suggest that 82% of cancer caused by toxic air pollutants is caused by diesel emissions. A recent expert elicitation found that carbonaceous combustion particles, either “elemental and organic carbon,” “diesel PM2.5” or “traffic PM2.5” were concluded to be the most harmful types of ambient PM2.5 (Cooke et al., Citation2007). What other evidence might there be for BC and biologically active co-emissions being particularly harmful components of PM2.5 pollution?
Some epidemiological studies make suggestive findings. Pope et al. (Citation2002), in a follow-up to the original ACS study, reported that there were no PM2.5 associations for people with better than high school education, for all-cause, CVD, lung cancer, and all-other-cause mortality. In the early 1980s, did people of high socioeconomic status (SES) live in polluted urban areas, or in less polluted suburbs? Jerrett et al. (Citation2007) reanalyzed data from the ACS study, examining how mortality associations changed over five different time periods, for sulfate and for PM2.5. The authors report that sulfate associations weakened in the most recent time periods, but that PM associations strengthened in the 1990s. Jerrett et al. (Citation2007) suggest that the reason for strengthening PM associations might be because “toxic mobile sources are now the largest contributors to PM in urban areas.” Lipfert et al. (Citation2006b) found long-term mortality associations with traffic indicators (traffic density, EC, and NO3) but not with PM2.5. Hoffmann et al. (Citation2007, Citation2009) found robust CVD risks associated with close proximity to major roads, but rarely with PM2.5.
Puett et al. (Citation2011) reported no mortality associations with PM2.5 in a cohort of well-educated males of high socioeconomic status; the authors reported that this finding contrasted with results of a study of nurses in the same states for whom there were PM2.5–mortality associations. The cohort from Puett et al. (Citation2011) differed from that in the Six Cities study, where up to 53% of the subjects were occupationally exposed to various dusts and fumes. Better health was also discussed as a reason for a lack of mortality associations with PM2.5. However, Puett et al. (Citation2011) did not pursue whether the different socioeconomic status of the nurses might be a marker for living in closer proximity to heavy traffic. Reynolds et al. (Citation2001) found that those with lower socioeconomic status were exposed to an order of magnitude greater traffic near their homes than were other groups.
Mills et al. (Citation2008) found that neither healthy individuals nor males with coronary heart disease exposed to 190 μg/m3 of concentrated PM2.5 containing little carbon taken from the relatively clean city of Edinburgh, Scotland, suffered effects similar to those found in people exposed to fresh diesel emissions of not-dissimilar concentrations. These effects included attenuation of vasodilation (Barath et al., Citation2010), impaired release of “clot-busting” tPA (Mills et al., Citation2005), and increased systolic blood pressure (Cosselman et al., Citation2012). Mills et al. (Citation2008) reiterate the toxicity of BC and co-emissions relative to other PM2.5 species. These studies are suggestive of the possibility that ambient air containing either little fresh urban emissions, or those from a relatively clean coastal city, may be less harmful than ambient air in larger urban areas, containing BC and other diesel emissions.
Some human panel studies with good subject exposure consistently show HRV changes associated with BC/EC (Schwartz et al., Citation2005a; Adar et al., 2007; Suh and Zanobetti, Citation2010). Suh and Zanobetti (Citation2010) found no associations between five different measures of HRV and personally monitored PM2.5, but did find associations with the five HRV measures with personally monitored BC. Adar et al. (2007) found consistent HRV associations with both BC and PM, most likely because both pollutants were highly correlated, as they rose significantly while subjects were on a diesel bus, but were otherwise not elevated. Schwartz et al. (Citation2005a) tested four different measures of HRV and two time periods (1 and 24 hours). The authors found that BC was associated with HRV reductions in seven of the eight tests, but that PM2.5 was associated in only three of the tests. The authors then used an algorithm to mathematically remove BC from PM2.5 on an hourly basis, calling the remainder “secondary PM2.5.” Secondary PM2.5 was not associated with HRV reduction in any of the eight tests. The authors plotted PM2.5 versus an HRV measure and found that PM2.5 was associated with HRV reduction only when it was highly correlated with BC (from near zero to about 18 μg/m3 PM2.5). However, as PM2.5 increased from about 20 to about 50 μg/m3, there was no further effect on HRV. The authors concluded that it was the BC that drove the PM2.5 associations, and that when PM was not correlated with BC, it caused no HRV changes ().
Figure 3. Smoothed plot of the percentage deviation from predicted SDNN (based on the model with all other covariates) versus PM2.5 concentrations in Boston. The association flattens out at high concentrations where the correlation between PM2.5 and black carbon disappears. From Schwartz et al. (2005). © BMJ Publishing Group Ltd. Reproduced by permission of BMJ Publishing Group Ltd. Permission to reuse must be obtained from the rightsholder.
A similar finding was made by Creason et al. (2001). The authors found a “U”-shaped function with increasing PM2.5 in a study of retirement-home residents 15 km north of Baltimore, MD. At first, HRV was reduced as PM2.5 increased, but as PM2.5 reached about the midpoint of indoor PM2.5 concentrations, the direction of change reversed upward, and at the highest PM2.5 levels, HRV had recovered to its initial level, when PM2.5 was virtually zero. The authors identified a 2-day period of high PM levels that were responsible for the “U”-shaped function, and using back-trajectory analysis, found these emissions did not come from urbanized locations or from the industrialized Midwest, but rather from a more rural trajectory. After removal of these 2 days of data, the relationship between increasing PM2.5 and reduced HRV became nearly linear. Thus, Creason et al. (2001) found that urban aerosols, even at low concentrations, reduced HRV, but that nonurban, nonindustrial aerosols, even at high concentrations, did not reduce HRV. Schwartz et al. (Citation2005a) came to the same conclusions, using a different study design. Both studies concur with that of Suh and Zanobetti (Citation2010) with regard to the associations of BC with reduced HRV, while Schwartz et al. (Citation2005a), Creason et al. (2001), and Adar et al. (2007) suggest that PM2.5 associations with HRV reduction are due to its BC or urban component. Gold et al. (Citation2005) found that BC, but not PM2.5, was associated with ST-segment depression.
To what extent might findings like these, which show effects from BC, or from urban air, but not from secondary or rural PM, be generalized to all-cause mortality and CVD mortality? The limited review of effects from BC, versus those of PM2.5, in the previous several paragraphs contains suggestive evidence that BC might be substantially responsible for effects that had previously been found for PM2.5, in studies where data regarding BC or traffic density or other vehicular emissions had not yet been included. BC, roadway proximity, and traffic density are increasingly specific indicators for physicochemical properties of pollution that contain toxic species and harm human health. However, none of these indicators, including BC, can yet be seen as causally responsible for the majority of mortality and morbidity effects attributed to PM2.5 exposure, without further substantiation. The studies reviewed herein appear to justify regulation of BC per se in a manner that also removes substantially all co-emissions. Future research should attempt to further parse out effects of biologically active carbonaceous material (and biologically active metals) to further elucidate the roles of different PM2.5 species in causing human mortality and CVD morbidity.
Health Effects of BC Using EPA Criteria for Judging Potential Causality
This section places the health effects of BC and co-emissions in the context of the assessment criteria that the U.S. EPA currently uses for PM2.5 mass. The first assessment criterion is “Consistency of the observed association.” Virtually every population-based epidemiology study reviewed herein that assessed BC (in Europe, BS) found associations with BC, despite studies being done in different countries in North America and Western Europe, with different study designs, with different mixes of pollutants, and with poorer exposure information for pollutants (such as BC) having greater local spatial variability. Several studies found BC associations, but did not find total PM2.5 mass associations, and associations with other PM2.5 species were not as consistent as were those for BC. In the Delfino et al. (Citation2010) human panel studies, SOC was rarely associated with endpoints studied, although POC and BC/EC almost always were so associated, suggesting that atmospheric processes might reduce the toxicity of organic gases, to the extent that such gases might be toxic in the first place. Results of most studies that assessed health effects in people living in close proximity to major roadways found significant adverse CVD findings similar to those associated with BC in many human panel studies, and similar to the findings in Stern et al. (Citation1988) when airborne vehicular pollution was reduced.
The second U.S. EPA criterion is “Coherence,” while the third is “Biological plausibility.” The CVD mortality and hospital admission findings are coherent with toxicology, controlled human exposure, and human panel studies as they relate to various endpoints that underlie cardiovascular disease (e.g., oxidative stress, inflammation, platelet aggregation, vasoconstriction, vascular function, endothelial dysfunction, lipoperoxidation, increases in atherosclerotic lesions). These studies suggest biological mechanisms whereby the CVD mortality and morbidity associations in population based epidemiology may be explained. Biological plausibility is thus established for many different CVD endpoints. Some endpoints found in controlled human exposure and human panel studies (e.g., ST-segment changes) are also consistent with CVD mortality and morbidity findings. Peters et al. (Citation2004) found a significant association between people having a first MI and being in traffic (car, bicycle, mass transit) an hour before the MI. Similarly, Albert et al. (Citation2007) found large associations for having an implantable cardoverter–defibrillator (ICD) shock, specific to ventricular tachycardia or ventricular fibrillation, with driving an hour previously. Both these findings add coherence to other studies finding adverse CVD morbidity or mortality effects associated with vehicular emissions. A further review of coherence among toxicology, human panel studies, and population-based epidemiology for CVD biological mechanisms is found in Grahame and Schlesinger (Citation2010).
Few studies reviewed herein attempted to examine a gradient of response, for example, a “dose-response function”, the fourth criterion. However, Kim et al. (Citation2012) found that DPM caused increasing oxidative stress in a dose-dependent manner in neonatal rat cardiomyocytes, and Li et al. (Citation2002) found a decreasing ability of macrophages and human lung epithelial cells to deal with oxidative stress with increasing exposure to DPM.
With regard to the fifth criterion, “Experimental evidence,” the E-ZPass study (Currie et al., Citation2009) is an intervention that resulted in fewer idling and accelerating vehicles at toll plazas, and a nearly immediate reduction in incidence of prematurity and low birth weight among mothers who lived within 2 km of the now less polluted toll plazas, versus those living near the highways but further than 2 km from the toll plazas. A similar intervention, from an earlier time frame, was reviewed in Stern et al. (Citation1988), which showed that after ventilation was added to tunnels in New York City circa 1970, a constant annual reduction in mortality from atherosclerotic disease was observed in tunnel officers, relative to bridge officers (who, being outdoors, had always had natural ventilation). Highway proximity studies themselves can be seen as a kind of intervention study, in that they examine the extent to which effects are elevated where highway pollutants are high, versus where they are not.
The evidence appears quite strong that BC (and associated carbonaceous co-emissions) is causally related to risks of all-cause and CVD mortality and of CVD morbidity, using the five U.S. EPA criteria. Although the literature regarding BC/EC/PAHs/diesel emissions as they affect birth outcomes, cognitive development, and other CNS effects is not nearly as extensive as that for many different CVD effects, the limited literature that is available suggests important adverse effects for which further study is crucial. It does appear, however, that diesel emissions are causally linked to the development of lung cancer (IARC, Citation2012).
The findings reviewed in this synthesis section contain our conclusions with regard to research results in the United States and Western Europe. Our review suggests that regulators worldwide might wish to consider establishing an ambient air quality standard for BC, as suggested by the WHO (EU branch). In the next section, we consider studies of mortality and morbidity related to ambient air pollution, as well as concentrations of emissions of BC, OC, and PAHs, in other parts of the world.
Characterization of Amounts, Selected Sources, and Health Effects of Carbonaceous Emissions in Developing Countries
Overview of BC, OC, and PAH emissions
As shown in , estimated emissions of BC and OC in Asia and Africa, and in selected countries such as India and China, are far higher than in the United States and Western Europe. The higher emissions are mainly from industry and residences, and less so from transport, with only small differences in emissions from the electricity generation sector, which in any case are quite low (8111 Gg/yr of worldwide BC emissions, of which 73 Gg/yr is estimated from electricity generation). Total BC emissions are in close agreement with the 7950 Gg/yr in Bond et al. (Citation2004), but BC from power is 10 times higher than the 7 Gg/yr in Bond et al. Despite the 10-fold difference in estimates of BC emissions from power generation, the near-complete combustion of carbonaceous material in power boilers seems to result in relatively little emissions of BC and OC.
One advantage of these emission estimates is that they were developed, worldwide, from inventories of fuels and assumptions about fuel quality and emissions control in different localities, enabling comparison among large geographic areas on a consistent basis. One possible unexpected finding from this table is the lack of major differences among East Asia, South Asia, the United States, and OECD Europe in BC emissions from transportation, considering the levels of pollution controls in the United States and Western Europe on vehicles of all types.
Despite the value of having a consistent basis for estimation across geographic areas, it is also crucial to have up-to-date knowledge about local conditions such as pollution controls in specific countries; actual emissions might on this basis differ considerably from estimates. Tables S11 (China) and S12 (India) from Lei et al. (Citation2011) and Kurokawa et al. (Citation2013), respectively, contain estimates using such knowledge, for 2000, 2005, and 2008, for PM2.5, BC, and OC, including several industrial sectors.
There are important differences between BC emissions among the tables. For example, estimates BC emissions from transport in 2010 in China as 104 Gg/yr, while Table S11 suggests BC emissions from transport in 2005 as 190 Gg/yr. estimates Chinese industrial BC emissions at 951 Gg/yr, higher than the 620 Gg/yr in Table S11 (from all the industrial activities combined). Residential BC emissions estimates aren't too far apart (627 Gg/yr in ; 700 Gg/yr in Table S11, total of residential coal and residential biomass).
Similarly, there are important differences in estimates of BC emissions in India, comparing (2010) and Table S12 (2008). In , transportation emissions of BC in India are 84 Gg/yr, while in Table S12, the estimate is 243 Gg/yr, almost 3 times higher. However, estimated BC emissions in Table S12 are lower for all industrial activities (88 Gg/yr) than in (128 Gg/yr). Thus, while there isn't yet clarity in pinpointing amounts of BC emissions from different sources in China and India, there is no question that the emissions, in toto, are quite high (1760 or 1510 Gg/yr in China, 528 or 713 Gg/yr in India, from , S11, and S12).
Major sources of BC and OC emissions in Asia, Africa, and Latin America were once major sources of such emissions in the United States and Western Europe. Primary among these are biomass (mainly for cooking) and coal (mainly for heating and industrial purposes), combusted in most of these cases without pollution control equipment and at ground level. Table S11 shows that residential biofuel BC emissions in China in 2005 were 590 Gg/yr, considerably more than the 321 Gg/yr of BC emitted by all sources in the United States in 2010 (). Chinese residential coal BC emissions in Table S11 are estimated to be 110 Gg/yr, more than three times higher than total residential BC emissions in the United States of 40 Gg/yr (). Yet the United States has less than one-fourth of the population of China, burns very little coal in residences, and has emission controls for woodstoves, which are a relatively small percentage of residential heating. Thus, given how much coal is burned for heating north of the Huai River in China (see following discussion), it is quite possible that the residential coal BC emissions estimate of 110 Gg/yr may be low. As shown in the following, uncontrolled coal and biomass burning can cause extremely high PM levels both within residences and in ambient air, with the potential for related mortality and morbidity.
Characteristics of emissions in China and elsewhere
Zhang et al. (Citation2012) report on chemical characteristics from burning of coal and wood in rural Chinese households. Carbonaceous PM dominate PM2.5 mass in each (41% for wood, 55% for coal). The OC/EC ratio was 10.8 for wood, 7.6 for coal, although it must be noted that these ratios can exhibit considerably variability. Peak values for particle size were under 100 nm, with PM from wood in smaller sizes (bimodal 10–20 nm and 40–50 nm for wood, unimodal 70–76 nm for coal). PAHs were not reported.
Pachauri et al. (Citation2013) report winter period EC and OC concentrations for 16 Chinese locations. Rural areas could have high levels of each (EC = 37 μg/m3, OC = 72.6 μg/m3, Miyun, China); the southern city of Guangzhou had lower, but still elevated concentrations (EC = 14.5 μg/m3, OC = 41.μg/m3).
Wu et al. (Citation2005) monitored winter and autumn concentrations of 16 particulate PAHs in several locations in the northern Chinese city of Tianjin, a city with extensive use of coal for heating. In the location with highest winter PAH levels, total PAH concentration was 2165.2 ng/m3 in winter but only 11.72 ng/m3 in autumn; in the location with lowest winter period values, differential concentrations were 69.3 ng/m3 (winter) and 33.6 ng/m3 (autumn). Wang et al. (Citation2011) report annual average PAH levels in three northern Chinese sites. “Background site” PAH concentrations were 39.4 ng/m3; “rural village” concentrations were 355 ng/m3; and “urban” PAHs were 1010 ng/m3. Gao et al. (Citation2013) used tracer-based source apportionment to estimate the proportion of PAHs from different sources in the southern coastal Chinese city of Guangzhou. Traffic, biomass, and coal were found to contribute 11%, 31%, and 58% of PAHs, respectively. For comparison, annual average PAH levels in the urban Roxbury section of Boston, MA, were 18 ng/m3 (Levy et al., Citation2003).
Fine et al. (Citation2001) characterize chemical emissions from fireplace burning of six hardwoods and softwoods from the northeastern United States. OC constituted more than 80% of PM2.5 emissions from each wood type. EC was generally between 3% and 7% of mass, but was considerably higher when woods high in hardened sap (eastern white pine), or with bark that produced high amounts of black smoke (paper birch) were used. OC/EC ratios were 3:1 or less for these last two woods, but roughly 15:1 to 20:1 for the other four. Many PAHs were consistently found, but were not major constituents of wood smoke mass.
Bari et al. (Citation2009) analyze particle composition and amounts in a part of Germany that uses wood for heating. In January, the authors found that wood smoke approached 60% of ambient PM10. By March, that percentage had declined to 20%. In a 5-month winter period, biomass burning contributed 93% of combustion-related PAHs in PM10, and the concentration of PAHs with carcinogenic properties was 49% total PAHs, indicating that wood smoke contributed significant amounts of PAHs, but that these PAHs on average had about half the carcinogenicity of other ambient PAHs.
A study from the highlands of Tanzania examined PM2.5 and PAH concentrations in households burning different fuels for cooking (Titcombe and Simcik, Citation2011). The fuels used were liquefied petroleum gas (LPG), a kerosene/charcoal mix, charcoal, and wood (open burning). PM2.5 concentrations associated with these fuels were 14, 88, 588, and 1574 μg/m3, respectively. PAH concentrations associated with these fuels were <1, 57, 334, and 5040 ng/m3, respectively (Titcombe and Simcik, Citation2011). Considering that large U.S. cities have annual average BC concentrations (central monitor) from 0.3 to 3.0 μg/m3 (U.S. EPA, 2012), PAH concentrations above 5 μg/m3 are exceptionally high. The authors also examined benzo[a]pyrene (BaP) equivalent exposures, which reflect potential carcinogenicity, finding equivalent exposures of 0, 8, 44, and 767 ng/m3, respectively, for the four fuels.
Saraswat et al. (Citation2013) used spatiotemporal land use regression models to establish a “map” of PM2.5, BC, and ultrafine PM in New Delhi, India. The authors found marked predicted peak levels of all three emissions in the morning. Morning versus afternoon concentration peaks were approximately 315 versus 100 μg/m3, 35 versus 8 μg/m3, and 90 versus 70 ultrafine particles/1000 cm3, respectively, as predicted on a 30-km transect. The PM2.5 to BC ratio was thus about 10 in the morning and about 12 in the afternoon. Monthly predicted BC and PM concentrations were higher in February (approximately 70 and 300 μg/m3, respectively), than in March, April, and May. Saraswat et al. (Citation2013) thus established that BC levels in New Delhi can be considerably elevated, relative to the United States and Western Europe.
Generally, as noted here and in particular in the wood-smoke toxicology section that follows, the characteristics of wood smoke and biomass emissions depend on the chemical makeup of the biomass or wood being burned, moisture content of the fuel, heat of combustion, and completeness of combustion. Thus, characteristics of emissions can vary widely. What emissions of wood smoke and biomass have in common is that much of the emissions is carbonaceous, either solid or gas, contains multiple PAHs, and has carcinogenic potential. The mix of BC, OC, and PAHs will differ from emissions from other inefficiently combusted solid fuels, but all will be present.
Overview of health effects
Health effects from the high pollution levels in places like Asia are substantial, as shown in the following, but as with data on pollution concentrations, considerably more work will be needed to refine risk estimates.
Yang et al. (Citation2013) reports that Chinese life expectancy rose considerably from 1990 through 2010, from 69.3 years to 75.7 years, despite the increases in air pollution in that time frame which reflected the unprecedented pace of China's economic growth and industrialization. Comparable figures for the United States for these same years were 75.2 and 78.2 years. At the same time, Yang et al. (Citation2013) report that ambient PM was the fourth largest cause of disability-adjusted life years (DALYs), and household pollution from all solid fuels was the fifth largest cause (approximately 15% of DALYs combined). Most of these DALYs were via cardiovascular diseases in each case. Both ambient PM and household pollution from solid fuels were each seen as causing DALYs roughly comparable to DALYs from smoking. Because the second largest cause of DALYs was high blood pressure, and because particulate pollution is associated with higher blood pressure in polluted Chinese cities (Langrish et al., Citation2009), it is quite possible that ambient PM might actually cause a higher percentage of DALYs via the mechanism of high blood pressure.
A different method of looking at air pollution's impact on mortality is found in Chen et al. (Citation2013a), which examined consequences of China's “Huai River policy.” Starting around 1950, the Chinese government gave free coal for heating homes and offices north of the Huai river, which bisects China between the Yangtze and Yellow Rivers. The coal-burning infrastructure is still in place, and still used.
Thus, another “natural” intervention: If burning substantial amounts of coal without any pollution control is particularly harmful, could increased harm be observed where coal was the prevalent source of heating versus areas where it was not? The authors used a regression discontinuity design that is common in disciplines other than air pollution (two-stage least squares), in which mortality and pollution were measured and then associated both north and south of the Huai River. In this study, TSP was the pollutant used, presumably because it was the most widely monitored form of PM.
Chen et al. (Citation2013a) reported the following findings:
Life expectancies are about 5.5 years lower north of the Huai River than south of it.
Total suspended particulate (TS)P levels are about 184 μg/m3 (55%) higher north of the river.
Long-term exposure to an additional 100 μg/m3 of TSP is associated with a reduction of life expectancy of ˜3 yr.
Chen et al. (Citation2013a) also used the conventional ordinary least-squares design, finding no significant differences in life expectancies north versus south of the Huai River. In comparison, using the regression discontinuity design, the authors reported that the estimate of 3 years loss of life associated with exposure to an added 100 μg/m3 of TSP annually is more than 5 times higher than was derived from ordinary least squares.
These findings are both striking and new, and as such will require replication. Two issues immediately come to mind: (A) What specific constituents of the coal burning might be most harmful, and (B) since heating degree days are over twice as high north of the Huai River than south of it (Chen et al., Citation2013a), how much difference in mortality might have been observed if biomass instead of coal had been used for heating north of the river?
Mestl et al. (Citation2007a) used Monte Carlo simulations, based upon published indoor air pollution (IAP) studies and population time activity information, to create generic estimates of PM10 exposure in urban and rural areas, in the north and the south of China, combining estimates of indoor exposure from solid fuels, and outdoor exposure. For rural populations, either in the north or south, IAP represented 80–90% of PM10 exposures. In urban areas, the comparable figures were 50–60%. Average estimated exposures in southern and northern cities were 340 and 440 μg/m3, respectively. Average rural population exposure was estimated to be 750 μg/m3 in the south and 680 μg/m3 in the north. Heaviest PM10 exposure was estimated to occur in areas relying on biomass. It is difficult to compare PM concentrations in this study versus those in Chen et al. (Citation2013a) because the latter study used measured TSP, while Mestl et al. (Citation2007a) used estimated PM10. Further, the time periods are not fully comparable.
Mestl et al. (Citation2007b) uses PM10 estimates in Mestl et al. (Citation2007a), further refined by fuel type. PM10 concentrations are broken down, in each of four generic areas (rural vs. urban, north vs. south), into separate contributions of outdoor air and indoor air, depending on indoor fuel used. In the rural south, residences using biomass are exposed to an average of ˜1000 μg/m3 from all sources, ˜90% of which is due to indoor exposure. Residences using biomass for cooking in urban areas, or in the rural north, are estimated to be exposed to only marginally less average emissions than in the rural south. In contrast, those using coal in the south are exposed to less than 600 μg/m3 in average exposure from all sources. Outdoor pollution in the south is less than 100 μg/m3, versus about 250 μg/m3 in the north, presumably reflecting widespread use of coal for heating, in many cases with ventilation, in the urban north. The authors report that solid fuels are used for cooking in 80–90% of rural households, and in 40–50% of urban households. The three most common fuels for cooking are biofuels (45%), coal (27%), and gas (27%). One result that stands out in these estimates is how much higher IAP is when biomass is used, than when coal is used. This may reflect preferential use of lower emitting coal briquettes in urban areas, especially in the north (personal communication, Dr. Kristen Aunan), in combination with greater use of chimneys in urban areas.
Mestl et al. (Citation2007b) use their estimates of pollution exposure to develop estimates of mortality from indoor use of solid fuels (assuming that PM from biomass and PM from coal are equally harmful). They estimate that 3.5 million people die prematurely due to IAP in China, which in their estimation accounts for 47% of all deaths in China. This estimate is considerably higher than a 2005 WHO estimate quoted in their study of about 420,000 premature deaths from IAP. The difference in estimates likely reflects that such analyses are still in early stages in places like China. Mestl et al. (Citation2007b) appear to base their analysis of premature deaths on the difference between actual IAP and assuming that every household in China met an air quality standard of 150 μg/m3. Mestl et al. (Citation2007b) suggest that the WHO estimate is low for specific technical reasons. Wherever the correct estimate lies, the effects are very substantial. Questions the authors raise include one highly pertinent to this review, namely, are uncontrolled emissions from biomass and from coal fairly equivalent in mortality effects?
At least two studies associate lung cancer mortality with residential coal burning of coal. Hosgood et al. (Citation2011) is a meta-analysis of 25 case-control studies, virtually all from China. Heightened risk of lung cancer mortality was associated with household coal use (OR = 2.15, CI = 1.61–2.89), but risks were considerably higher in southern China than elsewhere (mainly northern or middle China, or Taiwan). The authors found that lung cancer risks in Xuanwei could differ by as much as 25-fold, depending on the mine from which the coal came. Risks were similar when studies that did not account for smoking were excluded. Changing from unvented indoor combustion to one with a chimney, or living in a home with large openings for ventilation, both decreased risk of lung cancer death.
Barone-Adesi et al. (Citation2012) examined risks of lung cancer, associated with different types of coal, in a retrospective cohort study of lifetime coal users. The authors found that there were very small risks of using “smokeless” anthracite coal in Xuanwei (9,962 users), but very large risks of using “smoky” bituminous coals (27,310 users). Absolute risks of lung cancer death before age 70 years, for using smoky coal, were 18% and 20% for men and women, versus less than 0.5% for users of anthracite. Lung cancer accounted for ˜40% of all deaths before age 60 years for those using smoky coal. Smoking history was taken into account. Despite the much higher smoking rate of men than of women, absolute risks of death from lung cancer among those using “smoky” coal were virtually equivalent for men and women. The findings of Barone-Adesi et al. (Citation2012) suggest that the 25-fold difference in lung cancer risks found in the previous study (depending on the mine), also in Xuanwei, might be due to use of anthracite versus bituminous coals. Large et al. (Citation2009) find that smoky coals from Xuanwei have high quartz content, and suggest that the lethality of such coals might be due to interactions between PM10 silica particles and organics.
Mortality effects from wood burning are estimated in another “natural experiment,” from Tasmania, south of the Australian continent (Johnston et al., Citation2013). Launceston is a city of 67,000 people, located in a valley and subject to inversions. A multi-million-dollar program to replace wood heaters, and to simultaneously enforce environmental regulations in homes with high emissions, began in 2001. By 2004, the prevalence of wood stoves had declined from 66% to 30% of households, replaced by electricity (mostly from hydro); improved operation of wood heaters occurred in remaining homes. Wintertime outdoor PM10 declined from 44 μg/m3 during 1944–2000 to 27 μg/m3 during 2001–2007. Significant declines occurred for all-cause (11.4%) and CVD (17.9%) mortality among men (Johnston et al., Citation2013). In order to discern whether reductions might have been a product of widespread temporal improvements in health, results were compared to mortality trends for the same periods in the control city of Hobart, Tasmania, which did not experience a reduction in wood stove emissions. No significant improvements in mortality were observed in Hobart during the time frames analyzed for Launceston, suggesting that the improvements in Launceston were due not to secular mortality trends, but to the reductions in wood-smoke emissions there.
Riojas-Rodriguez et al. (2011) examined PAH content of biomass burning in Mexico, via urinary biomarkers before and after replacement of conventional open burning with lower emission Patsari stoves. The authors found significant reductions, post replacement, of between 34% and 42% for hydroxylated metabolites of several PAHs.
Bassani et al. (Citation2010) report that solid-fuel use in India (mostly biomass or dung) is associated with increased mortality in children aged 1–4 years, accounting for 20% of childhood deaths in this age group. Coal use was highly associated with lung cancer among all users and never-smokers, while wood use was associated with hypopharyngeal cancer among all individuals, but not among never-smokers, in a study of indoor solid fuel use in India (Sapkota et al., Citation2008). A study of Indian women who used biomass for cooking examined various endpoints associated with CVD, with controls being women who cooked with LPG (Dutta et al., Citation2010). Biomass users, who were exposed to three times the concentrations of PM in kitchens, had higher prevalence of hypertension, elevated oxidized low-density lipoprotein, increased platelet P-selectin expression, upregulation of ROS generation in leukocytes, and depletion of erythrocyte superoxide dismutase (SOD, which fights oxidative stress). All of these biological changes are factors in development of cardiovascular diseases. Delfino et al. (Citation2008, Citation2009) found elevated platelet P-selectin expression and depletion of erythrocyte superoxide dismutase (SOD) in a panel study of elderly residents of retirement centers in Los Angeles, in association with higher concentrations of BC, EC, and primary organic carbon (POC), suggesting commonality of effects from different sources of similar emissions. Increased ROS has been found in association with exposure to diesel emissions in workplaces (e.g., Sauvain et al., 2009, bus maintenance workers; Lee et al., Citation2011, diesel exhaust inspectors). The findings of similar effects in association with higher levels of biomass emissions raise the possibility that BC/EC and POC may cause these CVD effects, as common emissions from different sources.
Barone-Adesi et al. (Citation2012) showed lung cancer mortality in China to be strongly associated with “smoky” bituminous coal, but not with “smokeless” anthracite coal. One of the studies assessed in the meta-analysis of Hosgood et al. (2012) and finding highly elevated lung cancer mortality risks is a study (in Chinese) finding elevated lung cancer risks for those using smoky coal versus using anthracite. An explanation for such differences is found in Liu et al. (Citation2009), who evaluate the PAH content (sum of PAHs) and benzo[a]pyrene equivalent carcinogenicity (BaPE) of bituminous and anthracite coals and briquettes, from Beijing and Shanxi, in North China. Bituminous coals (including briquettes) had considerably higher PAH content than anthracite coals (and briquettes); the sum of 15 PAHs from anthracite ranged from 52.8 to 155.8 mg/kg (emission factors), whereas for bituminous coals, the emission factors ranged from 325.3 to 1095.6 mg/kg. The greatest difference between bituminous and anthracite coals, however, was in carcinogenicity potential, as measured by BaPE: Bituminous coals measured between 30.5 and 134.5 (depending on specific coal and on high vs. low heat), while the anthracite coals were never higher than 1.1. Since PAHs (and BaP specifically) are known risk factors for lung cancer, this information appears to largely explain the far higher lung cancer mortality found for those using “smoky” bituminous coals (Barone-Adesi et al., Citation2012).
Shen et al. (Citation2010) found that the PAH emission factor (16 PAHs) of the anthracite coal (briquette) was 6.25 mg/kg, and those of bituminous coals up to 253 mg/kg. Bituminous coal briquettes (14.3 mg/kg) had lower PAH emission factors than the “chunk” bituminous coals (140 and 253 mg/kg).
Chen et al. (Citation2005) analyzed emission factors for five types of coal briquettes used in China: one subbituminous, three bituminous, and one anthracite. BC emission factors for anthracite were tens to hundreds of times smaller than for the other four coals (0.004 g/kg for anthracite, between 0.064 to 0.675 g/kg for the other coals); OC emission factors were several hundred to thousands of times less (0.017 g/kg for anthracite, between 3.580 and 13.818 g/kg for the others). As in Liu et al. (Citation2009), emission factors for carcinogenic BaPE were smaller for the anthracite briquettes than the other briquettes. Thus, the anthracite coals emit not just less PAH emissions, but also less BC and OC. An important question for future study would be: Are anthracite coals also associated with considerably smaller CVD mortality risks, given their smaller emissions of PAHs, BC, and OC?
Biomass intervention studies
Biomass intervention studies almost uniformly show benefits of reduced emissions when cleaner-burning stoves are introduced, and/or when ventilation is improved. In a study of women in highland Guatemala, McCracken et al. (Citation2011) found that when chimney stoves were used instead of open fires, PM2.5 exposures declined from 266 μg/m3 to 102 μg/m3, and odds of ST-segment depression were concurrently reduced. McCracken et al. (Citation2007) also found that such an intervention, with similar reductions of PM, was associated with reductions in blood pressure (DBP reduction of 3.0 mm Hg, CI = –5.7, –0.4; SBP reduction of 3.7 mm Hg, CI = –8.1, 0.6). Another study of effects of poorly combusted biomass among women in rural China (Baumgartner et al., Citation2011) assessed pollution exposure using personal monitors. The study found that among women older than 50 years (but not among younger women), both SBP and DBP were increased in association with increases in PM2.5, in homes using biomass (increase of 4.1 mm Hg in SBP, 1.8 mm Hg in DBP, per 1-log- μg/m3 increase in PM exposure).
Birth outcomes are affected by levels of biomass emissions in several parts of the world. In Guatemala, Thompson et al. (Citation2011) found that pregnant women who used a chimney stove, versus open fires, gave birth to children weighing 89 g more (CI = –27 to 204 g). There were 174 total births (69 to mothers with chimney stoves). A larger study from rural Guatemala, with 1717 total births, found significantly lower birth weight (63 g lower) after adjustments for children born to women using open fires instead of chimney stoves or cleaner fuels (Boy et al., Citation2002). A considerably larger intervention involved the extensive California wildfires of the fall of 2003, which exposed most people in the Los Angeles basin to substantial amounts of smoke from burning vegetation for 19 days. Holstius et al. (Citation2012) compared birth outcomes in pregnant women not exposed to the smoke to outcomes in those exposed during each trimester. The authors found that relative to pregnancies before and after the wildfires, mean birth weight was estimated to be 7.0 g lower (CI = –11.8, –2.2, 38,739 births) when the wildfire occurred during the third trimester; 9.7 g lower weight (CI = –14.5, –4.8, 39,435 births) when it occurred during the second trimester; and 3.3 g (CI = –7.2, 0.6, 60,270 births) lower for first-trimester exposure. Cookstove intervention studies are different from that of Holstius et al. (Citation2012) in several ways: users of biomass for cooking in places like Central America receive wood-smoke exposures daily (not for just 19 days); exposure levels may be more certain (during the 19-day wildfire in California, PM levels were characterized as “heavy” or “light”); and perhaps diet and health of the mother might be better in California. Although the reduction in birth weight due to exposure was considerably smaller in California, it is nevertheless striking that the same low-birth-weight outcomes, after exposure to biomass smoke, were observed after the 19-day wildfire in mountains east of the Los Angeles basin as well as in places where daily biomass exposure can be very high.
Toxicology of Wood Smoke
The last seven entries in Table S8 list health effects (oxidative stress, vascular and cardiac effects, inflammation, lipoperoxidation, etc.) found in controlled human exposures to wood smoke. An overview of the toxicology of wood and biomass combustion is presented here.
One of the more commonly examined endpoints used in studies of wood smoke toxicology involves pulmonary immune defense against infectious agents. In various animal species, short-term exposure to wood smoke has produced reduction in both pulmonary macrophage-mediated bacterial phagocytosis and intracellular killing of certain pathogens with no evidence of pulmonary inflammation, indicating that wood-smoke exposure may result in subclinical effects in the absence of any apparent acute lung injury (Fick et al., Citation1984); suppressed pulmonary clearance of certain bacteria, again with no other evidence of pulmonary pathology (Zelikoff et al., Citation2000; Migliaccio et al., Citation2013); and exacerbation of some indices of allergic airway inflammatory response (Barrett et al., Citation2006). Effects on ROS production and the oxidative/reductive state, which can affect resistance to microbial infection, also appear to be a common result of wood-smoke exposure. This includes suppressed production of superoxide (Thomas and Zelikoff, Citation1999); reduction in glutathione peroxidase activity; increased catalase activity; enhanced HO-1 activity (Ramos et al., Citation2013); and increased free radical generation. While some effects may occur without inflammation, wood smoke is capable of producing an inflammatory response as evidenced by increased generation of inflammatory cytokines (Leonard et al., Citation2000; Matthew et al., Citation2001; Demling and LaLonde, Citation1990; Demling et al., Citation1994; LaLonde et al., Citation1994; Liu et al., Citation2005; Seagrave et al., 2005; LaLonde et al., 1997).
Controlled exposure studies with healthy human subjects are not consistent in terms of effects of wood smoke exposure. Some studies have shown no induction of oxidative stress (Sehlsedt et al., Citation2010) nor inflammation (Riddevold et al., Citation2012; Forchhammer et al., Citation2012). However, others have shown increased heart rate and decreased HRV, including the high-frequency domain indicating vagal inhibition, and an increase in central arterial stiffness, which was suggested as likely driven by alteration in vascular autonomic control, vascular endothelial dysfunction, or reduced bioavailability of NO (Unosson et al., Citation2013). However, others have found no change in exhaled NO due to wood-smoke exposure or any effect on arterial function (Riddervold et al., Citation2012; Barregard et al., Citation2008).
Wood smoke consists of emissions from a variety of biomass appliances, which differ in the fuel type, burning conditions, and combustion technology. In this regard the physicochemical properties of PM generated during wood combustion may vary considerably, depending upon combustion conditions, type of appliance, type and moisture content of the wood, and the combustion phase (e.g., Weimer et al., Citation2007; Kocbach et al., Citation2009). The potential for any wood-smoke-derived PM to produce adverse health effects is highly dependent upon these physical and chemical properties, such as particle size, surface area, chemical composition (mostly BC and OC by mass), any adsorbed gases, and so on. For example, Zelikoff et al. (Citation2002) noted a diminished immunotoxic response due to inhaled wood-smoke effluent in rats following exposure to effluent having the PM phase removed, and PM derived from different combustion conditions has been shown to induce differential proinflammatory responses (Jalava et al., Citation2010; Danielsen et al., Citation2011). Furthermore, PM derived from poor combustion conditions having elevated organic content has been shown to have a greater cytotoxic potential than PM derived from more complete combustion conditions (Jalava et al., Citation2010; Bolling et al., Citation2012). When effects of total PM with organic extracts of the PM and washed particles were compared, the response induced by the organic fraction was not linked to the content of PAHs, suggesting that other organic components (e.g., quinone-like compounds) were involved (Bolling et al., Citation2012). Studies that examined effects of combustion cycle phase have shown differences in response at the startup versus the burn-out phase (Stockfelt et al., Citation2012); there are differences in the organic chemical makeup between these two phases.
In addition to immune and inflammatory response, wood smoke has also been shown to alter lung mechanics in animal studies. Wood smoke derived from Douglas fir reduced pulmonary compliance in guinea pigs (Wong et al., Citation1984) and in dogs (Stephenson et al., Citation1975) following acute exposure, while increased dynamic compliance was observed in mice exposed for 4 or 12 wk to pine wood smoke (Tesfaifgzi et al., Citation2002). Wood smoke has also been shown to alter airway responsiveness, inducing airway hyperreactivity in response to bronchoconstrictor challenge (Hus and Kou, 2001; Lin et al., Citation2001). However, results in controlled human exposures examining other ventilatory parameters have found no effect (Sehlstedt et al., Citation2010; Riddervold et al., Citation2012). However, in humans, wood smoke also resulted in increased serum amyloid A (SSA), an acute-phase protein that is often found to increase under conditions associated with inflammation and is a risk factor for CV morbidity (Ridker et al. Citation2000) and may also play some role in development of atherosclerosis (Chait et al., Citation2005). Exposure was also associated with increased plasma levels of Factor VIII and the Factor VIII/von Willebrand Factor ratio, indicating an effect on the balance of coagulation factors involved in blood clotting
In summary, the bulk of toxicological evidence provides biological plausibility, in terms of potential toxicological mechanisms, for epidemiological observations of adverse health outcomes from exposure to wood smoke. While the biological effects are dependent upon the type of wood and the combustion conditions, short-term exposure has been shown to compromise pulmonary immune defense against infectious agents, and at least one of the specific targets implicated in this effect are the pulmonary macrophages. This suggests that exposure would make the host more susceptible to infectious disease. In addition, depending upon the condition, exposure can result in an inflammatory response. While such effects are most pronounced with high-dose exposures, these studies nevertheless can provide insight into potential mechanisms that may occur at lower exposure levels. Very few studies have examined biological responses from PM derived under varying combustion conditions and fuel types. In vitro assessments suggest that biological outcomes may vary with conditions of combustion, in that PM derived from poor combustion seems to have greater cytotoxic effect than PM derived from more complete combustion conditions (Bolling et al., Citation2009). When combustion is less complete, PM contains more BC and organic compounds (and brown carbon under smoldering conditions). It appears that these organics in wood-smoke PM may be the specific fraction most involved in the release of inflammatory mediators.
A potential concern related to wood smoke exposure is production of lung cancer. While specific components of these emissions are suggestive to be carcinogenic during in vitro studies, and PAHs are present in incompletely combusted wood and biomass smoke, neither epidemiological nor toxicological evidence is yet robust enough to come to any conclusive decision (Lim and Seow, Citation2011).
The relative toxicity of wood smoke compared with vehicle exhaust emissions is unclear. The database for DEE is more robust and consistent than that for wood smoke, especially related to controlled human exposures. However, some generalizations may be made. Epidemiological and observational studies have shown that BC contributes to cardiovascular and respiratory morbidity/mortality. While, as noted, the exact mechanisms remain somewhat unclear, the available database suggests that oxidative stress-induced inflammation from wood smoke is likely involved (Mudway et al., Citation2004). Furthermore, it is also likely that inflammatory changes produced in the respiratory tract can induce systemic inflammation, production of acute-phase proteins, alteration of coagulation factors in the liver, and destabilization of atheromatous plaques (Donaldson et al., 2001, Citation2005; Pope et al., 2004). Pulmonary inflammation has been shown to result in an increase in systemic cytokine levels (Ruckerl et al., Citation2007). Both DEE and wood smoke may alter autonomic control of cardiac rhythm, which, in turn, increases the risk for cardiac arrhythmia (Donaldson et al., Citation2005). Toxicology of wood-smoke emissions provides evidence for epidemiological associations observed, mainly in localities with substantial biomass combustion.
BC, Multipollutant Studies
Only limited studies assess mortality and morbidity associations in developing countries with BC and other PM2.5 species to date. Geng et al. (Citation2013) uses daily BC and PM2.5 concentrations to analyze associations with daily mortality (all-cause, CVD). An IQR of BC (2.7 μg/m3) was associated with significant increases in daily all-cause (2.3%) and CVD (3.2%) mortality; the size of the effects roughly doubled after adjustment for PM2.5, remaining significant, while PM2.5 effects (IQR for PM2.5 = 41.8 μg/m3) became negative and nonsignificant after adjustment for BC. Mortality associations were considerably larger in winter months. Geng et al. (Citation2013) noted the similarity between their results and those of Janssen et al. (Citation2011), who found that reducing a unit of BC would increase life expectancy 4 to 9 times more than reducing a unit of PM2.5 (roughly one-tenth of which is BC in the United States and western Europe). In Shanghai, “major sources of BC are motorized traffic, coal burning, shipping emissions, and industrial sources” (Geng et al., Citation2013).
Two multipollutant studies performed in Santiago, Chile, used 18 PM2.5 species, mostly metals but including EC, OC, and sulfur. Cakmak et al. (Citation2009a: daily all-cause and cardiac mortality, 2009b: daily hospital admissions) found association only with EC and OC, in single-pollutant models, and after full adjustments for long-term trends, day of the week, average humidity index on day of death and day prior to death, and other PM2.5 species. Associations for EC were larger than for OC (RR for mortality = 1.079 for an IQR increase in EC, 1.066 for OC; relative risk [RR] for emergency department [ED] visit = 1.115 for an IQR increase in EC, 1.093 for OC). Results of these two studies closely parallel results of several studies in the United States that use a large number of PM2.5 species, including Bell et al. (Citation2009a), Peng et al. (Citation2009), Lipfert et al. (Citation2006b, Citation2009), and Krall et al. (Citation2013), in that all of these studies associate BC with all-cause and/or CVD mortality or hospital admissions (of these studies that included OC, OC was also associated).
General Conclusions
In assessing (in the preceding Synthesis section) the health effects from BC, diesel exhaust, and co-emissions in the United States and Western Europe, we have found these emissions to be associated with multiple mortality and morbidity endpoints in both population-based epidemiology and human panel studies, and found that toxicological studies provide mechanistic plausibility. These associations were stronger when evaluated in studies using improved methods of assessing subject exposure. Furthermore, in models with multiple pollutants, BC/EC was more often associated with adverse health outcomes than were other PM2.5 species. Using the U.S. EPA rubric for judging causality, we conclude that there is a causal relationship between BC/EC for all-cause and various CVD mortality and morbidity endpoints.
When examining concentrations in rapidly developing countries having high levels of air pollution, it is clear that concentrations of BC and PAHs are considerably higher than in the United States and Western Europe, and that adverse health outcomes are also highly elevated in these countries. The incidence of lung cancer was extremely elevated when exposure was to residential emissions of smoky bituminous coals high in heavy molecular weight PAHs having a high benzo[a]pyrene equivalent. However, when exposure was to emissions of “smokeless” Chinese anthracite coals low in high molecular PAHs and having a low benzo[a]pyrene equivalent, lung cancer incidence was not elevated.
These findings suggest that other emissions with high-molecular-weight PAHs also cause lung cancer. Wood and biomass smoke have many deleterious effects, including premature mortality. Wood-smoke emissions contain BC, OC, and PAHs, but these vary substantially with types of biomass, completeness of combustion, and degree of pollution control. More research is needed to understand how best to reduce toxicity of biomass combustion in places where it cannot be replaced by cleaner burning fuels.
Thus, adverse health effects as well as levels of BC and PAHs occur to a great extent in the developing world. Reducing these specific emissions will benefit health worldwide. The U.S. EPA rubric for judging causality would be a useful tool in prioritizing which specific emissions and sources to control and regulate.
List of Acronyms
| ACS | = | American Cancer Society |
| AMI | = | acute myocardial infarction |
| ASHD | = | arteriosclerotic heart disease |
| B(a)P | = | benzo(a)pyrene, a PAH considered to be particularly carcinogenic among PAHs |
| BaPE | = | benzo(a)pyrene equivalent, a means of summing and expressing total carcinogenic potential of many PAHs emitted from a source |
| B(b)F | = | benzo(b)fluoranthene, a PAH |
| BC | = | black carbon, see text for definition |
| BDNF | = | brain-derived neurotrophic factor |
| BS | = | black smoke, a measure of PM used in Europe for decades |
| CAA | = | Clean Air Act of 1970, which established the ability of EPA to regulate pollutants such as PM under the National Ambient Air Quality Standards part of the Act |
| CAPs | = | concentrated ambient particles |
| CASAC | = | Clean Air Scientific Advisory Committee, a committee which advises EPA in considering new air quality standards for six pollutants, including PM |
| CB | = | carbon black (industrial product) |
| CHD | = | coronary heart disease |
| CI | = | confidence interval |
| CNT | = | carbon nanotube |
| CVD | = | cardiovascular disease |
| DBP | = | diastolic blood pressure |
| DEE | = | diesel engine exhaust |
| DPM | = | diesel particulate matter, also a descriptor of one of 33 specific air pollutants listed under EPA’s National Air Toxic Assessment (see http://www.epa.gov/ttn/atw/nata/34poll.html); also used as a synonym for DEP (diesel exhaust particulate) to avoid confusion |
| EC | = | elemental carbon, see text for definition |
| ED | = | emergency department |
| GIS | = | geographic information systems |
| HO-1 | = | heme oxygenase-1, an anti-oxidant enzyme |
| HRV | = | heart rate variability |
| HSPH | = | Harvard School of Public Health |
| IAP | = | indoor air pollution |
| IARC | = | International Agency for Research on Cancer, a unit of the WHO |
| ICD | = | implantable cardioverter-defibrillator, |
| IQR | = | Interquartile range, where pollutant concentrations are separated into quartiles of concentrations, enabling comparison of health effects, for example, between exposures at the 25% and 75% exposure values |
| IHD | = | ischemic heart disease |
| LDL | = | low density lipoprotein |
| LPG | = | liquefied petroleum gas |
| MAPKs | = | mitogen-activated protein kinases. MAPKs are involved in directing cellular responses to various stimuli |
| MI | = | myocardial infarction (heart attack) |
| NAAQS | = | National Ambient Air Quality Standards |
| NF-kB | = | a protein complex which controls various inflammatory and other reactions to stresses such as pathogenic stimuli |
| NLCS | = | Netherlands Cohort Study on Diet and Cancer |
| nm | = | nanometer |
| NMDA | = | N-methyl-D-aspartate |
| NRC | = | National Research Council, a body of the National Academy of Sciences which often assembles expert panels to advise US government agencies |
| NRDE | = | nanoparticle rich diesel exhaust |
| Nrf2 | = | a transcription factor which activates cellular anti-oxidant activity |
| OC | = | organic carbon |
| PAH | = | polycyclic aromatic hydrocarbon. These are multi-benzene ring compounds of various molecular weights and toxicities |
| PM | = | particulate matter |
| PM2.5 | = | particulate matter 2.5 microns in size or smaller |
| PM10 | = | particulate matter 10 microns in size or smaller |
| POC | = | primary organic carbon, e.g., emitted initially in particulate form, as opposed to secondary organic carbon (note: in other contexts, POC is an acronym for pyrolized organic carbon, but we do not adopt that usage here) |
| ppb | = | parts per billion |
| Pyr | = | pyrene, a PAH quasi-ultrafine: particles less than 250 nm in diameter |
| ROS | = | reactive oxygen species |
| RR | = | relative risk |
| S | = | sulfur |
| SBP | = | systolic blood pressure |
| SDNN | = | standard deviation of all normal to normal RR-intervals, a measure of HRV |
| SES | = | socioeconomic status |
| SOC | = | secondary organic carbon, a carbonaceous particulate compound that initially was a gaseous emission, but was transformed by atmospheric reactions into a particle |
| SVOC | = | semi-volatile organic compound |
| SWCNT | = | single walled carbon nanotube |
| TOC | = | total organic carbon |
| TSP | = | total suspended particulate, the initial measure of PM used by U.S. regulators |
| uf | = | ultrafine, particles 100nm or smaller in diameter |
| US EPA | = | the United States Environmental Protection Agency |
| VOC | = | volatile organic compound |
| WHO | = | World Health Organization |
Supplemental Material
Supplemental material for this article can be accessed at the publisher’s website. There are six sections in the supplemental material:
Discussion, Definitional Issues, and Selected Ambient Concentrations of BC/EC and OC.
Early Evaluation of PM Effects.
Tables S2 through S12.
Toxicology of Relevant PM2.5 Species: Cardiopulmonary Effects of Diesel Exhaust.
Neurological Effects: Cognition and Behavior.
Toxicological Effects of Biomass Smoke.
All of these sections, except the tables, are more detailed discussions of the comparable sections in the main text.
2014 Critical Review Presentation Slides
Download Zip (16 MB)Supplementary Material
Download PDF (468.4 KB)Disclaimer and Acknowledgment
The views expressed herein represent those of the authors alone, and do not purport to represent views of any U.S. government agency. The authors thank Dr. David Streets (Argonne National Laboratory), Dr. George Hidy, Ralph Freedman (U.S. Department of Energy), and JoAnn Yuill and Diana Hendershot (National Energy Technology Laboratory) for invaluable assistance.
Additional information
Notes on contributors
Thomas J. Grahame
Thomas J. Grahame has worked on economic and environmental issues related to energy and electricity for the past 37 years, including the past 35 at the U.S. Department of Energy (DOE). Prior to his DOE work, he analyzed energy issues for U.S. Senator John Durkin (D, NH), serving on the U.S. Senate Energy and Natural Resources Committee, where much of his time was involved with the Public Utility Regulatory Policies Act (PURPA), a law that began to open the door to competition in electricity generation in the early 1980s. Mr. Grahame has an undergraduate degree in Government, and a Master’s Degree in City and Regional Planning, both from Harvard University.
Mr. Grahame has authored or co-authored seven journal articles on the health impacts of different air pollutants (e.g., sulfate or black carbon); analyzing toxicological and epidemiological evidence with regard to which types of particulate matter appear to be most harmful; exploring whether study methodologies consistently cause studies to come to different conclusions; analyzing toxicological, human panel, and population-based epidemiological studies, concluding that black carbon emissions were likely causally related to eight different cardiovascular mortality or morbidity health effects; and the hypothesis that vehicular emissions marked by black carbon may cause people to die prematurely because these emissions cause oxidative stress, thus causing premature erosion of protective telomeres and quicker biological aging.
Mr. Grahame has also analyzed underlying studies and authored DOE comments to the U.S. Environmental Protection Agency (EPA) regarding the National Ambient Air Quality Standards for Particulate Matter, in 1995 and from 2002 through 2009. He initiated the most comprehensive federal government study of externalities (the field of attempting to value harm or benefit from pollution in economic terms, and then incorporate subsequently determined economic values in prices paid for relevant good and services) in 1990; authored DOE testimony on externalities before Massachusetts and Texas regulators; and defended DOE testimony in Massachusetts. Additionally, Mr. Grahame was the author of the 140-page annex on transmission access issues, part of the National Energy Strategy of the early 1990s, which led to the Energy Policy Act of 1992.
Rebecca Klemm
Richard B. Schlesinger, Ph.D., is Associate Dean for Academic Affairs and Research in the Dyson College of Arts and Sciences of Pace University, New York, NY, and Professor of Biology and Environmental Science. He received his B.A. degree in Biology from Queens College of the City University of NY, and his M.S. and Ph.D. degrees in Biology/Environmental Health Science, from New York University. Prior to his appointment at Pace University, he was Professor of Environmental Medicine and Director of the Systemic Toxicology Program in the Department of Environmental Medicine, NYU School of Medicine, as well as Director of the Graduate Program in Environmental Health Science at the NYU Graduate School of Arts and Sciences. He has developed and taught various courses at the graduate and undergraduate level in the areas of general toxicology, systemic toxicology, pulmonary toxicology, environmental health, and industrial hygiene at New York University.
Dr. Schlesinger has published extensively in the areas of respiratory toxicology of ambient air pollutants, especially related to the deposition of inhaled particles and the relationship of both particulate and gaseous air pollutant exposure to the pathogenesis of non-neoplastic pulmonary disease, and most recently in relation to overall morbidity and mortality. He is recipient of the Society of Toxicology Inhalation Specialty Section Career Achievement Award, the ILSI Morgareidge Award for Achievement in Inhalation Toxicology, and the Herbert Stokinger Award for contributions to the field of industrial and environmental toxicology. He is a Fellow of the Academy of Toxicological Sciences.
Dr. Schlesinger has served on numerous National Academy of Science committees, including the Committee on Research Priorities for Airborne Particulate Matter, the Committee on Gulf War and Health III, and the Committee on Acute Exposure Guideline Levels. He served as a member of the AIHA ERPG Committee. He has served as consultant to various governmental agencies, contributing to U.S. Environmental Protection Agency (EPA) Air Pollutant Criteria Documents and Integrated Science Assessments, and to WHO, contributing to the Clean Air for Europe Group air quality documents. He has served as a member of the EPA Clean Air Scientific Advisory Committee Review Panel for Sulfur Oxides and is currently a member of this Committee for Nitrogen Oxides. He is also a Science Advisor to the New York State Energy Research and Development Agency, and an Associate Editor of the journal, Inhalation Toxicology.
Richard B. Schlesinger
Rebecca Klemm, Ph.D., is president of Klemm Analysis Group, and directs her company’s projects to ensure the timely completion of every project within the budget allocated and adherence to strict quality control standards. Dr. Klemm has directed many projects in the area of health services research for the U.S. Departments of Veterans Affairs and of Health and Human Services, and commercial clients. She has also published extensively on the effects of air pollution on human health. Dr. Klemm has also specialized in analyses regarding public policy and regulation within a legal environment for the past 20 years. The range of projects for which she has served as an expert includes policy and regulation, employment (including Title VII, ADEA, and pay equity), Section 482 tax, quality control, product liability, and statistical auditing. She has combined her statistical expertise with her training in operations research and stochastic programming to a variety of subject matters, including the performance of econometric modeling for the U.S. Department of Energy.
Dr. Klemm served as Principal Investigator of the Coordinating Center for a large multi-center translational study of diabetes in managed care organizations for the Centers for Disease Control and Prevention (CDC). She has served as Technical Director of a contract to provide statistical, epidemiologic, and data management services to CDC for several years. Dr. Klemm has served two terms as one of the seven scientists of the National Conference of Lawyers and Scientists of the American Association for the Advancement of Science and the Science and Technology Section, American Bar Association.
References
- Adar S.D., D.R. Gold, B.A. Coull, et al. Focused exposures to airborne traffic particles and heart rate variability in the elderly. Epidemiology 18: 95–103. doi:10.1097/01.ede.0000249409.81050.46
- Albert, C.M., L. Rosenthal, H. Calkins, et al. 2007. Driving and implantable cardioverter-defibrillator shocks for ventricular arrhythmias. J. Am. Coll. Cardiol. 50:2233–40. doi:10.1016/j.jacc.2007.06.059
- Allen, J., C.A. Trenga, A. Peretz, et al. 2009. Effect of diesel exhaust inhalation on antioxidant and oxidative stress responses in adults with metabolic syndrome. Inhal. Toxicol. 21(13): 1061–67. doi:10.3109/08958370902721424
- Andreae, M.O., and A. Gelencser. 2006. Black carbon or brown carbon? The nature of light-absorbing carbonaceous aerosols. Atmos. Chem. Phys. 6:3131–48. doi:10.5194/acp-6-3131-2006
- Bai, N., K. Takashi, H. Suzuki, et al. 2011. Changes in atherosclerotic plaques induced by inhalation of diesel exhaust. Atherosclerosis 216:299–306. doi:10.1016/j.atherosclerosis.2011.02.019
- Barath S., N.L. Mills, M. Lundback, et al. 2010. Impaired vascular function after exposure to diesel exhaust generated at urban transient running conditions. Particle Fibre Toxicol. 7:19. doi:10.1186/1743-8977-7-19
- Bari, M.D., G. Baumbach, B. Kuch, et al. 2009. Wood smoke as a source of particle-phase organic compounds in residential areas. Atmos. Environ. 43:4722–32. doi:10.1016/j.atmosenv.2008.09.006
- Barregard, L., G. Sallsten, P. Gustafson, et al. 2006. Experimental exposure to wood-smoke particles in healthy humans: Effects on markers of inflammation, coagulation, and lipid peroxidation. Inhal. Toxicol. 18:845–53. doi:10.1080/08958370600685798
- Barregard, L., G. Sallsten, L. Andersson, et al. 2008. Experimental exposure to wood smoke: Effects on airway inflammation and oxidative stress. Occup. Environ. Med. 65:319–24. doi:10.1136/oem.2006.032458
- Barrett, E.G., R.D. Henson, S.K. Seilkop, et al. 2006. Effects of hardwood smoke exposure on allergic airway inflammation in mice. Inhal. Toxicol. 18: 33–43. doi:10.1080/08958370500282340
- Bartell, S. M., J. Longhurst, T. Tjoa, et al. 2013. Particulate air pollution, ambulatory heart rate variability, and cardiac arrhythmia in retirement community residents with coronary artery disease. Environ. Health Perspect. 121: 1135–41. doi:10.1289/ehp.1205914
- Bassani, D.G., P. Jha, N. Dhingra, et al. 2010. Child mortality from solid-fuel use in India: A nationally representative case-control study. BMC Public Health. 10:491 (open access).
- Baulig, A., M. Garlatti., V. Bonvallot, et al. 2003. Involvement of reactive oxygen species in the metabolic pathways triggered by diesel exhaust particle in human airway epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 285: L671–79.
- Baumgartner, J., J.J. Schauer, M. Ezzati, et al. 2011. Indoor air pollution and blood pressure in adult women living in rural China. Environ. Health Perspect. 119:1390–95. doi:10.1289/ehp.1003371
- Barone-Adesi, F., R.S. Chapman, D.T. Silverman, et al. 2012. Risk of lung cancer associated with domestic use of coal in Xuanwei, China: Retrospective cohort study. Br. Med J. (online access). doi:10.1136/bmj.e5414
- Becker, S., J.M. Soukup, M.I. Gilmour, et al. 1996. Stimulation of human and rat alveolar macrophages by urban air particulates: Effects on oxidant radical generation and cytokine production. Toxicol. Appl. Pharmacol. 141:637–48. doi:10.1006/taap.1996.0330
- Beckerman, B., M. Jerrett, M. Finkelstein, et al. 2012. The association between chronic exposure to traffic-related air pollution and ischemic heart disease. J. Toxicol. Environ. Health A 75:402–11. doi:10.1080/15287394.2012.670899
- Beelen, R., G. Hoek, D. Houthuijs, et al. 2009. The joint association of air pollution and noise from road traffic with cardiovascular mortality in a cohort study. Occup. Environ. Med. 66:243–50. doi:10.1136/oem.2008.042358
- Bell, M.L., K. Ebisu, R.D. Peng, et al. 2009a. Hospital admissions and chemical composition of fine particle air pollution. Am. J. Respir. Crit. Care Med. 179:1115–20. doi:10.1164/rccm.200808-1240OC
- Bell, M.L., K. Ebisu, R.D. Peng, et al. 2009b. Adverse health effects of particulate air pollution modification by air conditioning. Epidemiology 20(5): 682–86. doi:10.1097/EDE.0b013e3181aba749
- Beverland I. J., C. Yap, C. Robertson, et al. 2007. Department of Health research project (Ref: 0020015). Health Effects of Long-term Exposure to Air Pollution in Scotland. http://www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/@dh/@en/documents/digitalasset/dh_092831.pdf
- Beverland, I.J., G.R. Cohen, M.R. Heal, et al. 2012. A comparison of short-term and long-term air pollution exposure associations with mortality in two cohorts in Scotland. Environ. Health Perspect. 120:1280–85. doi:10.1289/ehp.1104509
- Billionnet, C. D. Sherrill, I. Annesi-Maesano, et al. 2012. Estimating the health effects of exposure to multi-pollutant mixture. Ann. Epidemiol. 22(2): 126–41. doi:10.1016/j.annepidem.2011.11.004
- Block, M.L., X. Wu, Z. Pei, et al. 2004. Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: the role of microglia, phagocytosis, and NADPH oxidase. FASEB J. 18:1618–20. doi:10.1096/fj.04-1945fje
- Block, M.L., and J.S. Hong. 2007. Chronic microglial activation and progressive dopaminergic neurotoxicity. Biochem. Soc. Trans. 35:1127–32.
- Block, M.L., L. Zecca, and J.S. Hong. 2007. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci 8:57–69. doi:10.1038/nrn2038
- Bolling, A.K., J. Pagels, K.E. Yttri, et al. 2009. Health effects of residential wood smoke particles: the importance of combustion conditions and physicochemical particle properties. Particle Fibre Toxicol. 6:29. doi:10.1186/1743-8977-6-29
- Bolling A.K., A. I. Totlandsdal, G. Sallsten, et al. 2012. Wood smoke particles from different combustion phases induce similar pro-inflammatory effects in a co-culture of monocyte and pneumocyte cell lines. Particle Fibre Toxicol. 9:45–59. doi:10.1186/1743-8977-9-45
- Bond, T.C., D.G. Streets, K.F. Yarber, et al. 2004. A technology-based global inventory of black and organic carbon emissions from combustion. J. Geophys. Res. 109:D14203. doi:10.1029/2003JD003697
- Bonvallot, V., A. Baeza-Squiban, A. Baulig, et al. 2001. Organic compounds from diesel exhaust particles elicit a proinflammatory response in human airway epithelial cells and induce cytochrome p450 1A1 expression. Am. J. Respir. Cell Mol. Biol. 25:515–21. doi:10.1165/ajrcmb.25.4.4515
- Boy, E., N. Bruce, and H. Delgado. 2002. Birth weight and exposure to kitchen wood smoke during pregnancy in rural Guatemala. Environ. Health Perspect. 110:109–14. doi:10.1289/ehp.02110109
- Brenneman, K.A., B.A. Wong, M.A. Buccellato, et al. 2000. Direct olfactory transport of inhaled manganese (54MnCl2) to the rat brain: Toxicokinetic investigations in a unilateral nasal occlusion model. Toxicol. Appl. Pharmacol 169:238–48. doi:10.1006/taap.2000.9073
- Brunekreef, B., R. Beelen, G. Hoek, et al. 2009. Effects of long-term exposure to traffic-related air pollution on respiratory and cardiovascular mortality in the Netherlands: The NLCS-AIR Study. Charleston, MA: Health Effects Institute.
- Burke, A.P., A. Farb, G.T. Malcom, et al. 1997. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N. Engl. J. Med. 336:1276–82. doi:10.1056/NEJM199705013361802
- Cakmak, S, R.E. Dales, and C. B. Vida. 2009a. Components of particulate air pollution and mortality in Chile. Int J Occup. Environ. Health 15:152–58. doi: 10.1179/107735209799195844; 10.1179/oeh.2009.15.2.152
- Cakmak, S, R.E. Dales, T. Gultekin, et al. 2009b. Components of particulate air pollution and emergency department visits in Chile. Arch. Environ. Occup. Health 64:148–55. doi:10.1080/19338240903240228
- Calderon-Garciduenas, L., B. Azzarelli, H. Acuna, et al. 2002. Air pollution and brain damage. Toxicol. Pathol. 30:373–89.
- Calderon-Garciduenas, L, R.R. Maronpot, R. Torres-Jardon, et al. 2003. DNA damage in nasal and brain tissues of canines exposed to air pollutants is associated with evidence of chronic brain inflammation and neurodegeneration. Toxicol. Pathol. 31:524–38. doi:10.1080/01926230390226645
- Campbell, A., M. Oldham, A. Becaria, et al. 2005. Particulate matter in polluted air increases inflammatory indices in the mouse brain. Neurotoxicology 26:133–40. doi:10.1016/j.neuro.2004.08.003
- Campbell, A., J.A. Araujo, H. Li, et al. 2009. Particulate matter induced enhancement of inflammatory markers in the brains of Apolipoprotein E knockout mice. J. Nanosci. Nanotechnol. 9:5099–104. doi:10.1166/jnn.2009.GR07
- Campen, M.J., A.K. Lund, T.L. Knuckles, et al. 2010. Inhaled diesel emissions alter atherosclerotic plaque composition in ApoE‐/‐ mice. Toxicol. Appl. Pharmacol. 424:310–26.
- Carvey, P.M., A.Punati, and M.B. Newman. 2006. Progressive dopamine neuron loss in Parkinson;s disease: the multiple hit hypothesis. Cell Transplant. 15:239–50. doi:10.3727/000000006783981990
- CASAC (Clean Air Scientific Advisory Committee). 1996. “Closure” letter from CASAC Chairman Dr. George T. Wolff to U.S. EPA Administrator Carol M. Browner, June 13.
- Cassee, F.R., M.-E. Heroux, M.E. Gerlofs-Nijland, et al. 2013. Particulate matter beyond mass: Recent health evidence on the role of fractions, chemical constituents, and sources of emission. Inhal. Toxicol. 25(14):802–12. doi:10.3109/08958378.2013.850127
- Chahine, T., A. Baccerelli, A. Litonjua, et al. 2007. Particulate air pollution, oxidative stress genes, and heart rate variability in an elderly cohort. Environ. Health Perspect. 115:1617–22. doi:10.1289/ehp.10318
- Chait, A, C.Y. Han, J.F. Oram et al. 2005. Thematic review series: the immune system and atherogenesis. Lipoprotein-associated inflammatory proteins: markers or mediators of cardiovascular disease? J. Lipid Res. 46: 389–403. doi:10.1194/jlr.R400017-JLR200
- Chan, R.C-F., W. Wang, N. Li, et al. 2006. Pro-oxidative diesel exhaust particle chemicals inhibit LPS-induced dendritic cell responses involved in T-helper differentiation. J. Allergy Clin. Immunol. 118:455–65. doi:10.1016/j.jaci.2006.06.006
- Chen, Y., A. Ebenstein, M. Greenstone, et al. 2013a. Evidence on the impact of sustained exposure to air pollution on life expectancy from China’s Huai River Policy. Proc. Natl. Acad. Sci. USA 110(32): 12936–41. doi:10.1073/pnas.1300018110
- Chen H., M.S. Goldberg, R.T. Burnett, et al. 2013b. Long-term exposure to traffic-related air pollution and cardiovascular mortality. Epidemiology 24:35–43. doi:10.1097/EDE.0b013e318276c005
- Chen, Y, G. Sheng, X. Bi, et al. 2005. Emission factors for carbonaceous particles and polycyclic hydrocarbons from residential coal combustion in China. Environ. Sci Technol. 39:1861–67. doi:10.1021/es0493650
- Chiu, J.-H.M., D.C. Bellinger, B.A. Coull, et al. 2013. Associations between traffic-related black carbon exposure and attention in a prospective birth cohort of urban children. Environ. Health Perspect. 121:859–64. doi:10.1289/ehp.1205940
- Choi, H., V. Rauh, R. Garfinkel, et al. 2008. Prenatal exposure to airborne polycyclic aromatic hydrocarbons and risk of intrauterine growth restriction. Environ. Health Perspect. 116:658–65. doi:10.1289/ehp.10958
- Chow, J.C., J.G. Watson, D. Crow, et al. 2001. Comparison of IMPROVE and NIOSH carbon measurements. Aerosol Sci. Technol. 34(1): 23–34. doi:10.1080/02786820119073; 10.1080/027868201300081923
- Chow, J.C., J.G. Watson, L.-W.A. Chen, et al. 2007. The IMPROVE_A temperature protocol for thermal/optical carbon analysis: Maintaining consistency with a long-term database. J. Air Waste Manage. Assoc. 57(9): 1014–23. doi:10.3155/1047-3289.57.9.1014
- Chow, J.C., J.G. Watson, D.H. Lowenthal, et al. 2011. PM2.5 source profiles for black and organic carbon emissions inventories. Atmos. Environ. 45:5407–14. doi:10.1016/j.atmosenv.2011.07.011
- Clarke, L, J. Edmonds, H. Jacoby, et al. 2007. Scenarios of greenhouse gas emissions and atmospheric concentrations. Synthesis and assessment product 2.1. Report by the U.S. Climate Change Science Program and the Subcommittee on Global Change Research. Washington, DC: Department of Energy, Office of Biological & Environmental Research.
- Cohen, A.J., and C.A. Pope III. 1995. Lung cancer and air pollution. Environ. Health Perspect. 103(S8): 219–25. doi:10.1289/ehp.95103s8219
- Cooke, R.M., A.M. Wilson, J.T. Tuomisto, et al. 2007. A probabilistic characterization of the relationship between fine particulate matter and mortality: Elicitation of European experts. Environ. Sci. Technol. 41:6598–605. doi:10.1021/es0714078
- Corsini, E., S. Budello, L. Marabini, et al. 2013. Comparison of wood smoke PM2.5 from the combustion of fir and beech pellets on inflammation and DNA damage in A549 and THP-1 human cell lines. Arch. Toxicol., 87(12): 2187–2199. doi:10.1007/s00204-013-1071-z
- Cosselman, K.E., R.M. Krishnan, A.P. Oron, et al. 2012. Blood Pressure Response to Controlled Diesel Exhaust Exposure in Human Subjects. Hypertension 59:943–48. doi:10.1161/HYPERTENSIONAHA.111.186593
- Cruts, B., L. vat Etten, H. Tornquist, et al. 2008. Exposure to diesel exhaust induces changes in EEG in human volunteers. Particle Fibre Toxicol. 5:4 (open access). doi:10.1186/1743-8977-5-4
- Currie, J., and Walker, R. 2009. Traffic congestion and infant health: Evidence from E-Z Pass. Working paper 15413. Cambridge, MA: National Bureau of Economic Research.
- Danielsen, P.H., P.Moller, K.A. Jensen, et al. 2011. Oxidative stress, DNA damage, and inflammation induced by ambient air and wood smoke particulate matter in human A549 and THP-1 cell lines. Chem. Res. Toxicol. 24: 168–84. doi:10.1021/tx100407m
- Dazert, P., Y. Suofu, M. Grube, et al. 2006. Differential regulation of transport proteins in the periinfarct regions following reversible middle cerebral artery occulusion in rats. Neuroscience 142:1071–79. doi:10.1016/j.neuroscience.2006.07.056
- Delfino, R.J., N. Staimer, T. Tjoa, et al. 2008. Circulating biomarkers of inflammation, antioxidant activity, and platelet activation are associated with primary combustion aerosols in subjects with coronary artery disease. Environ. Health Perspect. 116:898–906. doi:10.1289/ehp.11189
- Delfino, R.J., N. Staimer, T. Tjoa, et al. 2009. Air pollution and circulating biomarkers of effect in a susceptible population: Clues to potential causal component mixtures and mechanisms. Environ. Health Perspect. 117: 1232–38. doi:10.1289/ehp.0800194
- Delfino, R.J., T. Tjoa, D.L. Gillen, et al. 2010. Traffic-related air pollution and blood pressure in elderly subjects with coronary artery disease. Epidemiology. 21:396–404. doi:10.1097/EDE.0b013e3181d5e19b
- Demling, R.H., and C. LaLonde. 1990. Moderate smoke inhalation produces decreased oxygen delivery, increased oxygen demands, and systemic but not lung parenchymal lipid peroxidation. Surgery 108:544–52.
- Demling, R., C. LaLonde, L. Picard, et al. 1994. Changes in lung and systemic oxidant and antioxidant activity after smoke inhalation. Shock 1:101–7. doi:10.1097/00024382-199402000-00004
- Desai, B.S., A.J. Monahan, P.M. Carvey, et al. 2007. Blood-brain barrier pathology in Alzheimer’s and Parkinson’s disease: Implications for drug therapy. Cell Transplant. 16:285–99.
- Devouassoux, G., A. Saxon, D.D. Metcalfe et al. 2002. Chemical constituents of diesel exhaust particles inducee IL-4 production and histamine release by human basophils. J. Allergy Clin. Immunol. 109: 847–53. doi:10.1067/mai.2002.122843
- Diaz-Sanchez, D., A.R. Dotson, H. Takenaka, et al. 1994. Diesel exhaust particles induce local IgE production in vivo and alter the pattern of IgE messenger RNA isoforms. J. Clin. Invest. 94:1417–25. doi:10.1172/JCI117478
- Diaz-Sanchez, D., A. Tsien, J. Fleming, et al. 1997. Combined diesel exhaust particulate and ragweed allergen challenge markedly enhances human in vivo nasal ragweed specific IgE and skews cytokine production to a T helper cell 2-type pattern. J. Immunol. 158:2406–13.
- Diaz-Sanchez, D., M. Jyrala, M. Ng, et al. 2000. In vivo nasal challenge with diesel exhaust particles enhances expression of the CC chemokines Rantes, MIP-1α, and MCP-3 in humans. Clin. Immunol. 97:140–45. doi:10.1006/clim.2000.4921
- Dockery, D.W., C.A. Pope III, X. Xu, et al. 1993. An association between air pollution and mortality in six U.S. cities. N. Engl. J. Med. 329:1753–59. doi:10.1056/NEJM199312093292401
- Dominici, F., R.D. Peng, C.D. Barr, et al. 2010. Protecting human health from air pollution: Shifting from a single-pollutant to a multi-pollutant approach. Epidemiology 21(2): 187–94. doi:10.1097/EDE.0b013e3181cc86e8
- Donaldson, K., N. Mills, W. MacNee, et al. 2005. Role of inflammation in cardiopulmonary health effects of PM. Toxicol. Appl. Pharmacol 207:S483–88. doi:10.1016/j.taap.2005.02.020
- Dons, E., L.I Panis, M.Van Poppel, et al. 2011. Impact of time-activity patterns on personal exposure to black carbon. Atmos. Environ. 45:3594–602. doi:10.1016/j.atmosenv.2011.03.064
- Dutta, A., B. Mukherjee, D. Das, et al. 2011. Hypertension with elevated levels of oxidized low-density lipoprotein and anticardiolipin antibody in the circulation of premenopausal Indian women chronically exposed to biomass smoke during cooking. Indoor Air 21:165–76. doi:10.1111/j.1600-0668.2010.00694.x
- Dvonch, J.T., S. Kannan, A.J. Schulz, et al. 2009. Acute effects of ambient particulate matter on blood pressure. Hypertension 53:853–59. doi:10.1161/HYPERTENSIONAHA.108.123877
- Edwards, S.C., W. Jedrychowski, J. Butscher, et al. 2010. Prenatal exposure to airborne polycyclic aromatic hydrocarbons and children’s intelligence at 5 years of age in a prospective cohort study in Poland. Environ. Health Perspect. 118:1326–31. doi:10.1289/ehp.0901070
- Fick, R.B., Jr., E.S. Paul, W.W. Merrill, et al. 1984. Alterations in the antibacterial properties of rabbit pulmonary macrophages exposed to wood smoke. Am. Rev. Respir. Dis. 129:76–81.
- Fine, P.M., G.R. Cass, and B.R.T. Simoneit. 2001. Chemical characterization of fine particle emissions from fireplace combustion of woods grown in the northeastern United States. Environ. Sci Technol. 35:2665–75. doi:10.1021/es001466k
- Finkelstein, M., M. Jerrett, and M.R. Sears. 2004a. Traffic air pollution and mortality rate advancement periods. Am. J. Epidemiol 160:173–77. doi:10.1093/aje/kwh181
- Finkelstein, M., D.K. Verma, D. Sahai, et al. 2004b. Ischemic heart disease mortality among heavy equipment operators. Am. J. Ind. Med. 46:16–22. doi:10.1002/ajim.20036
- Finkelstein, M., M. Jerrett, M.R. Sears. 2005. Environmental inequality and circulatory disease mortality gradients. J. Epidemiol. Commun. Health 59:481–87. doi:10.1136/jech.2004.026203
- Forchhammer, L., P. Moller, I.S. Riddervold, et al. 2012. Controlled human wood smoke exposure: Oxidative stress, inflammation and microvascular function. Particle Fibre Technol. 9:7. doi:10.1186/1743-8977-9-7
- Frank-Cannon, T.C., L.T. Alto, F.E. McAlpine, et al. 2009. Does neuroinflammation fan the flame in neurodegenerative disease? Mol. Neurodegen. 4:47. doi:10.1186/1750-1326-4-47
- Gan, W.Q., L. Tamburic, H.W. Davies, et al. 2010. Changes in residential proximity to road traffic and the risk of death from coronary heart disease. Epidemiology 21:642–49. doi:10.1097/EDE.0b013e3181e89f19
- Gan, W.Q., M. Koehoorn, H.W. Davies, et al. 2011. Long-term exposure to traffic-related air pollution and the risk of coronary heart disease hospitalization and mortality. Environ. Health Perspect. 119:501–7. doi:10.1289/ehp.1002511
- Gao, B., H. Guo, X.-M. Wang, et al. 2013. Tracer-based source apportionment of polycyclic aromatic hydrocarbons in PM2.5 in Guangzhou, southern China, using positive matrix factorixation (PMF). Environ. Sci Pollut Res. 20:2398–409. doi:10.1007/s11356-012-1129-0
- Garshick, E., F. Laden, J.E. Hart, et al. 2008. Lung cancer and vehicle exhaust in trucking industry workers. Environ. Health Perspect. 116:1327–32. doi:10.1289/ehp.11293
- Garshick, E., F. Laden, J.E. Hart, et al. 2004. Lung cancer in railroad workers exposed to diesel exhaust. Environ. Health Perspect. 112:1539–43. doi:1289/ehp.7195
- Gehring, U., J. Heinrich, U. Kramer, et al. 2006. Long-term exposure to ambient air pollution and cardiopulmonary mortality in women. Epidemiology 17:545–51. doi:10.1097/01.ede.0000224541.38258.87
- Genc, S, Zadeoglulari, S.H. Fuss, et al. 2012. The adverse effects of air pollution on the nervous system. J. Toxicol. (open access). doi:10.1155/2012/782462
- Geng, F., J. Hua, Z. Mu, et al. 2013. Differentiating the associations of black carbon and fire particle with daily mortality in a Chinese city. Environ. Res. 120:27–32. doi:10.1016/j.envres.2012.08.007
- Gerlofs-Nijland, M.E., D. van Berlo, F.R. Cassee, et al. 2010. Effect of prolonged exposure to diesel engine exhaust on proinflammatory markers in different regions of the rat brain. Particle Fibre Toxicol. 7:12. doi:10.1186/1743-8977-7-12
- Ghio, A.J., J. Stonehuerner, L.A. Dailey, et al. 1999. Metals associated with both the water-soluble and insoluble fractions of an ambient air pollution particle catalyze an oxidative stress. Inhal. Toxicol. 11:37–49. doi:10.1080/089583799197258
- Glass, C.K., K. Saijo, B. Winner, et al. 2010. Mechanisms underlying inflammation in neurodegeneration. Cell 140:918–34. doi:10.1016/j.cell.2010.02.016
- Gold, D., A.A. Litonjua, A. Zanobetti, et al. 2005. Air pollution and ST-segment depression in elderly subjects. Environ. Health Perspect. 113:883–87. doi:10.1289/ehp.7737
- Goldberg, M., and R. Burnett. 2003. Revised analysis of the Montreal time series study. In Revised Analyses of Time-Series Studies of Air Pollution and Health, Health Effects Institute Special Report, 113–131. Charleston, MA: Health Effects Institute.
- Grahame, T., and G. Hidy. 2004. Using Factor Analysis to Attribute Health Impacts to Particulate Pollution Sources. Inhal. Toxicol. 16(suppl. 1): 143–52. doi:10.1080/08958370490443231
- Grahame, T., and G.M. Hidy. 2007a. Pinnacles and pitfalls for source apportionment of potential health effects from airborne particle exposure. Inhal. Toxicol. 19(9): 727–44. doi:10.1080/08958370701399687
- Grahame, T., and G.M. Hidy. 2007b. Secondary sulfate effects? Environ. Health Perspect. 115:A532. doi:10.1289/ehp.10293
- Grahame, T.J. 2009. Does improved exposure information for PM 2.5 constituents explain differing results among epidemiological studies? Inhal. Toxicol. 21(5): 381–93. doi:10.1080/08958370802380495
- Grahame, T.J., and R.B. Schlesinger. 2010. Cardiovascular health and particulate vehicular emissions: A critical evaluation of the evidence. Air Qual. Atmos. Health 3:3–27. doi:10.1007/s11869-009-0047-x
- Grahame, T.J., and R.B. Schlesinger. 2012. Oxidative stress-induced telomeric erosion as a mechanism underlying airborne particulate matter-related cardiovascular disease. Particle Fibre Toxicol. 9:21. doi:10.1186/1743-8977-9-21
- Hartz, A.M.S., B. Bauer, M.L. Block, et al. 2008. Diesel exhaust particles induce oxidative stress, proinflammatory signaling, and P-glypoprotein up-regulation at the blood-brain barrier. FASEB J. 22:2723–32. doi:10.1096/fj.08-106997
- Health Effects Institute. 2010. Special report 17: Traffic related air pollution: A critical review of the literature on emissions, exposure, and health effects. Boston, MA: Health Effects Institute.
- Heitzer, T., T. Schlinzig, K. Krohn, et al. 2001. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 104:2673–78. doi:10.1161/hc4601.099485
- Hiura, T.S., M.P. Kaszubowski, N. Li, et al. 1999. Chemicals in diesel exhaust particles generate reactive oxygen radicals and induce apoptosis in macrophages. J. Immunol. 163: 5582–91.
- Hjortenkrans, D.S.T., B.G. Bergback, and A.V. Haggerud. 2007. Metal emissions from brake linings and tires: Case studies of Stockholm, Sweden 1995/1998 and 2005. Environ. Sci Technol. 41:5224–30. doi:10.1021/es070198o
- Hoek, G., B. Brunekreef, S. Goldbohm, et al. 2002. Association between mortality and indicators of traffic-related air pollution in the Netherlands: A cohort study. Lancet 360:1203–9. doi:10.1016/S0140-6736(02)11280-3
- Hoffmann, B., S. Moebus, A. Stang, et al. 2006. Residence close to high traffic and prevalence of coronary heart disease. Eur. Heart J. 27:2696–702. doi:10.1093/eurheartj/ehl278
- Hoffmann, B., S. Moebus, S. Mohlenkamp, et al. 2007. Residential exposure to traffic is associated with coronary atherosclerosis. Circulation 116:489–96. doi:10.1161/CIRCULATIONAHA.107.693622
- Hoffmann, B., S. Moebus, K. Kroger, et al. 2009. Residential exposure to urban air pollution, Ankle–Brachial Index, and peripheral arterial disease. Epidemiology 20:280–88. doi:10.1097/EDE.0b013e3181961ac2
- Holstius, D.M., C.E. Reid, B. M. Jesdale, et al. 2012. Birth weight following pregnancy during the 2003 southern California wildfires. Environ. Health Perspect. 120:1340–45. doi:10.1289/ehp.1104515
- Hosgood, H.D. III, H. Wei, A. Sapkota, et al. 2011. Household coal use and lung cancer: Systematic review and meta-analysis of case-control studies, with an emphasis on geographic variation. Int. J. Epidemiol. 40:719–28. doi:10.1093/ije/dyq259
- Hsu, S.-I., K. Ito, and M. Lippmann. 2011. Effects of thoracic and fine PM and their components on heart rate and pulmonary function in COPD patients. J. Expos. Sci. Environ. Epidemiol. 21:464–72. doi:10.1038/jes.2011.7
- Hsu, T.H., and Y.R. Kou. 2001. Airway hyperresponsiveness to bronchoconstrictor challenge after wood smoke exposure in guinea pigs. Life Sci. 68:2945–56. doi:1016/S0024-3205(01)01088-8
- Ikeda, M., M. Shitashige, H. Yamasaki, et al. 1995. Oxidative modification of low density lipoprotein by diesel exhaust particles. Biol. Pharm. Bull. 18:866–71. doi:10.1248/bpb.18.866
- International Agency for Research on Cancer. 2012. IARC: Diesel engine exhaust carcinogenic. June. http://www.iarc.fr/en/media-centre/pr/2012/pdfs/pr213_E.pdf
- Jacobs, L. J. Emmerechts, M.F. Hoylaerts, et al. 2011. Traffic air pollution and oxidized LDL. PLoS One 6(1)(open online access). doi:10.1371/journal.pone.0016200
- Jalava, P.I., R.O. Salonen, K. Nuutinen, et al. 2010. Effect of combustion condition on cytotoxic and inflammatory activity of residential wood combustion particles. Atmos. Environ. 44:1691–98. doi:10.1016/j.atmosenv.2009.12.034
- Janssen, N.A.H., J. Schwartz, A. Zanobetti, et al. 2002. Air conditioning and source-specific particles as modifiers of the effect of PM10 on hospital admissions for heart and lung disease. Environ. Health Perspect. 110: 43–49. doi:10.1289/ehp.0211043
- Janssen, N.A.H., G. Hoek, M. Simic-Lawson, et al. 2011. Black carbon as an additional indicator of the adverse health effects of airborne particles compared with PM10 and PM2.5. Environ. Health Perspect. 119:1691–99. doi:10.1289/ehp.1003369
- Jerrett, M., K.B. Newbold, R.T. Burnett, et al. 2007. Geographies of uncertainty in the health benefits of air quality improvements. Stoch. Environ. Res. Risk Assess. 21:511–22. doi:10.1007/s00477-007-0133-2
- Jerrett, M, M.M. Finkelstein, J.R. Brook, et al. 2009. A cohort study of traffic-related air pollution and mortality in Toronto, Ontario, Canada. Environ. Health Perspect. 117:772–77. doi:10.1289/ehp.11533
- Johnston, F.H., I.C. Hanigan, S.B. Henderson, et al. 2013. Evaluation of interventions to reduce air pollution from biomass smoke on mortality in Launceston, Australia: Retrospective analysis of daily mortality, 1994–2007. Br. Med J. (open access). doi:10.1136/bmj.e8446
- Kan, H., G. Heiss, K.M. Rose, et al. 2008. Prospective analysis of traffic exposure as a risk factor for incident coronary heart disease: The Atherosclerosis Risk in Communities (ARIC) study. Environ. Health Perspect. 116:1463–68. doi:10.1289/ehp.11290
- Katsouyanni, K., G. Touloumi, C. Spix, et al. 1997. Short term effects of ambient sulphur dioxide and particulate matter on mortality in 12 European cities: Results from time series data from the APHEA project. Br. Med J. 314: 1658–1663. doi:10.1136/bmj.314.7095.1658
- Kim, J.-B., Kim, C., Choi, E., et al. 2012. Particulate air pollution induces arrhythmia via oxidative stress and calcium calmodulin kinase II activation. Toxicol. Appl. Pharmacol. 259:66–73. doi:10.1016/j.taap.2011.12.007
- Kim, R.B. 2002. Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug Metab. Rev. 34:47–54. doi:10.1081/DMR-120001389
- Kleinman, M.T., J.A Araujo, A. Nel, et al. 2008. Inhaled ultrafine particulate matter affects CNS inflammatory processes and may act via MAP kinase signaling pathways. Toxicol. Lett. 178:127–30. doi:10.1016/j.toxlet.2008.03.001
- Kleinman, M. 2013. Cardiopulmonary health effects: Toxicity of semi-volatile and non-volatile components of PM. Draft final report to California Air Resources Board and the California Environmental Protection Agency. http://www.arb.ca.gov/research/rsc/3-8-13/item1dfr07-307.pdf
- Kocbach, B.A., J. Pagels, K.E. Yttri, et al. 2009. Health effects of residential wood smoke particles: The importance of combustion conditions and physicochemical particle properties. Particle Fibre Toxicol. 6:29. doi:10.1186/1743-8977-6-29
- Krall, J.R., G.B. Anderson, F. Domenici, et al. 2013. Short-term exposure to particulate matter constituents and mortality in a national study of U.S. urban communities. Environ. Health Perspect. 121:1148–1153., doi:10.1289/ehp.1206185
- Kuenzli, N., M. Jerrett, R. Garcia-Esteban R, et al. 2010. Ambient air pollution and the progression of atherosclerosis in adults. PLoS One 5 (2)(open access online). doi:10.1371/journal.pone.0009096
- Kurokawa, J., T. Ohara, T. Morikawa, et al. 2013. Emissions of air pollutants and greenhouse gases over Asian regions during 2000–2008: Regional Emission inventory in ASia (REAS) version 2. Atmos. Chem. Phys. 13: 11019–58.See http://www.nies.go.jp/REAS
- Lack, D.A., H. Moosmueller, G.R. McMeeking, et al. 2014. Characterizing elemental, equivalent black, and refractory black carbon aerosol particles: A review of techniques, their limitations and uncertainties. Anal. Bioanal. Chem. 406(1): 99–122. doi:10.1007/s00216-013-7402-3
- LaGier, A.J., N.D. Manzo, and J.A. Dye. 2013. Diesel exhaust particles induce aberrant alveolar epithelial directed cell movement by disruption of polarity mechanisms. J. Toxicol. Environ. Health A 76:71–85. doi:10.1080/15287394.2013.738169
- Laden, F., L.M. Neas, D.W. Dockery, et al. 2000. Association of fine particulate matter from different sources with daily mortality in six U.S. cities. Environ. Health Perspect. 108:941–47. doi:10.1289/ehp.00108941
- Laden, F., J.E. Hart, T.J. Smith, et al. 2007. Cause-specific mortality in the unionized U.S. trucking industry. Environ. Health Perspect. 115:1192–96. doi:10.1289/ehp.10027
- LaLonde, C., L.Picard, C. Campbell, et al. 1994. Lung and systemic oxidant and antioxidant activity after graded smoke exposure in the rat. Circ. Shock 42:7–13.
- Langrish, J.P., N.L. Mills, J.K.K. Chan, et al. 2009. Beneficial cardiovascular effects of reducing exposure to particulate air pollution with a simple facemask. Particle Fibre Technol. 6:8. doi:10.1186/1743-8977-6-8
- Langrish, J.P., X. Li, S. Wang, et al. 2012. Reducing personal exposure to particulate air pollution improves cardiovascular health in patients with coronary heart disease. Environ. Health Perspect. 120:367–72. doi:10.1289/ehp.1103898
- Large, D.J., S. Kelly, B. Spiro, et al. 2009. Silica–volatile interaction and the geological cause of the Xuan Wei lung cancer epidemic. Environ. Sci. Technol. 43:9016–21. doi:1021/es902033j
- Laurent, S., P. Boutouyrie, R. Asmar, et al. 2001. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension 37:1236–41. doi:10.1161/01.HYP.37.5.1236
- Lee, M.-W., M.-L. Chen, S.-C.C. Lung, et al. 2011. Increase of urinary concentrations of 8-hydroxy-2’-deoxyguanosine in diesel exhaust emission inspector exposed to polycyclic aromatic hydrocarbons. Int. Arch. Occup. Environ. Health 85(3): 273–82.
- Lei, Y., Q. Zhang, K.B. He, et al. 2011. Primary anthropogenic aerosol emission trends for China, 1990–2005. Atmos. Chem. Phys. 11:931–54. doi:10.5194/acp-11-931-2011
- Leonard, S.S., S. Wang, X. Shi, et al. 2000. Wood smoke particles generate free radicals and cause lipid peroxidation, DNA damage, NFkappB activation and TNF-alpha release in macrophages. Toxicology 150:147–57. doi:10.1016/S0300-483X(00)00256-0
- Levesque, S., M.J. Surace, J. McDonald, et al. 2011. Air pollution and the brain: subchronic diesel exhaust exposure causes neuroinflammation and elevates early markers of neurodegenerative disease. J. Neuroinflam. 8:105–14. doi:10.1186/1742-2094-8-105
- Levy, J.I., D.H. Bennett, S.J. Melly, et al. 2003. Influence of traffic patterns on particulate matter and polycyclic aromatic hydrocarbon concentrations in Roxbury, Massachusetts. J. Expos. Anal. Environ. Epidiemiol. 13:364–71. doi:10.1038/sj.jea.7500289
- Li, N., M. I. Venkatesan, A. Miguel, et al. 2000. Induction of heme oxygenase-1 expression in macrophages by diesel exhaust particle chemicals and quinones via the antioxidant-responsive element. J. Immunol. 165:3393–401.
- Li, N., M. Wang, T.D. Oberley, et al. 2002. Comparison of the prooxidative and proinflammatory effects of organic diesel exhaust particle chemicals in bronchial epithelial cells and macrophages. J. Immunol. 169:4531–41.
- Li N., C. Sioutas, A. Cho, et al. 2003. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ. Health Perspect. 111(4):455–60. doi:10.1289/ehp.6000
- Li, N., R.F. Phalen, W.C. Hinds, et al. 2003. Particulate air pollutants and asthma. A paradigm for the role of oxidative stress in PM-induced adverse health effects. Clin Immunol. 109:250–65. doi:10.1016/j.clim.2003.08.006
- Li, N, J. Alam, M. I. Venkatesan, et al. 2004. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J. Immunol. 173:3467–81.
- Li, N., and A. Nel. 2006. The cellular impacts of diesel exhaust particles: Beyond inflammation and death. Eur. Respir. J. 27:667–68. doi:10.1183/09031936.06.00025006
- Li, Y-J., T. Kawada, and A. Azuma. 2013. Nrf2 is a protective factor against oxidative stresses induced by diesel exhaust particles in allergic asthma. Oxid. Med. Cell. Longevity (online). doi:10.1155/2013/323607
- Lim, W.-Y., and A. Seow. 2012. Biomass fuels and lung cancer. Respirology 17:20–31. doi:10.1111/j.1440-1843.2011.02088.x
- Lin, S., J.P. Munsie, S.-A. Hwang, et al. 2002. Childhood asthma hospitalization and residential exposure to state route traffic. Environ. Res. Sec. A 88:73–81. doi:10.1006/enrs.2001.4303
- Lin, Y.S., C.Y. Ho, G.J. Tang, et al. 2001. Alleviation of wood smoke-induced lung injury by tachykinin receptor antagonist and hydroxyl radical scavenger in guinea pigs. Eur. J. Pharmacol 425:141–48. doi:10.1016/S0014-2999(01)01184-0
- Lipfert, F.W., R.E. Wyzga, J.D. Baty, et al. 2006a. Traffic density as a surrogate measure of environmental exposures in studies of air pollution health effects: Long-term mortality in a cohort of U.S. veterans. Atmos. Environ. 40:154–69.
- Lipfert, F.W., J.D. Baty, R.E. Wyzga, et al. 2006b. PM2.5 constituents and related air quality variables as predictors of survival in a cohort of U.S. military veterans. Inhal. Toxicol. 18:645–57. doi:10.1080/08958370600742946
- Lipfert, F.W., R.E. Wyzga, J.D. Baty, et al. 2009. Air pollution and survival within the Washington University—EPRI Veterans Cohort: Risks based on modeled estimates of ambient levels of hazardous and criteria air pollutants. J. Air Waste Manage. Assoc. 59:473–89. doi:10.1080/10473289.2009.10465740
- Liu, W., Y. Wang, A. Russell, et al. 2005. Atmospheric aerosol over two urban–rural pairs in the southeastern United States: Chemical composition and possible sources. Atmos. Environ. 39:4453–70. doi:10.1016/j.atmosenv.2005.03.048
- Liu, W., Y. Wang, A. Russell, et al. 2006. Enhanced source identification of southeast aerosols using temperature resolved carbon fractions and gas phase components. Atmos. Environ. 40:S445–66. doi:10.1016/j.atmosenv.2005.11.079
- Liu, W.X, H. Dou, Z. C. Wei, et al. 2009. Emission characteristics of polycyclic aromatic hydrocarbons from combustion of different residential coals in North China. Sci. Total Environ. 407:1436–46. doi:10.1016/j.scitotenv.2008.10.055
- Lough, G.C., J.J. Schauer, J.-S. Park, et al. 2005. Emissions of metals associated with motor vehicle roadways. Environ. Sci Technol. 39:826–36. doi:10.1021/es048715f
- Lucking, A.J., M. Lundback, N.L. Mills, et al. 2008. Diesel exhaust inhalation increases thrombus formation in man. Eur. Heart J. 29: 3043–51. doi:10.1093/eurheartj/ehn464
- Lucking, A.J., M. Lundback, S.L. Barath, et al. 2011. Particle traps prevent adverse vascular and prothrombotic effects of diesel engine exhaust inhalation in men. Circulation. 123:1721–28. doi:10.1161/CIRCULATIONAHA.110.987263
- Lue, S.-H., G.A. Wellenius, E.H. Wilker, et al. 2013. Residential proximity to major roadways and renal function. J. Epidemiol. Commun. Health. 67:629–634., doi:10.1136/jech-2012-202307
- Lundback, M., N.L. Mills, A. Lucking, et al. 2007. Experimental exposure to diesel exhaust increases arterial stiffness in man. Particle Fibre Toxicol. 6:7. doi:10.1186/1743-8977-6-7
- Maciejczyk, P.B., J.H. Offenberg, J. Clemente, et al. 2004. Ambient pollutant concentrations measured by a mobile laboratory in South Bronx, NY. Atmos. Environ. 38:5283–94. 10.1016/j.atmosenv.2004.02.062
- Maseri, A., C. Newman, and G. Davies. 1989. Coronary vasomotor tone: A heterogeneous entity. Eur. Heart J. 10(Suppl. F): 2–5. doi:10.1093/eurheartj/10.suppl_F.2
- Matthew, E., G. Warden, and J. Dedman. 2001. A murine model of smoke inhalation. Am. J. Physiol. Lung Cell Mol. Physiol 280:L716–23.
- Mattson, M. 2001. Mechanisms of neuronal apoptosis and excitotoxicity. In Pathogenesis and Neurodegenerative Disorders, pp. 1–20. Baltimore, MD: Humana Press.
- Mauderly J., and J. Chow. 2008. Health effects of organic aerosols. Inhal. Toxicol. 20:257–88. doi:10.1080/08958370701866008
- Maynard D., B.A. Coull, A. Gryparis, et al. 2007. Mortality risk associated with short-term exposure to traffic particles and sulfates. Environ. Health Perspect. 115: 751–55. doi:10.1289/ehp.9537
- McCracken, J., K.R. Smith, A. Diaz, et al. 2007. Chimney stove intervention to reduce long-term wood smoke exposure lowers blood pressure among Guatemalan women. Environ. Health Perspect. 115:996–1001. doi:10.1289/ehp.9888
- McCracken, J., K.R. Smith, P. Stone, et al. 2011. Intervention to lower household wood smoke exposure in Guatemala reduces ST-segment depression on electrocardiograms. Environ. Health Perspect. 119:1562–68. doi:10.1289/ehp.1002834
- McDonald, J.D., K.S. Harrod, J. Seagrave, et al. 2004. Effects of low sulfur fuel and a catalyzed particle trap on the composition and toxicity of diesel emissions. Environ. Health Perspect. 112:1307–12. doi:10.1289/ehp.7059
- McDonald, J.D., M.J. Campen, K.S. Harrod, Et al. 2011. Engine-operating load influences diesel exhaust composition and cardiopulmonary and immune responses. Environ. Health Perspect. 119:1136–41. doi:10.1289/ehp.1003101
- Medina-Ramon, M., T. Goldberg, S. Melly, et al. 2008. Residential exposure to traffic-related air pollution and survival after heart failure. Environ. Health Perspect. 116:481–85. doi:10.1289/ehp.10918
- Mestl, H.E.S., K. Aunan, H.M. Seip, et al. 2007a. Urban and rural exposure to indoor air pollution from domestic biomass and coal burning across China. Science Total Environ. 377:12–26. doi:10.1016/j.scitotenv.2007.01.087
- Mestl, H.E.S., K. Aunan, and H.M. Seip. 2007b. Health benefits from reducing indoor air pollution from household solid fuel use in China—Three abatement scenarios. Environ. Int. 33(6): 831–40. doi:10.1016/j.envint.2007.03.012
- Migliaccio, C.T., E. Kobos, Q.O. King, et al. 2013. Adverse effects of wood smoke PM2.5 exposure on macrophage function. Inhal. Toxicol. 25:67–76. doi:10.3109/08958378.2012.756086
- Mills, N.L., H. Tornquist, S.D. Robinson, et al. 2005. Diesel exhaust inhalation causes vascular dysfunction and impaired endogenous fibrinolysis. Circulation 112:3930–36. doi:10.1161/CIRCULATIONAHA.105.588962
- Mills, N.L., H. Tornquist, M. Gonzalez, et al. 2007. Ischemic and thrombotic effects of dilute diesel-exhaust inhalation in men with coronary heart disease. N. Engl. J. Med. 357:1075–82. doi:10.1056/NEJMoa066314
- Mills, N.L., S.D. Robinson, P.H.B. Fokkens, et al. 2008. Exposure to concentrated ambient particles does not affect vascular function in patients with coronary heart disease. Environ. Health Perspect. 116:709–15. doi:10.1289/ehp.11016
- Mills, N.L., M.R. Miller, A.J. Lucking, et al. 2011. Combustion-derived nanoparticulate induces the adverse vascular effects of diesel exhaust inhalation. Eur. Heart J. 32:2660–71. doi:10.1093/eurheartj/ehr195
- Miyabara, Y., H. Takano, T. Ichinose, et al. 1998. Diesel exhaust enhances allergic airway inflammation and hyperresponsiveness in mice. Am. J. Respir. Crit. Care Med. 157:1138–44. doi:10.1164/ajrccm.157.4.9708066
- Mogi, M., M. Harada, P. Riederer, et al. 1994. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 165:208–10. doi:10.1016/0304-3940(94)90746-3
- MohanKumar, S.M., A.Campbell, M. Block, et al. 2008. Particulate matter, oxidative stress, and neurotoxicity. NeuroToxicol. 29:479–88. doi:10.1016/j.neuro.2007.12.004
- Morgan, T.E., D.A. Davis, N. Iwata, et al. 2011. Glutamatergic neurons in rodent models response to nanoscale particulate urban air pollutants in vivo and in vitro. Environ. Health Perspect. 119:1003–9. doi:10.1289/ehp.1002973
- Morello-Frosch, R., and B.M. Jesdale. 2006. Separate and unequal: Residential segregation and estimated cancer risks associated with ambient air toxics in U.S. metropolitan areas. Environ. Health Perspect. 114:386–93.
- Mudway, I.S., N. Stenfors, S.T. Duggan, et al. 2004. An in vitro and in vivo investigation of the effects of diesel exhaust on human airway lining fluid antioxidants. Arch. Biochem. Biophys. 423:200–12. doi:10.1016/j.abb.2003.12.018
- Muller, L., C.V.E. Chehrazi, M.W. Henderson, et al. 2013. Diesel exhaust particles modify natural killer cell function and cytokine release. Particle Fiber Toxicol. 10:16.
- Naeher, L.P., M. Brauer, M. Lipsett, et al. 2007. Woodsmoke health effects: A review. Inhal. Toxicol. 19:67–106. doi:10.1080/08958370600985875
- National Research Council. 1998. Research priorities for airborne particulate matter: I: Immediate priorities and a long-range research portfolio. Washington, DC: National Academy Press
- Neupane, B., M. Jerrett M, R.T. Burnett, et al. 2010. Long-term exposure to ambient air pollution and risk of pneumonia in older adults. Am. J. Respir Crit. Care Med. 181: 47–53. doi:10.1164/rccm.200901-0160OC
- Newby, D.E., R.A. Wright, C. Jabinjoh, et al. 1999. Endothelial dysfunction, inpaired endogenous fibrinolysis, and cigarette smoking: A mechanism for arterial thrombosis and myocardial infarction. Circulation 99:1411–15. doi:10.1161/01.CIR.99.11.1411
- Newby, D.E., A. L. McLeod, N.G. Uren, et al. 2001. Impaired coronary tissue plasminogen activator release is associated with coronary atherosclerosis and cigarette smoking: Direct link between endothelial dysfunction and atherothrombosis. Circulation 103:1936–41. doi:10.1161/01.CIR.103.15.1936
- Ni, M., W.Q. Chen, and Y. Zhang. 2009. Animal models and potential mechanisms of plaque destabilisation and disruption. Heart 95:1393–98. doi:10.1136/hrt.2008.143461
- Oliver, J.J., and D.J. Webb. 2003. Noninvasive assessment of arterial stiffness and risk of atherosclerotic events. Arterioscler. Thromb. Vasc. Biol. 23:554–66. doi:10.1161/01.ATV.0000060460.52916.D6
- Ostro, B., W.-Y. Feng, R. Broadwin, et al. 2007. The effects of components of fine particulate air pollution on mortality in California: Results from CALFINE. Environ. Health Perspect. 115:13–19. doi:10.1289/ehp.9281
- Ostro, B., M. Lipsett, P. Reynolds, et al. 2010. Long-term exposure to constituents of fine particulate matter air pollution and mortality: Results from the California Teachers Study. Environ. Health Perspect. 118:363–69.
- Ostro, B., P. Reynolds, D. Goldberg, et al. 2011. Erratum: Assessing long-term exposure in the California Teachers Study. Environ. Health Perspect. 119:A242–43.
- Ozkaynak, H. and G.D. Thurston. 1987. Associations between 1980 U.S. mortality rates and alternative measures of airborne particle concentration. Risk Anal. 7:449–61. doi:10.1111/j.1539-6924.1987.tb00482.x
- Pachauri, T., A. Satsangi, V. Singla, et al. 2013. Characteristics and sources of carbonaceous aerosols in pm2.5 during wintertime in Agra, India. Aerosol Air Qual. Res. 13:997–91. doi:10.4209/aaqr.2012.10.0263
- Park, J.S., G.R. Hong, S.W. Baek, et al. 2002. Expression and regulation of endothelial nitric oxide synthase by vascular endothelial growth factor in ECV 304 cells. J. Korean Med. Sci. 17:161–67.
- Park, S.K., M.S. O’Neill, B.J.B. Stunder, et al. 2007. Source location of air pollution and cardiac autonomic function: Trajectory cluster analysis for exposure assessment. J. Expos. Sci. Environ. Epidemiol. 17:488–97. doi:10.1038/sj.jes.7500552
- Peng, R.D., M.L. Bell, A.S. Geyh, et al. 2009. Emergency admissions for cardiovascular and respiratory diseases and the chemical composition of fine particle air pollution. Environ. Health Perspect. 117:957–63. doi:10.1289/ehp.0800185
- Perera, F.P., V. Rauh, S.-Y. Tsai, et al. 2003. Effects of transplacental exposure to environmental pollutants on birth outcomes in a multiethnic population. Environ. Health Perspect. 111:201–5. doi:10.1289/ehp.5742
- Perera, F.P., D. Tang, R. Whyatt, et al. 2005. DNA damage from polycyclic aromatic hydrocarbons measured by benzo(a)pyrene–DNA adducts in mothers and newborns from northern Manhattan, the World Trade Center Area, Poland, and China. Cancer Epidemiol. Biomarkers Prevent. 14(3): 709–14. doi:10.1158/1055-9965.EPI-04-0457
- Perera, F.P., V. Rauh, R.M. Whyatt, et al. 2006. Effect of prenatal exposure to airborne polycyclic aromatic-hydrocarbons on neurodevelopment in the first 3 years of life among inner-city children. Environ. Health Perspect. 114:1287–92. doi:10.1289/ehp.9084
- Perera F.P., Z. Li, R. Whyatt, et al. 2009. Prenatal airborne polycyclic aromatic hydrocarbon exposure and child IQ at age 5 years. Pediatrics 124(2):e195–202. doi:10.1542/peds.2008-3506
- Perera, F.P., S. Wang, J. Vishnevetsky, et al. 2011. Polycyclic aromatic hydrocarbons–aromatic DNA adducts in cord blood and behavior scores in New York City children. Environ. Health Perspect. 119:1176–81. doi:10.1289/ehp.1002705
- Perera, F.P., D. Tang, S. Wang, et al. 2012. Prenatal polycyclic aromatic hydrocarbon (PAH) exposure and child behavior at age 6-7 years. Environ. Health Perspect. 120:921–26. doi:10.1289/ehp.1104315
- Peretz, A., E.C. Peck, T.K. Bammler, et al. 2007. Diesel exhaust inhalation and assessment of peripheral blood mononuclear cell gene transcription effects: An exploratory study of healthy human volunteers. Inhalation Toxicol. 19: 1107–19. doi:10.1080/08958370701665384
- Peretz, A., J.D. Kaufman, C.A. Trenga, et al. 2008. Effects of diesel exhaust inhalation on heart rate variability in human volunteers. Environ. Res. 107:178–84. doi:10.1016/j.envres.2008.01.012
- Peters A., S. von Klot, M. Heier, et al. 2004. Exposure to traffic and onset of myocardial infarction. N. Engl. J. Med. 351:1721–30. doi:10.1056/NEJMoa040203
- Peters, A., B. Veronesi, L. Calderon-Garciduenas, et al. 2006. Translocation and potential neurological effects of fine and ultrafine particles: A critical update. Particle Fibre Toxicol. 3:13. doi:10.1186/1743-8977-3-13
- Peterson, J.D., L.A. Herzenberg, K. Vasquez, et al. 1998. Glutathione levels in antigen-presenting cells modulate Th1 and Th2 response patterns. Proc. Natl. Acad. Sci. USA 95:3071–76. doi:10.1073/pnas.95.6.3071
- Pope, C A. III, and D. W. Dockery. 2006. Health effects of fine particulate air pollution: Lines that connect. J. Air Waste Manage. Asoc. 56:709–42. doi:10.1080/10473289.2006.10464485
- Pope, C.A., M.J. Thun, M.M. Namboodiri, et al. 1995. Particulate air pollution as a predictor of mortality in a prospective study of U.S. adults. Am. J. Crit. Care Med. 151:669–74. doi:10.1164/ajrccm.151.3.7881654
- Pope, C.A., R.T. Burnett, M.J. Thun, et al. 2002. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. J. Am. Med. Assoc. 287(9):1132–41. doi:10.1001/jama.287.9.1132
- Poss, J., D. Lorenz, C. Werner, et al. 2013. Diesel exhaust particles impair endothelial progenitor cells, compromise endothelial integrity, reduce neoangiogenesis, and increase atherogenesis in mice. Cardiovasc. Toxicol. 13: 290–300. doi:10.1007/s12012-013-9208-0
- Provoost, S., T. Maes, M.A.M. Willart, et al. 2010. Diesel exhaust particles stimulate adaptive immunity by acting on pulmonary dendritic cells. J. Immunol. 184: 426–32. doi:10.4049/jimmunol.0902564
- Puett, R.C., J.E. Hart, H. Suh, et al. 2011. Particulate matter exposures, mortality, and cardiovascular disease in the Health Professionals Follow-Up Study. Environ. Health Perspect. 119:1130–35. doi:10.1289/ehp.1002921
- Quincey, P. 2007. A relationship between Black Smoke Index and black carbon concentration. Atmos. Environ. 41:7964–68. doi:10.1016/j.atmosenv.2007.09.033
- Rahn, K.A., and D.H. Lowenthal. 1985. Pollution aerosol in the Northeast: Northeastern-Midwestern contributions. Science 228:275–84. doi:10.1126/science.228.4697.275
- Ramos, C., J. Pedraza-Chaverri, C. Becerril, et al. 2013. Oxidative stress and lung injury induced by short-term exposure to wood smoke in guinea pigs. Inhal. Toxicol. 23: 711–22. doi:10.3109/15376516.2013.843113
- Ren, C., S. Fang, R.O. Wright, et al. 2011. Urinary 8-hydroxy-2’-deoxyguanosine as a biomarker of oxidative DNA damage induced by ambient pollution in the Normative Aging Study. Occup. Environ. Med. 68:562–69. doi:10.1136/oem.2010.056358
- Reponen, T., S.A. Grinshpun, S. Trakumas, et al. 2003. Concentration gradient patterns of aerosol particles near interstate highways in the Greater Cincinnati airshed. J. Environ. Monit. 5:557–62. doi:10.1039/b303557c
- Restrepo, C., R. Zimmerman, G. Thurston, et al. 2004. A comparison of ground-level air quality data with New York State Department of Environmental Conservation monitoring stations data in South Bronx, New York. Atmos. Environ. 38:5295–304. doi:10.1016/j.atmosenv.2004.06.004
- Reynolds, P., E. Elkin, R. Scalf, et al. 2001. A case-control pilot study of traffic exposures and early childhood leukemia using a geographic information system. Bioelectromagnetics Suppl. 5:S58–68. doi:10.1002/1521-186X(2001)22:5+<::AID-BEM1024>3.0.CO;2-9
- Riddervold, I.S., J.H. Bonlokke, A.-C. Olin, et al. 2012. Effects of wood smoke particles from wood-burning stoves on the respiratory health of atopic humans. Particle Fibre Technol. 9:12. doi:10.1186/1743-8977-9-12
- Riediker, M., R. Devlin, T. Griggs, et al. 2004. Cardiovascular effects in patrol officers are associated with fine particulate matter from brake wear and engine emissions. Particle Fibre Technol. 1:2. doi:10.1186/1743-8977-1-2
- Ridker, P.M, C.H. Hennekens, J.E. Buring, et al. 2000. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N. Engl. J. Med. 342:836–43. doi:10.1056/NEJM200003233421202
- Risom, L., P. Moller, and S. Loft. 2005. Oxidative stress-induced DNA damage by particulate air pollution. Mutat. Res. 592:119–37. doi:10.1016/j.mrfmmm.2005.06.012
- Ristovski, A., B. Miljevic, N.C. Surawski et al. 2011. Respiratory health effects of diesel particulate matter. Respirology 17: 201–12. doi:10.1111/j.1440-1843.2011.02109.x
- Rivest, S. 2001. How circulating cytokines trigger the neural circuits that control the hypothalamic-pituitary-adrenal axis. Psychoneuroendocrinology 26: 761–88. doi:10.1016/S0306-4530(01)00064-6
- Rock, R.B., G. Gekker, S. Hu et al. 2004. Role of microglia in central nervous system infections. Clin. Microbiol. Rev. 17: 942–64. doi:10.1128/CMR.17.4.942-964.2004
- Roorda-Knape, M.C., N.A.H. Janssen, J.J. de Hartog, et al. 1998. Air pollution from traffic in city districts near major motorways. Atmos. Environ. 32(11): 1921–30. doi:10.1016/S1352-2310(97)00496-2
- Ross, R. 1999. Atherosclerosis-an inflammatory disease. N. Engl. J. Med. 340:115–26. doi:10.1016/S0002-8703(99)70266-8
- Ruckerl, R., S. Greven, P. Ljungman, et al. 2007. Air pollution and inflammation (interleukin-6, C-reactive protein, fibrinogen) in myocardial infarction survivors. Environ. Health Perspect. 115:1072–80. doi:10.1289/ehp.10021
- Salvi, S., C. Nordenhall, A. Blomberg et al. 2000. Acute exposure to diesel exhaust increases IL-8 and GRO-alpha production in healthy human airways. Am. J. Respir. Crit. Care Med. 161: 550–57. doi:10.1164/ajrccm.161.2.9905052
- Sapkota, A., V. Gajalakshmi, D.H. Jetli, et al. 2008. Indoor air pollution from solid fuels and risk of hypopharyngeal/laryngeal and lung cancers: A multicentric case-control study from India. Int. J. Epidemiol. 37:321–28. doi:10.1093/ije/dym261
- Saraswat, A., J.S. Apte, M. Kandilakar, et al. 2013. Spatiotemportal land use regression models of fine, ultrafine, and black carbon particulate matter in New Delhi, India. Environ. Sci Technol. 47:12903–11. doi:10.1021/es401489h
- Sauvain, J.-J., A. Setyan, P. Wild, et al. 2011. Biomarkers of oxidative stress and its association with the urinary reducing capacity in bus maintenance workers. J. Occup. Med. Toxicol. 6:1–13. doi:10.1186/1745-6673-6-18
- Sawyer, K, S. Mundandhara, A.J. Ghio, et al. 2010. The effects of ambient particulate matter on human alveolar macrophage oxidative and inflammatory responses. J. Toxicol. Environ. Health A 73:41–57. doi:10.1080/15287390903248901
- Schwartz J., A. Litonjua, H. Suh, et al. 2005a. Traffic related pollution and heart rate variability in a panel of elderly subjects. Thorax 60:455–61. doi:10.1136/thx.2004.024836
- Schwartz, J., S.K. Park, M.S. O’Neill, et al. 2005b. Glutathione-S-transferase M1, obesity, statins, and autonomic effects of particles. Am. J. Respir. Crit. Care Med. 172:1529–33. doi:10.1164/rccm.200412-1698OC
- Shukla, A., C. Timblin, K. Berube, et al. 2000. Inhaled particulate matter causes expression of nuclear factor (NF)-B related genes and oxidant-dependent NF-B activation in vitro. Am. J. Respir. Cell Mol. Biol. 23:182–87. doi:10.1165/ajrcmb.23.2.4035
- See, S.W., Y.H. Wang, and R. Balasubramian. 2007. Contrasting reactive oxygen species and transition metal concentrations in combustion aerosols. Environ. Res. 103:317–24. doi:10.1016/j.envres.2006.08.012
- Sehlstedt, M., R. Dove, C. Boman, et al. 2010. Antioxidant airway responses following experimental exposure to wood smoke in man. Particle Fibre Technol. 7:21. doi:10.1186/1743-8977-7-21
- Shen, Q., W. Wang, Y. Yang, et al. 2010. Emission factors and particulate matter size distribution of polycyclic aromatic hydrocarbons from residential coal combustions in rural northern China. Atmos. Environ. 44(39): 5237–5243.
- Smith, S.J., and T.M.L. Wigley. 2006. Multi-gas forcing stabilization with the MiniCAM. Energy J. special issue 3: 373–91. doi:10.5547/ISSN0195-6574-EJ-VolSI2006-NoSI3-19
- Somers, C.M., B. McCarry, F. Malek, and J.S. Quinn. 2004. Reduction of particulate air pollution lowers the risk of heritable mutations in mice. Science 304:1008–10. doi:10.1126/science.1095815
- Stephenson, S.F., B.C. Esrig, H.C. Polk, Jr., et al. 1975. The pathophysiology of smoke inhalation injury. Ann. Surg. 182:652–60. doi:10.1097/00000658-197511000-00020
- Stern, F.B., W.E. Halperin, R.W. Nornung, et al. 1988. Heart disease mortality among bridge and tunnel officers exposed to carbon monoxide. Am. J. Epidemiol. 128(6): 1276–88.
- Stockfelt, L., G. Sallsten, A.-C. Olin, et al. 2012. Effects on airways of short-term exposure to two kinds of wood smoke in a chamber study of healthy humans. Inhal. Toxicol. 24(1): 47–59. doi:10.3109/08958378.2011.633281
- Suglia, S.F., A. Gryparis, R.O. Wright, et al. 2008. Association of black carbon with cognition among children in a prospective birth cohort study. Am. J. Epidemiol. 167:280–86. doi:10.1093/aje/kwm308
- Suh, H.H., and A. Zanobetti. 2010. Exposure error masks the relationship between traffic-related air pollution and heart rate variability. J. Occup. Environ. Med. 52:685–92. doi:10.1097/JOM.0b013e3181e8071f
- Svedas, E. K.B. Islam, H. Nisell, et al. 2003. Vascular endothelial growth factor induced functional and morphologic signs of endothelial dysfunction in isolated arteries from normal pregnant women. Am. J. Obstet. Gynecol. 188:168–76. doi:10.1067/mob.2003.110
- Takano, H., T. Yoshikawa, T. Ichinose, et al. 1997. Diesel exhaust particles enhance antigen-induced airway inflammation and local cytokine expression in mice. Am. J. Respir. Crit. Care Med. 156:36–42. doi:10.1164/ajrccm.156.1.9610054
- Taylor, E.M. 2002. The impact of efflux transporters in the brain on the development of drugs for CNS disorders. Clin. Pharmacokinet. 41:81–92. doi:10.2165/00003088-200241020-00001
- Tesfaigzi, Y., S.P. Sinsgh, J.E. Foster, et al. 2002. Health effects of subchronic exposure to low levels of wood smoke in rats. Toxicol. Sci 65:115–25. doi:10.1093/toxsci/65.1.115
- Thomas, P.T., and J.T. Zelikoff. 1999. Air pollutants: Modulators of pulmonary host resistance against infection. In Air Pollution and Health, ed. S.T. Holgate, J.M. Samet, H.S. Koren, H.S. Maynard, 357–79, San Diego,CA: Academic Press
- Thompson, L.M., N. Bruce, B. Eskenazi, et al. 2011. Impact of reduced maternal exposures to wood smoke from an introduced chimney stove on newborn birth weight in rural Guatemala. Environ. Health Perspect. 119:1489–94. doi:10.1289/ehp.1002928
- Tong, H., A.G. Rappold, M. Caughey, et al. 2014. Cardiovascular effects caused by increasing concentrations of diesel exhaust in middle-aged healthy GSTM1 null human volunteers. Inhal Toxicol. 26(6):319–329.
- Tonne, C., S. Melly, M. Mittleman, et al. 2007. A case-control analysis of exposure to traffic and acute myocardial infarction. Environ. Health Perspect. 115:53–57. doi:10.1289/ehp.9587
- Tonne, C., J. Yanosky, A. Gryparis, et al. 2009. Traffic particles and occurrence of acute myocardial infarction: A case-control analysis. Occup. Environ. Med. 66:797–804. doi:10.1136/oem.2008.045047
- Totlandsdal, A.I., J. I Herseth, A.K. Bolling, et al. 2012. Differential effects of the particle core and organic extracat of diesel exhaust particles. Toxicol. Lett. 208:262–68. doi:10.1016/j.toxlet.2011.10.025
- Unosson, J., A. Blomberg, T. Sandstrom, et al. 2013. Exposure to wood smoke increases arterial stiffness and decreases heart rate variability in humans. Particle Fibre Toxicol. 10:20. doi:10.1186/1743-8977-10-20
- U.S. Environmental Protection Agency. 2009. Integrated science assessment for particulate matter. Washington, DC: U.S. EPA.
- U.S. Environmental Protection Agency. 2012. Report to Congress on Black Carbon (Chapter 4). http://www.epa.gov/blackcarbon/
- U.S. Environmental Protection Agency Scientific Advisory Board. 1975. Scientific and technical issues relating to sulfates, by an ad hoc panel of the Science Advisory Board. Washington, DC: U.S. EPA.
- van Berlo, D., C. Albrecht, A. M. Knaapen, et al. 2012. Comparative evaluation of the effects of short-term inhalation exposure to diesel engine exhaust on rat lung and brain. Arch. Toxicol. 84:553–62.
- van der Gon, H.A.C.D., M.E. Gerlofs-Nijland, R. Gehrig, et al. 2013. The policy relevance of wear emissions from road transport, now and in the future—An international workshop report and consensus statement. J. Air Waste Manage. Assoc. 63(2):136–49. doi: 10.1080/10962247.2012.741055
- Van Hee, V.C., S.D. Adar, A.A. Szpiro, et al. 2009. Exposure to traffic and left ventricular mass and function. Am. J. Respir Crit Care Med. 179:827–34. doi:10.1164/rccm.200808-1344OC
- Van Rijt, L.S., N. Vos., M. Willart et al. 2004. Essential role of dendritic cell CD80/CD86 costimulation in the induction, but not reactivation, of TH2 effector responses in a mouse model of asthma. J. Allergy Clin. Immunol. 114: 166–73. doi:10.1016/j.jaci.2004.03.044
- Verma, V., P. Pakbin, and K.L. Cheung, et al. 2011. Physiochemical and oxidative characteristics of semi-volatile components of quasi-ultrafine particles in an urban atmosphere. Atmos. Environ. 45:1025–33. doi:10.1016/j.atmosenv.2010.10.044
- Veronesi, B., O. Makwana, M. Pooler, et al. 2005. Effects of subchronic exposure to concentrated air pollution in ApoE-/- mice: VII. Degeneration of dopaminergic neurons. Inhal. Toxicol. 17:235–41. doi:10.1080/08958370590912888
- Vivier, E., D.H. Raulet, A. Moretta, et al. 2011. Innate or adaptive immunity? The example of natural killer cells. Science 331:44–49. doi:10.1126/science.1198687
- Wang, M., G.G. Xiao, N. Li, et al. 2005. Use of a fluorescent phosphoprotein dye to characterize oxidative stress-induced signaling pathway components in macrophage and epithelial cultures exposed to diesel exhaust particle chemicals. Electrophoresis 26: 2092–108. doi:10.1002/elps.200410428
- Wang, W., S. Simonich, B. Giri, et al. 2011. Atmospheric concentrations and air-soil gas exchange of polycyclic aromatic hydrocarbons (PAHs) in remote, rural village, and urban areas of Beijing–Tianjin region, North China. Science Total Environ. 409:2942–50.doi:10.1016/j.scitotenv.2011.04.021
- Watson, J.G., J.C. Chow, and L.-W. Antony Chen. 2005. Summary of organic and elemental carbon/black carbon analysis methods and intercomparisons. Aerosol Air Qual. Res. 5(1): 65–102.
- Watson, J.G., and J.C. Chow. 2005. Receptor models. In Air Quality Modeling—Theories, Methodologies, Computational Techniques, and Available Databases and Software, Vol. II—Advanced Topics, ed. P. Zannetti, 455–501. Pittsburgh, PA: Air & Waste Management Association and EnviroComp Institute.
- Wauters A., C. Dreyfuss, S. Pochet, et al. 2013. Acute exposure to diesel exhaust impairs nitric oxide-mediated endothelial vasomotor function by increasing endothelial oxidative stress. Hypertension 62:352–58. doi:10.1161/HYPERTENSIONAHA.111.00991
- Weimer, S., M.R. Alfarra, D. Schreiber, et al. Organic aerosol mass spectral signatures from wood-burning emissions: Influence of burning conditions and wood type. J. Geophys. Res. 113: D10304. doi:10.1029/2007JD009309
- Weldy, C.S., I.P. Luttrell, C.C. White, et al. 2013. Glutathione (GSH) and the GSH synthesis gene Gclm modulate plasma redox and vascular responses to acute diesel exhaust inhalation in mice. Inhal. Toxicol. 25:444–54. doi:10.3109/08958378.2013.801004
- Weldy, C.S., C.C. White, H.-W. Wilkerson, et al. 2011a. Heterozygosity in the glutathione synthesis gene Gelm increases sensitivity to diesel exhaust particulate induced lung inflammation in mice. Inhal. Toxicol. 23:724–35. doi:10.3109/08958378.2011.608095
- Weldy, C.S., H-W Wilkerson, T.V. Larson et al. 2011b. Diesel particulate exposed macrophages alter endothelial cell expression of eNOS, iNOS, MCP1 and glutathione synthesis genes. Toxicol. In Vitro 25: 2064–73. doi:10.1016/j.tiv.2011.08.008
- Wellenius, G.A., M.R. Burger, B.A. Coull, et al. 2012. Ambient air pollution and the risk of acute ischemic stroke. Arch. Intern. Med. 172:(3): 229–34. doi:10.1001/archinternmed.2011.732
- Wise, M.A., K.V. Calvin, A.M. Thomson, et al. 2009. Implications of limiting CO2 concentrations for land use and energy. Science 324:1183–1186. doi:10.1126/science.1168475
- Wong, K.L., M.F. Stock, D.E. Malek, et al. 1984. Evaluation of the pulmonary effects of wood smoke in guinea pigs by repeated CO2 challenges. Toxicol. Appl. Pharmacol 75:69–80. doi:10.1016/0041-008X(84)90077-2
- World Health Organization, IPCS. 2009. Principles for Modelling Dose-Response for the Risk Assessment of Chemicals. Geneva, Switzerland: WHO.
- World Health Organization (European Office). 2012. Health Effects of Black Carbon. Copenhagen, Denmark: WHO.
- Wu, S.-P., S. Tao, Z.-H. Zhang, et al. 2005. Distribution of particle-phase hydrocarbons, PAHs and OCPs in Tianjin, China. Atmos. Environ. 39:7420–32. doi:10.1016/j.atmosenv.2005.08.031
- Xiao, G.G., M. Wang, N. Li., et al. 2003. Use of proteomics to demonstrate a hierarchical oxidative stress response to diesel exhaust particle chemicals in a macrophage cell line. J. Biol. Chem. 278: 50781–90. doi:10.1074/jbc.M306423200
- Xu, X., N. Kherada, X. Hong, et al. 2009. Diesel exhaust exposure induces angiogenesis. Toxicol. Lett. 191:57–68. doi:10.1016/j.toxlet.2009.08.006
- Yang, G, Y. Wang, Y. Zeng, et al. 2013. Rapid health transition in China, 1990–2010: Findings from the Global Burden of Disease Study 2010. Lancet 381:1987–2015. doi:10.1016/S0140-6736(13)61097-1
- Zeiher, A.M., H. Drexler, H. Wollschlager, et al. 1991. Modulation of coronary vasomotor tone in humans. Progressive endothelial dysfunction with different stages of coronary atherosclerosis. Circulation 83:391–401. doi:10.1161/01.CIR.83.2.391
- Zelikoff, J.T. 2000. Woodsmoke, kerosene heater emission, and diesel exhaust. In Pulmonary Immunotoxicology, ed. M.D. Cohen, J.T. Zelikoff, and R.B. Schlesinger, 369–87. Boston, MA: Kluwer Academic. doi:10.1007/978-1-4615-4535-4_15
- Zelikoff, J.T., L.C. Chen, M.D. Cohen, et al. 2002. The toxicology of inhaled woodsmoke. J. Toxicol. Environ. Health A 85:269–82. doi:10.1080/10937400290070062
- Zhang, H., S. Wang, J. Hao, et al. 2012. Chemical and size characteristics of particles emitted from the burning of coal and wood in rural households in Guizhou, China. Atmos. Environ. 51:94–99. doi:10.1016/j.atmosenv.2012.01.042
- Zhu, Y., W.G. Hinds, S. Kim, et al. 2002. Concentration and size distribution of ultrafine particles near a major highway. J Air Waste Manage Assoc. 52:1032–42. doi:10.1080/10473289.2002.10470842
- Titcombe, M.E., and M. Simcik. 2011. Personal and indoor exposure to PM and polychclic aromatic hydrocarbons in the southern highlands of Tanzania: A pilot-scale study. Environ. Monit. Assess. 180:461–76. doi:10.1007/s10661-010-1799-3