
ABSTRACT
The carcinogenicity of perfluorooctanoic acid (PFOA) has been reviewed previously by several different regulatory agencies and researchers, with contradictory conclusions–especially regarding epidemiological findings on kidney cancer and testicular cancer. In addition, previous dose-response assessments have focused primarily on evidence from animal studies. This critical review summarizes peer reviewed epidemiological studies on PFOA and cancers of the kidneys and testes, using modified Hill’s criteria to assess the evidence for causation. We converted exposures to a common scale based on serum PFOA concentrations and applied meta-analysis to estimate the average increase in cancer risk reported by the studies with sufficient information to estimate serum PFOA. Using random effects meta-analysis, we found that the average relative increase in cancer risk per 10 ng/mL increase in serum PFOA for these studies is 16% (95% CI: 3%, 30%) for kidney cancer and 3% (95% CI: 2%, 4%) for testicular cancer. These associations are most likely causal, but results are limited by the small number of studies for testicular cancer, the overlapping study populations for several studies, and the lack of measured or modeled serum PFOA concentrations for several studies.
Implications: Our review meta-analysis indicates an average increase in cancer risk per 10 ng/mL increase in serum PFOA for kidney and testicular cancers. These associations are most likely causal, but results are limited by the small number of studies for testicular cancer, the overlapping study populations for several studies, and the lack of measured or modeled serum PFOA concentrations for several studies. The weight of evidence could be even stronger with the addition of future studies conducted in large cohorts.
Introduction
Scott M. Bartell

Verónica M. Vieira

Per- and polyfluoroalkyl substances (PFAS) are a complex class of emerging man-made contaminants with varying chemical properties, and rapidly changing production and use. Over 4000 individual PFAS chemicals have been produced, and used in a variety of products including (but not limited to) food packaging, contact paper, cookware, jackets, waterproof gear, furniture, carpeting, upholstery, industrial surfactants, cleansers, polishes, paints, wire insulation, ski waxes, and aqueous film forming foams (AFFF) used in firefighting (Sunderland et al. Citation2019). Although U.S. production dates back to the early 1950s (Shin et al. Citation2011a; ITRC Citation2021; 3M Citation2021), largely centered on perfluorooctanoate (PFOA) and perfluorooctanesulfonate (PFOS), historical production amounts and product formulations are generally not publicly available. Moreover, the PFAS chemicals produced and used in the U.S. have changed over time, particularly after 8 large producers phased out U.S. production of PFOA, PFOS, and their precursors between 2000 and 2015, shifting toward shorter chain homologues, fluorotelomer-based products, per- and polyfluoroalkyl ether substances, and other replacements (ITRC Citation2021; Strynar et al. Citation2015). Although production has changed, PFAS are extremely stable and persistent in the environment due to their strong fluorine-carbon bonds, remaining in soil and groundwater for decades or longer (ATSDR Citation2018).
The relative contributions of various exposure pathways and their impacts on biomonitoring results for the general population are not well understood, but some relevant studies are available for the most common PFAS chemicals (Sunderland et al. Citation2019). PFOA, historically one of the most widely used PFAS chemicals, is widespread and persistent in the environment, bioaccumulative in humans, and associated with toxicity in some animal experiments and human studies (ATSDR Citation2018). PFOA has been reported at concentrations of 20 ng/L or higher in about 2% of public water supplies in the U.S. (Hu et al. Citation2016), and the proportion testing above the analytical limit of detection has been estimated to be substantially higher (Andrews Citation2018). For communities with PFOA-contaminated water supplies, often due to proximity to PFAS-related industrial sites, military fire training areas, AFFF-certified airports, or wastewater treatment plants (Hu et al. Citation2016), water consumption is often the primary exposure route (Shin et al. Citation2011b; Bartell Citation2017; Sunderland et al. Citation2019). In some of those communities, water PFOA concentrations have exceeded 10,000 ng/L (Shin et al. Citation2011a; Michigan PFAS Action Response Team Citation2021), resulting in exposures and serum PFOA concentrations comparable to those of PFOA production workers (Steenland and Woskie Citation2012). Wastewater treatment plants can also act as an exposure source due to transport and use of treated sewage sludge on farmland (ATSDR Citation2018). Food ingestion is thought to be the predominant PFOA exposure route for most adults, and breast milk is thought to be the predominant exposure pathway for breastfed infants. Inhalation is a primary exposure route for production workers, and household dust ingestion and inhalation of PFOA precursors in indoor air may be major contributors to exposure for some individuals with stain-resistant carpeting and/or upholstery (Haug et al. Citation2011; IARC Citation2016; Raleigh et al. Citation2014). Dermal exposures to PFOA are believed to be low, even in occupationally exposed individuals, though some models suggest potential for dermal absorption from PFAS in cosmetics (de Silva et al. Citation2021; Sunderland et al. Citation2019). Exposure routes are thought to be similar for other legacy PFAS (Sunderland et al. Citation2019).
However, because environmental sampling for PFAS has been sporadic, largely based on convenience sampling rather than systematic approaches, and subject to limited understanding of precursor chemistry and analytical chemistry limitations, the relative importance of different exposure routes for the general population remains uncertain (de Silva et al. Citation2021). Recognizing these difficulties and the challenges posed by the multitude and ubiquity of PFAS chemicals, some authorities and researchers use class-based measurement techniques such as the Total Oxidizable Precursor Assay, particle-induced gamma-ray emission, or combustion ion chromatography to measure total perfluoroalkyl acid oxidizable precursors or total organoflourine content, instead of or as an adjunct to measuring individual PFAS chemicals (ITRC Citation2021). A detailed review and recent guidance on analytical methods for traditional targeted and untargeted measurement of PFAS are also provided by the Interstate Technology and Regulatory Council (ITRC Citation2021).
Cancer studies for PFAS in animals and humans were recently reviewed by the Agency for Toxic Substances and Disease Registry (ATSDR Citation2018). That review noted that ingestion of PFOA or PFOS in animals has been reported in various studies to cause adenomas of the liver, pancreas, testes, and thyroid, but struggled to relate these findings to humans due to “profound differences in the toxicokinetics of perfluoroalkyls between humans and experimental animals” and questions about the mode(s) of action for PFAS, which include peroxisome proliferator-activated receptor-α (PPAR-α) activation, for which humans are less affected than rodents. However, as pointed out elsewhere in the ATSDR report and in another recent review (Temkin et al. Citation2020), knockout studies indicate that PFAS toxicity also occurs via mechanisms other than PPAR-α, including increased oxidative stress, changes in gene expression, and immune suppression, all of which are relevant in humans and have been described as key characteristics of carcinogens.
Other reviews of the evidence for carcinogenicity of PFOA have reached different conclusions, even when examining similar bodies of evidence. For example, before much of the current body of evidence was available, the US EPA Science Advisory Board (Citation2006) recommended classifying PFOA as a “likely human carcinogen,” based on findings of multi-site carcinogenicity in two separate rodent studies, rather than the characterization “suggestive evidence of carcinogenicity” originally proposed by the US EPA in its draft risk assessment. A decade later, after consideration of additional studies including new epidemiological evidence associating PFOA exposure and cancer, the US EPA returned to its original characterization of “suggestive evidence of carcinogenic potential” of PFOA in humans in its Drinking Water Health Advisory for PFOA (US EPA Citation2016).
A similar conclusion was reached by a monograph workgroup for the International Agency for Research on Cancer (IARC), which classified PFOA as “possibly carcinogenic to humans” based on limited evidence in experimental animals, limited evidence in humans, and moderate evidence for mechanisms of carcinogenesis (IARC Citation2016). The IARC review noted that there was credible evidence of kidney cancer and testicular cancer with PFOA exposure in humans, but that chance, bias, and confounding could not be ruled out with reasonable confidence.\After its own subsequent independent review, the European Food Safety Authority agreed with IARC (Citation2016) assessment (EFSA Citation2018).
In contrast, two reviews conducted during the last decade concluded that epidemiological evidence does not support a causal relationship between PFOA exposure and cancer (Australia Expert Health Panel for PFAS Citation2018; Chang et al. Citation2014). These reviews raised concerns about perceived inconsistency of findings across studies, small numbers of studies and small numbers of cancer cases, lack of evidence of a monotonic exposure-response relationship, risk of confounding or other types of bias, and discordant cancer types for the human and animal studies. Somewhat confusingly, one report noted that there is suggestive evidence relating PFOA to kidney and testicular cancer, and that it is “possible there is increased risk,” but ultimately concluded that those results might be due to chance and that “there is no current evidence that suggests an increase in overall cancer risk” (Australia PFAS Expert Health Panel Citation2018).
Also in contrast, after reviewing human and animal studies, a panel of epidemiologists tasked with obtaining and reviewing evidence on the health effects of PFOA under the terms of a class-action settlement determined in 2012 that there was a probable link (i.e., a scientific determination that it was more likely than not that PFOA exposure had caused disease or death) between PFOA exposure and testicular cancer, and between PFOA exposure and kidney cancer, in exposed communities in West Virginia and Ohio (C8 Science Panel Citation2012). This conclusion was based primarily on evidence from their large epidemiological studies in that population, the results of which have been published in peer-reviewed scientific journals (Barry, Winquist, and Steenland Citation2013; Steenland and Woskie Citation2012; Vieira et al. Citation2013). These epidemiologists recently revisited those assessments, and reaffirmed their original conclusions regarding carcinogenicity, noting that recent evidence provides stronger support for kidney cancer than was available at the time of their original assessment (Steenland et al. Citation2020).
Because previous reviews of the evidence for carcinogenicity of PFOA disagree on interpretation of the quality and consistency of the epidemiological evidence, this critical review focuses on that issue. In particular, we review and evaluate the human studies on PFOA with kidney cancer and testicular cancer, the two cancer types highlighted as having possible or probable causal associations in most of the previous reviews. First we discuss a modified version of Hill’s criteria, a formal set of criteria used by epidemiologists to assess causation, and then we apply the criteria to evaluate the evidence for a causal association between PFOA and cancers of the kidneys and testes. In addition, we employ formal meta-analysis to compare, combine, and evaluate the results of the kidney and testicular cancer studies that used comparable quantitative exposure metrics. Because meta-analysis includes statistical tools for evaluating heterogeneity across studies and combining effect sizes across studies, it is particularly well suited for addressing questions regarding consistency and elucidation of overall patterns among multiple studies. We are not aware of any previous meta analyses on this topic.
Although lack of understanding of the precise mode(s) of action for carcinogenicity is also an issue of concern raised in previous reviews, we do not address that topic further here, other than to note that lack of identification of the precise mechanism for observed carcinogenicity of PFOA in animals is not dispositive regarding causation.
Epidemiological study designs for investigating carcinogenicity
For obvious ethical reasons, human studies of the potential health effects of exposure to toxic substances cannot use random assignment of study participants to treatment or control groups, even though randomization is generally regarded as the most reliable study design (Chandler et al. Citation2020). Instead, epidemiologists use a variety of observational study designs, assessing the absence/presence or the degree of exposure to a toxicant experienced “naturally” (not under control of the researcher) by each participant in a study, as well as the resulting health outcomes. Rates of disease or other health outcome measures are then compared between those exposed and not exposed to the toxicant, or across multiple groups categorized by the amount of exposure. Common study designs used for cancer investigations include (1) cohort studies, in which individuals who are free of the disease of interest are assessed for exposure and followed up over time to determine health outcomes, (2) case-control studies, in which exposures are assessed retrospectively among individuals who developed the disease of interest (“cases”) and among a representative sample of those who did not develop the disease (“controls”), and (3) ecological studies, in which disease rates are compared between exposed and unexposed groups of people (Szklo and Nieto Citation2019).
However, because some toxicant exposures can be affected by or associated with demographic characteristics, socioeconomic status, dietary choices, and other behaviors which might also affect the risk of disease, it is important for epidemiologists to also consider and account for these confounding variables when comparing disease rates across exposure groups, to ensure that any observed differences are not due simply to these common causes of exposure and disease, rather than being caused by the exposure itself. A variety of statistical methods are available and widely used to account for confounding variables in observational studies, including matching, stratification, and multiple regression (Szklo and Nieto Citation2019), and a strong theoretical framework is available for determining which variables should be treated as confounders when other disease risk factors and their relationships to exposure and to each other are well characterized (Hernán et al. Citation2002). However, difficulties may arise in confounder adjustment when risk factors are not well understood, or when they cannot be measured or obtained from the participants, in which case observed associations may be unintentionally biased as a result of inadequate adjustment.
Observational studies may also be affected by other sources of unintentional bias, including selection bias if study participants are not representative of the target population with respect to the exposure-disease relationship, information bias if variables are not measured accurately (a particular challenge for epidemiologic studies of environmental exposures to toxicants), aggregation bias if data can only be collected at the group level rather than the individual level, and reporting bias if results are selectively reported based on statistical significance or other characteristics related to study outcomes, so that published findings are not representative of all relevant investigations (Sterne et al. Citation2020).
Because of the variety of potential threats to validity in observational studies, it can be difficult to ascertain whether an epidemiological finding really indicates a causal effect of exposure on the disease, or merely a statistical correlation. Epidemiologists therefore emphasize careful consideration of the relative strengths and limitations of each study, replication or triangulation of findings with multiple investigations using different study designs or different populations, and evaluation of epidemiological study results in light of experimental toxicology findings and any other relevant lines of evidence (Bartell Citation2019; Sterne et al. Citation2020). Here we apply Hill’s criteria, an influential and longstanding methodology for evaluating causation (Rothman and Greenland Citation2005), to the body of peer-reviewed epidemiological evidence on PFOA and cancer of the kidney and testes.
Modified Hill’s criteria for causation
We use a modified version of Hill’s criteria to evaluate the weight of evidence for causation of PFOA with kidney cancer and testicular cancer, focusing on the strength of the association, consistency, temporality, biological gradient, and biological plausibility/coherence. Sir Austin Bradford Hill originally proposed 9 criteria for assessing whether or not epidemiological associations between environmental exposures and disease are likely to be causal: 1. strength, 2. consistency, 3. specificity, 4. temporality, 5. biological gradient, 6. plausibility, 7. coherence, 8. experimental evidence, and 9. analogy (Hill Citation1965). These criteria are widely recognized, debated, and cited by health scientists in making professional assessments about the weight of evidence for disease causation in all areas of epidemiology (Rothman and Greenland Citation2005; van Reekum, Streiner, and Conn Citation2001). Notably, Hill warned that this set of criteria should not be used as a required checklist before concluding that a causal relationship exists (Hill Citation1965):
What I do not believe – and this has been suggested – that we can usefully lay down some hard-and-fast rules of evidence that must be obeyed before we can accept cause and effect. None of my nine viewpoints can bring indisputable evidence for or against the cause-and-effect hypothesis and none can be required as a sine qua non. What they can do, with greater or less strength, is to help us to make up our minds on the fundamental question– is there any other way of explaining the set of facts before us, is there any other answer equally, or more, likely than cause and effect?
Thus, Hill’s criteria are intended as a list of issues to consider before making an informed scientific judgment based on the weight of evidence; not every criterion needs to be fulfilled to conclude that causation is present. Hill also wrote that “no formal tests of [statistical] significance can answer those questions. Such tests can, and should, remind us of the effects that the play of chance can create, and they will instruct us in the likely magnitude of those effects. Beyond that they contribute nothing to the ‘proof’ of our hypothesis” (Hill Citation1965).
Strength of association
Various statistical measures are used to assess the strength of association, including hazard ratios and odds ratios. A ratio is the quotient of two measurements; for hazard ratios and odds ratio the denominator is a measure of disease risk among a “referent group.” The referent group is a group that is not exposed, or a group that is less exposed, if the study does not include a group that is not exposed. Thus, each ratio indicates the relative degree of disease risk, comparing a group with more exposure to a group with less or no exposure. For example, a ratio of 2 indicates that the exposure is associated with double the risk of disease, compared to the referent group in the study. Higher values indicate stronger associations. In human studies of PFAS exposure, the referent group is often those in the first quintile or quartile of exposure. Thus, if PFAS is a cause of the disease under study, the reported relative risks likely would have been larger, and the association stronger, if the referent group had been completely unexposed.
Hill’s widely accepted argument for using strength of association as a criterion for causation is predicated on the fact that weak or moderate confounding or bias cannot explain strong associations (Rothman and Greenland Citation2005). It is therefore easier to rule out these alternative explanations for a strong association than it would be for a weak association.
Consistency
Consistency refers to repeated observations of an exposure-disease association using different study designs, different populations, and/or different circumstances. Sir Hill and others take pains to warn that the absence of this criterion is less informative than its presence, due to sampling variation, differences in study designs, and other factors that could result in different effect estimates even if the exposure causes the disease (Hill Citation1965; Rothman and Greenland Citation2005). Nonetheless, similar study findings under different circumstances is a strong indication that observed associations are not a result of a particular bias, flaw, or quirk in any single analysis (Bartell Citation2019; Hill Citation1965; Munafò and Davey Smith Citation2018).
Statistical significance is not a consideration in Hill’s criterion of consistency. As explained by Rothman and Greenland (Citation2005),
It is sometimes claimed that a literature or set of results is inconsistent simply because some results are “statistically significant” and some are not. This sort of evaluation is completely fallacious even if one accepts the use of significance testing methods: The results (effect estimates) from the studies could all be identical even if many were significant and many were not, the difference in significance arising solely because of differences in the standard errors or sizes of the studies.
Specificity
Specificity is a controversial criterion originating from an outdated theory that every disease has only one cause, and that an exposure can cause only one disease. Sir Hill himself expressed some doubts about that theory, since replaced by multi-causation, and modern authorities often dismiss the specificity criterion as invalid (van Reekum, Steiner, and Conn Citation2001; Rothman and Greenland Citation2005). Thus, this criterion is not addressed further in this review.
Temporality
An exposure that did not precede the disease cannot have been its cause. Studies measuring exposures that occurred before the disease occurs, rather than at the same time or after the disease, are necessary to demonstrate temporality (van Reekum, Streiner, and Conn Citation2001). Temporality can be demonstrated using prospective or retrospective studies, if reliable indicators of exposure are available for the period preceding the disease. For example, occupational health studies often rely on job records as proxies for past exposures in the workplace, and community studies often rely on residential histories as proxies for past exposures to air pollutants or water pollutants.
Biological gradient
This criterion refers to evidence of increasing risk of disease with increasing exposure. Although the absence of such evidence is not a refutation of causality, and might be a consequence of a causal relationship with a non-monotonic or threshold dose-response pattern (Hill Citation1965; Rothman and Greenland Citation2005), or simply random variation with an underlying monotonic dose-response relationship (Kim, Bartell, and Gillen Citation2015), an average trend of increasing risk across several increasing levels of exposure is consistent with causation.
Plausibility and coherence
As Sir Hill (Citation1965) wrote, “it will be helpful if the causation we suspect is biologically plausible. But this is a feature I am convinced we cannot demand. What is biologically plausible depends upon the biological knowledge of the day.” Coherence is similar to biological plausibility, but refers more broadly to preexisting evidence about the natural history and biology of the disease (Hill Citation1965; Rothman and Greenland Citation2005; van Reekum, Streiner, and Conn Citation2001). Thus, if a toxicant has been demonstrated to affect a biological mechanism or mode of action that is a known component of the etiology for a particular disease, it is more likely that observed epidemiological associations between that toxicant and that disease are causal. Similarly, animal experiments showing that a similar disease or disease process can be induced by exposure to the toxicant, provide evidence in support of biological plausibility and coherence.
The absence of a proposed mechanism or other corroborating biological information is not dispositive with regard to plausibility or coherence, as the study and knowledge of biological mechanisms typically develops over time after exposure-disease relationships are reported, if at all, rather than beforehand.
Experiment
Because it is usually unethical to conduct research intentionally exposing humans to toxic chemicals, controlled experiments in humans are generally lacking (Rothman and Greenland Citation2005). It is sometimes possible to perform intervention studies, e.g., observe the health effects of removing an exposure source, such as studies of the health effects of smoking cessation. Such studies are rarely performed for toxic chemicals and chronic diseases, due to practical difficulties, but they provide a powerful test of causality when available. No human experimental evidence is available for PFAS exposure and the health outcomes in this review.
Analogy
Sir Hill described analogy to different exposures causing similar diseases as a potential consideration for causation, but this criterion is now believed to be unnecessary and possibly misleading (Rothman and Greenland Citation2005; van Reekum, Streiner, and Conn Citation2001). Thus, this criterion is not addressed further in this review.
PFOA and cancer in humans
Summary of studies
Human studies of PFAS and cancer were summarized in a recent scoping review, which identified 28 published primary studies on the topic (Steenland and Winquist Citation2021). Among these studies, those that examined PFOA and kidney cancer are listed in , and those that examined PFOA and testicular cancer are listed in . Because it is a major determinant of the statistical precision and power of each study, as well as the ability to evaluate dose-response trends, the studies were listed in order by the number of cancer cases included, with the largest case numbers first. Some studies used additional data from external groups for comparison, but only those cases used in internal dose-response analyses were included here. This ordering reflects only the likely impacts of case counts on sampling variability, and should not be confused with study quality or reliability; for example, the study listed first in (Mastrantonio et al. Citation2018) had the largest number of kidney cancer deaths, but relatively weak ascertainment of exposure, and an ecological study design allowing only group-level assessment of most variables. Three cohort studies reported positive but non-significant associations between PFOA and testicular cancer mortality with only 1 testicular cancer death (Gilliland and Mandel Citation1993; Leonard et al. Citation2008; Steenland and Woskie Citation2012), and were excluded from due to an insufficient sample size.
Table 1. Peer-reviewed epidemiological studies on PFOA and kidney cancer
Table 2. Peer-reviewed epidemiological studies on PFOA and testicular cancer
The studies described in used various measures of association. Hazard ratios, risk ratios, and odds ratios are approximately equal for rare diseases such as cancer, and are comparative measures of the risk of developing the particular disease. Mortality ratios are comparative measures of the risk of death due to the particular disease. Additional analyses using selected subsets of participants, alternative exposure metrics such as time-lagged exposure, annual serum concentrations, (continuous) log exposure, or water district, and alternative referent groups are also described in several of these papers; most of those results were qualitatively similar to the primary analyses and are not reproduced here. Some occupational studies also present additional analyses comparing workers to the general population. Those results usually differ from those in the table, often showing less cancer risk among exposed workers than the general population, but are not reproduced here because they are less informative than comparisons to other workers or internal dose-response analyses due to healthy worker bias (Steenland and Winquist Citation2021). For the occupational studies presented in these tables (Consonni et al. Citation2013; Raleigh et al. Citation2014; Steenland and Woskie Citation2012), the lowest PFOA groups have rate ratios that differ from 1 due to their selection of non–exposed workers as the referent group. For studies in which both mortality ratios and incidence measures were provided (e.g., Raleigh et al. Citation2014), we present the incidence measures as the preferred analysis in due to the larger number of cases than deaths and the goal of understanding potential effects of PFOA on both fatal and non-fatal cancers.
The two community-based C8 Science Panel studies included in the tables (Barry, Winquist, and Steenland Citation2013; Vieira et al. Citation2013) both reported increases in kidney cancer and testicular cancer incidence in the groups with the highest exposures to PFOA. The Barry et al. study population consumed PFOA–contaminated water in West Virginia (WV) or Ohio, due to emissions from a local industrial facility (Frisbee et al. Citation2009). Participants in this study consumed water with PFOA concentrations ranging from about 1 to 20,000 ppt (Shin et al. Citation2011a) for at least one year, up to many decades, resulting in serum PFOA concentrations measured during 2005-2006 that ranged from below the limit of detection (<0.5 ng/mL) to 4,752 ng/mL (Barry, Winquist, and Steenland Citation2013; Frisbee et al. Citation2009; Shin et al. Citation2011b). Although many study participants were highly exposed, with serum PFOA concentrations comparable to those with occupational exposures in some cases, this study also included thousands of community members consuming water containing less than 70 ppt PFOA, and with serum PFOA concentrations within the range of general population exposures. The fact that study participants spanned such a wide range of exposures allowed researchers to compare health outcomes across a gradient of exposures, from serum concentrations typical of the general population to many times higher concentrations, increasing statistical power and providing clear distinctions between different exposure categories.
Strengths of the Barry, Winquist, and Steenland (Citation2013) study included a large number of participants, individual-level exposure reconstruction using detailed residential histories for each individual who consented to participate in the research (Shin et al. Citation2011a, Citation2011b), large exposure differences resulting from a “natural experiment” (a situation in which people are not randomly assigned to different exposure levels, but the exposure is viewed as pseudo-random because people did not choose their exposure and were unaware of it), medical records verification, and careful control for potential confounding by time-varying smoking, time-varying alcohol consumption, sex, education, and 5-year birth year period. However, because individuals who died before 2005 could not participate in the community study, some pre-2005 kidney cancer patients may have been excluded from the study. As pointed out by the authors, any bias resulting from those exclusions would likely be in the direction of underestimation of the health risks from PFOA exposure (Barry, Winquist, and Steenland Citation2013).
The Vieira et al. (Citation2013) case-control study used the cancer registry for the entire state of Ohio, but not WV, and therefore included some, but not all, of the cases included in the Barry et al. study. It also included cancer cases in Ohio who lived outside of the area with PFOA-contaminated water, and were therefore not included in Barry et al. study. Analyses were adjusted for age, sex, diagnosis year, smoking status (current, past, unknown, or never), and insurance provider (government-insured Medicaid, uninsured, unknown, or privately insured). Although it relied on a similar exposure assessment approach and the same fate and transport model as the Barry et al. study for those participants with PFOA-contaminated water, residential addresses were only available at the time of diagnosis for the Vieira et al. study, so the researchers assumed that participants had been living at the same address for decades prior to diagnosis. This likely resulted in some nondifferential misclassification of exposures. Their use of registry-based controls (excluding kidney, pancreatic, testicular, and liver cancers) rather than population-based controls could also introduce bias if PFOA exposure is related to any of cancer types that were selected for comparison. However, any bias resulting from these limitations would likely be in the direction of underestimation of the health risks from PFOA exposure (Vieira et al. Citation2013).
The recent nested case-control study by Shearer et al. (Citation2020) also found an increase in kidney cancer with PFOA exposure and is influential due to its strong study design and inclusion of the largest number of kidney cancer cases among all the studies listed in . Because it was nested in a large cohort of approximately 150,000 participants with measured PFOA in stored pre–diagnostic serum samples that has been followed for over 20 years, there are few or no concerns regarding control selection, survivor bias, temporality, or other potential threats to validity. Potential confounding was controlled by individually matching on age at enrollment (55–59 years, 60–64 years, 65-69 years, or ≥70 years), sex, race and ethnicity (non-Hispanic white, non-Hispanic Black, Hispanic, Asian, or Native American), study center, and study year of blood draw. One limitation of this study is exposure classification based on only a single blood sample for each participant; however, as the authors point out, the relatively long half-life of PFOA and evidence from repeated measurements in another general population study (Sun et al. Citation2018) suggest that a single measurement can be a good proxy for a longer term exposure in the general population (Shearer et al. Citation2020).
The ecological mortality study by Mastrantonio et al. (Citation2018) had the largest number of kidney cancer deaths and the second largest number of testicular cancer deaths among the studies in , but relatively weak ascertainment of exposure, only sorting participants into two groups: those in regions with PFAS-contaminated water and those without known PFAS in their regional water supply. In addition, though PFOA was the primary PFAS found in these drinking water supplies, PFOS was also common and other PFAS were detected as well, making it difficult to isolate PFOA as the causative factor. Analyses were adjusted for sex and age. Regional socioeconomic status and smoking differed slightly but not significantly between the exposed and unexposed regions, apparently with a slightly higher smoking prevalence and socioeconomic status for the exposed regions. Although the mortality rate ratio for testicular cancer (1.86) is strong in this study, the rate ratio for kidney cancer is weaker (1.07), and cigarette smoke is a known carcinogen, making it difficult to rule out potential confounding by smoking as a threat to validity in this study. It is also well understood that ecological (group-level) analyses may not produce the same results as individual-level analyses, which introduces another potential source of bias. The 3 occupational studies of PFOA exposure and kidney cancer listed in (Consonni et al. Citation2013; Raleigh et al. Citation2014; Steenland and Woskie Citation2012) have smaller numbers of cases, but benefit from the long-term workplace records used to classify each participant’s job and likely PFOA exposure over time. Although each of these analyses used a cumulative exposure metric, the exact metric varied by study: cumulative modeled serum PFOA concentration for Steenland and Woskie (Citation2012), cumulative air ammonium PFOA concentration for Raleigh et al. (Citation2014), and cumulative arbitrary unit-years for Consonni et al. (Citation2013). Steenland and Woskie (Citation2012) used a combination of measured worker serum PFOA concentrations and modeled serum PFOA concentrations from residential ingestion of contaminated water to develop an integrated workplace and community exposure model, whereas the other two studies relied solely on characterizations of PFOA in workplace air to classify exposures. Although each of these studies adjusted for age, there was little ability to adjust for other potential confounding variables.
Although some researchers have an instinctive preference for studies that use measured biomarkers to assess exposures rather than environmental measurements and exposure models, methodologists have recently pointed out that modeled exposures are resistant to physiological confounding and reverse causation, making it easier to rule out these potential types of bias compared to studies using only measured serum concentrations (Weisskopf and Webster Citation2017). With the exception of Shearer et al. (Citation2020), all of the studies in are based on modeled exposures and are therefore resistant to physiological confounding and reverse causation. Shearer et al. (Citation2020) is also somewhat resistant to these threats to validity, because the blood samples were collected at the outset of the study, before several decades of follow-up, making it unlikely that preclinical disease or undetected conditions caused changes in both serum PFOA concentrations and the likelihood of eventual kidney cancer. Shearer et al. (Citation2020) also determined that pre–diagnostic measures of kidney function were comparable for their cases and controls, further demonstrating that physiological confounding was an unlikely explanation for their findings.
We also identified but did not include in several additional unpublished ecological analyses comparing cancer incidence rates in counties with water PFOA contamination to other counties without PFOA in the water supply. For example, an analysis of 2005-2014 cancer registry data for the State of New Hampshire found that residents of Merrimack, a town with PFAS-contaminated drinking water, had a 24% higher incidence rate for kidney cancer and several other cancer types than the rest of the state (New Hampshire Department of Health and Human Services Citation2018). Testicular cancer was not more common in Merrimack. However, some comparisons for both of these cancers were based on very small numbers of cases (<10), there was no quantitative assessment of exposure or adjustment for potential confounders, and none of the increases were statistically significant.
Some of the studies performed formal statistical trend tests to determine if there was an increasing (or decreasing) dose-response. Such tests typically rely on statistical regression, using individual-level continuous dose metrics and/or inverse-variance weighted regression across exposure categories (e.g., Barry, Winquist, and Steenland Citation2013; Shearer et al. Citation2020; Steenland and Woskie Citation2012). Individual-level regression is more powerful but requires access to raw data, whereas inverse-variance weighted regression can be performed using typical summaries of results and is expected to produce similar estimates of the average trend in dose-response (van Wijngaarden and Hertz-Picciotto Citation2004). For studies that included an individual-level regression trend test, we listed those p-values in . Some studies presented point estimates and confidence intervals for the dose-response slope or difference between exposed and unexposed groups, rather than p-values (e.g., Mastrantonio et al. Citation2018; Shearer et al. Citation2020); we converted those results to trend test p-values using standard two-sided z-tests of the null hypothesis that the log rate ratio was 0. For studies in which no formal trend test was reported, we used the reported point estimates and confidence intervals to perform inverse-variance weighted regression of the log rate ratio vs. the exposure category rank (0 for the referent group, 1 for the next highest exposed group, etc.), using 0-intercept models for the studies using internal referents and regression-estimated intercepts for those with external referents (see Supplementary Material).
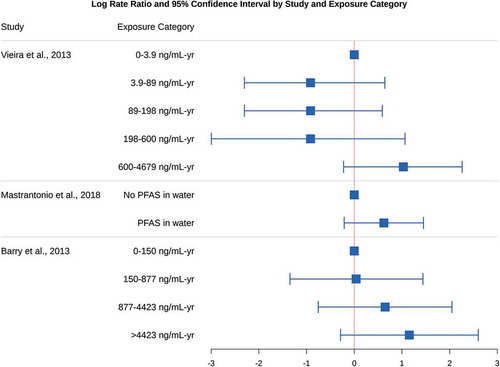
show the point estimates and confidence intervals for the log (base e) rate ratios in each study and exposure category. A log rate ratio of 0 indicates that the incidence or mortality for that type of cancer does not differ between that exposure group and the referent group, and a positive value indicates that cancer incidence or mortality is more common in that exposure group than the referent group. In one study and exposure category (Steenland and Woskie Citation2012), the point estimate and lower bound for the rate ratio was reported as 0.00; we substituted 0.005 for this value in our plots and subsequent analyses involving log rate ratios, because the log of 0 is undefined. Also in that study, cumulative exposure units were incorrectly reported as ppm-yr, and should have been listed as ng/mL–yr (Kyle Steenland, personal communication, January 10, 2021); the corrected units are shown in and used in our subsequent analyses.
Figure 1. Log rate ratios for human studies of PFOA and kidney cancer
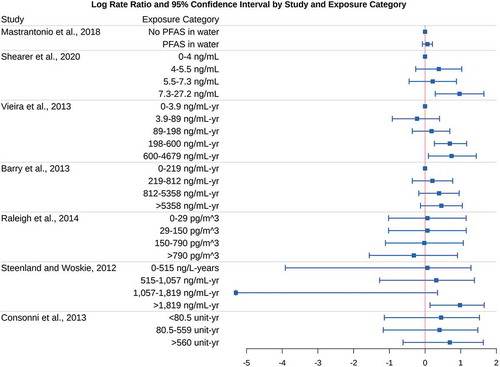
Figure 2. Log rate ratios for human studies of PFOA and testicular cancer
Weight of evidence
The effect of PFOA on kidney and testicular cancer incidence or mortality is strong in most of these studies, often in the range of 2-3 fold higher risk for the highest exposed groups compared to less exposed groups. The strength of association criterion is clearly met for these two types of cancer, as it would require a high correlation with even stronger unmeasured risk factors to explain those results by uncontrolled confounding.
The criterion of consistency is fulfilled for kidney and testicular cancer, as most results in show patterns of increased cancer with increased exposure, and nearly all exposed categories have higher cancer rates than their referents (i.e., and log rate ratios greater than 0). The study by Raleigh et al. (Citation2014) is an exception, with no apparent increase in cancer among the highest exposed groups, but is based on relatively small numbers of cases. Although not all of the trend tests were statistically significant in , that is of little relevance to judging consistency, as previously explained. Nearly all of the individual–level studies reported similar 2-3 fold higher cancer rates in at least one of the two highest exposure groups, and studies comparing PFOA-exposed to unexposed workers reported about twice as many cancer cases/fatalities among those who were exposed. Considering sampling variability and the overlapping confidence intervals, this is a remarkable degree of consistency across studies. A simple binomial sign test of the point estimates in , though not dispositive, indicates that chance alone is a highly unlikely explanation for the 18 positive associations out of 22 non-referent categories among the kidney cancer studies (p = .004). For the testicular cancer studies, 5 of 8 non-referent categories yielded positive associations, resulting in an inconclusive sign test (p = .73).
With the exception of the case-control study by Vieira et al. and the ecological study by and the Mastrantonio et al., which relied on the residence at time of diagnosis or death only to categorize PFOA exposure, each of the studies summarized in can be used to establish temporality. The cohort studies used generally reliable methods to reconstruct past PFOA exposures from residential histories and/or job histories, modeling PFOA exposures that preceded cancer diagnosis or death. That being said, due to the known history of PFOA emissions in these regions, the slow movement of PFOA in groundwater, and the expectation that any residential mobility would likely be non-differential with respect to PFOA exposure, it is reasonable to assume that residential location at the time of diagnosis or death is a reasonable but imperfect proxy for past exposure.
The largest and most precise individual-level studies of PFOA reported clearly increasing dose-response relationships between PFOA exposure and the risk of kidney cancer (Barry, Winquist, and Steenland Citation2013; Shearer et al. Citation2020; Vieira et al. Citation2013). We believe these findings constitute sufficient evidence of biological gradient for PFOA exposure and kidney cancer. Additional support for a biological gradient for kidney cancer is provided by Steenland and Woskie (Citation2012), supported marginally by Consonni et al. (Citation2013), and contradicted by Raleigh et al. (Citation2014), but those three studies have fewer cases and wider confidence intervals.
Due to the smaller number of cases and deaths from testicular cancer, fewer studies reported on these cancers or were able to assess the biological gradient across multiple exposure categories for that cancer type. Barry, Winquist, and Steenland (Citation2013) reported a clear and statistically significant monotonic increase in testicular cancer incidence across four exposure categories. Although this study had the fewest number of cases among the 3 testicular cancer studies, it also had the highest quality exposure assessment, using detailed residential histories for individual–level exposure reconstruction (Shin et al. Citation2011a, Citation2011b). In contrast, Vieira et al. (Citation2013) reported a decrease in testicular cancer incidence in the middle 3 exposure categories compared to the lowest exposure category, followed by highly elevated incidence in the highest exposure group. It is worth noting that the referent category for Barry, Winquist, and Steenland (Citation2013) ranged from 0-150 ng/mL–yr, which equated to approximately the first 3 exposure categories in the Vieira et al. (Citation2013) study (0-189 ng/mL-yr). Mastrantonio et al. (Citation2018) only compared mortality for those exposed and non-exposed, preventing any analysis of gradient. Together, these studies provide suggestive but limited evidence in support of a dose-response gradient for PFOA exposure and testicular cancer.
Regarding biological plausibility and coherence, ingestion of PFOA or PFOS has been reported to cause adenomas or carcinomas of the liver, pancreas, testes, kidney, and thyroid in In controlled experiments in animals (ATSDR Citation2018; NTP Citation2020), providing clear empirical evidence of biological plausibility for induction of cancers in general, but also some evidence for concordance of human and animal findings at specific cancer sites for the kidney and testes. However, as discussed previously, the relevance of some proposed mechanisms of carcinogenicity to humans is disputed, and PFOA doses in the animal studies are far higher than those typically experienced by humans.
The available studies on PFOA and kidney cancer clearly meet Hill’s criteria of strength of the association, consistency, temporality, biological gradient, and biological plausibility/coherence. Thus, the evidence is sufficient to indicate that PFOA is most likely a cause of kidney cancer in humans.
The available studies on PFOA and testicular cancer clearly meet Hill’s criteria of strength of the association, temporality, and biological plausibility/coherence. Although the 3 studies in are consistent in finding increases of testicular cancer among the highest exposed groups, two of these studies rely on overlapping populations (Barry, Winquist, and Steenland Citation2013; Vieira et al. Citation2013). The ecological study results (Mastrantonio et al. Citation2018) could not be used to investigate gradient, and only 1 of the 2 studies with dose-response information provides unambiguous evidence of a dose-response gradient (Barry, Winquist, and Steenland Citation2013). Overall, the limited available evidence indicates that PFOA is most likely a cause of testicular cancer in humans.
Dose-response analysis and meta-analysis
Not all human health studies can or should be used to perform quantitative dose-response modeling or risk assessment. In particular, one authority recommends (Hertz-Picciotto Citation1995) that the following four criteria must be met when choosing the best epidemiological study for performing dose-response modeling: 1. a moderate to strong positive association is present, 2. strong biases are ruled out or unlikely, 3. confounding is controlled or likely to be limited, and 4. the study performed quantification of exposures linked to individuals.
In recent years, due in part to concerns about the potential for bias from selecting only the strongest study, some authorities have recommended performing dose-response analysis for all high-quality studies with sufficient exposure quantification, and possibly combining those dose-response results using meta-analysis and formal Bayesian methods (Samet et al. Citation2014). In order to combine the effect estimates of multiple studies, they must be reported using the same exposure metrics and outcomes, or exposure metrics that can be converted to comparable units. In , the majority of studies use single serum PFOA concentration or cumulative serum concentration (i.e., the sum of annual serum concentrations over time, or the area under the curve for serum concentration vs. time) as the primary exposure metric. Shearer et al. (Citation2020) used a single pre-diagnostic serum PFOA concentration for each participant, collected at the start of several decades of follow-up. Vieira et al. (Citation2013) used cumulative serum PFOA over 10 years, ending 10 years prior to cancer diagnosis. Steenland and Woskie (Citation2012) used cumulative serum PFOA from the time of workplace entry until the end of follow-up, averaging 30 years in duration, and Barry, Winquist, and Steenland (Citation2013) used cumulative serum PFOA from birth until the end of follow-up. Because the duration differs substantially across these studies, and is a strong determinant of cumulative serum, we believe it is more appropriate to convert them to estimated average serum PFOA concentrations for comparisons across studies. By the mean value theorem, the average serum concentration is equal to the cumulative serum concentration divided by the duration. Thus, we divide the reported cumulative serum PFOA concentrations for each exposure category by 10 for Vieira et al. (Citation2013), by 30 for Steeland and Woskie (Citation2012), and by 64 (the average age of diagnosis for kidney cancer; ACS Citation2020a) or 33 (the average age of diagnosis for testicular cancer, ACS Citation2020b) for Barry, Winquist, and Steenland (Citation2013) to estimate the time-averaged serum concentration for each exposure category. In addition, we assume that the single time-point serum PFOA concentrations measured by Shearer et al. (Citation2020) are reasonable surrogates for each participant’s time-averaged serum PFOA concentration.
Due to lack of quantitative exposure assessment, we were unable to convert results from the Mastrantonio et al. (Citation2018) or Consonni et al. (Citation2013) studies to this common exposure scale. Although Raleigh et al. (Citation2014) provide quantitative exposure estimates, conversion of cumulative air concentrations to cumulative serum concentrations would require additional data not provided by the authors and difficult to approximate, such as average hours worked per year, average breathing rates, and duration of employment. We do not include this study in our meta-analysis, but note its potential importance as an outlier and discuss the potential impacts of its inclusion, if sufficient information becomes available to estimate the average serum PFOA concentrations corresponding to these exposure categories.
Some studies fit continuous dose-response models to the individual-level data and reported cancer rate ratios per log serum PFOA concentration or per log cumulative serum PFOA concentration (Barry, Winquist, and Steenland Citation2013; Shearer et al. Citation2020). We report those values in , converting log rate ratios per log2 serum PFOA (Shearer et al. Citation2020) to log rate ratios per loge serum PFOA using the change of base formula, multiplying by log2(e). Conversion from cumulative to average serum PFOA concentration for the Barry, Winquist, and Steenland (Citation2013) study, by dividing by 64 or 33 years, does not change the effect estimate per log unit exposure because that slope is invariant to multiplicative changes in the exposure scale.
Table 3. Rate ratio for kidney cancer incidence or mortality using continuous exposure metrics
Table 4. Rate ratio for testicular cancer incidence using continuous exposure metrics
We also converted each set of study results into an average effect size per 10 ng/mL increase in serum PFOA concentration, by using inverse-variance weighted regression of reported log rate ratios vs. the midpoints of the time-averaged serum PFOA categories within each study. 10 ng/mL is the estimated steady-state serum PFOA concentration after long-term consumption of water containing 70 ppt PFOA, the current EPA health advisory value. In two studies, the maximum value of the highest exposure quartile was not reported (Barry, Winquist, and Steenland Citation2013; Steenland and Woskie Citation2012); in these studies we estimated the maximum value as four times the 75th percentile, consistent with the ratio reported by Shearer et al. (Citation2020). R code for all calculations is provided in the Supplementary Material, and results are shown in .
Results are fairly consistent across the C8 Science Panel studies (Barry, Winquist, and Steenland Citation2013; Steeland and Woskie Citation2012; Vieira et al. Citation2013), though this may not be entirely unexpected given their overlapping populations. The results of Shearer et al. (Citation2020) indicate a substantially higher risk of kidney cancer from PFOA exposure than the other studies, using an independent study population. A formal measure of heterogeneity that estimates the percentage of variation in effect estimates not caused by sampling variability (Higgins and Thompson Citation2002) indicates no substantial discrepancies in effect estimates per 10 ng/mL serum PFOA across the two testicular cancer studies (I2 = 0%) but substantial heterogeneity across the four kidney cancer studies (I2 = 94%), likely due to the outlying results from Shearer et al. (Citation2020). Although all four kidney cancer studies show increasing risk with PFOA exposure, the range of exposure is much smaller in Shearer et al. (Citation2020), which may influence the results if the rate ratio per unit serum PFOA is higher at lower exposure than it is at higher exposures. Alternatively, the higher rate ratio might be due to better exposure classification via the use of pre-diagnostic measured serum, whereas the other studies relied on modeled serum, which may have reduced the apparent rate ratios due to nondifferential exposure misclassification (Avanasi et al. Citation2016).
Formal meta-analysis estimates combining the log rate ratios across all studies in are also reported. Meta-analysis is a statistical technique for combining effects estimates across multiple studies, in order to obtain a single unified effect estimate and confidence interval that summarizes the entire body of evidence, weighting the results of each study according to the precision of its effect estimates (Samet et al. Citation2014). We present results from two common versions of meta-analysis here: fixed effects meta-analysis and random effects meta-analysis. Although there is substantial debate about which version has a better theoretical underpinning, random effects meta-analysis is arguably more appropriate when there is substantial heterogeneity across studies (Samet et al. Citation2014), as it does not assume that every study population has the same underlying response.
Results from all meta–analysis models show increases in kidney cancer risk and testicular cancer risk with increasing PFOA serum concentrations, with an estimated 15–49% increase in the risk of kidney cancer for each loge unit increase in serum PFOA, an estimated 4-16% increase in the risk of kidney cancer for each 10 ng/mL increase in serum PFOA, and an estimated 3% increase in the risk of testicular cancer for each 10 ng/mL increase in serum PFOA. All meta-analysis risk estimates except the fixed effects meta-analysis for loge serum were statistically significant, indicating that the balance of studies indicate an increase in kidney cancer and testicular cancer with serum PFOA that cannot be explained by random chance.
Although it is somewhat unusual to combine incidence and mortality rate ratios together for the meta-analysis as we have done in , we note that the mortality and incidence rate ratios in are similar, and combination of these disparate outcomes is only inappropriate if fatality among those who develop cancer is differentially impacted by PFOA exposure, in addition to any effects of PFOA on cancer incidence.
These results are tempered somewhat by the overlapping study populations for the C8 Science Panel studies (Barry, Winquist, and Steenland Citation2013; Steenland and Woskie Citation2012; Vieira et al. Citation2013), as meta-analysis methods assume independent studies, and by the exclusion of three kidney cancer studies due to lack of quantitative exposure estimates that could be converted to serum PFOA concentrations (Consonni et al. Citation2013; Mastrantonio et al. Citation2018; Raleigh et al. Citation2014). Although based on relatively small numbers of cases, one of these three excluded studies (Raleigh et al. Citation2014) appears to have a slightly negative dose-response, which could have decreased the meta-analysis dose-response estimate if included. On the other hand, another excluded study with a similar number of cases (Consonni et al. Citation2013) showed a strong trend of increasing dose-response, potentially balancing the effects of exclusion to some extent. Because of the overlap in cases in the C8 Science Panel studies, we also excluded the smaller two of these studies in an additional meta-analysis of kidney cancer per 10 ng/mL increase in serum PFOA, which produced slightly larger rate ratios than shown in (1.07 for fixed effects model and 1.33 for the random effects model), without changing the statistical significance of either result.
R code for all of our calculations is included in the appendix.
Discussion
Evaluation using the modified Hill’s criteria indicates that PFOA is a likely cause of both kidney cancer and testicular cancer in humans. Although the evidence for kidney cancer is stronger, sufficient evidence from a limited number of human studies, supported by evidence in animal studies and considerations of key characteristics of carcinogens, also implicate PFOA as a likely cause of testicular cancer.
Because the associations with cancer are quite strong in many of these studies, two of which investigated PFOA exposures resulting from essentially a natural experiment, it is unlikely that the pattern of results can be explained by unmeasured or unidentified confounders. However, if such a risk factor is identified and shown to be strongly associated with PFOA exposure in the general population, natural experiment, and occupational settings included in these studies, it could overturn our interpretation of these associations.
Several of the studies reviewed here rely on exposure models that estimate the serum concentrations and cumulative serum concentrations for each participant over time. Although a variety of pharmacokinetic models have been proposed to characterize the relationship between PFOA exposures and serum concentrations, one-compartment pharmacokinetic models are widely used in the scientific literature (Norman Citation1992), and have been applied successfully for PFOA in humans by a variety of investigators (Olsen et al. Citation2007; Bartell et al. Citation2010; Lorber and Egeghy Citation2011; Shin et al. Citation2011b; Bartell Citation2017; Post et al. Citation2017; Kotlarz et al. Citation2020).
The one-compartment pharmacokinetic models in the Steenland and Woskie (Citation2012), Vieira et al. (Citation2013), and Barry, Winquist, and Steenland (Citation2013) studies used an elimination half-life of 3.5 years (Olsen et al. Citation2007). More recent models for PFOA (Bartell Citation2017; Lu and Bartell Citation2019) use an elimination half-life of 2.3 years (Bartell et al. Citation2010). The half-life value of 2.3 years was obtained by observing the rate of decline in serum PFOA among 200 participants from the C8 Science Panel studies, after water treatment plants were upgraded to remove PFOA from the water supply (Bartell et al. Citation2010). It is consistent with most other recent estimates of the half–life for PFOA in humans, which largely fall in the 2-3 year range (Gomis et al. Citation2016; Li et al. Citation2019; Russell, Waterland, and Wong Citation2015; Zhang et al. Citation2013). Although exposure reconstruction models using 2.3 vs. 3.5 years for the half-life produced highly correlated serum concentrations among participants with PFOA–contaminated water, and were both well correlated with measured 2015-2016 serum concentrations (Shin et al. Citation2011b), changing the half-life from 3.5 years to 2.3 years implies slightly faster excretion and would produce lower cumulative serum concentrations than originally reported in these cancer studies, and therefore slightly higher risk estimates per unit serum PFOA concentration than implied by our dose-response analyses.
Although future studies might address specific mechanisms of carcinogenicity in animal experiments and their relevance to humans, we find it difficult to attribute consistent observations of increased cancer risk in humans exposed to PFOA to chance, bias, or confounding, especially for kidney cancer, and additional mechanistic information is unlikely to refute these findings. Additional investigations could include longer-term follow up to identify additional cancer cases for previously studied populations, which we and others have proposed for the C8 Science Panel studies, as well as new studies using large cohorts or targeted and well powered case-control studies with pre-diagnostic serum samples or other pre-diagnostic exposure characterization. Notably, because several large studies are already available and included in our analyses, additional studies using small numbers of cases are unlikely to affect these findings substantially.
In summary, our meta-analysis showed an average increase in cancer risk per 10 ng/mL increase in serum PFOA for kidney and testicular cancers. These associations are most likely causal, but results are limited by the small number of studies for testicular cancer, the overlapping study populations for several studies, and the lack of measured or modeled serum PFOA concentrations for several studies. The weight of evidence could be even stronger with the addition of future studies conducted in large cohorts.
Supplemental Material
Download PDF (69.7 KB)Acknowledgments
The authors thank Yachen Zhu at the University of California, Irvine, for providing example R code that was helpful in producing our figures.
Disclosure statement
SMB serves as an expert witness for plaintiffs in two PFAS medical monitoring lawsuits, and receives compensation for those services; the terms of this arrangement were reviewed and approved by the University of California Irvine in accordance with its conflict of interest policies. SMB and VMV were members of the C8 Science Panel study team that produced several of the health studies reviewed here. The C8 Science Panel research was funded by the C8 Class Action Settlement Agreement (Circuit Court of Wood County, WV) between DuPont and plaintiffs, with funds administered by an agency that reported to the court.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
Notes on contributors
Scott M. Bartell
Scott M. Bartell is Professor of Environmental and Occupational Health and Statistics at the University of California, Irvine (UCI). His research interest is environmental health methodology, with applications in environmental epidemiology, exposure science, and risk assessment. Since 2006, much of his research has focused on per- and polyfluoroalkyl substances (PFAS), including linkage of fate and transport models and a pharmacokinetic model for exposure reconstruction and epidemiological analyses in the C8 Studies, development of Bayesian statistical methods for biomarker-based pharmacokinetic calibration in exposure reconstruction, and assessment of the potential impacts of exposure measurement error on previous epidemiological findings for PFAS. Dr. Bartell currently serves as Principal Investigator for the UCI PFAS Health Study, the California site in the CDC/ATSDR Multi-Site PFAS Study.
Verónica M. Vieira
Verónica M. Vieira is Professor and Interim Chair of the Department of Environmental and Occupational Health at the University of California, Irvine. Her research involves spatiotemporal analyses of health data for examining the contributions of known risk factors and environmental exposures to the underlying geographic pattern of disease risk including cancer and birth outcomes. She works extensively with reconstructing historic environmental exposures using GIS and has an extensive knowledge of groundwater modeling and perfluorooctanoic acid (PFOA).
References
- 3M. 3M’s commitment to PFAS stewardship. Accessed March 1, 2021. https://www.3m.com/3M/en_US/pfas-stewardship-us/pfas-history/
- Agency for Toxic Substances and Disease Registry (ATSDR). 2018. Toxicological profile for perfluoroalkyls. (Draft for public comment). Atlanta, GA, U.S.: Department of Health and Human Services, Public Health Service.
- American Cancer Society. 2020b.Key statistics for testicular cancer. January 8. Accessed January 18, 2021. https://www.cancer.org/cancer/testicular-cancer/about/key-statistics.html
- American Cancer Society (ACS). 2020a. Key statistics about kidney cancer. American Cancer Society, January 8. Accessed January 18, 2021. https://www.cancer.org/cancer/kidney-cancer/about/key-statistics.html
- Andrews, D. 2018. Report: Up to 110 million Americans could have PFAS-contaminated drinking water. Environmental Working Group, May 22. Accessed March 1, 2021. https://www.ewg.org/research/report-110-million-americans-could-have-pfas-contaminated-drinking-water
- Australia Expert Health Panel for PFAS. 2018. Australian government, department of health. Combined Report, May 7. https://www1.health.gov.au/internet/main/publishing.nsf/Content/ohp-pfas-expert-panel.htm
- Avanasi, R., H. M. Shin, V. M. Vieira, and S. M. Bartell. 2016. Variability and epistemic uncertainty in water ingestion rates and pharmacokinetic parameters, and impact on the association between perfluorooctanoate and preeclampsia in the C8 health project population. Environ. Res. 146:299–307. doi:10.1016/j.envres.2016.01.011.
- Barry, V., A. Winquist, and K. Steenland. 2013. Perfluorooctanoic acid (PFOA) exposures and incident cancers among adults living near a chemical plant. Environ. Health Perspect. 121:1313–18. doi:10.1289/ehp.1306615.
- Bartell, S. M. 2017. Online serum PFOA calculator for adults. Environ. Health Perspect. 125:104502. doi:10.1289/EHP2820.
- Bartell, S. M. 2019. Understanding and mitigating the replication crisis, for environmental epidemiologists. Curr. Environ. Health Rep. 6:8–15. doi:10.1007/s40572-019-0225-4.
- Bartell, S. M., A. M. Calafat, C. Lyu, K. Kato, P. B. Ryan, and K. Steenland. 2010. Rate of decline in serum PFOA concentrations after granular activated carbon filtration at two public water systems in Ohio and West Virginia. Environ. Health Perspect. 118 (2):222–28. doi:10.1289/ehp.0901252.
- C8 Science Panel. 2012. C8 science panel probable link reports [website]. updated October 29. Accessed January 18, 2021. http://www.c8sciencepanel.org/prob_link.html
- Chandler, J., M. Cumpston, J. Thomas, J. P. T. Higgins, J. J. Deeks, M. J. Clarke, and I. Chapter. 2020. Introduction. In ed.. J. P. T. Higgins, J. Thomas, J. Chandler, M. Cumpston, T. Li, M. J. Page, and V. A. Welch, Cochrane handbook for systematic reviews of interventions version 6.1. Cochrane. The Cochrane Collaboration. Accessed September 2020. www.training.cochrane.org/handbook
- Chang, E. T., H. O. Adami, P. Boffetta, P. Cole, T. B. Starr, and J. S. Mandel. May 2014. A critical review of perfluorooctanoate and perfluorooctanesulfonate exposure and cancer risk in humans. Crit. Rev. Toxicol. 44(Suppl 1):1–81. doi: 10.3109/10408444.2014.905767.
- Consonni, D., K. Straif, J. M. Symons, J. A. Tomenson, L. G. P. M. van Amelsvoort, A. Sleeuwenhoek, J. W. Cherrie, P. Bonetti, I. Colombo, D. G. Farrar, et al. 2013. Cancer risk among Tetrafluoroethylene synthesis and polymerization workers. Am. J. Epidemiol. 178(3):350–58. doi:10.1093/aje/kws588.
- de Silva, A. O., J. M. Armitage, T. A. Bruton, C. Dassuncao, W. Heiger–Bernays, X. C. Hu, A. Kärrman, B. Kelly, C. Ng, A. Robuck, et al. 2021. PFAS exposure pathways for humans and wildlife: A synthesis of current knowledge and key gaps in understanding. Environ. Toxicol. Chem. 40:631–57. doi:10.1002/etc.4935.
- European Food Safety Authority (EFSA), Panel on Contaminants in the Food Chain. 2018. Scientific opinion on the risk to human health related to the presence of perfluorooctane sulfonic acid and perfluorooctanoic acid in food. Efsa J. 16(12):5194.
- Frisbee, S. J., A. P. Brooks Jr, A. Maher, P. Flensborg, S. Arnold, T. Fletcher, K. Steenland, A. Shankar, S. S. Knox, C. Pollard, et al. 2009. The C8 health project: Design, methods, and participants. Environ. Health Perspect. December 117(12):1873–82. doi: 10.1289/ehp.0800379.
- Gilliland, F. D., and J. S. Mandel. September 1993. Mortality among employees of a perfluorooctanoic acid production plant. J. Occup. Environ. Med. 35(9):950–54. doi: 10.1097/00043764-199309000-00020.
- Gomis, M. I., R. Vestergren, H. Nilsson, and I. Cousins. 2016. Contribution of direct and indirect exposure to human serum concentrations of perfluorooctanoic acid in an occupationally exposed group of ski waxers. Environ. Sci. Technol. 50:7037–46. doi:10.1021/acs.est.6b01477.
- Haug, L. S., S. Huber, G. Becher, and C. Thomsen. 2011. Characterisation of human exposure pathways to perfluorinated compounds — Comparing exposure estimates with biomarkers of exposure. Environ. Int. 37:687–93. doi:10.1016/j.envint.2011.01.011.
- Hernán, M. A., S. Hernández-Díaz, M. M. Werler, and A. A. Mitchell. 2002. Causal knowledge as a prerequisite for confounding evaluation: An application to birth defects epidemiology. Am. J. Epidemiol. 155:176–84. doi:10.1093/aje/155.2.176.
- Hertz-Picciotto, I. 1995. Epidemiology and quantitative risk assessment: A bridge from science to policy. Am. J. Public Health 85 (4):484–91. doi:10.2105/ajph.85.4.484.
- Higgins, J. P. T., and S. G. Thompson. 2002. Quantifying heterogeneity in a meta-analysis. Stat. Med. 21 (11):1539–58. doi:10.1002/sim.1186.
- Hill, A. B. 1965. The environment and disease: Association or causation? Proc. R. Soc. Med. 58:295–300.
- Hu, X. C., D. Q. Andrews, A. B. Lindstrom, T. A. Bruton, L. A. Schaider, P. Grandjean, R. Lohmann, C. C. Carignan, A. Blum, S. A. Balan, et al. 2016. Detection of poly- and perfluoroalkyl substances (PFASs) in U.S. Drinking water linked to industrial sites, military fire training areas, and wastewater treatment plants. Environ. Sci. Technol. Lett. 3:344–50. doi:10.1021/acs.estlett.6b00260.
- International Agency for Research on Cancer (IARC) Working Group. 2016. IARC monographs on the evaluation of carcinogenic risks to humans. Vol. 110. Lyon, France: IARC. Some Chemicals Used as Solvents and in Polymer Manufacture.
- Interstate Technology and Regulatory Council (ITRC). PFAS technical and regulatory guidance document and fact sheets PFAS-1. Washington, DC: ITRC PFAS Team. Accessed March 1, 2021. https://pfas-1.itrcweb.org
- Kim, S. B., S. M. Bartell, and D. L. Gillen. 2015. Estimation of a benchmark dose in the presence or absence of hormesis using posterior averaging. Risk Anal. 35 (3):396–408. doi:10.1111/risa.12294.
- Kotlarz, N., J. McCord, D. Collier, C. S. Lea, M. Strynar, A. B. Lindstrom, A. A. Wilkie, J. Y. Islam, K. Matney, P. Tarte, et al. 2020. Measurement of novel, drinking water-associated PFAS in blood from adults and children in Wilmington, North Carolina. Environ. Health Perspect. July 128(7):77005. doi: 10.1289/EHP6837.
- Leonard, R. C., K. H. Kreckmann, C. J. Sakr, and J. M. Symons. 2008. Retrospective cohort mortality study of workers in a polymer production plant including a reference population of regional workers. Ann. Epidemiol. 18 (1):15–22. doi:10.1016/j.annepidem.2007.06.011.
- Li, Y., Y. Xu, K. Scott, C. Lindh, K. Jakobsson, and T. Fletcher. 2019. Half-lives of PFOA, PFPeS, PFHxS, PFHpS and PFOS after end of exposure to contaminated drinking water. Environ. Epidemiol. 3:237. doi:10.1097/01.EE9.0000608476.06577.16.
- Lorber, M., and P. P. Egeghy. 2011. Simple intake and pharmacokinetic modeling to characterize exposure of Americans to perfluoroctanoic acid, PFOA. Environ. Sci. Technol. October 1. 4519: 8006–14.doi: 10.1021/es103718h
- Lu, S., and S. M. Bartell 2019. Serum PFAS calculator for adults, version 1.1. Accessed May 25, 2020. www.ics.uci.edu/~sbartell/pfascalc.html
- Mastrantonio, M., E. Bai, R. Uccelli, V. Cordiano, A. Screpanti, and P. Crosignani. 2018. Drinking water contamination from perfluoroalkyl substances (PFAS): An ecological mortality study in the Veneto region, Italy. Eur. J. Public Health 28 (1):180–85. doi:10.1093/eurpub/ckx066.
- Michigan PFAS Action Response Team. 2021. PFAS response/investigations: Rockford Tannery, Rockford, Kent County. Michigan Department of Environment, Great Lakes, and Energy. February 22. Lansing, Michigan. Accessed March 1, 2021. https://www.michigan.gov/pfasresponse/0,9038,7-65-86511_82704-488796–,00.html.
- Munafò, M. R., and G. Davey Smith. 2018. Robust research needs many lines of evidence. Nature January 25. 5537689: 399–401.doi: 10.1038/d41586-018-01023-3
- National Toxicology Program (NTP). Technical report on the toxicology and carcinogenesis studies of perfluorooctanoic acid (CASRN 335-67-1) administered in feed to Sprague Dawley Rats. Technical Report 598, North Carolina, USA: National Toxicology Program, Public Health Service, U.S. Department of Health and Human Services, Research Triangle Park. May 2020.
- New Hampshire Department of Health and Human Services. 2018. Cancer Incidence Report: Merrimack, NH. Division of Public Health Services, January. Accessed April 21, 2021. https://www.dhhs.nh.gov/dphs/pfcs/documents/merrimack-cancer-012018.pdf.
- Norman, J. 1992. One-compartment kinetics. Br. J. Anaesth. 69:387–96. doi:10.1093/bja/69.4.387.
- Olsen, G. W., J. M. Burris, D. J. Ehresman, J. W. Froehlich, A. M. Seacat, J. L. Butenhoff, L. R. Zobel. 2007. Half-life of serum elimination of perfluorooctanesulfonate, perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environ. Health Perspect. 115(9):1298–305. doi:10.1289/ehp.10009.
- Post, G. B., J. A. Gleason, K. R. Cooper, and L. S. Birnbaum. 2017. Key scientific issues in developing drinking water guidelines for perfluoroalkyl acids: Contaminants of emerging concern. PLoS Biol. December 20. 1512: e2002855. doi: 10.1371/journal.pbio.2002855
- Raleigh, K. K., B. H. Alexander, G. W. Olsen, G. Ramachandran, S. Z. Morey, T. R. Church, P. W. Logan, L. L. F. Scott, E. M. Allen. 2014. Mortality and cancer incidence in ammonium perfluorooctanoate production workers. Occup. Environ. Med. 71:500–06. doi:10.1136/oemed-2014-102109.
- Rothman, K. J., and S. Greenland. 2005. July 1. Causation and causal inference in epidemiology. Am. J. Public Health 95S1: S144–S150. doi: 10.2105/AJPH.2004.059204
- Russell, M. H., R. L. Waterland, and F. Wong. 2015. Calculation of chemical elimination half-life from blood with an ongoing exposure source: The example of perfluorooctanoic acid (PFOA). Chemosphere 129:210–16. doi:10.1016/j.chemosphere.2014.07.061.
- Samet, J. M., S. Bartell, L. Bero, A. Bostrom, K. Dickersin, D. C. Dorman, D. L. Eaton. 2014. Review of EPA’s integrated risk information system (IRIS) process. Washington, D.C.: National Academies Press.
- Shearer, J. J., C. L. Callahan, A. M. Calafat, W. Y. Huang, R. R. Jones, V. S. Sabbisetti, N. D. Freedman, J. N. Sampson, D. T. Silverman, M. P. Purdue. et al. Serum concentrations of per- and polyfluoroalkyl substances and risk of renal cell carcinoma. JNCI 2020. djaa143, doi:10.1093/jnci/djaa143. corrected proof.
- Shin, H. M., V. M. Vieira, P. B. Ryan, K. Steenland, and S. M. Bartell. 2011b. Retrospective exposure estimation and predicted versus observed serum perfluorooctanoic acid concentrations for participants in the C8 health project. Environ. Health Perspect. 119 (12):1760–65. doi:10.1289/ehp.1103729.
- Shin, H. M., V. M. Vieira, P. B. Ryan, R. Detwiler, B. Sanders, K. Steenland, and S. M. Bartell. 2011a. Environmental fate and transport modeling for perfluorooctanoic acid emitted from the Washington works facility in West Virginia. Environ. Sci. Technol. 45 (4):1435–42. doi:10.1021/es102769t.
- Steenland, K., and A. Winquist. 2021. PFAS and cancer, a scoping review of the epidemiologic evidence. Environ. Res. 194:110690. doi:10.1016/j.envres.2020.110690.
- Steenland, K., and S. Woskie. 2012. Cohort mortality study of workers exposed to perfluorooctanoic acid. Am. J. Epidemiol. November 15. 17610: 909–17.doi: 10.1093/aje/kws171
- Steenland, K., T. Fletcher, C. R. Stein, S. M. Bartell, L. Darrow, M. J. Lopez-Espinosa, P. B. Ryan, and D. A. Savitz. 2020. Commentary: Evolution of evidence on PFOA and health following the assessments of the C8 science panel. Environ. Int. 145:106125. doi:10.1016/j.envint.2020.106125.
- Sterne, J. A. C., M. A. Hernán, A. McAleenan, B. C. Reeves, and J. P. T. Higgins. 2020. Chapter 25: Assessing risk of bias in a non-randomized study. ed.. J. P. T. Higgins, J. Thomas, J. Chandler, M. Cumpston, T. Li, M. J. Page, and V. A. Welch, Cochrane handbook for systematic reviews of interventions version 6.1. Cochrane. The Cochrane Collaboration. Accessed September 2020. www.training.cochrane.org/handbook
- Strynar, M., S. Dagnino, R. McMahen, S. Liang, A. Lindstrom, E. Andersen, L. McMillan, M. Thurman, I. Ferrer, and C. Ball. 2015. Identification of novel perfluoroalkyl ether carboxylic acids (PFECAs) and sulfonic acids (PFESAs) in natural waters using accurate mass time-of-flight mass spectrometry (TOFMS). Environ. Sci. Technol. 49:11622–30. doi:10.1021/acs.est.5b01215.
- Sun, Q., G. Zong, D. Valvi, F. Nielsen, B. Coull, and P. Grandjean. 2018. Plasma concentrations of perfluoroalkyl substances and risk of type 2 diabetes: A prospective investigation among U.S. women. Environ. Health Perspect. 126 (3):037001. doi:10.1289/EHP2619.
- Sunderland, E. M., X. C. Hu, C. Dassuncao, A. K. Tokranov, C. C. Wagner, and J. G. Allen. 2019. A review of the pathways of human exposure to poly- and perfluoroalkyl substances (PFASs) and present understanding of health effects. J. Expo. Sci. Environ. Epidemiol. March 29(2):131–47. doi: 10.1038/s41370-018-0094-1.
- Szklo, M., and F. J. Nieto. 2019. Epidemiology: Beyond the basics. 4th ed. Boston, MA: Jones & Bartlett Learning.
- Temkin, A. M., B. A. Hocevar, D. Q. Andrews, O. V. Naidenko, and L. M. Kamendulis. 2020. Application of the key characteristics of carcinogens to per and polyfluoroalkyl substances. Int. J. Environ. Res. Public Health March 4. 175: 1668.doi: 10.3390/ijerph17051668
- U.S. Environmental Protection Agency. 30 May 2006. Report of the perfluorooctanoic acid review panel. Washington, DC: Science Advisory Board.
- US Environmental Protection Agency. 2016. Health effects support document for perfluorooctanoic acid (PFOA). Office of Water, May. EPA 822-R-16-003.
- van Reekum, R., D. L. Streiner, and D. K. Conn. 2001. Applying Bradford Hill’s criteria for causation to neuropsychiatry. J. Neuropsychiatry Clin. Neurosci. 13 (3):318–25. doi:10.1176/jnp.13.3.318.
- van Wijngaarden, E., and I. Hertz-Picciotto. 2004. A simple approach to performing quantitative cancer risk assessment using published results from occupational epidemiology studies. Sci. Total Environ. 332:81–87. doi:10.1016/j.scitotenv.2004.04.005.
- Vieira, V. M., K. Hoffman, H. M. Shin, J. M. Weinberg, T. F. Webster, and T. Fletcher. 2013. Perfluorooctanoic acid exposure and cancer outcomes in a contaminated community: A geographic analysis. Environ. Health Perspect. 121:318–23. doi:10.1289/ehp.1205829.
- Weisskopf, M. G., and T. F. Webster. 2017. Trade-offs of personal versus more proxy exposure measures in environmental epidemiology. Epidemiology (Cambridge, Mass.) 28 (5):635–43. doi:10.1097/EDE.0000000000000686.
- Zhang, Y., S. Beesoon, L. Zhu, and J. W. Martin. 2013. Biomonitoring of perfluoroalkyl acids in human urine and estimates of biological half-life. Environ. Sci. Technol. 47 (18):10619–27. doi:10.1021/es401905e.