
ABSTRACT
Lignocellulose constitutes a major component of discarded wastes from various industries viz. agriculture, forestry and municipal waste treatment. The potential use of lignocellulose from such types of biomass can be maximized by enzymatic degradation using glycoside hydrolases (GHs) and oxidative enzymes to produce renewable fuels. Nonetheless, besides the slow rate of degradation and low yields, lignocellulose is also physicochemically recalcitrant and costly to process, further limiting its mass utilization. Therefore, bioprospecting for micro-organisms producing efficient lytic polysaccharide monooxygenases (LPMOs) to overcome these drawbacks may prove beneficial. The use of GHs and LPMOs can potentially help to circumvent some limitations in the conversion of lignocellulosic biomass into fermentable sugars. LPMOs are classified as family GH61 or family 33 carbohydrate-binding module (CBM33), whose unusual surface-exposed active site is bound to a copper (II) ion. To date, there are more than 20 known genes encoding cellulose-active LPMOs in bacteria and fungi, with diverse biological activities. Only by thorough comprehension of the diversity, enzymology and role of primary GHs, i.e. celullases and their oxidative machinery can the degradation of lignocellulosic biomass be improved. This review provides insight into the diversity, structure and mechanisms, structural and functional aspects of the oxidative breakdown of cellulose by LPMOs of the cellulose-active GH family.
Introduction
Lignocellulose offers the largest inexpensive and renewable source of potentially degradable carbohydrate on earth [Citation1,Citation2]. Unfortunately, the full potential of such biomass is normally underutilized and mostly wasted in the form of pre- and post-harvest agricultural losses and wastes from the food-processing industry [Citation3]. According to the review of literature, the available lignocellulosic feedstock yielded from agriculture and other sources amounts to an approximate 180 million tons per year [Citation4]. The lignocellulosic biomass primarily consists of polysaccharide polymers, cellulose and hemicellulose, and the phenolic polymer lignin [Citation5,Citation6]. However, cellulose, being the most abundant constituent, is structurally entrapped by the other cell-wall components, which hinders its enzymatic breakdown [Citation7].
The structural robustness of cellulose can be attributed to the way the biopolymer is organized in large crystals containing tens of thousands of glucose molecules. The complex hydrogen-bonding (H-bonding) network found within and between glucan chains combined with the degree of polymerization involving thousands of glucose monomers further add to the limited accessibility of the glycolytic linkages to the hydrolytic enzymes [Citation8]. Since the cellulose crystals are embedded in (but not covalently linked to) a matrix of hemicellulose and lignin, this renders the access of biodegrading enzymes very difficult. Nevertheless, the recent discoveries of a diverse glycoside hydrolase family of enzymes and their oxidative auxiliaries promise huge potential applications of such enzymes for depolymerization of lignocellulosic biomass for bioconversion into value-added products.
In this review, we focus on the current standing of glycoside hydrolase families, their new class of copper-dependent lytic polysaccharide monooxygenases (LPMOs), highlighting their discovery, protein structure, predicted role and mechanism of catalysis. Their potential applications in crop waste biomass degradation for biofuel production are also discussed.
Enzymatic hydrolysis of cellulosic biomass
Cellulose is a very stable molecule and it has been estimated that the uncatalysed hydrolytic degradation of such component would result in a half-life as long as 5 million years [Citation9,Citation10]. Thus, cellulose-degrading systems mediated by cellulases are necessary for industrial cellulose breakdown as well as to sustain the natural global carbon cycle. Cellulases are a class of enzymes produced mainly by fungi, bacteria and protozoans specifically for the cellulolysis (or hydrolysis) of biomass, i.e. cellulose [Citation11]. As a matter of fact, the role of cellulases in the breakdown of cellulose has been investigated for both complex enzyme mixtures and individual components [Citation12]. So far, the accepted model for enzymatic degradation of cellulose has been based on hydrolytic cellulase enzymes [Citation13]. The revised model of Elwyn et al. [Citation14] has shown that cellulose hydrolysis takes place in three concurrent steps: (1) physical and chemical changes in the yet unhydrolysed solid substrate; (2) primary hydrolysis in which soluble cello-oligomers are released from the solid cellulose to the hydrolysate and (3) secondary hydrolysis, in which the dissolved oligomers are hydrolysed to glucose [Citation15].
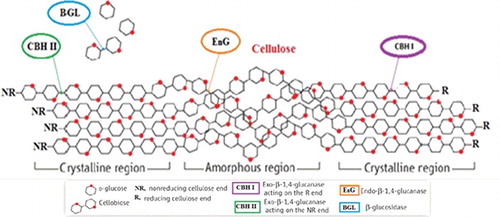
Cellulases are members of the glycoside hydrolase (GH) families of enzymes that catalyse the hydrolysis of β-1,4-glycosidic bonds of cellulose to glucose [Citation13,Citation16]. The cellulose-degrading enzymes are further divided into three major groups: endo-glucanases (EG), exoglucanases (cellobiohydrolases, CBH) and β-glucosidases (BGL), which belong to the EC 3.2.1.X class. They include endo-β-(1,4)-glucanases (EC 3.2.1.4), exo-β-(1,4)-glucanase (EC 3.2.1.91), exo-β-(1,4)-glucosidase (EC 3.2.1.74) and β-glucosidase (EC 3.2.1.21) [Citation17–19] (). depicts the hydrolytic breakdown of cellulose by cellulases. The active sites of cellulases are found as a cleft or tunnel shape lined with aromatic residues whose role is to enhance the release of a glucan chain from cellulose [Citation22,Citation23]. It is hypothesized that the limiting step in the cellulase-catalysed breakdown of crystalline cellulose is the detachment of the glucan chain from the strong H-bonding network in cellulose into the active site grove [Citation24].
Table 1. Cellulases, functions and their corresponding glycosyl hydrolase enzyme families.
Figure 1. Enzymatic cellulose degradation model.
Glycosyl hydrolase enzyme diversity
Glycosyl hydrolases (GH), also known as glycosidases, are a group of enzymes that hydrolyse glycosidic bonds between two or more sugars or a sugar and a non-sugar moiety within carbohydrates or oligosaccharides [Citation25]. The enzymes within this family are widely distributed across prokaryotic, eukaryotic and archaea species [Citation26–30] and have been reported to demonstrate interesting functional diversity as well as variation in copy number among organisms. To date, a total of 115 GH families have been identified based on their modes of action and amino-acid sequence. However, in recent times, due to the availability of more information on the protein structure and functions of such enzymes, it became clear that classification simply based on substrate-specificity was unsuitable. This is in light of the fact that similar protein folds may often exhibit several types of substrate specificities [Citation30]. The recommendation of Cantarel et al. [Citation28] on a classification method based on the effect of protein folding guided by the amino-acid sequence was found more suitable in assigning enzymes into their families and sub-families.
Carbohydrate-binding module
Generally, the use of GHs enzymes alone is inadequate for the breakdown of insoluble polysaccharides owing to difficulties in the enzymes’ access to the specific position of the substrate during catalysis. To overcome this problem, the GH catalytic modules are usually appended to one or more carbohydrate-binding modules (CBMs) capable of degrading insoluble polysaccharides. CBMs are the non-catalytic part of polysaccharide-degrading enzymes, such as cellulases and hemicellulases, that bind to the cell-wall polymers. In fact, the term CBM was suggested as a more inclusive term to designate all of the non-catalytic sugar-binding modules derived from GHs. Although many of these modules target components of the plant cell wall, several CBM families contain proteins that bind to insoluble storage polysaccharides, such as starch and glycogen. The role of CBMs has been known for enhancing the binding of the enzymes to the cellulose substrates [Citation31], an aspect pertinent in efficient catalysis and degradation of unwanted biomass, such as agricultural-based biomass.
According to the review of literature, in recent years, the carbohydrate-active enzymes (CAZy) database groups the CBMs into 81 different families [Citation29,Citation32], based on amino-acid sequence similarity. Similar to the catalytic modules of GHs, 54 of the CBM families have been classified into Types A, B and C. CBMs of Type A, perhaps, are the most distinct among the CBMs, as this class only specifically binds to the surface of insoluble highly crystalline polysaccharides, such as cellulose and/or chitin [Citation33]. The Type B CBMs function to identify internal glycan chains (endo-type) and interact only with single polysaccharide chains that bind to polysaccharides which constitute the substrates for the cognate catalytic module of the enzyme. Examples of enzymes appended to Type B CBMs are cellulases, xylanases and mannanases. In contrast, Type C CBMs link to the termini of glycans (exo-type). This unique class of CBMs has a ‘lectin-like’ feature that binds optimally to mono-, di- or tri-saccharides [Citation34]. This is in agreement with their ability to recognize small sugars; hence their well-known ‘small-sugar-binding’ capacity, unlike those seen in Type B CPMs, which exhibit higher binding affinity towards longer oligosaccharide ligands [Citation32,Citation35].
CBMs and glycoside hydrolase linkers
Thorough comprehension of the structural biology by which CBMs bind to their target ligands may provide invaluable insights into the mechanisms of carbohydrate–protein recognition. Currently, there are several mechanisms that elucidate the ligand-binding between CBMs, enzymes as well as linkers. An earlier mechanism proposed by Creagh et al. [Citation36] described that such ligand-binding involves entropically driven expulsion of water molecules from hydrophobic surfaces of cellulose and protein, typically shown by the binding between a CBM33 from the β-1,4-exoglucanase Cex of Cellulomonas fimi and insoluble bacterial microcrystalline cellulose. However, the molecular basis of the thermodynamically driven forces to explain the protein–carbohydrate-binding remains highly contentious.
Another mechanism, on the other hand, proposed that the cooperative-binding between the CBM and hydrolase was essential for carbohydrate recognition. An example can be seen in the modular enzyme Xyn10B from Clostridium thermocellum consisting of an N-terminal family 22 CBM (CBM22-1), a family 10 GH catalytic domain (GH10), another CBM22 module (CBM22-2), a dockerin sequence and a C-terminal family 1 carbohydrate esterase (CE1) catalytic domain. Residues from helix H4 of the GH10 module initiate the main contacts by binding into the minor groove of the CBM22-1 module [Citation37]. The CBM22-1 orientates in such a way that permits the substrate to bind loosely and be subsequently conveyed to the active site progressively [Citation37], while another study reported that, by binding to a family 1 CBM, the thermostability of the GH10 xylanase from Talaromyces cellulolytica was improved [Citation38]. It has been suggested that the family 10 CBM in the C-terminus of thermostable endo-β-1,4-xylanase (Xyl10A) plays two major roles in the synergistic hydrolysis of lignocellulose by Xyl10A and cellulases. The binding of Xyl10A to family 10 CBM was seen to enhance lignocellulosic xylan hydrolysis via attachment to cellulose, as well as facilitating efficient removal of xylan obstacles that impede cellulase activity (because of the similar binding target of CBM1) [Citation39]. Consequently, the combination of CBM-containing cellulases and xylanases in fungal systems could contribute to reduction of the enzyme loading in the hydrolysis of pretreated lignocellulose [Citation38].
Likewise, CBMs have been found useful in the non-hydrolytic substrate disruption (amorphogenesis) [Citation40] as well as in assisting the amorphogenesis of non-hydrolytic proteins [Citation39]. Interestingly, CBMs can be multi-modulated with several lignocellulose-degrading catalytic domains by tethering to flexible glycosylated linkers [Citation41]. Certain linkers have been shown to substantially improve binding as compared to only CBM. This results in effective binding of the substrate, an aspect that, presumably, improves the enzyme activity [Citation42].
Cellulosomes: multienzyme complexes
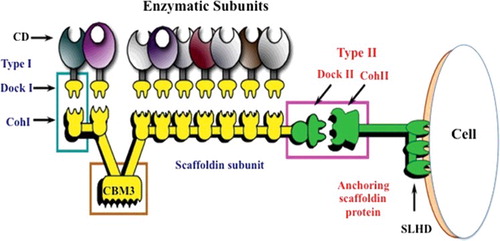
Bioprospecting for effective cellulose degrading micro-organisms led to the discovery of cellulosomes in the early 1980s. Cellulosomes are multienzyme complexes [Citation43] produced mostly by anaerobic bacteria and fungi such as Clostridium and Ruminococcus spp., and Chytridomycetes [Citation44], respectively. It is apparent that these proteins, or nanomachines [Citation45], are important for efficient degradation of cellulose and hemicellulose [Citation46]. The architecture of cellulosomes includes enzyme subunits such as endoglucanases, xylanases and cellobiohydrolases. These enzymes display varying substrate specificities and catalytic mechanisms coordinated by the scaffoldin protein.
Apart from the catalytic module, the dockerin module modulates enzyme interaction alongside the scaffoldin protein. Scaffoldins are multidomain and multifunctional proteins that boost interactions between catalytic proteins with the GH dockerin domains, consequently improving the affinity of the enzyme complex and its catalytic efficiency via CBMs [Citation47] (). The cohesion module sited on the scaffoldin binds to a dockerin module on each enzymatic subunit [Citation48,Citation49]. This architecture enables synergistic and well-coordinated enzymatic interactions, making cellulosomes about the most adequate biochemical system for the degradation of cellulose [Citation20]. The past decade has seen artificial cellulosomes being constructed for the sole purpose of improving enzymatic functions for the saccharification reaction [Citation50]. The construction was carried out via a chemical approach whereby a multicellulase conjugate was assembled on a double-stranded DNA scaffoldin. The resulting complex DNA–(endoglucanase)n conjugate exhibits a unique hydrolytic activity on crystalline cellulose (Avicel). The activity of the enzyme conjugate is dependent on the cellulase/DNA ratio of the DNA-based artificial cellulosome [Citation51].
Figure 2. General schematic diagram of the cellulosome and structural units.
Reaction steps in enzymatic lignocellulosic breakdown
Enzymatic hydrolysis that converts lignocellulosic biomass to fermentable sugars involves complex steps. It has been extensively described that the enzymatic hydrolysis process is influenced by both the structural features of cellulose and the mode of enzyme action. Complete degradation of lignocellulosic biomass generally involves different sets of hydrolytic enzymes, such as cellulases, hemicellulases and other accessory enzymes [Citation52]. To improve the lignocellulosic biomass breakdown for maximum biomass conversion, research efforts have been directed mainly on substrate-related factors, which include crystallinity, degree of polymerization, accessibility, preparation and properties of model substrates, and pretreated lignocellulosic materials [Citation53].
Substrate-related factors
Structurally, the cellulose polymer is highly heterogeneous, which is why the hydrolysis of this biopolymer by cellulases incurs a highly complex process. Hence, establishing rational models based on mechanistic steps can be rather problematic due to the inherent complexities in both the substrate and enzyme [Citation54,Citation55]. The structure of cellulose consists of sugar rings in different chains, aligned on the same plane interacting with other layers of cellulose chains, forming the intra- and inter-chain hydrogen bond network [Citation56] that gives cellulose its highly organized and stable, tightly packed structure, i.e. ‘crystalline’ [Citation57]. Additionally, cellulose components include amorphous regions at varying sizes and accessibility as well as degradation rates. While the above-mentioned factors may differ from one carbohydrate source to another, the structurally relevant parameters, viz. chain length, crystallinity and number of accessible binding sites, can change with the progression of degradation [Citation54,Citation55].
It has been described that the efficiency of enzymatic hydrolysis of lignocellulose is affected by: (1) accessibility, (2) availability of surface area, (3) crystallinity, (4) degree of polymerization, (5) lignin and hemicellulose content, (6) changes in feature during degradation and, finally, (7) the pretreatment process [Citation15]. Aside from adsorption, other factors such as diffusion, desorption and unproductive binding of different enzymes on different heterogeneous substrates should also be considered [Citation58–60].
Enzyme-related factors
The exploitation of microbial cellulases for agricultural and industrial processes has been reported since the 1990s [Citation21]. In general, cellulases are comprised of a mixture of several enzymes, three being most common in the hydrolysis process: (1) endoglucanase (EG, endo-1,4-D-glucanohydrolase, or EC 3.2.1.4.), which attacks regions of low crystallinity in the cellulose fibre, creating free chain-ends; (2) exoglucanase, or cellobiohydrolase (CBH, 1,4-b-D-glucan cellobiohydrolase, or EC 3.2.1.91.), which degrades the molecule further by removing cellobiose units from the free chain-ends and (3) β-glucosidase (EC 3.2.1.21), which hydrolyses cellobiose to produce glucose as the substrate for a myriad of manufacturing processes [Citation61]. Studies that focus on bioprospecting for new micro-organisms have highlighted that both bacteria and fungi are good producers of cellulases, which includes Clostridium, Cellulomonas, Bacillus, Thermomonospora, Ruminococcus, Bacteriodes, Erwinia, Acetovibrio, Microbispora, Streptomyces, Cellulomonas fimi and Thermomonospora fusca, as well as the cellulolytic anaerobes such as C. thermocellum and Bacteroides cellulosolvens [Citation62].
However, depending on the type of micro-organisms producing cellulases, the two most common cellulase systems are: (1) non-complexed cellulase system (usually associated with aerobic bacteria and fungi) and (2) complexed cellulase system (usually associated with anaerobic micro-organisms) [Citation63]. The cellulases produced by the genus Trichoderma have received intensive attention due to the high levels of secreted cellulase; thus, the most fully investigated non-complexed cellulase system is the Trichoderma reesei model. Trichoderma reesei (teleomorph Hypocrea jecorina) is a saprobic fungus, known as an efficient producer of extracellular enzymes [Citation64], which includes two cellobiohydrolases, at least seven endoglucanases and several glucosidases. The three hydrolytic processes to degrade cellulose carried out by T. reesei occur simultaneously and ultimately produce glucose as the final product [Citation41]. On the other hand, the production of cellulosomes is usually associated with anaerobic bacteria (complexed cellulose system), which give some advantages: (1) synergism of the cellulases; (2) absence of unspecific adsorption [Citation58].
Enzymatic lignocellulosic breakdown
Enzyme diffusion into (void of) solid substrates has been investigated and modelled, and the premise on which the model [Citation59] was formed has been solved. The solved conditions include that (1) the enzymatic hydrolysis of lignocellulose depends on particle size tunable to a preferred size by pretreatment as well as (2) dramatic decrease in reaction rate after the initial burst. Thus, the influence of diffusion is downplayed.
The adsorption of cellulases is mediated by CBMs, type-specific for crystalline or amorphous regions only. Crystalline-specific CBMs are inclined to clustering on ridges, linear regions of glucan chains aligned parallel to one another. An example is the Trichoderma reesei cellobiohydrolase I (TrCBHI) that binds preferentially to the hydrophobic (‘planar’) face of crystalline cellulose microfibrils [Citation65]. The CBHI-binding sites are limited on crystalline cellulose due to the tight packing of chains as well as inaccessible chain-ends buried within the crystals. In contrast, amorphous-specific CBMs bind more uniformly across the surface, binding tightly to exposed glycan chains [Citation65]. Likewise, endoglucanases bind specifically to more amorphous regions [Citation54,Citation55] of cellulose.
GH families involved in lignocellulosic biomass degradation
A diverse number of GH families contain the majority of enzymes that can catalyse LGC biomass degradation. shows the cellulase enzymes and their respective GH families. Some of the largest groups are discussed below.
GH family 3
The GH family 3 (GH3) is one of the most abundant ones in the CAZy database, comprising over 6000 enzymes extensively distributed in plants, fungi and bacteria. The family exhibits various activities such as exo-acting β-D-glucosidases, α-L-arabinofuranosidases, β-D-xyloparanosidases and N-acetyl-β-D-glucosaminidases [Citation66], all of which use a retaining glycosidase mechanism [Citation20,Citation67–69]. In addition to hydrolytic activities, some GH3 enzymes can catalyse glycosidic bond formation either via thermodynamically controlled reverse hydrolysis, or kinetically controlled transglycosylation [Citation70,Citation71]. In all, GH3 enzymes catalyse a range of functions, including cellulosic biomass degradation, plant and bacterial cell-wall remodelling, energy metabolism and pathogen defence [Citation67,Citation72]. These enzymes also have important roles in many other biological processes such as synthesis of functional glycosides from glycoside precursors [Citation73] and cyanide-based biological defence mechanisms in plants [Citation74].
With regards to substrate specificities, the GH3 family has extensive substrate-specificity with respect to monosaccharide residues, linkage position and chain length of the substrate. GH3 β-D-glucan glucohydrolases are also broadly specific exohydrolases that remove single glucosyl residues from the non-reducing ends of a range of β-D-glucans, (1,6)-β-D-glucosides and aryl β-D-glucosides. They include 1,3-β-D-glucans, 1,4-β-D-glucans, (1,3:1,4)-β-D-glucans and (1,6)-β-D-glucans, 4-nitrophenyl-β-D-glucoside, certain cyanogenic β-D-glucosides and some β-D-oligoxylglucosides [Citation75]. In contrast to the broad substrate-specificity of the GH3 enzymes described above, there are exceptions such as the GH3 N-acetyl-β-D-glucosamine (GlcNAc) of Cellulomonas fimi Nag3 [Citation76,Citation77].
GH3 glycoside hydrolases act via a classical Koshland double-displacement mechanism, in which glycosyl residues are singly removed from the non-reducing ends of their substrates [Citation78]. For several enzymes, the released glycose has been experimentally shown to retain its anomeric configuration. The active site of a GH3 enzyme has two glucosyl-binding subsites, designated as −1 and +1; the junction of these two subsites is the location where the enzymic nucleophile and general acid/base residue are found [Citation79]. Detailed studies to further comprehend the kinetics and mechanism of the GH3 enzymes have been carried out encompassing the β-glucosidases from the Gram-negative bacteria Thermotoga neopolitana [Citation80], GH3 glucosylceramidase from the Gram-positive bacteria Paenibacillus sp. TS12 [Citation81] and fungi, Aspergillus wentii [Citation82], Flavobacterium meningosepticum [Citation83] and Aspergillus niger [Citation84,Citation85]. The kinetics of the ‘bifunctional’ β-D-glucan glucohydrolases and α-L-arabinofuranosidase/β-D-xylopyranosidases from barley have been described [Citation86]. Similarly, kinetic and mechanistic analyses of N-acetyl-β-D-glucosaminidases from Gram-negative microbes such as Salmonella typhimerium [Citation87], Vibrio cholerea [Citation88] and Vibrio furnisii [Citation89] as well as the Gram-positive Bacillus subtilis [Citation90] also corroborate reports describing the catalytic nucleophile of the GH3 enzymes as being conserved, while the location and identity of the general acid/base residue as non-conserved.
GH family 5
GH family 5 (GH5), formerly known as cellulase family A, is a huge GH family that belongs to Clan GH-A. This family includes an array of enzymes found only in prokaryotes, eukaryotes and viruses, but not in humans. In fact, more than 3000 GHs enzyme sequences have been successfully identified in the CAZy database. The current 51 sub-families were grouped from 80% of the known sequences (GH5-1 to GH5-53), excluding GH5-3 and GH5-6, which have been merged into GH5-4 and GH5-5, respectively [Citation91].
To date, there are 51 reported GH5 three-dimensional structures. The enzymes of GH5 consist of an amino-acid chain which forms a (β/α)8 fold, creating an open groove surrounding a conserved active site which harbours the catalytic nucleophile Glu and acid/base Glu at the C-terminus of β-strand 7 and β-strand 4, respectively. During catalysis, the carbohydrate substrate binds to the substrate-binding site from the non-reducing end (−subsites) to the reducing end (+ subsites). Typically, the GH5 enzymes have a conserved amino-acid residue (glutamic acid), which is also the catalytic residue [Citation92].
GH family 9
It is the second largest cellulase family and encompasses mainly endoglucanases with a small number of processive ones [Citation92]. Pertinently, the processive endoglucanases all contain a CBM of the 3C family naturally strongly attached to the C-terminus of the catalytic domain [Citation93]. The cellulases in the GH family 9 (GH9) are mainly divided into two major sub-groups, namely, the EI and EII. The former contains only bacterial cellulases, of both aerobes and anaerobes, while the latter comprises cellulases of bacterial and non-bacterial origin [Citation94]. All common plant cellulases are grouped under GH9, while the remaining members include cellulases that are of eubacterial, archaeal, arthropod, Echinodermata, earthworm, chordate and mollusk origin. Characteristically, the GH9 endoglucanases display appreciable activity on soluble cellulose derivatives such as carboxymethylcellulose, plant polysaccharides and phosphoric acid swollen non-crystalline cellulose, but little or no activity on crystalline cellulose [Citation95,Citation96].
The mechanism by which the GH9 enzymes catalysis is achieved is by the inversion of anomeric stereochemistry [Citation92]. The catalysis is mediated by three amino acids that form the catalytic triad of the GH9 enzymes, consisting of a conserved Glu residue as the general catalytic acid and two Asp residues. One of the Asp residues, E424, functions to bind the catalytic water, while two Glu residues, D55 and D58, act as the general catalytic bases and a Tyr residue, Y318, binds the crystalline cellulose substrates. Mutation studies confirm that the conserved Glu to Gly, Ala or Gln residues are essential in carbohydrate hydrolysis [Citation97]. The activity of the mutant enzyme is reduced to less than 0.5% of the wild-type (WT) [Citation97], whereas mutation of the Asp that binds the catalytic water to Asn or Ala results in reduction in enzyme activity by less than 2% of WT on all cellulose substrates [Citation98]. It is known that all catalytic domain structures of GH9 have an (a/b)6 barrel fold that features an open active site groove consisting of at least six sugar-binding subsites −4 to +2 [Citation99].
Lytic polysaccharide monooxygenase (LPMO)
Classical cellulases form a major part of the enzymes in different glycoside hydrolase (GH) families, which hydrolyse the glycosidic bonds in glucose polymers. However, the conventional hydrolytic model of cellulose depolymerization has been challenged for the past few years. There have been questions on the occurrence and functional relevance of a novel class of glycolytic oxidative enzymes of both fungal and bacterial origin [Citation100,Citation101]. Several studies have shown that the secreted enzymes are capable of catalysing the cleavage of glycosidic bonds of glucose polymers through oxidative mechanism instead of the hydrolytic route [Citation102–104]. These enzymes have since been reclassified as LPMOs [Citation105], as their oxidation reaction for cleaving the glycosidic bonds is copper-dependent, producing oxidized chain-ends of either aldonolactone or a 4-ketoaldose [Citation106–109]. The requirements for such reactions include molecular oxygen and an extracellular electron source, which could be supplied by cellobiose dehydrogenase (CDH) or small molecule reductants present in the lignocellulosic biomass. This fundamentally unique mechanism of cellulose chain-cleavage is assumed to circumvent the energetically difficult removal of glucan chain from highly crystalline cellulose, thereby creating new accessible ends for exoglucanase action. The oxidized carbon position in the glycan chain can also vary, as some LPMOs solely act at positions C1 or C4, and a third group oxidizes either the C1 or C4 sites [Citation110–114].
The discovery of LPMOs
The first fungal LPMOs were identified as secreted enzymes that degrade cellulose, during bioprospecting works carried out in the early 1990s [Citation115,Citation116]. At the beginning, these enzymes were reported as hydrolases and were classified as family 61 glycoside hydrolase (GH61) enzymes until late 2011. These enzymes were named PMOs (polysaccharide monooxygenases), and later, LPMOs within the auxiliary activity families [Citation104,Citation107,Citation117–119].
In 2001, a cellulase TrCel61 produced by T. reesei was reported to have four types of endoglucanases that showed hydrolytic activity on cellulose [Citation120], although this activity was hundred-fold lower than that of other known T. reesei endoglucanases. It was only later that researchers found that the very low cellulolytic activity of TrCel61 on all polysaccharide substrates was due to contamination. In 2008, the first crystal structure of the TrCel61 cellulase showed that the enzyme protein has a highly conserved flat surface, unlike the tunnel or cleft active sites typically found in cellulases. The TrCel61 cellulase also noticeably lacked the conserved carboxylate residues that catalyse hydrolysis. The enzyme does show weak structural similarities with CBP21, a chitin-binding protein (CBP) from the bacterium Serratia marcescens that is believed to improve the efficiency of the bacteria to degrade chitin. Chitin is another example of crystalline polysaccharide, similar to cellulose, consisting of β-1,4-linked N-acetylglucosamine [Citation121,Citation122].
Expression and secretion of GH61 in response to cellulose was initially observed in T. reesei [Citation123] and subsequently, in some other fungi [Citation124–127]. A surprising number of GH61 genes in some fungal genomes have been found, as many cellulolytic fungal species have significantly more genes for GH61 than for cellulases. An in-depth biochemical characterization of GH61s by Harris et al. [Citation106] showed that the activity of cellulases on acid-pretreated corn stover could be enhanced, but not on pure cellulose, using an unknown mechanism. Later in 2010, the bacterial CBP was reported to be the enzyme responsible for catalysing the oxidative degradation of chitin in the presence of molecular oxygen and a chemical reductant [Citation102]. The chitinolytic activity was reported to be attributed to Mg2+ and Zn2+ metal ions that cannot generate an oxidant from molecular oxygen. A similar reaction has also been proposed to occur in fungal GH61 cells [Citation102].
Soon findings from several major experiments linked the bacterial CBP21 oxidative cleavage reaction to the GH61 fungal enzymes. This reaction occurs through extracellular electron sources that reductively activate the GH61s [Citation103,Citation107,Citation108]. This was likely due to most fungi expressing CDH. It is an extracellular hemoflavoenzyme from the glucose-methanol-choline oxidoreductase superfamily that catalyses the oxidation of cellobiose to cellobionolactone [Citation128,Citation129]. Langston et al. [Citation103] suggested that the CDHs were important in activating functions of the GH61s. Consequently, a genetic study proved this theory by deleting the key CDH isoform in Neurospora crassa to result in a twofold decrease in secreted cellulase activity on pure cellulose substrate in the mutant enzyme [Citation107].
The mechanism of enzyme activation was thought to begin with copper as the functional active site metal with the products of GH61s catalysis being generated at both the reducing and non-reducing ends of the glucan chain. Initially, the crystal structures of GH61 and bacterial family 33 carbohydrate-binding modules (CBM33) were solved with nickel [Citation130], magnesium and zinc [Citation106] in the binding site. In 2011, Quinlan et al. [Citation108] solved the first crystal structure of Thermoascus aurantiacus GH61A (TaGH61A) and confirmed that the GH61 was bound to copper. Their finding was consistent with a report that natively purified N. crassa GH61s contained copper and only copper facilitated the catalysis of GH61 INCU01050 secreted by N. crassa [Citation107]. Most importantly, the two studies provided evidence that GH6 could oxidize the non-reducing end of sugars. In contrast, studies by Quilan et al. [Citation108] and Phillips et al. [Citation107] indicated that GH61 enzymes could also oxidize sugars at positions C6 and C4, respectively. In 2012, Beeson et al. [Citation110] presented experimental evidence for C4 oxidation and, following the observed common catalysed reactions, they suggested the name of the enzymes from GH61s and CBM33s to be changed to PMOs. They also suggested classifying PMOs as type 1 and type 2 PMOs based on their ability to oxidize both the reducing and non-reducing ends of the glucan chain.
LPMO structure
The first indication that GH61 enzymes were not glycoside hydrolases came with the establishment of the GH61 crystal structure of TrCel61B in 2008 [Citation130]. The structure of TrCel61B was found to lack the identifying active site cleft of the acid/base residues, unlike those seen in glycoside hydrolases. Instead, the active site of the enzyme was revealed to be a flat surface with a supposed metal-binding site. Afterwards, structural studies on fungal cellulose-active LPMOs between 2010 and 2013 shed more light on the LPMO function. These studies focused on the active site residues internal electron transfer, substrate-binding interactions and the regioselectivity of oxidation [Citation106,Citation108,Citation111,Citation131]. Similar structural studies on bacterial LPMOs, especially on the chitin-active ones have further contributed to the present knowledge on the active site of such enzymes [Citation111,Citation121,Citation131]. So far, most structural studies on LPMO have utilized X-ray crystallography, while the structures of CBP21 were determined using nuclear magnetic resonance (NMR) [Citation132], and in some cases assisted with computational software to further enhance structural analyses [Citation111,Citation131]. Advancements in protein analysis technologies have resulted in successful visualization and resolution of structures from a new family of chitin-active fungal LPMOs [Citation118] and bacterial LPMOs with cellulose-related activity [Citation104].
Copper-catalysed monooxygenase activity
Vaaje-Kolstad et al. [Citation102] first reported the oxygen- and reductant-dependent oxidative activity from the chitinolytic bacterium S. marcescens, which secretes CBP21. The oxidative activity of this enzyme was formerly accredited to a redox divalent metal ion like Mg2+ or Zn2+ in the active site. Although the metal ions were not associated to any oxidative activity, prior reports have indicated that divalent metal ions are required for stimulating cellulose breakdown by some GH61 proteins [Citation106]. The finding of that study provided the first clear explanation on the chemistry of LPMOs. Subsequently, studies on fungal cellulose-active LPMOs involving reconstructed apo-LPMOS with different metal ions further revealed that copper is the native metal co-factor of LPMOs [Citation102]. As a matter of fact, copper was later found to exist in chitin- and cellulose-active bacterial LPMOs [Citation104,Citation105,Citation118,Citation131–133] and chitin-active fungal LPMOs [Citation118].
While there were difficulties in analysing the reaction products, which made determination of the substrate oxidation site in cellulases challenging, several researchers managed to identify key oxidation sites. Vaaje-Kolstad et al. [Citation102], using oxygen atoms labelled with isotopes either as H218O or 18O2, showed that the C1 position of the N-acetyl-glucosamine unit in chitin is oxidized to the corresponding carboxylic acid. A similar observation was later described in other cellulose-active bacterial and fungal LPMOs [Citation104,Citation107,Citation108,Citation110,Citation113,Citation114,Citation131,Citation134–136], while certain cellulases were found to oxidize C4 and/or specifically oxidize only the C4 position of cellulose.
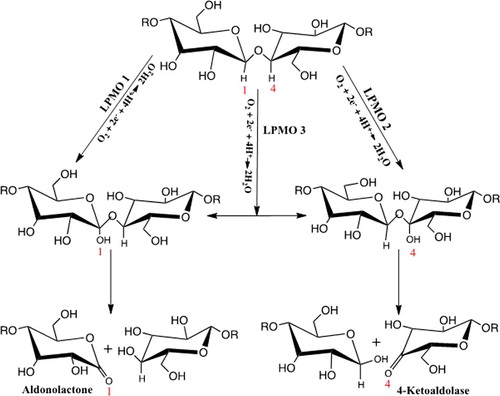
In view of such findings, a mechanism for the oxidative cleavage of glycosidic bonds was proposed (). In this mechanism, the LPMOs attach to the hydroxyl groups of either the C1 or C4 of the glycosidic bond in cellulose to form unstable hemiketal intermediates. These intermediates undergo elimination to produce either aldonolactones (C1 oxidation) or 4-ketoaldoses (C4 oxidation), in which the former undergo either spontaneous or enzyme-catalysed hydrolysis to form aldonic acid products [Citation137]. A combination of mass spectrometry and chemical derivation [Citation111] ascertained that 4-ketoaldoses were the products of C4 oxidation. This finding was later confirmed by Isaksen et al. [Citation113] using two-dimensional NMR. Similar studies using mass spectrometric analyses of reaction products suggested that oxidation at C6 by LPMOs was possible [Citation107,Citation135]. However, Vu et al. [Citation114], who studied the regioselectivity of phylogenetically diverse fungal cellulose-active LPMOs, reported otherwise. Both the fungal and bacterial LPMOs could not oxidize the C6 of cellulose, presumably due to inherent differences in their active site. This discrepancy could be linked to variances in conserved catalytic residues, H-bonding and angles of orientation, inferring the use of different mechanisms by LPMOs to activate oxygen for catalysis [Citation24].
Figure 3. The lytic polysaccharide monooxygenase (LPMO) reaction showing the regioselective hydroxylation of cellulose by LPMOs.
LPMO auxiliary activity families
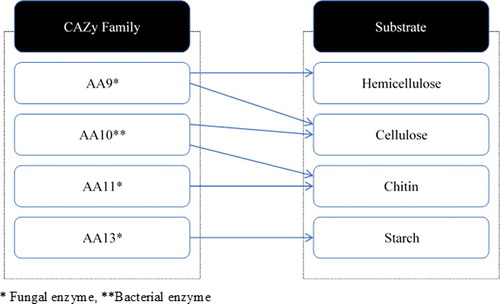
Four families of LPMOs that fall under auxiliary activity families in the CAZy database have been identified, so far [Citation117]. These enzyme families act on cellulose as auxiliaries to the hydrolytic cellulases [Citation102,Citation106, Citation107, Citation108,Citation134], thus playing a significant role in reducing the cellulase dosage for total hydrolysis [Citation103,Citation106]. They include: fungal AA9 LPMOs (previously GH61) that act on cellulose [Citation106,Citation107,Citation110,Citation108]; bacterial AA10 LPMOs (previously CBM33), active either on cellulose or on chitin [Citation102,Citation133,Citation134]; fungal AA11 LPMOs that act on chitin [Citation118] and fungal AA13 LPMOs that hydrolyse starch [Citation119,Citation136] (). Many AA9 LPMOs act in concert with electron donors [Citation103] such as CDHs that are co-secreted by fungi in effecting redox-mediated glycosidic bond cleavage in cellulose [Citation103,Citation107,Citation138]. An interesting work by Langston et al. [Citation103] combined a GH61 from T. aurantiacus with Humicola insolens CDH which hydrolysed cellulase to produce a mixture that contains reducing-oxidized and non-reducing end modified cellooligosaccharides [Citation103]. The same group also demonstrated that Thielavia terrestis GH61 and CDH of T. terrestris could synergistically hydrolyse microcrystalline cellulose [Citation103]. Interestingly, novel aldonolactonases have been discovered in the supernatant of M. thermophilia capable of catalysing the hydrolysis of glucono-δ-lactone, a by-product of enzymatic oxidation of cellulose [Citation110]. Sugar lactones, i.e. glucono-δ-lactone, have been known to be potent inhibitors of glycosyl hydrolases [Citation139], especially β-glucosidase [Citation20]. The oxidative cleavage by LPMOs can also be affected by reducing agents such as glutathione or gallate ascorbic acid [Citation102,Citation108,Citation109].
Figure 4. CAZy auxiliary enzymes and their substrates.
In line with the major role of auxiliary enzymes in cleaving glycosidic bonds, the presence of AA9 proteins is thought to enhance the activity of other cellulases [Citation106,Citation140] by attacking the crystalline surface on the cellulose before the action of hydrolases, creating more accessible sites for other cellulases to act [Citation141]. Pertinently, a remarkable quality of AA9 LPMOs is the extreme expansion in the genes encoding these proteins in fungal genomes. On average, the genomes of cellulose-degrading fungi harbour as many as 10 AA9 LPMOs, and those of Aspergilli, eight ones [Citation142–144].
Borisova et al. [Citation145] exploited the structural basis of the unique functional properties of NcLPMO9C, a C4 oxidising AA9LPMO (LPMO9) from N. crassa, also known as NcU02916 or NcGH61-3, and found that the enzyme acts both on cellulose and on non-cellulose β-glucans such as cellodextrins and xyloglucan. The catalytic domain crystal structure of the NcLPMO9C revealed an expanded, high polar substrate-binding surface suitable for interaction with a variety of sugar substrates. Electron spin resonance studies showed that the Cu2+ centre environment in NcLPMO9C is altered upon substrate binding, although isothermal titration calorimetry analysis attributed the binding affinities in the low micro-molar range for polymeric substrates, in part, to the presence of a carbohydrate-binding module (CBM1). Further comparative analysis showed that the oxidative region-selectivity of LPMO9s (C1, C4 or both) correlates with the specific structural features of the copper coordination sphere. Access to the solvent-facing axial coordination position is restricted in C1-oxidising LPMOs due to a conserved tyrosine residue, but seemingly not in C4-oxidising LPMOs. Cellulases producing a mixture of C1- and C4-oxidized products suggest adoption of an intermediate state [Citation145].
A rather recent work by Arfi et al. [Citation146] involving several dockerin-fused LPMOs based on enzymes from the bacterium T. fusca revealed resulting chimeras having activity levels on microcrystalline cellulose, similar to that of the WT. The complexes showed a 1.7-fold and a 2.6-fold increase in the release of soluble sugars from cellulose as compared to the free enzymes (with LPMO enhancement) and without LPMO enhancement, respectively. Hence, the suggestion that it is feasible for LPMOs to convert to the cellulosomal mode, benefitting from the proximity effects generated from the cellulosome architecture [Citation146].
Application of cellulases and potential application of LPMOs in industrial biofuel production
Due to the alarming level of environmental pollution through the release of green house and toxic gases from fossil fuels, the global community is now focused on biofuels, especially bioethanols. Biofuels are expected to replace 20% of the fossil fuel used by 2020. Initially, the focus was on the production of first-generation biofuels (e.g. corn bioethanol) from the bioconversion of crops such as corn grain (starch), melon seeds (fatty acids), sugarcane (sucrose), etc. However, due to the competitiveness of these grains for food to man, cost implications as well as relative abundant availability, there has been a shift to the use of lignocellulosic biomass as feedstock for the production of bioethanol. Lignocelulose can be majorly sourced from agricultural residues such as wheat, rice, corn straw, sugarcane bargasse, to biofuels (second generation) [Citation147,Citation148]. There are two main processes involved in the conversion; hydrolysis of cellulose in the lignocellulosic biomass to yield reducing sugars, and fermentation of the sugars to ethanol [Citation62]. Fungal cellulases from Trichoderma, Aspergillus and Penicillum spp. play pivotal roles in the hydrolysis process [Citation149–153].
As is the case with most newly discovered catalysts, there will be challenges in determining the full commercial applications of LPMO enzymes. The results presented by Harris et al. [Citation106] showed that the addition of LPMOs to (commercial) cellulase cocktails can reduce the required enzyme dose for conversion of pretreated corn stover by as much as twofold. Despite these initial auspicious results, there may be limitations in using oxygen-dependent catalysts for biomass conversion [Citation154]. An example is the simultaneous saccharification and fermentation process for production of cellulosic ethanol that is frequently used in the production of bioethanol [Citation155]. Under anaerobic or microaerobic conditions required for fermentation, there may be lack of oxygen for oxidative cleavage reactions catalysed by LPMOs [Citation156,Citation157]. If separate hydrolysis and fermentation approaches are used industrially, the LPMOs will oxidize a fraction of carbohydrate extracellulary, thus the enzymes will be no longer available for sugar fermentation. A balance between the energy losses due to oxidation by LPMOs, in feedstock, and the savings in cost from lower enzyme doses or reduced processing time must be strategically planned to maximize profits [Citation24].
Conclusions
Cellulases occupy a central position in the degradation and efficient utilization of lignocellulosic biomass. From the previous discussion, it is clear that cellulases are not just fascinating proteins from an agricultural and industrial perspective, but are also of fundamental scientific interest. As a matter of fact, the ever-increasing demand for natural and sustainable products has further elevated the significance of these enzymes, especially in biofuel production, and has greatly changed our view of the importance of microbial cellulose degradation. In this perspective, further studies into the structure-related function of cellulases, fundamental mechanisms of their activity and protein engineering merit scientific attention. While there have been a significant accumulation of data in documenting structural features of cellulase enzyme components, research in the area of structural modelling of cellulase enzyme systems remains limited due to constrains in technological advancements for predicting protein function from structure. Hence, developing the area of functional modelling of proteins would effectively expedite the progress of informed functional models for cellulases to improve the understanding of their structure-related functions. It is a more productive means for future research into tailoring catalytic properties of various cellulases and cellulase systems for effective utilization of (ligno)cellulosic biomass for improved production of renewable chemicals and fuels.
Acknowledgments
The authors would like to acknowledge valuable help and suggestions provided by their colleagues.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Mtui G, Nakamura Y. Bioconversion of lignocellulosic waste from selected dumping sites in Dares Salaam, Tanzania. Biodegradation. 2005;16(6):493–499.
- Singh S, Moholkar VS, Goyal A. Isolation, identification, and characterization of a cellulolytic Bacillus amyloliquefaciens Strain SS3S from Rhinocerus dung. ISRN Microbiol. 2013 [ cited 2017 Apr 3]; Article ID 728134. DOI:10.1155/2013/728134.
- Demain A, Newcomb M, Wu JHD. Cellulase, Clostridia and ethanol. Microbiol Mol Biol R. 2005;69(1):124–154.
- Lynd LR, Jin H, Michels JG, et al. Bioenergy: background, potential and policy. A policy briefing prepared for the Centre for Strategic and International Studies. Washington (DC): Centre for Strategic and International Studies; 2003.
- Kumar A, Gautam A, Dutt D. Biotechnological transformation of lignocellulosic biomass in to industrial products: an overview. Adv Biosci Biotechnol. 2016;7:149–168.
- Kumar P, Barrett DM, Delwiche MJ, et al. Methods for pretreatment of lignocellulosic biomass for efficient hydrolysis and biofuel production. Ind Eng Chem Res. 2009;48(8):3713–3729.
- Wang H, Squina F, Segato F, et al. High temperature enzymatic breakdown of cellulose. Appl Environ Microb. 2011;77(15):5199–5206.
- Himmel ME, Ding SY, Johnson DY, et al. Biomass recalcitrance: engineering plants and enzymes for biofuel production. Science. 2007;315(5813):804–807.
- Wolfenden R, Lu X, Young G. Spontaneous hydrolysis of glycosides. J Am Chem Soc. 1998;120:6814–6815.
- Wahlstrom R, Rovio S, Suurnakki A. Partial enzymatic hydrolysis of microcrystalline cellulose ionic liquids by Trichderma reesei endoglucanases. RSC Adv. 2012;2:4472–4480.
- Golan AE. Cellulase: types and action, mechanism, and uses. New York (NY): Nova Science Publishers; 2011.
- Wahlstrom R. Enzymatic hydrolysis of cellulose in aqueous ionic liquids [dissertation]. Espoo: Aalto University School of Chemical Technology; 2004.
- Lynd LR, Weimer PJ, van Zyl WH, et al. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev. 2002;66:506–577.
- Elwyn TR, Ralph GHS, Hillel SL. The biological degradation of soluble cellulose derivatives and its relationship to the mechanism of cellulose hydrolysis. J Bacteriol. 1950;59:485–497.
- Zhang YH, Lynd LR. Toward an aggregated understanding of enzymatic hydrolysis of cellulose: noncomplexed cellulase systems. Biotechnol Bioeng. 2004;88:797–824.
- Medie FM, Davies GJ, Drancourt M. Genome analyses highlight the different biological roles of cellulases. Nat Microbiol. 2012;10:227–234.
- Shewale JG. Glucosidases: its role in cellulase synthesis and hydrolysis of cellulose. Int J Biochem. 1982;14(16):435–443.
- Woodward J, Wiseman A. Fungal and other β-D-glucosidases: their properties and applications. Enz Microbiol Technol. 1983;4(2):73–79.
- Saini JK, Saini R, Tewari L. Lignocellulosic agriculture wastes as biomass feedstocks for second-generation bioethanol productions concepts and recent developments. 3 Biotech. 2014;5(4):337–353.
- Xiros C, Topakas E, Christakopoulos P. Hydrolysis and fermentation for cellulosic ethanol production. In: Lund DE, Byrne J, Berndes G, Vasalos LA, editors. Advances in bioenergy: the sustainability challenge. New York (NY): Wiley; 2016. p. 11–31.
- Gilbert HJ, Hazlewood GP. Bacterial cellulases and xylanases. J Gen Microbiol. 1993;139:187–194.
- Payne CM, Bomble YJ, Taylor CB, et al. Multiple functions of aromatic-carbohydrate interactions in a processive cellulase examined with molecular simulation. J Biol Chem. 2011;286(47):41028–41035.
- Taylor CB, Payne CM, Himmel ME. Binding site dynamics and aromatic–carbohydrate interactions in processive and non-processive family 7 glycoside hydrolases. J Biol Chem. 2014;117:4924–4933.
- Beeson WT, Vu VV, Span EA, et al. Cellulose degradation by polysaccharide monooxygenases. Annu Rev Biochem. 2015;84:923–946.
- Lehninger A, Nelson DL, Cox M. Principles of biochemistry. 3rd ed. New York (NY): Worth Publishers; 2003.
- Henrissat BA. Classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J. 1991;280(Pt2):309–316.
- Henrissat B, Bairoch A. New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J. 1993;293(Pt2):781–788.
- Cantarel BL, Coutinho PM, Rancurei C, et al. The carbohydrate-active enzymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37:D233–238.
- Lombard V, Golaconda RH, Drula E, et al. The carbohydrate-active enzyme database (CAZy) in 2013. Nucleic Acid Res. 2014;42:D490–D495.
- Sathya A, Khan M. Diversity of glycosyl hydrolase enzymes from metagenome and their application in food industry. J Food Sci. 2014;79(11):2149–2156.
- Bornscheuer U, Buchholz K, Seibel J. Enzymatic degradation of (lingo)cellulose. J Gesellschaft Deutscher Chemiker. 2014;53(41):10876–10893.
- Boraston AB, Bolan DN, Gilbert HJ, et al. Carbohydrate-binding odules: fine-tuning polysaccharide recognition. Biochem J. 2004;382(3):769–781.
- McLean BW, Bray MR, Boraston AB, et al. Analysis of binding of the family 2a carbohydrate-binding module from Cellulomonas fimi xylanase 10A to cellulose: specificity and identification of functionally important amino acid residues. Protein Eng. 2000;13:801–809.
- Boraston AB, Revett TJ, Boraston CM, et al. Structural and thermodynamic dissection of specific mannan recognition by a carbohydrate binding module, TmCBM27. Structure. 2003;11:665–675.
- Notenboom V, Boraston AB, Chiu P, et al. Recognition of cello-oligosaccharides by a family 17 carbohydrate-binding module: an X-ray crystallographic, thermodynamic and mutagenic study. J Mol Biol. 2001;314:797–806.
- Creagh AL, Ong E, Jervis E, et al. Binding of the cellulose-binding domain of exoglucanase Cex from Cellulomonas fimi to insoluble microcrystalline cellulose is entropically driven. Proc Natl Acad Sci USA. 1996;93:12229–12234.
- Najmudin BA, Pinheiro JA, Prates HJ, et al. Putting an N-terminal end to the Clostridium thermocellum xylanase Xyn10B story: crystal structure of the CBM22–1–GH10 modules complexed with xylohexaose. J Struct Biol. 2010;72:353–362.
- Inoue H, Kishishita S, Kumagai A, et al. Contribution of a family 1 carbohydrate-binding module in a thermostable glycoside hydrolase 10 xylanase from Talaromyces cellulolyticus toward synergistic enzymatic hydrolysis of lignocellulose. Biotechnol Biofuels. 2015 [ cited 2017 May 3];8:77. DOI:10.1186/s130–015–0259–2.
- Jager G, Girfoglio M, Dollo F, et al. How recombinant swollenin from Kluyveromyces lactis affects cellulosic substrates and accelerates their hydrolysis. Biotechnol Biofuels. 2011 [ cited 2017 May 3];4:33. DOI:10.1186/1754–6834–433.
- Arantes V, Saddler JN. Access to cellulose limits the efficiency of enzymatic hydrolysis: the role of amorphogenesis. Biotechnol Biofuels. 2010 [ cited 2017 May 3];3:4. DOI:10.1186/1754–6834–3–4.
- Ting CL, Makarov DE, Wang ZG. A kinetic model for the enzymatic action of cellulase. J Phys Chem. 2009;13(14):4970–4977.
- Payne CM, Resch MG, Chen L, et al. Glycosylated likers in multi modular ligno cellulose-degrading enzymes dynamically bind to cellulose. Proc Natl Acad Sci USA. 2013;110:14646–14651.
- Bayer EA, Kenig R, Lamed RJ. Adherence of Clostridium thermocellum to cellulose. Bacteriology. 1983;156:818–827.
- Blumer-Schuette SE, Brown SD, Sander KB, et al. Thermophilic lignocellulose deconstruction. FEMS Microbiol Rev. 2014;38:393–448.
- Zhou F, Olman V, Xu Y. Large-scale analyses of glycosylation in cellulases. Genomics Proteomics Bioinformatics. 2009;7(4):194–199.
- Fontes CM, Gilbert HJ. Cellulosomes: highly efficient nanomaches designed to deconstruct plant cell wall complex carbohydrates. Annu Rev Biochem. 2010;79:665–681.
- Bayer EA, Belaich J, Shoham Y, et al. The cellulosomes: multienzyme machines for degradation of plant cell wall Polysaccharides. Annu Rev Microbiol. 2004;58:521–554.
- Noach I, Levy-Assaraf M, Lamed R, et al. Modular arrangement of a cellulosomal scaffoldin subunit revealed from the crystal structure of a cohesin dyad. J Mol Biol. 2010;399:294–305.
- Hamberg Y, Ruimy-Isreali V, Dassa B, et al. Elaborate cellulosome architecture of Acetivibrio cellulolyticus revealed by selective screening of cohesion-dockerin interactions. Peer J. 2014 [ cited 2017 May 3];2:e636. DOI:10.7717/peerj.636.
- Gefen G, Anbar M, Morag E, et al. Enhanced cellulose degradation by targeted integration of a cohesin-fused β-glucosidase into the Clostridium thermocellum cellulosome. Proc Natl Acad Sci USA. 2012;109:10298–10303.
- Mori Y, Ozasa S, Kitaoka M, et al. Aligning an endoglucanase Cel5A from Thermobifida fusca on a DNA scaffold: potent design of an artificial cellulosome. Chem Commun. 2013;49:6971–6973.
- Himmel ME, Karplus PA, Sakon J, et al. Polysaccharide hydrolase folds diversity of structure and convergence of function. Appl Biochem Biotechnol. 1997;63–65:315–325.
- Yang B, Dai Z, Ding SY, et al. Enzymatic hydrolysis of cellulosic biomass. Biofuels. 2011;2(4):421–450.
- Ganner T, Bubner P, Eibinger M, et al. Dissecting and reconstructing synergism in situ visualization of cooperativity among cellulases. J Biol Chem. 2012;287:43215–43222.
- Bubner P, Plank H, Nidetzky B. Visualizing cellulase activity. Biotechnol Bioeng. 2013;110:1529–1549.
- Sturcova A, His I, Apperley DC, et al. Structural details of crystalline cellulose from higher plants. Biomacromolecules. 2004;5:1333–1339.
- Habibi Y, Lucia LA, Rojas OJ. Cellulose nanocrystals: chemistry, self-assembly, and applications. Chem Rev. 2010;110(6):3479–3500.
- Fox JM, Jess P, Jambusaria RB, et al. A single-molecule analysis reveals morphological targets for cellulase synergy. Nat Chem Biol. 2013;9:356–361.
- Luterbacher JS, Walker LP, Moran-Mirabal JM. Observing and modeling BMCC degradation by commercial cellulase cocktails with fluorescently labeled Trichoderma reesei Cel7A through confocal microscopy. Biotechnol Bioeng. 2013;110:108–117.
- Luterbacher JS, Parlange JY, Walker LP. A pore-hindered diffusion and reaction model can help explain the importance of pore size distribution in enzymatic hydrolysis of biomass. Biotechnol Bioeng. 2013;110:127–136.
- Coughlan MP, Ljungdahl LG. Comparative biochemistry of fungal and bacterial cellulolytic enzyme system. In: Aubert, JP, Beguin P, Millet J, editors. FEMS Symposium: Biochemistry and Genetics of Cellulose Degradation. London: Academic Press; 1988. p. 11–30.
- Sun Y, Cheng J. Hydrolysis of lignocellulosic materials for ethanol production: a review. Biores Technol. 2002;83:1–11.
- Elkins JG, Ramen B, Keller M. Engineered microbial systems for enhanced conversion of lignocellulosic biomass. Curr Opin Biotechnol. 2010;21(5):657–662.
- Bayer EA, Chanzy H, Lamed R, et al. Cellulose, cellulases and cellulosomes. Current Opin Struct Biol. 1998;8(5):548–557.
- Chanzy H, Henrissat B, Vuong R. Colloidal gold labeling of 1,4-β-D-glucan cellobiohydrolase adsorbed on cellulose substrates. FEBS Lett. 1984;172:193–197.
- Harvey AJ, Hrmova M, De Gori R, et al. Comparative modeling of the three-dimensional structures of family 3 glycoside hydrolases. Proteins. 2000;41(2):257–269.
- Xiao WP, Clarkson WW. Acid solubilization of lignin and bioconversion of treated newsprint to methane. Biodegradation. 1997;8:61–66.
- Hamelinck CN, van Hooijdonk G, Faaij APC. Ethanol from lignocellulosic biomass: techno-economic performance in short-, middle- and long-term. Biomass Bioenerg. 2005;28:384–410.
- Pan X, Xie D, Kang KY, et al. Effect of organosolv ethanol pretreatment variables on physical characteristics of hybrid poplar substrates. Appl Biochem Biotechnol. 2007;137:367–377.
- Lee JW, Gwak KS, Park JY, et al. Biological pretreatment of softwood Pinus densiflora by three white rot fungi. J Microbiol. 2007;45:485–491.
- Mansfield SD, Mooney C, Saddler JN. Substrate and enzyme characteristics that limit cellulose hydrolysis. Biotechnol Progr. 1999;15:804–816.
- Cheng G, Varanasi P, Li CL, et al. Transition of cellulose crystalline structure and surface morphology of biomass as a function of ionic liquid pretreatment and its relation to enzymatic hydrolysis. Biomacromolecules. 2011;12:933–941.
- Bhat MK. Cellulases and related enzymes in biotechnology. Biotechnol Adv. 2000;18:355–383.
- Zhang MJ, Su RX, Qi W, et al. Enhanced enzymatic hydrolysis of lignocellulose by optimizing enzyme complexes. Appl Biochem Biotechnol. 2010;160:1407–1414.
- Hrmova A, MacGregor EA, Biely P, et al. Substrate binding and catalytic mechanism of a barley β-D-glucosidase/(1,4) –β–D–glucan exohydrolase. J Biol Chem. 1998;273:11134–11143.
- Alvira P, Tomas-Pejo E, Ballesteros M, et al. Pretreatment technologies for an efficient bioethanol production process based on enzymatic hydrolysis: a review. Bioresour Technol. 2010;101(13):4851–4861.
- Chitlaru E, Roseman S. Molecular cloning and characterization of a novel beta-N-acetylglucosaminidase from Vibrio furnissii. J Biol Chem. 1998;271(52):33433–33439.
- Fincher G, Mark B, Brumer H. Glycoside hydrolase family 3 [Internet]. In: Henrissat B, editor. CAZypedia. [ cited 2017 May 4]. Available from: http://www.cazypedia.org/
- Davies GJ, Wison KS, Henrissat B. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem J. 1997;321(Pt2):557–559.
- Pozzo T, Pasten JL, Karlsson EN, et al. Structural and functional analyses of beta-glucosidase 3B from Thermotoga neapolitana: a thermostable three-domain representative of glycoside hydrolase 3. J Mol Biol. 2010;397(3):724–739.
- Paal K, Ito M, Withers SG. Paenibacillus sp. Ts12 glucosylceramidase: kinetic studies of a novel sub-family of family glycosidases and identification of the catalytic residues. Biochem J. 2004;378:141–149.
- Bause E, Legler G. Isolation and amino acid sequence of a hexadocapeptide from the active site of beta-glucosidaseA3 from Aspergilllus wentii. Hoppe-Seyler's Z Physiol Chem. 1974;355(4):438–442.
- Chir J, Withers S, Wan CF, et al. Identification of the two essential groups in the family 3 beta-glucosidase from Flavobacterium meningosepticum by labeling and tandem mass spectrometric analysis. Biochem J. 2002;365(Pt 3):857–863.
- Dan S, Marton I, Dekel M, et al. Cloning, expression, characterization and nucleophile identification of family 3 Aspergillus niger beta-glucosidase. J Biol Chem. 2000;275(7):4973–4980.
- Thongpoo P, Mckee LS, Araujo AC, et al. Identification of the acid/base catalyst of a glycoside hydrolase family 3 (GH3) beta-glucosidase from Aspergillus niger ASKU28. Biochim Biophys Acta. 2013;1830(3):2739–2749.
- Lee RC, Hrmova M, Burton RA, et al. Bifunctional family 3 glycoside hydrolases from barley with alpha-L-arabinofuranosidase and beta-D-xylosidase activity, characterization, primary structures and CooH-terminal processing. J Biol Chem. 2003;278(7):5377–5387.
- Bacik JP, Whitworth GE, Stubbs KA, et al. Active site plasticity within the glycoside hydrolase NagZ underlies a dynamic mechanism of substrate distortion. Chem Biol. 2012;19:1471–1482.
- Stubbs KA, Balcewich M, Mark BL, et al. Small molecule inhibitors of a glycoside hydrolase attenuate inducible Amp C-mediated beta-lactam resistance. J Biol Chem. 2007;284(29):21382–21391.
- Vocadlo DJ, Withers SG. Detailed comparative analysis of the catalytic mechanisms of beta-N-acetyglucosaminidases from families 3 and 20 of glycoside hydrolase. Biochemistry. 2005;44(38):12809–12818.
- Litzinger S, Duckworth A, Nitzsche K, et al. Muropeptide rescue in Bacillus subtilis involves sequential hydrolysis by beta-N-acetylglucosaminidase and N-acetyl-muranyl-L-alanine amidase. J Bacteriol. 2010;192(12):3132–3143.
- Aspeborg H, Coutinho PM, Wang Y, et al. Evolution, substrate specificity and subfamily classification of glycoside hydrolase family 5 (GH5). BMC Evol Biol. 2012 [ cited 2017 May 3]. DOI:10.1186/1471–2148–12–186.
- Wilson DB, Urbanowicz B. Glycoside hydrolase family 9. In: Wilson DB, editor. CAZypedia. [ cited 2017 May 4]. Available from: http://www.cazypedia.org/
- Py B, Bortoli-German I, Haiech J, et al. Cellulase EGZ of Erwinia chrysanthemi, structural organization and importance of His-98 abd Glu-133 residues for catalysis. Protein Eng. 1991;4:325–333.
- Sakon J, Irwin D, Wilson DB, et al. Structure and mechanism of endo/exocellulase E4 from Thermomonospora fusca. Nat Struct Biol. 1997;44:810–818.
- Sathya TA, Khan M. Diversity of glycosyl hydrolase enzymes from metagenome and their applications in food industry. J Food Sci. 2014;79(11):R2149–R2156.
- Urbanowicz BR, Catala C, Irwinm D, et al. A tomato endo-beta-1,4,glucanase, SICel9C1, represents a distinct subclass with a new family of carbohydrate-binding modules (CBM49). J Biol Chem. 2007;282(16):12066–12074.
- Zhou W, Irwin DC, Escovar-Kousen J, et al. Kinetic studies of Thermofida fusca Cel9A active site mutant enzymes. Biochemistry. 2004;43(30):9655–9663.
- Li Y, Irwin DC, Wilson DB. Processivity, substrate binding and mechanism of cellulose hydrolysis by Thermobifida fusca Cel9A. Appl Environ Microbiol. 2007;73(10):3165–3172.
- Guerin DM, Lascombe MB, Costabel M, et al. Atomic (0.94A) resolution structure of an inverting glycosidase in complex with substrate. J Mol Biol. 2002;316(5):1061–1069.
- Klemm D, Heublein B, Fink HP, et al. Cellulose: fascinating biopolymer and sustainable raw material. Angew Chem Int Ed. 2005;44:3358–3393.
- Ding S, Himmel ME. The maize primary cell wall microfibril: a new model derived from direct visualization. J Agric Food Chem. 2006;54:597–606.
- Vaaje-Kolstad G, Westereng B, Horn SJ, et al. An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science. 2010;330:219–222.
- Langston JA, Shaghasi T, Abbate E, et al. Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl Environ Microbiol. 2011;77:7007–7015.
- Forsberg Z, Mackenzie AK, Sorlie M, et al. Structural and functional characterization of a conserved pair of bacterial cellulose–oxidizing lytic polysaccharidemonooxygenases. Proc Natl Acad Sci USA. 2014;111:8446–8451.
- Horn SJ, Vaaje-Kolstad G, Westereng B, et al. Novel enzymes for the degradation of cellulose. Biotechnol Biofuels. 2012;5:45–56.
- Harris PV, Welner D, McFarland KC, et al. Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry. 2010;49:3305–3316.
- Phillips CM, Beeson WT, Cate JH, et al. Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem Biol. 2011;6:1399–1406.
- Quinlan RJ, Sweeney MD, Lo Leggio L, et al. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc Natl Acad Sci USA. 2011;108:15079–15084.
- Westereng B, Ishida T, Vaaje-Kolstad G, et al. The putative endoglucanase PcGH61D from Phanerochaete chrysosporium is ametal-dependent oxidative enzyme that cleaves cellulose. PLoS One. 2012 [ cited 2017 May 3];6(11):e27807. DOI:10.1371/journal.pone.0027807.
- Beeson WT, Phillips CM, Cate JHD, et al. Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J Biol Chem. 2012;134:890–892.
- Li X, Beeson WT, Phillips CM et al. Structural basis for substrate targeting and catalysis by fungal polysaccharide monooxygenases. Structure. 2012;20:1051–1061.
- Hemsworth GR, Davies GJ, Walton PH. Recent insights into copper-containing lytic polysaccharide mono-oxygenases. Curr Opin Struc Biol. 2013;23:660–668.
- Isaksen T, Westereng B, Aachmann FL, et al. A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J Biol Chem. 2014;289:2632–2642.
- Vu VV, Beeson WT, Phillips CM, et al. Determinants of regioselective hydroxylation in the fungal polysaccharide monooxygenases. J Am Chem Soc. 2014;136:562–565.
- Raguz S, Yague E, Wood DA, et al. Isolation and characterization of a cellulose-growth specific gene from Agaricus bisporus. Gene. 1992;119:183–190.
- Armesilla AL, Thurston CF, Yague E. CEL1: a novel cellulose binding protein secreted by Agaricus bisporus during growth on crystalline cellulose. FEMS Microbiol Lett. 1994;116:293–299.
- Levasseur A, Drula E, Lombard V, et al. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels. 2013 [ cited 2017 May 3];6(1):41. DOI:10.1186/1754–6834–6–41.
- Hemsworth GR, Henrissat B, Davies GJ, et al. Discovery and characterization of a new family of lytic polysaccharide monooxygenases. Nat Chem Biol. 2014;10:122–126.
- Lo Leggio L, Simmons TJ, Poulen JN, et al. Structure and boosting activity of a starch-degrading lytic polysaccharide monooxygenase. Nat Commun. 2015 [ cited 2017 May 3];6:5961. DOI:10.1038/ncomms6961.
- Karlsson J, Saloheimo M, Siika-Aho M, et al. Homologous expression and characterization of Cel61A (EG IV) of Trichoderma reesei. Eur J Biochem. 2001;268:498–507.
- Vaaje-Kolstad G, Horn SJ, van Aalten DMF, et al. The non-catalytic chitin binding protein CBP21 from Serratia marcescens is essential for chitin degradation. J Biol Chem. 2005;280:28492–28497.
- Vaaje-Kolstad G, Houston DR, Riemen AHR, et al. Crystal structure and binding properties of the Serratia marcescens chitin-binding protein CBP21. J Biol Chem. 2005;280:11313–11319.
- Foreman PK, Brown D, Dankmeyer L, et al. Transcriptional regulation of biomass-degrading enzymes in the filamentous fungus Trichoderma reesei. J Biol Chem. 2003;278:31988–31997.
- Tian C, Beeson WT, Iavarone AT, et al. Systems analysis of plant cell wall degradation by the model filamentous fungus Neurospora crassa. Proc Natl Acad Sci USA. 2009;106:22157–22162.
- Martinez D, Challacombe J, Morgenstern I, et al. Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion. Proc Natl Acad Sci USA. 2009;106:1954–1959.
- Vanden WA, Gaskell J, Mozuch M, et al. Comparative transcriptome and secretome analysis of wood decay fungi Postia placenta and Phanerochaete chrysosporium. Appl Environ Microbiol. 2010;76:3599–3610.
- Eastwood DC, Floudas D, Binder M, et al. The plant cell wall decomposing machinery underlies the functional diversity of forest fungi. Science. 2011;333:762–765.
- Henriksson G, Johansson G, Pettersson G. A critical review of cellobiose dehydrogenases. J Biotechnol. 2000;78:93–113.
- Zamocky M, Ludwig R, Peterbauer C, et al. Cellobiose dehydrogenase – a flavocytochrome from wood-degrading, phytopathogenic and saprotropic fungi. Curr Protein Pept Sci. 2006;7:255–280.
- Karkehabadi S, Hansson H, Kim S, et al. The first structure of a glycoside hydrolase family 61 member, Cel61B from Hypocrea jecorina, at 1.6A˚ resolution. J Mol Biol. 2008;383:144–154.
- Wu M, Beckham GT, Larsson AM, et al. Crystal structure and computational characterization of the lytic polysaccharide monooxygenase GH61D from the Basidiomycota fungus Phanerochaete chrysosporium. J Biol Chem. 2013;288:12828–12839.
- Aachmann FL, Sørlie M, Skja˚k-Bræk G, et al. NMR structure of a lytic polysaccharide monooxygenase provides insight into copper binding, protein dynamics, and substrate interactions. Proc Natl Acad Sci USA. 2012;109:18779–18784.
- Forsberg Z, Røhr AK, Mekasha S, et al. Comparative study of two chitin-active and two cellulose-active AA10-type lytic polysaccharide monooxygenases. Biochemistry. 2014;53:1647–1656.
- Forsberg Z, Vaaje-Kolstad G, Westereng, B, et al. Cleavage of cellulose by a CBM33 protein. Protein Sci. 2011;20:1479–1483.
- Bey M, Zhou S, Poidevin L, et al. Cello-oligosaccharide oxidation reveals differences between two lytic polysaccharide monooxygenases (Family GH61) from Podospora anserine. Appl Environ Microbiol. 2013;79:488–496.
- Vu VV, Beeson WT, Span EA, et al. A family of starch-active polysaccharide monooxygenases. Proc Natl Acad Sci USA. 2014;111:13822–13827.
- Beeson WT, Iavarone AT, Hausmann CD, et al. Extracellular aldonolactonase from Myceliophthora thermophila. Appl Environ Microbiol. 2011;77:650–656.
- Sygmund C, Kracher D, Scheiblbrandner S, et al. Characterization of the two Neurospora crassa cellobiose dehydrogenases and their connection to oxidative cellulose degradation. Appl Environ Microbiol. 2012;78(17):6161–6171.
- Conchie J, Levvy GA. Inhibition of glycosidases by aldonolactones of corresponding configuration. Biochem J. 1957;65:389–395.
- Vaaje-Kolstad G, Bohle LA, Gaseidnes S, et al. Characterization of the chitinolytic machinery of Enterococcus faecalis V583 and high resolution structure of its oxidative CBM33 enzyme. J Mol Biol. 2012;416(2):239–254.
- Eibinger M, Ganner T, Bubner P, et al. Cellulose surface degradation by lytic polysaccharide monooxygenase and its effect on cellulase hydrolytic efficiency. J Biol Chem. 2014;289:35929–35938.
- Glass NL, Schmoll M, Cate JHD, et al. Plant cell wall deconstruction by ascomycete fungi. Annu Rev Microbiol. 2013;67:477–498.
- Segato F, Damasio AR, de Lucas RC, et al. Genomics review of holocellulose deconstruction by aspergilli. Microbiol Mol Biol Rev. 2014;78:588–613.
- Busk PK, Lange L. Classification of fungal and bacterial lytic polysaccharide monooxygenase. BMC Genomics. 2015 [ cited 2017 May 3];16:368. DOI:10.1186/s12864-015-1601-6.
- Borisova AS, Isaksen T, Dimarogona M, et al. Structural and functional characterization of a lytic polysaccharide monoxygenase with Broad substrate specificity. J Biol Chem. 2015;290:22955–22969.
- Arfi Y, Shanshoum M, Rogachev I, et al. Integration of bacterial lytic polysaccharide monoxygenases into designer cellulosomes promotes enhanced cellulose degradation. PNAS. 2014;111(25):9109–9114.
- Gray KA, Zhou L, Emptago M. Bioethanol Curr Opin Chem Biol. 2006;10(2):141–146.
- Sajith S, Priji P, Sreedevi S, et al. An overview of fungal cellulases with an industrial perspective. J Nutr Food Sci. 2016 [ cited 2017 May 3];4:461. DOI:10.4172/2155-9600.1000461.
- Zhou J, Wang YH, Chu J, et al. Identification and purification of the main components of cellulases from a mutant strain of Trichoderma viride T 100-14. Bioresour Technol. 2008;99:6826–6833.
- Sukumaran RK, Singhania RR, Mathew GM, et al. Cellulase production using biomass feed stock and its application in lignocellulose saccharification for bio-ethanol production. Renew Energy. 2009;34(2):421–424.
- Binod P, Sindhu R, Singhania RR, et al. Bioethanol production from rice straw: an overview. Bioresour Technol. 2010;101:4767–4774.
- Chen H, Qiu W. Key technologies for bioethanol production from lignocellulose. Biotechnol Adv. 2010;28:556–562.
- Sajith S, . Investigations on the lignocellulolytic activities of certain fungi with special reference to cellulase production [dissertation]. Kerala, India: University of Calicut; 2015.
- Cannella D, Hsieh CC, Felby C, et al. Production and effect of aldonic acids during enzymatic hydrolysis of lignocellulose at high dry matter content. Biotechnol Biofuels. 2012;5:26–35.
- Olofsson K, Bertilsson M, Liden GA. Short review on SSF – an interesting process option for ethanol production from lignocellulosic feedstocks. Biotechnol Biofuels. 2008 [ cited 2017 May 3];1:7. DOI:10.1186/1754-6834-1-7.
- Cannella D, Jørgensen H. Do new cellulolytic enzyme preparations affect the industrial strategies for high solids lignocellulosic ethanol production? Biotechnol Bioeng. 2014;111:59–68.
- Beguin P, Aubert J. The biological degradation of cellulose. FEMS Microbiol Rev. 1994;13(1):25–28.