
Abstract
Because the genomic information of lingonberry (Vaccinium vitis-idaea L.) is not available, the mechanism underlying the flavonoid metabolism in the fruits of this species is still unknown. The objective of this study was to obtain a comprehensive transcriptome assembly during lingonberry fruit development and characterize the key genes associated with the flavonoid metabolism in lingonberry. Our transcriptome data of lingonberry was obtained with next-generation sequencing technology. The expression patterns of key genes were analysed by quantitative reverse transcription polymerase chain reaction (qRT-PCR). The results showed that the total flavonoid content and the total anthocyanin content gradually increased during the lingonberry fruit development process. In total, about 169 million sequencing raw reads were obtained and de novo assembled into 67,836 unigenes, and 38,460 unigenes had been annotated from public databases. The average size of unigenes was 1040 nt, and the N50 value was 1718 nt. Among the unigenes mapped to KEGG pathways, 2697 (10.95%) were assigned to the category of ‘Biosynthesis of secondary metabolites’, 1533 unigenes were predicted to be associated with flavonoid biosynthesis, transport and regulation. We also found 10,022 candidate genes expressed differentially between green and white fruits, with 5335 differentially expressed genes uncovered between white and red fruits and 14,364 ones between green and red fruits. A comprehensive transcriptome assembly of lingonberry was generated, with data representing gene expression profiles throughout fruit development and ripening processes in lingonberry. Our results provide further insights into the molecular mechanisms of flavonoid metabolism.
Introduction
The genus Vaccinium L., which comprises approximately 450 species worldwide [Citation1, Citation2], includes three recently domesticated crops: lingonberries (V. vitis-idaea L.), blueberries and cranberries. Lingonberries are widely distributed dwarf woody shrubs found mainly in Europe and Asia, with a few species inhabiting arctic-montane regions. Lingonberry, also known as cowberry, foxberry, rock cranberry, redberry and partridgeberry, is thought to be a circumboreal cranberry–blueberry intermediate [Citation3].
From time immemorial, humans have gathered wild lingonberry fruits. These fruits have served as a source of juice, ice wine, jelly and jam, and have also been preserved and used as a condiment with meat. Both lingonberry fruits and leaves are full of phenolic compounds, such as flavonoids and aromatic acids [Citation4]. Because of their nutritional properties, lingonberries have been considered as potential materials for nutraceuticals and have been used to treat inflammatory diseases and wounds [Citation5–8]. As a result of increasing demand for lingonberry products, commercial cultivation and breeding programmes have been established in Europe, North America and China. Finally, lingonberries grow naturally under a wide range of ecological conditions, thus allowing Vaccinium crops to be cultivated further north than previously possible [Citation9].
Flavonoids as a kind of secondary metabolite are major secondary metabolites. Flavonoids, such as anthocyanins, flavonols and proanthocyanidins are particularly abundant in various berries [Citation10–13]. According to literature reports, lingonberry fruits are rich in flavonoids and have strong antioxidant capacity [Citation14–16]. The research on lingonberry flavonoids has been mainly in phytochemicals measurement, but the molecular mechanisms of the biosynthesis of flavonoids in lingonberry remain largely unknown.
The transcriptome sequencing technologies have been widely used in non-model plants for genetic study and molecular marker development [Citation17,Citation18]. RNA sequencing (RNA-seq) provides a powerful approach for investigating full-scale transcriptional profiles and revealing essential functional genes involved in agronomic traits. Transcriptome and genome databases have recently been published for Vaccinium crops, such as blueberry and cranberry [Citation19–23]. In contrast, the genetic information on lingonberry is still limited.
In this study, we carried out RNA-seq on lingonberry fruits at three development stages. To reveal the biological metabolic processes occurring during lingonberry berry development, we analysed the expression patterns of transcripts among the three fruit development stages. Our objective was to discover and characterize the key genes and regulatory elements associated with the flavonoid metabolism in lingonberry.
Materials and methods
Plant materials
All samples were taken from 6-year-old ‘Sunna’ lingonberry trees growing in acidic peat and well-drained soil. Fruits were sampled at three ripening stages from lingonberry trees, namely, green (30 d after full bloom, G), white (60 d after full bloom, W) and red (120 d after full bloom, R) fruit stages. Twenty representative fruits were harvested from each tree at each developmental stage and were sliced. The pooled samples were sunk into liquid nitrogen and preserved at −80 °C.
Cultivation location and conditions
The trees were grown in the small berry orchard of Jilin Agricultural University, Changchun, Jilin Province, China (43°79′54″N, 125°24′55″E); the altitude was 259 m; from May to September 2017 (growing season of crops), the mean temperature was 19.6 °C, the mean precipitation was 273.9 mm, and the mean light hours were 1233.1 h. Fertilization and irrigation were applied on the lingonberry trees to make the growth environment suitable and basically consistent according to the meteorological situation.
Determination of total flavonoids and total anthocyanins
Samples of bioactive substances were prepared using the Folin–Ciocalteu method [Citation24]. Total flavonoid content was determined using rutin as a standard substance (Rutin equivalent [RE]) by the NaNO2-Al (NO3)3 colorimetric method [Citation25]. The content of total anthocyanins was determined using the dual-wavelength pH-differential method with Cyanidin-3-glucoside as a standard substance [Citation26]. All indicators were measured in three parallel samples. The Design-Expert version 8.0.6 software (Stat Ease Inc., USA) was used for regression analysis and analysis of variance on the data. The SPSS version 19.0 (SPSS Inc., Chicago, IL) statistical software was used to analyse data one-way analysis of variance (ANOVA).
RNA sequencing
In order to obtain a representative biological selection of transcripts at each fruit stage, total RNA for sequencing was isolated from three replicates. The Invitrogen Trizol method (Carlsbad, CA) was used for isolating total RNA from lingonberry fruits. The cDNA libraries were prepared by Illumina protocol (San Diego, CA). Beijing Genomics Institute (BGI) was responsible for completing the RNA-seq with the Illumina HiSeq 2000 platform.
Sequence assembly and processing
Adaptor sequences, sequences containing ≥5% unknown nucleotides and low-quality sequences were removed from the sequencing raw data by filter-fq software. The clean read data were assembled with Trinity software [Citation27,Citation28]. As gene family clustering, the unigenes were classified as clusters and singletons, which were named with the prefixes ‘CL’ and ‘Unigene’, respectively.
Gene functional annotation
All-unigene sequences were aligned using BLASTn and BLASTx against the above databases with a cutoff E-value of 1 × 10−5 [Citation29,Citation30]. The direction and coding region of sequences were predicted by ESTScan software [Citation31]. GO analysis was performed using Blast2GO [Citation32], with additional GO functional classification conducted using WEGO to understand the distribution of gene functions [Citation33]. Metabolic pathway mapping of transcripts was carried out using Path_finder [Citation34].
Differential expression analysis
To analyse the expression pattern of transcripts from samples from different developmental stages, gene expression levels were measured by the fragments per kilobase per million (FPKM) method [Citation33]. Differentially expressed genes were predicted using a previously reported algorithm [Citation35]. Significantly different transcript expression levels were identified with the |log2 fold change|≥2 and adjusted p-value ≤ 10−3 criteria [Citation36].
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analysis
qRT-PCR analysis was performed with Takara SYBR Premix Ex Taq kit (Dalian City, Liaoning, China) following the manufacturer’s instructions. Applied Biosystems (ABI) Primer Express version 3.0.1 (Foster City, CA) was used for designing the qRT-PCR primers (Supplemental File 1). qRT-PCR reactions were performed on ABI StepOnePlus™ Real-time PCR system (Foster City, CA). The biological and technical replicates of qRT-PCR experiments were done in triplicate. The relative quantification expression levels were calculated with the 2−ΔΔCt method [Citation37]. The reference gene was glyceraldehyde-3-phosphate dehydrogenase (GAPDH, CL8364. Contig2_All) from the lingonberry transcriptome.
Results and discussion
Changes in total flavonoid content and total anthocyanin content during lingonberry fruit development
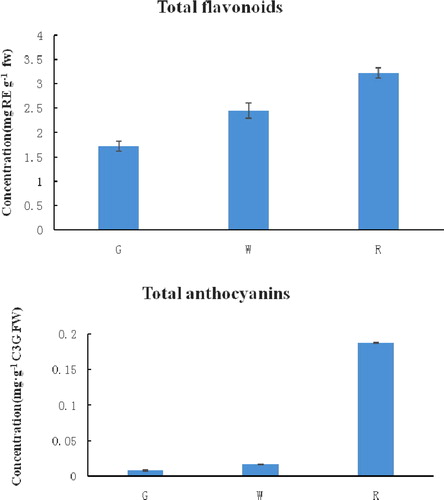
The total flavonoid content and the total anthocyanin content during lingonberry fruit development are shown in . The total flavonoid and total anthocyanin content during the fruit development process ranged from 1.72 to 3.23 mg RE g−1 FW and 0.008 to 0.18 mg C3G g−1 FW, respectively. The content of total flavonoids gradually increased from the green fruit (30 d after full bloom, G) period to the red fruit (120 d after full bloom, R) period, but the anthocyanin content increased sharply from the white fruit (60 d after full bloom, W) to the red fruit period. According to the previous data for red lingonberries, our results for the total flavonoid and total anthocyanin content are different with the lingonberry fruits collected in Canada and Poland [Citation38,Citation39]. These discrepancies may be caused by geographical location, the genotype of the plants, fruit ripening conditions, etc.
Figure 1. Changes of total flavonoids and total anthocyanins content during fruit development in lingonberry. Three fruit developmental stages were examined in this study as follows: green stage (G), white stage (W) and red stage (R). Error bars show the standard error calculated from three biological replicates.
RNA-seq and assembly
To obtain gene expression profiles during lingonberry fruit development, three cDNA libraries were constructed from fruits at green (G), white (W) and red (R) stages. Sequencing of the G, W and R libraries generated 54,616,730, 58,762,360 and 55,781,080 raw reads, respectively (). After trimming adapters and removing low-quality data, 51,182,530, 55,229,970 and 52,271,498 clean data were generated for G, W and R libraries, respectively. In total, 67,836 unigenes (ca. 70 Mb total length) were produced from all clean reads using Trinity (Supplemental File 2). The average size of unigenes was 1040 nt, and the N50 value was 1718 nt. The N50 value generated in this study is greater than the previous reports on transcriptomes of blueberry (1100 nt) [Citation19] and cranberry (1209 nt) [Citation22] fruits. The length distribution of unigenes was as follows: 62.5% (42,418) between 300 and 1000 nt, 32.5% (22,024) between 1000 and 3000 nt and 5.0% (3394) greater than 3000 nt ().
Figure 2. Sequence length distribution of All-Unigenes in ‘Sunna’ lingonberry fruit transcriptomes.
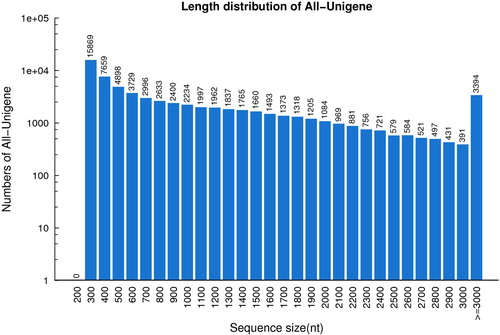
Table 1. Summary of the sequencing reads and assembly result.
Functional annotation based on similarity searching
A total of 42,620 unigenes (62.8%) obtained annotation from the public databases (). The databases included NCBI NT and non-redundant (NR), Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), Clusters of Orthologous Groups (COG) and Gene Ontology (GO). The remaining 37.2% corresponded to genes of unknown function, which indicates that fruit development in lingonberry is an unexpectedly complex process. Gene identities and distributions by species are presented in . Based on the E-value distribution analysis, 61.2% of unigenes were close homologs (E-value <1 × 10−45), whereas 38.9% were more distant homologs (E-value of 1 × 10−5 to 1 × 10−45) (). In regard to the similarity distribution analysis, 65.9% of unigenes had similarities higher than 60%, whereas 34.2% exhibited similarities between 17% and 60% (). As shown in , the closest matches of lingonberry unigenes were sequences from Vitis vinifera (43.1%), Lycopersicon esculentum (10.2%), Amygdalus persica (9.7%), Populus balsamifera subsp. trichocarpa (7.7%), Ricinus communis (7.6%), Fragaria vesca subsp. vesca (4.6%) and Glycine max (3.1%). The best matches to the remaining 14.1% sequences were with other species. According to the species distribution of matching sequences, approximately 86% were from dicotyledonous plants.
Figure 3. E-value distribution (A), similarly distribution (B) and species distribution (C) of the Blastx results in NCBI NR database.
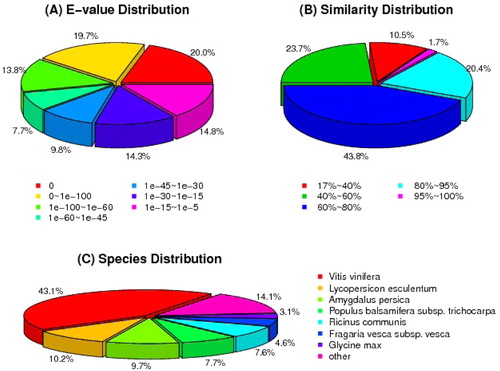
Table 2. Functional annotations of lingonberry All-Unigenes.
Functional annotation and classification
GO and COG functional classifications
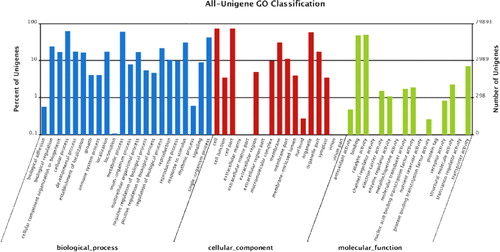
To facilitate categorization of the lingonberry transcriptome, GO was used to functionally annotate identified genes. Based on the GO analysis, 29,893 unigenes (44.1%) were assigned to GO ontologies (). Among them, 114,247, 86,664 and 34,403 sequences were assigned to the biological process (BP), cellular component (CC) and molecular function (MF) categories, respectively (). These sequences were further subdivided into 55 functional terms. Among BPs, ‘cellular process’ represented the most abundant subcategory followed by ‘metabolic process’ and ‘single-organism process’. In the MF category, ‘catalytic activity’ and ‘binding’ were the most represented GO terms. Regarding CCs, the major GO terms were ‘cell’, ‘cell part’ and ‘organelle’.
Figure 4. GO functional analysis of lingonberry All-Unigenes.
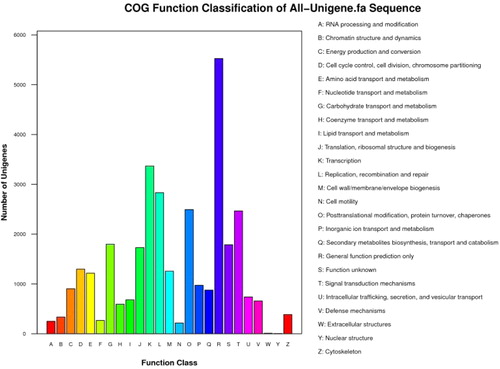
COG annotation can be used to phylogenetically evaluate the completeness and validity of unigene sequences. On the basis of COG analysis, 16,013 unigenes (23.6%) were annotated to 26 categories (). ‘General function prediction only’ (5526; 16.9%), ‘Transcription’ (3368; 10.3%) and ‘Replication, recombination and repair’ (2834; 8.7%) were the largest COG categories. ‘Extracellular structures’ (10; 0.03%) and ‘Nuclear structure’ (4; 0.01%) were shown as the smallest COG clusters. A total of 877 unigenes (2.7%) were matched into the ‘Secondary metabolites biosynthesis, transport and catabolism’ cluster, which suggests that the biosynthesis of such secondary metabolites has essential functions during the fruit development processes in lingonberry.
Figure 5. COG classification of lingonberry All-Unigenes.
KEGG enrichment analysis
KEGG analysis can aid the understanding of the biological functions of genes. As provided from and Supplemental File 3, 24,637 (36.3%) unigenes were annotated from the KEGG database and further categorized into 128 KEGG pathways. The metabolic pathway (5441; 22.08%) was the largest category. Secondly, 2697 unigenes (10.95%) were assigned to ‘Biosynthesis of secondary metabolites’. The most heavily represented processes under this category were ‘Phenylpropanoid biosynthesis’ (340), ‘Flavonoid biosynthesis’ (309), ‘Stilbenoid, diarylheptanoid and gingerol biosynthesis’ (234), ‘Flavone and flavonol biosynthesis’ (148), ‘Benzoxazinoid biosynthesis’ (80), ‘Isoflavonoid biosynthesis’ (63), ‘Isoquinoline alkaloid biosynthesis’ (57), ‘Tropane, piperidine and pyridine alkaloid biosynthesis’ (45), ‘Anthocyanin biosynthesis’ (37), ‘Glucosinolate biosynthesis’ (34), ‘Indole alkaloid biosynthesis’ (25), ‘Betalain biosynthesis’ (7) and ‘Caffeine metabolism’ (4). The KEGG enrichment analysis suggests that the biosynthesis pathway of flavonoids is active during lingonberry fruit development.
Putative genes related to flavonoid biosynthesis and transport
Flavonoids are a big class of polyphenolic phytochemicals with various physiological functions in plants [Citation40]. At least 42 phenolic compounds, including anthocyanidins, flavonols, phenolic acids, iridoids and proanthocyanidins, have been previously identified from lingonberry with liquid chromatography (LC) time-of-flight mass spectrometry (MS) and LC-MS/MS methods. Three cyanidin-derived anthocyanins have been identified, whereas quercetin-3-o-(4′′-HMG) -α-rhamnoside has been found to be a typical constituent of flavonols [Citation4]. Lingonberry flavonoid metabolites appear to involve quercetin in connection with cyanidin-derived anthocyanins, similar to the situation in apple, rosehips and flowers and callus cultures of bilberry [Citation41].
Flavonoids (in particular anthocyanins and proanthocyanidins) are biosynthesized by a cytosolic multienzyme complex via the upper part of the phenylpropanoid pathway [Citation42]. Candidate flavonoid structural genes (Supplemental File 4) in the lingonberry transcriptome included genes coding for phenylalanine ammonia lyase (PAL, 9), cinnamate 4-hydroxylase (C4H, 4), 4-coumarate CoA ligase (4CL, 9), chalcone synthase (CHS, 8), chalcone isomerase (CHI, 2), flavonoid 3-hydroxylase (F3H, 2), flavonoid 3′-hydroxylase (F3′H, 3), flavonoid 3′,5′-hydroxylase (F3′5′H, 7), dihydroflavonol 4-reductase (DFR, 12), anthocyanidin synthase (ANS, 2), flavonol synthase (FLS, 2), leucoanthocyanidin reductase (LAR, 5) and anthocyanidin reductase (ANR, 2). To be stably accumulated in vacuoles, free anthocyanins must first undergo various modification reactions, such as glycosylation, methylation and acylation [Citation43]. We found the following flavonoids in lingonberry: UDP-glucose flavonoid 3-O-glucosyl transferase (27), O-methyltransferase (8) and anthocyanin acyltransferase (13) (). Taken together, these results confirm that most flavonoid structural genes were expressed in lingonberry fruits. The exception was the expression of F3′5′H. Because no delphinidin derivatives have been detected in lingonberry, we assumed that F3′5′H is not expressed in lingonberry; however, several candidate F3′5′H transcripts were found in lingonberry transcriptome data. One possibility is that the F3′5′H enzyme is not expressed post-transcriptionally or post-translationally at detectable levels.
Figure 6. Lingonberry All-Unigenes involved in flavonoids metabolism emphasizing the anthocyanins, proanthocyanidins, and flavonols.
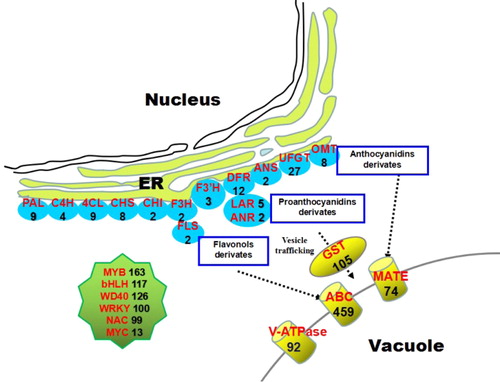
After completing the biosynthesis processes in the endoplasmic reticulum (ER), flavonoids are stored into vacuoles or other destinations [Citation44,Citation45]. Previous studies have suggested that flavonoid transport and storage involves three mechanisms. The mechanisms not only involve vesicle trafficking and a series of membrane transporters, but also involve glutathione S-transferase (GST) aided transport [Citation45,Citation46]. These flavonoid transport mechanisms do not exist wholly independently, but may instead be collaboratively integrated. In the lingonberry transcriptome (Supplemental File 4), 459 unigenes encoding ATP-binding cassette (ABC) transporter, 74 encoding MATE transporter, 105 encoding GST and 92 encoding V-ATPases were identified (). We also found 7, 38 and 8 unigenes encoding H+-PPases, SNAREs and vacuolar sorting receptors (VSRs), respectively. The result suggests that the three storage mechanisms may be present simultaneously but with different roles in lingonberry (i.e. transporting vesicles, GST or membrane transporters). In Arabidopsis, GREEN FLUORESCENT SEED9 (GFS9) is a Golgi-localized membrane protein with connecting function in vesicle trafficking mediated proanthocyanidin transport [Citation47]. Although two unigenes encoding GFS were identified in blueberry and cranberry in a previous study [Citation18,Citation21], we found no transcripts encoding GFS in lingonberry transcriptome. This absence indicates that the transport mechanisms in lingonberry may differ from those in blueberry and cranberry.
Putative transcription factors related to flavonoids biosynthesis and transport
TFs are important regulatory elements in flavonoid biosynthesis and transport [Citation48]. In most plants, the bHLH, MYB and WD transcription factors (TFs) regulate the flavonoid biosynthesis and transport at the transcriptional level, and these TFs could act as MBW ternary complexes [Citation49–52]. Among Vaccinium plants, putative R2R3MYB TFs have been found in bilberry, blueberry and bog bilberry [Citation18,Citation19,Citation53,Citation54]. Although the regulation of flavonoid biosynthesis is relatively well characterized in model plants, the overall regulatory mechanism of flavonoid accumulation is poorly understood in lingonberry. In our lingonberry transcriptome data set (Supplemental File 4), 163, 117 and 126 unigenes were annotated as MYB, bHLH and WD40 proteins, respectively. In addition, 100 WRKY, 99 NAC and 13 MYC TFs were uncovered (). The generated transcriptome data can thus provide an important tool to reveal regulation networks involved in the flavonoid metabolites and other metabolites in lingonberry. The exact functions of these putative TFs should be investigated in further studies.
Analysis of the DEGs throughout fruit development
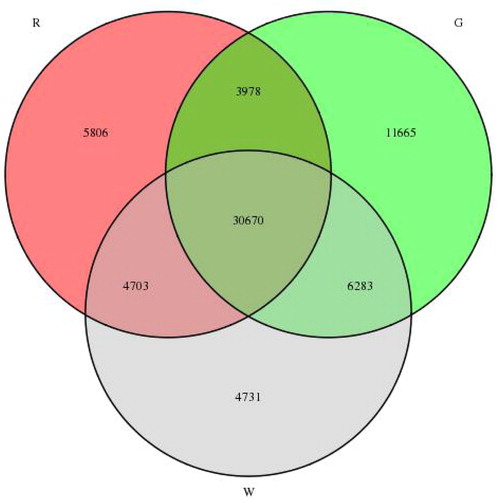
To study the gene expression patterns during lingonberry fruit development, clean reads from the cDNA libraries were compared with the assembled transcriptome. As indicated in , 30,670 unigenes (45.2%) were expressed throughout the three stages of fruit development. Furthermore, 22,202 unigenes (32.7%) were only stage-specifically expressed genes. Of these exclusively expressed unigenes, 11,665, 4731 and 5806 unigenes were specifically expressed at G, W and R, respectively.
Figure 7. Veen diagram showing the number of commonly and uniquely expressed genes at different fruit development stages.
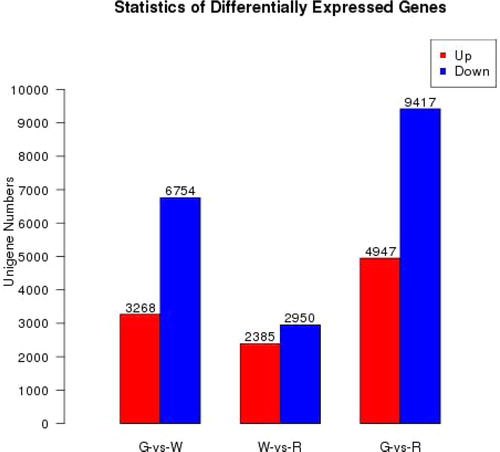
A comparison of the three libraries revealed 10,022 genes differentially expressed between G and W, with 5335 DEGs identified between W and R and 14,364 between G and R (). Overall, 29,721 (43.8%) unigenes showed differential expression during lingonberry fruit maturation, which indicates that a series of metabolic transitions took place between G, W and R stages of berry development. The fact that more DEGs were detected between G and W suggests that the process of fruit de-greening involves greater changes than that of the later red-colouring stage. Notably, downregulated genes predominated among these DEGs of three comparison combinations, which indicates that more genes related to the metabolism pathways are downregulated during fruit ripening.
Figure 8. The number of up- and down-regulated genes in G vs. W, W vs. R and G vs. R.
Annotation analysis of DEGs
To better understand the function of the DEGs and further reveal the mechanism regulating lingonberry flavonoids biosynthesis, all DEGs at three time points were successfully annotated using GO, COG, Pfam, KEGG, SwissProt and NR databases (Supplemental File 5).
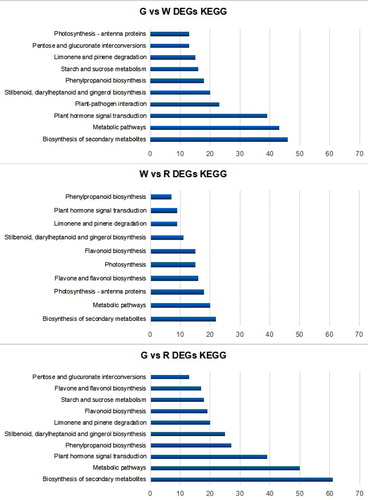
The transcriptome profile of the KEGG pathways of lingonberry fruit is valuable for understanding the physiological processes of lingonberry flavonoid biosynthesis. To evaluate the metabolic differences of the lingonberry fruit at three different stages, we also performed KEGG analysis to categorize DEG biological functions. The top 10 DEG-enriched pathways between library pairs are shown in . Comparisons of G, W and R libraries revealed that most DEGs were distributed in KEGG pathways associated with ‘Biosynthesis of secondary metabolites’, ‘Metabolic pathways,’ and ‘Plant hormone signal transduction’. With respect to G vs. W, fruit cells were characterized by high levels of DEGs involved in ‘Biosynthesis of secondary metabolites’, ‘Metabolic pathways,’ and ‘Plant hormones signal transduction’. A comparison of W vs. R indicated that fruit cells between those two stages had many DEGs related to flavonoid biosynthesis such as phenylpropanoid, flavone, flavonol and flavonoid biosynthesis pathways. Taken together, these results imply that lingonberry fruit development involves a series of changes to secondary metabolites and plant hormone signalling.
Figure 9. KEGG analysis of DEGs in G vs. W, W vs. R and G vs. R. The horizontal axis shows the negative log10 of p-value, the vertical axis represents KEGG metabolism pathway category.
Expression patterns analysis by qRT-PCR
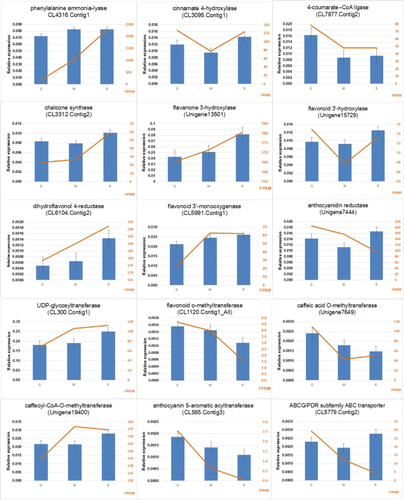
To further analyse the flavonoid biosythesis genes in lingonberry, we performed quantitative RT-PCR (qRT-PCR) analysis of their expression patterns with 15 unigenes (Supplemental File 1). As can be seen in , the expression profiles of most of these unigenes were similar to the patterns suggested by the transcriptome analysis. Therefore, the lingonberry transcriptome generated from the study presents reliable data, and provides a gene expression profile of flavonoid metabolism for further studies. The expression of genes involved in the flavonoid biosynthetic pathway generally increases during fruit development, especially the key genes in the middle and lower parts of the pathway are up-regulated strongly, which may imply a high accumulation of flavonoids in the fruit in the course of fruit development. The expression profiles of CHS, F3H, FLS, DFR, ANS and CCo AOMT were consistent with the trend of the total flavonoid content and the total anthocyanin content. These results further confirmed the reliability of the transcriptome data and that the candidate genes may be involved in the flavonoid synthesis pathway at the fruit development period. In addition, the analysis of the transcriptome data showed that the role of PAL genes in the anthocyanin biosynthesis pathway is not critical, and the up-regulated expression of CHS, F3H, DFR and ANS genes is the most critical link to increase the total anthocyanin content in lingonberry.
Figure 10. Expression pattern analysis of differentially expressed transcripts from RNA-seq and qRT-PCR. Line graphs represent RNA-seq expression data. Bars represent the qRT-PCR result.
Conclusions
We generated a comprehensive transcriptome assembly representing gene expression profiles throughout the fruit development and ripening processes in lingonberry. The RNA-seq data allowed us to identify many transcripts related to flavonoid biosynthesis, modification, transport and regulation, and to analyse their expression levels on the transcriptome level. Fifteen key enzyme genes related to flavonoids biosynthesis pathways were identified. The unigene sequences generated from this study could not only serve as a foundation to provide insights into molecular mechanisms underlying flavonoids accumulation in lingonberry, but also represent a genomics resource for development of molecular-assisted markers for lingonberry breeding programmes.
Consent for publication
Not applicable.
Supplemental Material
Download PDF (84.7 KB)Supplemental Material
Download MS Word (0)Supplemental Material
Download PDF (148.4 KB)Supplemental Material
Download PDF (103.1 KB)Supplemental Material
Download PDF (82.5 KB)Acknowledgements
We would like to thank Jian Xu, Hainan Liu and Yushan Liu for experimental materials and methods assistance.
Disclosure statement
Haiyue Sun and Yadong Li conceived the study; Youwen Tian contributed to analyse the data and wrote the article; Zhili Ma and Haohao Ma contributed to the experiments; Yu Gu prepared plant samples and reagents. The authors declare that they have no competing interests.
Availability of data and materials
The lingonberry RNA-Seq raw data files are available in NCBI SRA database (SAMN073054 17, SAMN07305418 and SAMN07305419).
Additional information
Funding
References
- Galletta GJ, Ballington JR. Blueberries, cranberries, and lingonberries. In: Janick J, Moore JN, editors. Vol. 2. Fruit breeding, vine and small fruits. Hoboken (NJ): Wiley; 1996. p. 65–83.
- Karppinen K, Zoratti L, Nguyenquynh N, et al. On the developmental and environmental regulation of secondary metabolism in Vaccinium spp. berries. Front Plant Sci. 2016;7:655.
- Vander Kloet SP. The genus Vaccinium in North America. In:Molly W, Sharon R, Frances S, editors. Monograph. Wolfville, Nova Scotia, Canada: Biology Department of Acadia University, Wolfville; 1988.
- Hokkanen J, Mattila S, Jaakola L, et al. Identification of phenolic compounds from lingonberry (Vaccinium vitis-idaea L.), bilberry (Vaccinium myrtillus L.) and hybrid bilberry (Vaccinium x intermedium Ruthe L.) leaves. J Agric Food Chem. 2009;57(20):9437–9447.
- Jungfer E, Zimmermann BF, Ruttkat A, et al. Comparing procyanidins in selected Vaccinium species by UHPLC-MS(2) with regard to authenticity and health effects. J Agric Food Chem. 2012;60(38):9688–9696.
- Heyman L, Axling U, Blanco N, et al. Evaluation of beneficial metabolic effects of berries in high-fat Fed C57BL/6J Mice. J Nutr Metab. 2014;2014:403041.
- Isaak CK, Petkau JC, O K, et al. Manitoba lingonberry (Vaccinium vitis-idaea) bioactivities in ischemia-reperfusion injury. J Agric Food Chem. 2015;63(23):5660–5669.
- Hossain MZ, Shea E, Daneshtalab M, et al. Chemical analysis of extracts from Newfoundland berries and potential neuroprotective effects. Antioxidants. 2016;5(4):36.
- Hall IV, Shay JM. The biological flora of Canada, 3: Vaccinium vitis-idaea L. var. minus. Lodd. Supplementary account. Can Field Nat. 1981;95:434–464.
- Del Valle JC, Alcalde-Eon C, Escribano-Bailón MT, et al. Stability of petal color polymorphism: the significance of anthocyanin accumulation in photosynthetic tissues. BMC Plant Biol. 2019;19(1):496.
- Garzón GA, Narváez CE, Riedl KM, et al. Chemical composition, anthocyanins, non-anthocyanin phenolics and antioxidant activity of wild bilberry (Vaccinium meridionale Swartz) from Colombia. Food Chem. 2010;122(4):980–986.
- Ieri F, Martini S, Innocenti M, et al. Phenolic distribution in liquid preparations of Vaccinium myrtillus L. and Vaccinium vitis idaea L. Phytochem Anal. 2014;24:467–475.
- Mikulic-Petkovsek M, Schmitzer V, Slatnar A, et al. A comparison of fruit quality parameters of wild bilberry (Vaccinium myrtillus L.) growing at different locations. J Sci Food Agric. 2015;95(4):776–785.
- Bujor OC, Tanase C, Popa ME. Phenolic antioxidants in aerial parts of wild Vaccinium species: towards pharmaceutical and biological properties. Antioxidants. 2019;8(12):649.
- Vilkickyte G, Raudonis R, Motiekaityte V, et al. Composition of sugars in wild and cultivated lingonberries (Vaccinium vitis-idaea L.). Molecules. 2019;24(23):4225.
- Račkauskienė I, Pukalskas A, Fiore A, et al. Phytochemical-Rich antioxidant extracts of Vaccinium Vitis-idaea L. leaves inhibit the formation of toxic maillard reaction products in food models. J Food Sci. 2019;84(12):3494–3503.
- Strickler SR, Bombarely A, Mueller LA. Designing a transcriptome next-generation sequencing project for a nonmodel plant species. Am J Bot. 2012;99(2):257–266.
- Mutz K-O, Heilkenbrinker A, Lönne M, et al. Transcriptome analysis using next-generation sequencing. Curr Opin Biotechnol. 2013;24(1):22–30.
- Li X, Sun H, Pei J, et al. De novo sequencing and comparative analysis of the blueberry transcriptome to discover putative genes related to antioxidants. Gene. 2012;511(1):54–61.
- Zifkin M, Jin A, Ozga JA, et al. Gene expression and metabolite profiling of developing highbush blueberry fruit indicates transcriptional regulation of flavonoid metabolism and activation of abscisic acid metabolism. Plant Physiol. 2012;158(1):200–202.
- Polashock J, Zelzion E, Fajardo D, et al. The American cranberry: first insights into the whole genome of a species adapted to bog habitat. BMC Plant Biol. 2014;14:165.
- Sun H, Liu Y, Gai Y, et al. De novo sequencing and analysis of the cranberry fruit transcriptome to identify putative genes involved in flavonoid biosynthesis, transport and regulation. BMC Genomics. 2015;16:652.
- Walworth AE, Chai B, Song GQ. Transcript profile of flowering regulatory genes in VcFT-Overexpressing Blueberry Plants. PLoS One. 2016;11(6):e0156993.
- Wootton-Beard PC, Moran A, Ryan L. Stability of the total antioxidant capacity and total polyphenol content of 23 commercially available vegetable juices before and after in vitro digestion measured by FRAP, DPPH, ABTS and Folin-Ciocalteu methods. Food Res Int. 2011;44(1):217–224.
- Wen-Fang MA, Hui-Lian D, Yi C, et al. Research on the determination of total flavonoids content and colour reaction system in Scoparia dulcis. Chin J Exp Tradition Med Form. 2013;19(03):112–115.
- Song IK, Rahim MA, Afrin KS. Expression of anthocyanin biosynthesis-related genes reflects the peel color in purple tomato. Hortic Environ Biotechnol. 2018;59(3):435–445.
- Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652.
- Pertea G, Huang X, Liang F, et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19(5):651–652.
- Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 19901;215(3):403–410.
- Gish W, States DJ. Identification of protein coding regions by database similarity search. Nat Genet. 1993;3(3):266–272.
- Iseli C, Jongeneel CV, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol. 1999;99:138–148.
- Conesa A, Götz S, García-Gómez JM, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676.
- Ye J, Fang L, Zheng H, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:W293–W 297.
- Kanehisa M, Araki M, Goto S, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–D48 4.
- Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7(10):986–995.
- Kim KI, van de Wiel MA. Effects of dependence in high-dimensional multiple testing problems. BMC Bioinformatics. 2008;9:114.
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45.
- Bhullar KS, Rupasinghe HP. Antioxidant and cytoprotective properties of partridgeberry polyphenols. Food Chem. 2015;168:595–605.
- Dróżdż P, Šėžienė V, Pyrzynska K. Phytochemical Properties and Antioxidant Activities of Extracts from Wild Blueberries and Lingonberries. Plant Foods Hum Nutr. 2017;72(4):360–364.
- Buer CS, Imin N, Djordjevic MA. Flavonoids: new roles for old molecules. J Integr Plant Biol. 2010;52(1):98–111.
- Jaakola L, Määttä K, Pirttilä AM, et al. Expression of genes involved in anthocyanin biosynthesis in relation to anthocyanin, proanthocyanidin, and flavonol levels during bilberry fruit development. Plant Physiol. 2002;130(2):729–739.
- Petrussa E, Braidot E, Zancani M, et al. Plant flavonoids-biosynthesis, transport and involvement in stress responses. Int J Mol Sci. 2013;14(7):14950–14973.
- Springob K, Nakajima J, Yamazaki M, et al. Recent advances in the biosynthesis and accumulation of anthocyanins. Nat Prod Rep. 2003;20(3):288–303.
- Yazaki K. Transporters of secondary metabolites. Curr Opin Plant Biol. 2005;8(3):301–307.
- Zhao J. Flavonoid transport mechanisms: how to go, and with whom. Trends Plant Sci. 2015;20(9):576–585.
- Zhao J, Dixon RA. The ‘ins’ and ‘outs’ of flavonoid transport. Trends Plant Sci. 2010;15(2):72–80.
- Ichino T, Fuji K, Ueda H, et al. Hara-Nishimura I. GFS9/TT9 contributes to intracellular membrane trafficking and flavonoid accumulation in Arabidopsis thaliana. Plant J. 2014;80(3):410–423.
- Hichri I, Barrieu F, Bogs J, et al. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J Exp Bot. 2011;62(8):2465–2483.
- Koes R, Verweij W, Quattrocchio F. Flavonoids: a colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005;10(5):236–242.
- Li S. Transcriptional control of flavonoid biosynthesis: fine-tuning of the MYB-bHLH-WD40 (MBW) complex. Plant Signal Behav. 2014;9(1):e27522.
- Xu W, Dubos C, Lepiniec L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 2015;20(3):176–185.
- Chezem WR, Clay NK. Regulation of plant secondary metabolism and associated specialized cell development by MYBs and bHLHs. Phytochemistry. 2016;131:26–43.
- Jaakola L, Poole M, Jones MO, et al. A SQUAMOSA MADS box gene involved in the regulation of anthocyanin accumulation in bilberry fruits. Plant Physiol. 2010;153(4):1619–1629.
- Primetta AK, Karppinen K, Riihinen KR, Jaakola L. Metabolic and molecular analyses of white mutant Vaccinium berries show down-regulation of MYBPA1-type R2R3 MYB regulatory factor. Planta. 2015;242(3):631–643.