
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
We have established a highly efficient 96-well format based strategy to characterize nanobody (Nb) from an expressed alpaca Nb repertoire by combining heavy-chain antibody variable region gene cloning with Nb expression and reactivity profiling at the single cell level. Individual alpaca B lymphocytes were isolated based on B cell surface marker and membrane type antigen specific antibodies expression by fluorescence activated cell sorting (FACS). Single cell reverse transcription and polymerase chain reaction (PCR) amplification of CH1 and CH2 domain enable the identification and exclusion of B cell clones that express regular antibodies. The corresponding heavy-chain antibody variable region gene transcripts, which are CH1-CH2 domain negative, are then amplified from single B cell clones by nested PCR. Then Nbs were expressed in Escherichia coli cells and four Nbs showed high affinity against Pseudomonas aeruginosa Exotoxin A (PEA). Moreover, two PEA Nbs that recognize unique epitopes on PEA have been successfully applied to protect human cells from PEA induced apoptosis. In summary, our FACS and single cell reverse transcription polymerase chain reaction (RT-PCR) based strategy for identifying recombinant Nbs from single alpaca B cells allows highly efficient and unbiased characterization of the expressed Nb repertoires by sequence analysis, expression and parallel antibody reactivity testing.
Introduction
The single-domain antibody (sdAb) is the smallest antibody naturally existing in camelidae and sharks. This unique antibody in camelidae lacks light chains and has only one variable region of heavy chain (VHH) and two constant regions, CH2 and CH3 [Citation1]. Compared to many other formats of antibodies, the sdAb has numerous advantages due to its natural characteristics. The variable region of the sdAb is known as nanobody (Nb). The comparatively low molecular mass (∼15 kD) of the Nb renders a better tissue penetration compared to common antibodies (150 kD −160 kD) or single-chain variable fragment (ScFv, 30 kD). Of the three complementary determining regions (CDRs) in the Nb, the extended CDR3 loop plays an important role in the antigen-binding specificity and affinity and could form a convex shape which has superior cryptic cleft accessibility than conventional antibodies [Citation2,Citation3]. The Nb can be easily expressed in bacteria, yeast or plants for production and modification [Citation4,Citation5]. In addition, the less lipophilic residues of the Nb increase its solubility in an aqueous environment [Citation6]. An efficient refolding ability ensures that the Nb regains its functionality after chemical and thermal denaturation and thus the Nb shows great heat resistance and stability. These unique features of the Nb make it a promising alternative to conventional antibodies for research, diagnosis and therapeutic application [Citation7].
Phage display is widely used for screening and isolating novel target-specific nanobody or antibody fragments [Citation8]. In general, the putative Nb sequences are cloned into a phage-display phagemid vector and transformed into Escherichia coli to generate an Nb library. Antigen binding immune repertoires could be isolated after several rounds of panning from the library. However, this method is often restricted by the library size and sequence diversity, and the panning of the library is a time-consuming process and could be limited by the coupling capability of the antigen to the solid phase [Citation9].
Fluorescence activated cell sorting (FACS) has been applied for rapid cloning of monoclonal antibodies from human, mouse and rabbit B cells [Citation10,Citation11]. In this setting, the fluorescence labelled antigen and antibodies against B cell surface specific markers were co-stained with the B cells, then the double positive cells are individually sorted into 96 well plates, single cell reverse transcription polymerase chain reaction (RT-PCR) is used to amplify light chain and heavy chain variable regions (VL and VH) to identify the positive immune repertoires. Theoretically, the same approach could be applied to Nb isolation. However, the FACS method has failed to clone Nb sequences thus far. Few camelidae B cell surface marker antibodies are available; only antigen binding camelidae peripheral blood mononuclear cells will be sorted. After sorting, both types of B cells that express regular antibodies and heavy chain antibodies as well as other antigen-binding cells including T cells, macrophages are all collected. To identify the Nb sequences from all cell collections, sequence analysis is required in addition to single-cell RT-PCR. Although Next Generation Sequencing (NGS) may be applied to identify Nb sequences from a B cell pool or library, this process is time consuming and costly [Citation12].
In the present study, we developed a rapid method to obtain Nb immune repertoires. An anti-llama pan B antibody conjugated with phycoerythrin (PE) labelled secondary antibody was employed to select alpaca B cells and a fluorescein-isothiocyanate (FITC) labelled antigen (PEA) for isolating the B cells which produce antigen specific antibodies. When applying fluorescence labelled pan B antibody combined with fluorescence labelled antigen to screen alpaca peripheral blood lymphocytes (PBLs), the double positive single B cells could be sorted into 96 well plates, and cDNA could then be synthesized from the single cell by reverse transcription. B cells that express regular antibodies (VH-B cells) were excluded by PCR using the primer located in the CH1 region that is absent in Nb. The specific Nbs could then be amplified from cDNA samples of single B cells that expressed heavy chain antibodies (VHH-B cells). The PCR products were cloned for subsequent sequence analysis and Nbs against PEA were expressed in E. coli. PEA, a member of the mono-ADP-ribosyltransferase family that can inactive elongation EF2 factor of the host cell and kill the cell, is one of the virulence factors produced by Pseudomonas aeruginosa, enabling it to adhere to tissue surfaces, to damage tissue for dissemination and nutrition supply [Citation13]. For successful treatment of P. aeruginosa infection, not only the colonization and proliferation of P. aeruginosa should be inhibited, but the PEA toxicity should also be neutralized to protect the patients from cell apoptosis and subsequent organ failure. Therefore, neutralization antibodies, especially Nbs for PEA, were essential for the effective treatment of P. aeruginosa infection. Herein, compared with the Phage display technology, single alpaca B cells to clone Nbs could be a rapid procedure for isolation of antigen-specific Nbs for various medical diagnostic applications.
Materials and methods
Llama immunization
To obtain PEA-specific binding heavy chain-only antibodies (VHHs), a healthy 2-year-old male alpaca provided by Qingdao Veterinary Research Institute was immunized subcutaneously with high purity PEA (400 μg) (Santa Cruz Biotech, CA, USA) mixed with an equal volume of Freund's complete adjuvant (Sigma, USA) for the first time, and Freund's incomplete adjuvant mixed with 400 μg PEA (1:1) was injected for the following 4 times at 2-week intervals. Ten days after the final immunization, 10 mL of blood was collected into tubes containing ethylenediaminetetraacetic acid (EDTA) as anticoagulant for later peripheral blood lymphocytes isolation.
Alpaca serum IgG purification and titration
To detect the antibody titres after each immunization, 0.4 mL of serum was diluted 2-fold in phosphate-buffered saline (PBS), IgG subclasses were obtained by successive affinity chromatography on 1 mL HiTrap Protein G and Protein A columns (GE) using an NGC Chromatography System (Bio-Rad, USA). The diluted serum was loaded onto the HiTrap protein G column (GE Healthcare, USA) at 0.2 mL/min, the flow through fraction (IgG2 subclass) was collected and labelled. The IgG3 subclass was then eluted from the protein G column using 0.15 mol/L NaCl, 0.1 mol/L acetic acid at pH 3.5. Then the IgG1 subclass was eluted from the column with 0.1 mol/L glycine–HCl, pH 2.7. The flow-through was loaded on the Protein A column to recover the IgG2 subclass. The IgG2 subclass was eluted using 0.15 mol/L NaCl, 0.1 mol/L acetic acid at pH 3.5. All the eluted fractions were neutralized using 2 mol/L Tris–HCl, pH 9.0 and dialysed against PBS [Citation14]. The IgG fractions were stored at –20 °C after assessed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE).
The 96-well plate was coated with 10 μg/mL PEA in Bicarbonate/carbonate coating buffer (100 mmol/L, pH 9.6) overnight at 4 °C. The plate was blocked with 200 µL blocking buffer (5% non-fat dry milk/PBS) per well. Then serially diluted alpaca serum and purified IgG2/3 were added to the wells and incubated for 2 h at room temperature. After washing, 100 μL of 1:5000 diluted horseradish peroxidase (HRP) labelled goat anti-llama antibody was added to each well and incubated for 1 h at room temperature. Finally, 100 μL of 3,3′-diaminobenzidine (DAB) substrate solution with H2O2 was added to each well for colour development.
Fluorescence labelled antigen preparation
The PEA antigen was labelled with FITC according to the protocol supplied by the ReadiLink™ Rapid FITC Antibody Labeling Kit (AAT Bioquest Inc., USA). Briefly, 50 μg of protein was diluted in 50 μL of labelling buffer in a filtration tube, 15 μL of FITC was added, mixed thoroughly and incubated at 37 °C for 60 min. After the incubation, the tube was centrifuged at 12,000 g for 10 min and repeated one more time after adding 50 μL of labelling buffer to remove uncoupled FITC molecules, followed by adding 0.2 mL labelling buffer to the filtration tube and mixing thoroughly. The filtration tube was inverted and placed on another clean centrifugal tube and FITC labelled PEA antigen eluted from the filtration tube by centrifugation at 12,000 g for 10 min.
Flow cytometry single cell sorting and cDNA preparation from single B cells
Ten millilitres blood collected from PEA immunized alpaca was diluted 1:1 with PBS and gently poured on top of equal volume of lymphocyte separation medium (Tianjin Hao Yang Biological Manufacture Co., Ltd., China) in a 50-mL tube, followed by centrifugation at 500 g for 20 min at room temperature. The ring of PBLs formed in the interface was aspirated carefully by a pipet from the tube and transferred into a new 15-mL conical tube, and the cells were washed twice with PBS and resuspended in 2 mL of PBS. Cells of 107 in 1 mL PBS were co-stained with both PEA antigen conjugated with FITC and anti-llama pan B cell antibody (Kingfisher Biotech Inc., USA), then a secondary antibody coupled with PE (Southern Biotech, USA) was used to label the anti-llama pan B cell antibody. Dead cells were excluded with 7-aminoactinomycin D (7-AAD) (eBioscience, USA). Single cells were sorted by a FACSAria II (BD) instrument (BD sciences, USA) into 96-well PCR plates containing 4 μL/well of ice-cold 0.2% Triton X-100 supplemented with 2 U/μL RNAsin, 1 μL oligo-dT, and 1 μL dNTP mix for direct cell lysis. For subsequent cDNA preparation, the plate was sealed with CyclerSeal Sealing Film and incubated at 72 °C for 3 min, then the plate was placed on ice immediately. Six microlitres/well of reverse transcriptase mix (Promega, USA) was added, and the plate was sealed again and incubated at 42 °C for 30 min to synthesize cDNA from each cell [Citation15].
Cloning of Nb gene by nested PCR and sequencing analysis
Five microlitres of the cDNA from each of the 96-well plates was transferred to a new 96-well PCR plate containing 15 μL of PCR master mix I with CALL001 and CALL002 primers (). The PCR conditions (Hangzhou Bio-gener Technology Co., Ltd., China) were: 94 °C 1 min, 58 °C 1 min and 72 °C 1 min for 30 cycles, both the VH and VHH fragments would be amplified from this step. After the first round of PCR, 1 μL of the PCR product was transferred to another 96 well PCR plate, 19 μL of PCR master mix II with CHPCitation1 and CHPCitation2 primers was added to identify VH fragments. Only VH fragments from VH-B cell cDNA would be amplified by CHPCitation1 and CHPCitation2 primers in this step. The PCR products were visualized in a 2.5% agrose electrophoresis. Finally, 1 μL of PCR product from the first round PCR that was negative for CHPCitation1 and CHPCitation2 amplification was used for VHH fragment amplification by PCR master mix III containing VHH-Back and VHH-For primers. The PCR products were harvested, digested by Pst I (NEB, USA) and BstE II (NEB, USA) and cloned into pMES4 vector for sequence analysis and periplasmic expression of the nanobodies. Recombinant plasmid was confirmed by digestion with appropriate restriction enzymes (NEB, USA) and subjected to sequence analysis with the primers pMES-F and pMES-R.
Table 1. List of primers used in this study.
Nbs expression, purification and identification of PEA nanobodies
After confirmation of the Nb sequences by sequencing analysis, the recombinant plasmid was directly transformed into E. coli BL21 (DE3) cells for Nb expression. The expression of Nb protein was induced with 1 mmol/L IPTG (Sigma, USA) at 30 °C overnight, cells collected by centrifugation at 5,000 g for 30 min, pellets resuspended in TES buffer (500 mmol/L sucrose, 200 mmol/L Tris-HCl, pH 8.0 and 0.5 mmol/L EDTA) on ice for 10 min, and then mixed with an equal volume of 1/4 concentration TES shocking buffer on ice for 10 min to burst the cells and release the periplasmic proteins [Citation16]. To purify the Nb protein, the supernatant was collected and loaded onto a Ni2+-NTA resin chromatography column (GE Healthcare, USA), the column was washed sequentially with 20 volumes of washing buffer containing 20 mmol/L imidazole and 50 mmol/L imidazole and then the Nb protein was eluted with elution buffer containing 500 mmol/L imidazole. Target protein fractions were pooled and dialysed against PBS to remove imidazole. The purity of the Nb protein was examined by SDS-PAGE.
Affinity determination by surface plasmon resonance
The affinity values (KD) of the obtained Nbs against PEA were determined by SPR technology using Biacore T100 system (GE Healthcare, USA). First, a commercial anti-His tag antibody was immobilized on CM5 sensor chip. Second, the Nbs of 10 μg/mL were captured onto the chip surface to obtain a proper response signal. Third, five different concentrations (80 μg/mL, 40 μg/mL, 20 μg/mL, 10 μg/mL, 5 μg/mL) of PEA were injected with a flow rate of 30 μL per minute for 60 s and then dissociated for 600 s. Afterward, the binding signals and dissociation signals were recorded and used to calculate the ka and kd values for affinity KD values (KD).
In vitro neutralization assay
A431 cells were maintained in DMEM (Dulbecco's Modified Eagle Medium) supplemented with 10% fetal bovine serum (FBS) at 37 °C in a CO2 incubator. Prior to the experiment, cells were seeded into 96-well culture plates and incubated for 4–6 h, 6000 cells per well. PEA and PEA/nanobody mixtures were incubated at 37 °C in cell culture media for 1 h before being added to the 96-well plates in triplicate wells; the plate was then incubated for 48 h at 37 °C in a CO2 incubator. The final concentration of PEA was 5 μg/mL, the concentrations of the nanobodies were: 200, 160, 80, 40, 20, 10, 5, 2.5 μg/mL. After 48 h, the culture medium was discarded and 10 μL of MTT and 100 μL of fresh medium were added to each well and incubated for 4 h. The culture medium was then discarded, 110 μL of Formazan solvent was added per well and the plate was spun for 10 min at low speed. Finally, the absorbance of each well was measured by a microplate reader at 570 nm.
Epitope mapping of the PEA nanobodies
The epitopes that were recognized by the two nanobodies were mapped by Western blotting using truncated PEA protein. The DNA fragment for N-terminal domain I and domain II (NP_249839.1: 27-409aa, tPEA1) and the DNA fragment for domain II and C-terminal domain (NP_249839.1: 277-638aa, tPEA2) were optimized for E. coli expression and synthesized The DNA fragments were cloned into pET28a vector. The recombinant proteins tPEA1 and tPEA2 expressed in the E. coli cells were purified and transferred to a PVDF (polyvinylidene difluoride) membrane. The membrane was blocked and incubated with the nanobody in the blocking buffer as the primary antibody; the membrane was then incubated with 1:5000 diluted secondary antibody (peroxidase-conjugated goat anti-llama IgG). The proteins on the membrane were detected with DAB developing solution.
Results and discussion
Confirmation of immune response in alpaca immunized with PEA
To generate maximal immune response, a 2-year-old healthy male alpaca was immunized with PEA over a period of 7 weeks. The total antigen-specific IgG and heavy chain antibodies IgG2/3 titres in the serum of the alpaca were tested after each immunization. The total IgG titre reached 1:1,000,000 and IgG2/3 titre 1:300,000 after the fourth immunization, indicating that the immunization with PEA raised a strong immune response in the animal. In order to identify the immune repertoires of Nb specifically produced in the B cells, the PBLs were isolated from blood of the immunized alpaca by gradient density centrifugation. In total, 2 × 107 PBLs were harvested from 10 mL blood for subsequent experiments.
Sorting of single B cells producing the immune repertoires of Nb
A number of single B cell technologies using flow cytometry as a sorting method allow the efficient cloning of the B cell repertoires from immunized animals, especially for human and mouse B cells, because of the availability of a large number of antibody reagents that enable both positive identification of B cells and negative elimination of non-B cell populations including T cells, macrophages and neutrophils. These technologies have shown significant advantages over surface display and hybridoma approaches. However, due to the lack of antibody reagents for camelids immune cells as well as reagents for distinguishing VHH-B cells from VH-B cells, the single B cell technologies have not been successfully applied to cloning Nb coding sequences from camelids B cell populations. To clone genes encoding Nbs, we tested a strategy as depicted in . A llama pan B antibody which could bind to all the B cells was used to isolate B cells from other cell populations such as T cells, macrophages, dendritic cells. Due to the lack of a fluorescence molecule conjugated product, a secondary antibody labelled with PE was used to bind the pan B antibody. PEA antigen, which is labelled with FITC, could bind to the B cells that only produced antibodies against PEA. Dual positive lymphocyte cells for the two labelled molecules contain the immune repertoires of Nb. As shown in , a positive signal was confirmed individually by staining PBLs with pan B antibody or PEA-FITC. Dead cells were sorted out by 7-AAD staining; live B cells positively stained with both FITC labelled PEA antigen and anti-llama pan B cell antibody (PE) were individually sorted into a 96-well plate (Q2 of ). Approximately 6.7% of the total PBLs were dual positive and harvested for subsequent investigation.
Figure 1. Schematic depictions of strategies to sort single alpaca antigen specific memory B cells.
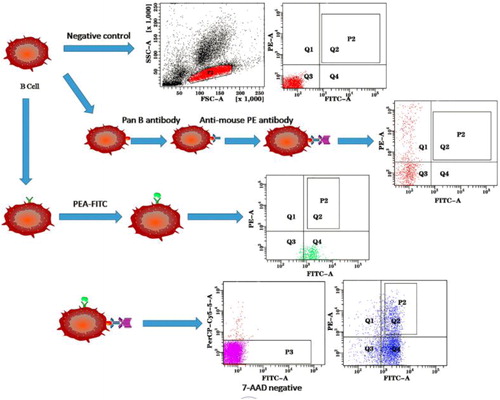
In addition, the phage display technology depends on fixing the antigen on the ‘solid phase’, which not only wastes large amounts of antigens, but also inevitably blocks part of the antigenic determinants, which will negatively affect the screening of antibodies, resulting in the missing of positive clones or the selection of false positive clones [Citation17,Citation18]. However, using of FACS allowed the maximal maintenance of the integrity of antigen epitopes, greatly increasing the interaction of antigen with B cells and thus the screening efficiency of positive cells.
Identifying Nb genes and their subcloning
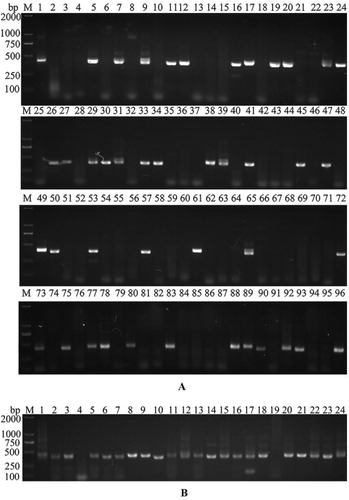
There are 3 types of IgG antibody produced by B cells in the blood of camelids, IgG1, IgG2 and IgG3. IgG1 is the regular antibody with light chain and regular VH-CH1-CH2-CH3 heavy chain, IgG2 and IgG3 are heavy chain only antibodies comprising VHH-CH2-CH3 heavy chain, lacking the light chain and the heavy chain CH1 domain. So two new primers, CHPCitation1 and CHPCitation2 were designed at the CH1 and CH2 regions of the IgG1 constant region to identify the Fc fragment amplified from IgG1 cDNA. cDNA was first synthesized from the single cells in 96-well plate, then the VH fragments of IgG1 or VHH fragments of IgG2/3 from single B cell cDNA samples were amplified using CALL001 and CALL002 primers as aforementioned [Citation19]. Fc fragments were next identified by a second round of PCR using CHPCitation1 and CHPCitation2 primers. Finally, VHH fragments were specifically amplified using VHH-Back and VHH-For primers from these cDNA samples that were negative from the second round of PCR. Forty-three out of 96 sorted single cells gave positive amplifications for CHPCitation1-CHPCitation2 PCR (), indicating that 48% of the analysed cells were VH-B cells producing regular antibody. Fifty-three negative clones were chosen for VHH fragment amplification; 45 VHH fragments were successfully amplified (). The PCR products were cloned into a bacterial expression vector. After sequence analysis, 39 fragments were confirmed as VHH sequences, 2 were regular VH sequences and 4 were unknown sequences without a long open reading frame. All 39 colonies (defined as Nb1 to Nb39) contained correct (in frame) VHH fragments representing 22 individual VHH sequences with diverse complementary determining region 3 (CDR3) sequences. Based on specific PCR analysis, we have demonstrated that about 90% of single sorted cells showed B cell origin, indicating that this dual parameter approach is a very efficient method to eliminate non-B cell origin cells.
Figure 2. Amplification of Nb genes by nested PCR. (A) The second PCR amplification using the CHPCitation1 and CHPCitation2 primer pair to distinguish the conventional antibody from Nb, with all the 96 amplification results shown. (B) Amplification of Nb genes in target samples by PCR with VHH primers and 24 amplification results shown.
For a standard phage display library construction and screening, 21–24 days would be required to obtain Nb sequences. For the method developed in this study, it took only 1 day for blood collection and cell sorting, with additional 1–2 days for single cell RT-PCR to directly clone the Nb sequences. Thus we have provided a proof of concept for rapid cloning of antigen specific Nbs from single alpaca B cells in 2–3 days. The method can be further optimized. For example, an antibody could be developed to distinguish VHH-B cells from VH-B cells for the purpose of obviating the first PCR step to identify VH-B cells. The procedure can also be automated for high throughput cloning of Nbs.
Functional and biophysical characterization of Nbs
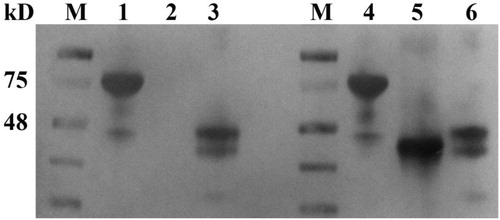
To determine whether Nbs isolated from single-cell sorting would function as antibodies, 12 VHH fragments inserted into the bacterial expression vector were transformed into BL21 cells for protein expression. The recombinant cells were induced by IPTG and periplasmic proteins were extracted and purified using a Ni2+–NTA resin chromatography column. Then the purified proteins were analysed by SDS-PAGE after dialysis against PBS and concentration with ultrafiltration. SDS-PAGE analysis indicated that the purified Nbs had an estimated molecular weight of around 15 kD () with purity of greater than 90%. The binding of the purified Nbs to the PEA antigen was confirmed by SPR. Four of the 12 Nbs displayed strong binding affinity to the antigen. The KD determined by ka and kd of the 4 Nbs was plotted in , The equilibrium dissociation constants (KD) of these 4 Nbs were 3.48 × 10−8, 4.98 × 10−9, 4.85 × 10−9 and 1.22 × 10−8, respectively (). Other Nbs had KD values ranging from 10−6 to 10−7(data not shown).
Figure 3. Purification and SDS-PAGE analysis of 12 Nbs expressed in E. coli.
Note: 12 Nbs were expressed in E. coli BL21 (DE3) and purified via Ni2+–NTA resin chromatography column and characterized by SDS-PAGE. The molecular weight of Nb is around 15 kD.
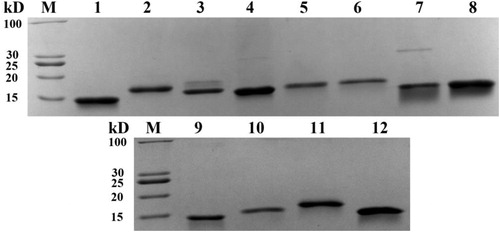
Figure 4. Affinity assay of Nbs binding to PEA.
Note: KD data between PEA and Nbs was obtained using Biacore T100 instruments. Four of the 12 Nbs showed strong binding activity to PEA antigen. Nb6, Nb8, Nb10 and Nb11 were immobilized on the chip surface and PEA dissolved in PBS at respective concentrations of 80, 40, 20, 10 and 5 μg/mL (C1–C5) were injected.
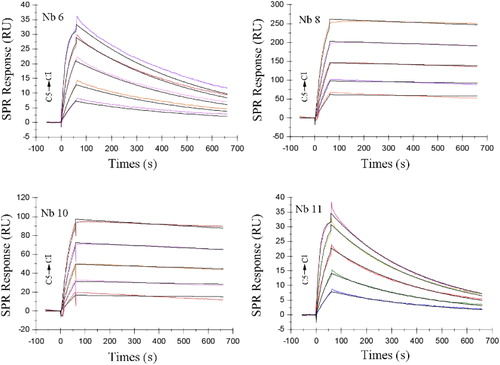
Table 2. Affinity of the 4 PEA-specific nanobodies with high affinity.
In vitro PEA toxicity neutralization assay
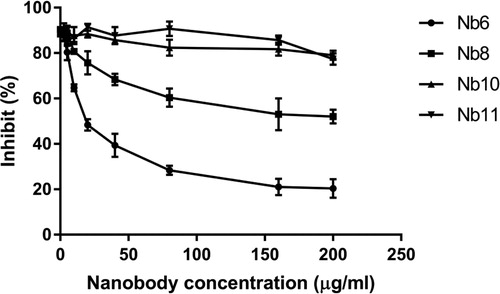
PEA is the most toxic substance produced by gram-negative Pseudomonas aeruginosa [Citation20], which can cause either serious acute infections in humans, resulting in endophthalmitis, endocarditis, meningitis and septicemia [Citation21] or chronic lung infections in cystic fibrosis patients [Citation22]. It is reported that PEA catalyses the ADP ribosylation of the eukaryotic elongation factor 2 (eEF-2) and therefore significantly affects the protein synthesis of the host cells [Citation23], thus blocking the binding of the toxin to its receptor is the first choice for neutralization antibody selection. The PEA cell toxicity and nanobody neutralization assays were conducted using the MTT method. The proliferation of A431 cells was inhibited by 95% by 5 μg/mL of PEA; the toxicity of PEA could be neutralized by adding Nb6 and Nb8 nanobodies and Nb6 showed high neutralization capability, with an IC50 28.5 nmol/L, while Nb8 showed only mild neutralization activity, and the inhibition of A431 cells by PEA could not be fully neutralized even at the maximum concentration used ().
Figure 5. The half inhibitory concentration of four nanobodies to PEA toxin.
Note: PEA concentration of 5 μg/mL used.
Epitopes mapping of the nanobodies
As a toxic protein, PEA contains three domains. Domain I (aa 1–252) is composed of antiparallel ß-sheets that can recognize CD91 on the host cell surface. Domain II (aa 253–364), with six consecutive α-helices, enables the toxin to translocate across cell membranes [Citation24]. Domain III (aa 400–613) is the catalytic subunit of the toxin with ADP-ribosyltransferase activity to inactive eEF-2 factor [Citation25,Citation26]. As shown in , the two truncated PEA proteins were successfully expressed in the E. coli cells. Nb6 nanobody could detect all the three proteins including PEA, tPEA1 and tPEA2; Nb8 could only detect PEA and tPEA2. This indicated that the epitope of Nb6 was on domain II of the PEA protein, the epitope of Nb8 was on the C-terminus domain III of the PEA proteins. Based on these epitope mapping results, the epitope of Nb6 was mapped to domain II, which enables the transmembrane transportation of the toxin into the host cells, demonstrating that blocking the translocation domain of the toxin can also neutralize the toxicity of PEA and therefore confer protection. The Nb8 had the highest affinity among all the binding nanobodies that we cloned, but it showed very mild neutralization ability. The epitope of Nb8 was mapped to domain III of PEA; the binding of the nanobody might cause its dissociation during the translocation process or inside the cell, thus rendering PEA catalytically inactive.
Figure 6. Western blot to detect the epitopes of the nanobodies. Lanes 1–3 with Nb8 as the primary antibody and PEA, tPEA1 and tPEA2 as respective antigens. Lanes 4–6 with Nb6 as the primary antibody and PEA, tPEA1 and tPEA2 as respective antigens.
Conclusions
The classical phage display method is an important technique for selecting antibody fragments and has been widely used for Nb isolation. However, the process of library construction and consecutive rounds of bio-panning is time-consuming and laborious. Here we developed an accurate, rapid, comprehensive and inexpensive screening method to obtain Nb immune repertoires at the single-cell level as demonstrated by obtaining Nb6 with high neutralization effect on PEA within 4 days. Thus we have provided a proof of concept for rapid cloning of antigen specific Nbs from single alpaca B cells. Moreover, the use of FACS allowed the maximal maintenance of the integrity of antigen epitopes, greatly increasing the interaction of antigen with B cells and thus the screening efficiency of positive cells.
Disclosure statement
The authors declare no commercial or financial conflicts of interest.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82:775–797.
- Desmyter A, Transue TR, Ghahroudi MA, et al. Crystal structure of a camel single-domain VH antibody fragment in complex with lysozyme. Nat Struct Biol. 1996;3(9):803–811.
- Muyldermans S, Cambillau C, Wyns L. Recognition of antigens by single-domain antibody fragments: the superfluous luxury of paired domains. Trends Biotechnol. 2001;26(4):230–235.
- Muyldermans S. Single domain camel antibodies: current status. Trends Biotechnol. 2001;74(4):277–302.
- De Meyer T, Muyldermans S, Depicker A. Nanobody-based products as research and diagnostic tools. Trends Biotechnol. 2014;32(5):263–270.
- Saccodossi N, De Simone EA, Leoni J. Structural analysis of effector functions related motifs, complement activation and hemagglutinating activities in Lama glama heavy chain antibodies. Vet Immunol Immunopathol. 2012;145(1–2):323–331.
- Li G, Zhu M, Ma L, et al. Generation of small single domain nanobody binders for sensitive detection of testosterone by electrochemical impedance spectroscopy. ACS Appl Mater Interfaces. 2016;8(22):13830–13838.
- Dufner P, Jermutus L, Minter RR. Harnessing phage and ribosome display for antibody optimisation. Trends Biotechnol. 2006;24(11):523–529.
- Pellis M, Pardon E, Zolghadr K, et al. A bacterial-two-hybrid selection system for one-step isolation of intracellularly functional nanobodies. Arch Biochem Biophys. 2012;526(2):114–123.
- Tiller T, Busse CE, Wardemann H. Cloning and expression of murine Ig genes from single B cells. J Immunol Methods. 2009;350(1–2):183–193.
- Kurosawa N, Yoshioka M, Fujimoto R, et al. Rapid production of antigen-specific monoclonal antibodies from a variety of animals. BMC Biol. 2012;10:80–93.
- Turner KB, Naciri J, Liu JL, et al. Next-generation sequencing of a single domain antibody repertoire reveals quality of phage display selected candidates. PLoS One. 2016;11(2):e0149393.
- Gray GL, Smith DH, Baldridge JS, et al. Cloning, nucleotide sequence, and expression in Escherichia coli of the exotoxin A structural gene of Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 1984;81(9):2645–2649.
- Cook DAN, Samarasekara CL, Wagstaff SC, et al. Analysis of camelid IgG for antivenom development: immunoreactivity and preclinical neutralisation of venom-induced pathology by IgG subclasses, and the effect of heat treatment. Toxicon. 2010;56(4):596–603.
- Picelli S, Faridani OR, Bjorklund AK, et al. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9(1):171–181.
- Wang P, Li G, Yan J, et al. Bactrian camel nanobody-based immunoassay for specific and sensitive detection of Cry1Fa toxin. Toxicon. 2014;92:186–192.
- Ahmadi S, Pucca MB, Jurgensen JA, et al. An in vitro methodology for discovering broadly-neutralizing monoclonal antibodies. Sci Rep. 2020;10(1):10765.
- Hanyu Y, Kato M. Screening antibody libraries with colony assay using scFv-alkaline phosphatase fusion proteins. Molecules. 2020;25(12):2905.
- Pardon E, Laeremans T, Triest S, et al. A general protocol for the generation of nanobodies for structural biology. Nat Protoc. 2014;9(3):674–693.
- Zanjani LS, Shapouri R, Dezfulian M, et al. Exotoxin A-PLGA nanoconjugate vaccine against Pseudomonas aeruginosa infection: protectivity in murine model. World J Microbiol Biotechnol. 2019;35(6):94.
- Zou G, de Leeuw E. Neutralization of Pseudomonas aeruginosa Exotoxin A by human neutrophil peptide 1. Biochem Biophys Res Commun. 2018;501(2):454–457.
- Javanmardi F, Emami A, Pirbonyeh N, et al. A systematic review and meta-analysis on exo-toxins prevalence in hospital acquired Pseudomonas aeruginosa isolates. Infect Genet Evol. 2019;75:104037.
- Balasubramanian V, Sellegounder D, Suman K, et al. Proteome analysis reveals translational inhibition of Caenorhabditis elegans enhances susceptibility to Pseudomonas aeruginosa PAO1 pathogenesis. J Proteomics. 2016;145:141–152.
- Muller F, Cunningham T, Beers R, et al. Domain II of Pseudomonas exotoxin is critical for efficacy of bolus doses in a xenograft model of acute lymphoblastic leukemia. Toxins (Basel). 2018;10(5):210.
- Allured VS, Collier RJ, Carroll SF, et al. Structure of exotoxin A of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc Natl Acad Sci USA. 1986;83(5):1320–1324.
- Santajit S, Seesuay W, Mahasongkram K, et al. Human single-chain antibodies that neutralize Pseudomonas aeruginosa-Exotoxin A-mediated cellular apoptosis. Sci Rep. 2019;9(1):14928.