
Abstract
Drought is one of the most severe environmental stressors hampering maize (Zea mays L.) production worldwide. Therefore, revealing the molecular mechanisms underpinning maize drought tolerance is critical for guiding drought resistance breeding and ensuring global food security. Herein, we performed a data-independent acquisition (DIA) proteomics-based comparative analysis of the proteome responses of two contrasting maize hybrids (drought-tolerant NongDan 476 and drought-sensitive ZhongXin 978) exposed to 12 days of water-deficit (drought) treatment at the kernel filling stage. Resultantly, our DIA-based analysis approach identified a total of 826 differentially abundant proteins (DAPs), among which 7 DAPs were specifically accumulated in tolerant genotype ND 476 under drought treatment conditions. Moreover, our results showed that there were significant differences in drought stress responses between the two hybrids at the proteomic level. Predominantly, our analysis revealed that maize drought tolerance is attributed to some key proteins such as chaperones, DNA replication related enzymes, vicilin and ABA-responsive RAB17 protein, participating in several metabolic networks including starch and sucrose metabolism, terpenoid backbone biosynthesis, cyanoamino acid metabolism and carbon metabolism pathways. Our findings provide further insights into the drought stress tolerance mechanisms in maize at the grain filling period, in addition to useful reference data for maize drought resistance breeding.
Introduction
Adverse environmental factors, of which drought stress represents the most severe constraint to agriculture, account for approximately 70 percent of potential yield loses across the world [Citation1, Citation2]. Global warming and climate change are also predicted to affect most severely arid climates, where agricultural ecosystems are most susceptible to climatic condition variations and where marginal increases in environmental temperature are very harmful to productivity [Citation3–5]. The development of crop cultivars with increased drought stress tolerance, by both conventional breeding and genetic engineering approaches, is central in meeting global food demands for the growing human population under such resource constrained environment [Citation6, Citation7]. More precisely, understanding how plants respond to drought stress at the molecular level is useful for developing improved genotypes which would perform well under water limited conditions [Citation8].
Maize (Zea mays L.), one of the most important food crops in the world, is very sensitive to water-deficit stress, especially during flowering, pollination and embryo development [Citation9, Citation10]. Prominently, the yield loss during the grain filling period in maize is usually greater than that occurring during the vegetative phase [Citation11]. Severe water stress during the maize grain filling period results in premature formation of the kernel black layer [Citation12, Citation13]. Therefore, revealing the mechanism of maize drought response during the grain-filling stage remains important for breeding drought-tolerant maize cultivars.
In response to drought stress, a wide range of physiological responses are activated at varying levels of plant organization. Activation of these physiological responses would eventually affect the transcription factor regulation and downstream stress responsive genes expression [Citation14, Citation15]. Examples of such responses to drought stress include but are not limited to the wilting, rolling and abscission of foliage; modification of the root to shoot ratio and antioxidant enzyme activities activation [Citation16, Citation17].
Meanwhile, there have been many studies on the analysis of crop resistance to drought stress, but most of them have mainly focused on physiology and biochemistry [Citation18–20] and transcriptomic analysis [Citation21–24]. For instance, Guo et al. [Citation25] examined the transcriptomic changes induced by drought and salt stress in 8-week-old seedlings of Picea wilsonii via RNA-seq analysis. Previously, we conducted comparative RNA-seq transcriptome studies [Citation26–28] and identified several candidate genes and metabolic pathways for drought tolerance in maize, including secondary metabolite biosynthesis related enzymes, such as probable O-methlytransfarase 2 (OMT2; Zm00001d019613) [Citation28]; transcription factors (TF) such as MYBs, NAC and WRKYs; detoxification and stress responsive proteins including aquaporin PIP2-4 (Zm00001d017288), HSPs and GSTs [Citation26–28]. However, transcriptome analysis of drought tolerance mechanisms is limited because mRNA translation levels are not always related to the corresponding protein levels and proteins are the direct bearers of biological function [Citation29, Citation30]. Since proteins are directly involved in plant stress responses, proteomic studies can eventually contribute to dissecting the possible relationships between protein changes and plant stress tolerance [Citation31, Citation32]. Therefore, proteomic analysis of the mechanisms underlying maize drought stress tolerance becomes essential [Citation33, Citation34].
Earlier, we employed the isobaric tags for absolute quantification (iTRAQ) strategy to perform proteomics analysis and identify key proteins and metabolic pathways regulating the drought stress response in maize [Citation35–38]. We observed diverse key drought responsive proteins, including chlorophyll a-b binding proteins (Lhcb5-1 and 542320/Lhcb5-2) associated with the light-harvesting complex; lipid metabolism related non-specific lipid transfer proteins [Citation35]; histone H2A proteins (Zm00001d006547, 100216750), carbohydrate metabolism-related protein malate dehydrogenase (MDH; Zm00001d009640) and several peroxidases [Citation36]; histone deacetylase (HDAC), HSPs, MDH, GSTs, seed storage proteins 50kD γ-zein, Z1D alpha zein [Citation37]; peroxidase (Zm00001d008898), stress responsive protein (100283176) and oxidoreductase family proteins [Citation38]. Notably, these genes and proteins were mainly involved in photosynthesis antenna proteins, nitrogen metabolism, secondary metabolism related pathways, especially in the tolerant maize genotype ND476 under drought stress [Citation28, Citation35, Citation38]. This has given us some insights into maize drought tolerance at different growth stages, although gaps in our knowledge still persist.
Besides iTRAQ proteomics, data-independent acquisition (DIA) has become a powerful tool for performing large-scale studies and comprehensive identification of drought responsive proteins in plants [Citation39]. The DIA approach is another strategy used in mass spectrometry to determine molecular structures [Citation40]. Unlike the data-dependent acquisition (DDA) method, which selects a fixed number of precursor ions for analysis by tandem mass spectrometry (TMS), the DIA approach alternatively uses all the ions within a selected m/z range, which undergoes fragmentation before analysis at the second phase of TMS [Citation41, Citation42]. Compared with the DDA method, the DIA strategy has the advantages of high data utilization, high repeatability and high quantitative accuracy [Citation43]. Therefore, DIA technology can obtain more accurate and rich results, suitable for protein detection of large sample size and complex systems.
Here, in order to study the response mechanisms of maize filling- kernel drought stress at the protein level, we used a DIA-based quantitative strategy to perform proteome analysis on two contrasting maize hybrids. In addition, we also evaluated some physiological indices of these two hybrids under drought stress, and the results of this research provide further insights into the drought stress tolerance mechanisms in maize.
Materials and methods
Plant materials and treatment
Two maize hybrids with dissimilar drought tolerance (drought-tolerant NongDan 476 and drought-sensitive ZhongXin 978) were used in this study [Citation26]. The materials were provided by the laboratory (North China Key Laboratory for Crop Germplasm Resources of Education Ministry, Hebei Agricultural University, Baoding, China). The field experiment was carried out at Qing Yuan Experimental Station (115 E, 38 N) of Hebei Agricultural University. The experiment was set up under a fully automated rain-proof shelter, and a randomized complete block design was used to plant maize hybrid lines. The size of each plot is 5 m × 5 m, and the plant spacing is 0.6 m × 0.3 m. We planted maize on 15th June 2018, and the two hybrid lines were irrigated with adequate water before grain filling. One week before the flowering period, we performed strict bagging in order to prevent maize plants from self-pollination. The maize in the experimental group was subjected to drought treatment on 23rd August 2018, about 13 days after pollination, and continued the artificial drought for 12 days until the afternoon of 4th September 2018, while the control group received sufficient watering. The intermediate seeds of the ears were selected for proteomic analysis. During the sample collection process, the collected samples were immediately frozen in liquid nitrogen, and then stored at −80 °C for future use.
Phenotypic characteristics measurement
Under well-watered and drought stress conditions, the phenotypic characteristics of the two hybrids at the late grain-filling stage were measured. Ten ears were randomly selected from each group and used to measure the four phenotypic data of each ear’s ear length, barren tip length, kernel rows per ear and kernels per row. Then we calculate the average and use SPSS software for significance analysis.
Protein extraction
The sample was ground to a powder in liquid nitrogen, then dissolved in 2 mL lysis buffer containing 8 mol/L urea, 2% sodium dodecyl sulphate (SDS), and 1× Protease Inhibitor Cocktail (Roche Ltd. Basel, Switzerland), followed by sonication on ice for 30 min and centrifugation at 15,000 g for 30 min at 4 °C. The supernatant was transferred to a new tube. For each sample, proteins were precipitated with ice-cold acetone at −20 °C overnight. The precipitations were cleaned with acetone three times and re-dissolved in 8 mol/L urea by sonication on ice. BCA Protein Assay Kit (Genedenovo Biotechnology Co., Ltd, Guangzhou, China) was used to determine the protein concentration of the supernatant. The protein extract quality was examined with SDS polyacrylamide gel electrophoresis (SDS-PAGE) [Citation44].
Protein digestion and high PH reverse phase separation
For each sample, 50 μg proteins extracted from cells were suspended in 50 μL solution, reduced by adding 1 μL of 1 mol/L dithiotreitolat 55 °C for 1 h, alkylated by adding 5 μL of 20 mmol/L iodoacetamide in the dark at 37 °C for 1 h. Then the sample was precipitated using 300 μL prechilled acetone at −20 °C overnight. The precipitate was washed twice with cold acetone and then re-suspended in 50 mmol/L ammonium bicarbonate. Finally the proteins were digested with sequence-grade modified trypsin (Promega, Madison, WI) at a substrate/enzyme ratio of 50:1 (w/w) at 37 °C for 16 h.
The peptide mixture was re-dissolved in buffer A (buffer A: 20 mmol/L ammonium formate in water, pH 10.0, adjusted with ammonium hydroxide), and then fractionated by high pH separation using Ultimate 3000 system (ThermoFisher scientific, MA, USA) connected to a reverse phase column (XBridge C18 column, 4.6 mm × 250 mm, 5 μm, (Waters Corporation, MA, USA). High pH separation was performed using a linear gradient, starting from 5% B to 45% B in 40 min (B: 20 mmol/L ammonium formate in 80% ACN, pH 10.0, adjusted with ammonium hydroxide). The column was re-equilibrated at the initial condition for 15 min. The column flow rate was maintained at 1 mL/min and the column temperature was maintained at 30 °C. Ten fractions were collected; each fraction was dried in a vacuum concentrator for the next step.
DIA: nano-HPLC-MS/MS analysis
The peptides were re-dissolved in 30 μL solvent A (A: 0.1% formic acid in water) and analyzed by on-line nanospray liquid chromatography with tandem mass spectrometry (LC-MS-MS) on an Orbitrap Fusion Lumos coupled to an EASY-nLC 1200 system (Thermo Fisher Scientific, MA, USA). Briefly, 3 μL peptide sample was loaded onto the analytical column (Acclaim PepMapC18, 75 μm × 25 cm) with a 120-min gradient, from 5% to 35% B (B: 0.1% formic acid in ACN). The column flow rate was maintained at 200 nL/min, and the column temperature at 40 °C. The electrospray voltage of 2 kV versus the inlet of the mass spectrometer was used.
The mass spectrometer was run under data independent acquisition mode, and automatically switched between MS and MS/MS mode. The parameters were: (1) MS: scan range (m/z) = 350–1200; resolution = 120,000; AGC target = 1e6; maximum injection time = 50 ms; (2) HCD-MS/MS: resolution = 30,000; AGC target = 1e6; collision energy = 32;stepped CE = 5%. (3) DIA was performed with a variable isolation window; each window overlapped 1 m/z, and the window number was 60.
Protein identification and quantification
Raw data of DIA were processed and analyzed by Spectronaut X (Biognosys AG, Switzerland) with default parameters. Retention time prediction type was set to dynamic iRT. Data extraction was determined by Spectronaut X based on the extensive mass calibration. Spectronaut Pulsar X determines the ideal extraction window dynamically depending on iRT calibration and gradient stability. False discovery rate (FDR) cutoff on precursor and protein level was applied at 1%. Decoy generation was set to mutated, which is similar to scrambled but will only apply a random number of AA position swamps (min = 2, max = length/2). All selected precursors passing the filters were used for quantification. The average top 3 filtered peptides which passed the 1% FDR cutoff were used to calculate the major group quantities. After Student’s t-test, differentially expressed proteins were filtered if their Q value was Q < 0.05 and the absolute AVG log2 ratio > 0.58.
Functional annotation and enrichment analysis of DAPs
We performed gene ontology (GO) annotation to assign GO terms to the DAPs using the Blast2GO web-based program (<seurld>https://www.blast2go.com/</seurld>). Further, the DAPs were searched against GO (Gene Ontology, http://www.geneontology.org/) and KEGG (Kyoto Encyclopedia of Genes and Genomics, http://www.genome.jp/kegg/) databases to obtain their biological functions. The GO analysis [Citation45] functionally categorized the identified DAPs into their BP, MF and CC involvement in response to drought stress. Moreover, the DAPs were assigned to various metabolic pathways using the KEGG pathway analysis. Further, significant GO functions and KEGG pathways enrichment analysis were examined within DAPs with the hypergeometric test, Q (Bonferroni-corrected p-value) < 0.05 set as the statistically significant threshold.
RNA extraction, cDNA synthesis and qRT-PCR analysis
We used the M5 Quickspin (Mei5 Biotechnology, Beijing, China) to extract total RNA. Total RNA was extracted from non-stressed and stressed kernels of the two hybrids (ND 476 and ZX 978) and prepared for quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis. To generate cDNA templates, 2 µg of total RNA was reverse-transcribed in a total volume of 20 µL, using the PrimerScriptTM RT reagent Kit (TAKARA, Dalian, China) according to the manufacturer’s instructions. We randomly selected 15 genes and designed gene-specific primers () for qRT-PCR using Primer Premier 5 Designer software. We used LightCycler® 96 for qRT-PCR, using TB Green ® Premix Ex TaqTM II (Tli RNaseH Plus) (TAKARA, Dalian, China). Each total 20 µL qRT-PCR reaction mixture comprised 1 µL of template cDNA, 0.8 µL of each primer, and 10 µL of 2 × TB Green Premix Ex Taq II (TAKARA, Dalian, China). The amplification program was as follows: 95 °C for 30 s followed by 40 cycles of 95 °C for 5 s and 60 °C for 20 s, 65 °C for 15 s. The maize gene GAPDH (accession no. X07156), with forward (GAPDH-F: 5′-ACTGTGGATGTCTCGGTTGTTG-3′), and reverse (GAPDH-R: 5′-CCTCGGAAGCAGCCTTAATAGC-3′) primers, was used as an internal control. We performed three biological replicates on each sample. The relative mRNA abundance was estimated using Livak and Schmittgen’s 2−ΔΔCt method [Citation46].
Table 1. Drought-responsive DAPs observed specifically in ND 476.
Results and discussion
Phenotypic differences between two hybrids
To validate the previous observations that ZX 978 is drought-sensitive and ND 476 is drought-tolerant, the two maize hybrids were treated with or without moisture deficit stress in a fully automated rain-proof shelter. We then recorded the phenotypic data of sample ears from two treatment groups of ND 476 and ZX 978 (). From the phenotypic perspective, before and after drought treatment, the four phenotypic indicators including ear length, barren tip length, kernel rows per ear and kernels per row did not change significantly in ND 476, which indicated that ND 476 is a comparatively drought-tolerant hybrid line (). Although the kernel rows per ear of ZX 978 did not change, the ear length was significantly reduced, the barren tip was significantly increased, and the kernels per row were significantly reduced, which indicated that ZX 978 is a drought-sensitive hybrid line ().
Figure 1. Phenotypic characterization of the two maize hybrid lines (drought-tolerant ND 476, T; and drought-sensitive ZX 978, S) ears’ responses to drought stress. Observations/measurements were made after 12 days of water-sufficient (control, C) or water-deficit (drought, D) conditions. (a-b) Ear phenotypes; (c) ear length; (d) ear bare tip length; (e) kernel rows per ear; (f) number of kernels per row. Data are presented as mean values ± standard errors (n = 3). Different letters on error bars indicate significant differences at p < 0.05; For c-f, each replication is an average for the measurement of 10 ears.
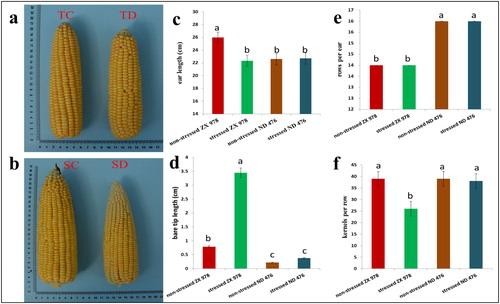
Phenotypic charactersitics could not show obvious changes in ND476 genotype before and after drought treatment, suggesting that the perception of the stress by ND476 could have been mild, which could not warrant significant phenotypic modifications. This is consistent with the expected results of ND 476 as a drought-tolerant hybrid line and ZX 978 as a drought-sensitive hybrid line. Therefore, we can infer that under drought conditions, water deficit will lead to short ear length and slow growth of maize; increased barren tip length, insufficient grain filling and reduced number of kernels per row.
Inventory of maize proteins identified by DIA-based analysis approach
The 830,311 spectra obtained by DIA proteomic strategy were matched to known spectra using Spectronaut software, and 50,587 precursors and 36,717 peptides were identified. Consequently, 5276 protein groups and 13,659 proteins were identified from the 36,717 peptides. Further analysis showed that of those 36,717 peptides, 2941 (8.01%) were < 1000 Da, 23,814 (64.86%) were 1000–2000 Da, 8049 (21.92%) were 2000–3000 Da, and 1913 (5.21%) were > 3000 Da in molecular weight (Supplemental Figure S1a). The distribution of the peptide lengths indicated that over 80% of the peptides had lengths between 7 and 20 amino acids (Supplemental Figure S1b). In terms of number of peptides defining each protein, our analysis showed that among the 5276 protein groups, 83.95% (4429 protein groups) had proteins composed of at least two peptides, whereas the remaining 16.05% (847 protein groups) were composed of only one identified peptide (Supplemental Figure S1c). Additionally, we used the local normalization method in Pulsar software to normalize the peak intensity of the whole sample. From the distribution of quantized values of normalized peptides, the signal intensity of most samples reached the same response intensity (Supplemental Figure S1d). Therefore, the DIA sequencing results qualified for use in the subsequent analyses.
Analysis of DAPs observed in different comparisons
Similar to the approach used previously [Citation38], comparative proteomic analysis was used to investigate the changes of maize filling-kernel protein profiles in ND 476 (drought-tolerant, T) hybrid and ZX 978 (drought-sensitive, S) hybrid under drought stress condition. A pairwise comparison of before and after treatments (drought, D, and control, C) was performed in ND 476 (TD_TC) and ZX 978 (SD_SC) individually. In addition, a comparative study on the drought stress proteome was performed between the tolerant hybrid and sensitive hybrid, under drought (SD_TD) and under water-sufficient (control) (SC_TC) conditions, giving four comparison groups (). In the drought-tolerant hybrid line, 36 proteins showed differential abundance before and after drought treatment (TD_TC), 8 of these DAPs were up-regulated, 28 were down-regulated (). In the drought-sensitive hybrid line, we identified 130 DAPs before and after drought treatment (SD_SC), 28 of these DAPs were up-regulated whilst 102 were down-regulated (). In total, 826 DAPs were found among the four comparison groups ().
Figure 2. Analysis of differentially abundant proteins (DAPs) identified in four experimental comparisons. (a) Total number of DAPs identified in each experimental comparison group. Red (up-regulated) means DAPs with increased differential abundance. Green (down-regulated) means DAPs with decreased differential abundance; (b) Venn diagram analysis of DAPs identified in the four comparisons. DAPs uniquely expressed in TD_TC (I), SD_TD (II), SD_SC (III) are indicated with arrows. Area IV shows 18 overlapping DAPs within line. Overlapping regions of the Venns indicate DAPs shared between/among corresponding groups. (c) Clustering analysis of DAPs in SD_TD comparison. Each row represents a significantly abundantly expressed protein. ZXD1-3 (drought-sensitive ZX 978, S; drought, D) refer to the biological replicate number for ZX 978, whilst NDD1-3 (drought-tolerant ND 476, T; drought, D) refer to the replicate number for ND 476. The DAPs were clustered based on the differentially expressed levels. The scale bar indicates the logarithmic value (log2 fold change) expression of the DAPs. Red (log2 fold change > 0) indicates up-regulated DAPs and blue (log2 fold change < 0) indicates down-regulated DAPs.
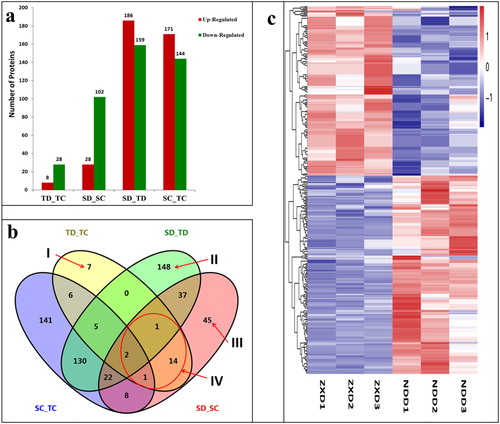
According to , it can be seen that area I encompasses the 7 specific DAPs of drought-tolerant hybrid ND 476 (); area II represents 148 specific DAPs of SD_TD and analysis of DAPs revealed that under drought stress, the number of DAPs in ZX 978 was significantly higher than that in the drought-tolerant hybrid ND 476 ( and Supplemental Figure S2); area III is a specific DAPs of the sensitive hybrid line ZX 978, with 45 (). Area IV represents the 18 DAPs shared by TD_TC and SD_SC, that is, the overlapping drought responsive DAPs within the two hybrid lines. Of these 18 common drought responsive DAPs, all were down-regulated in tolerant hybrid ND 476; whereas 2 were up-regulated and 16 down-regulated in sensitive hybrid ZX 978 ().
Table 2. Drought-responsive DAPs observed specifically in ZX 978.
Table 3. Overlapping drought-responsive DAPs between ZX 978 and ND 476.
Expression of ubiquitin associated proteins in ND 476
Among these drought-responsive proteins whose abundance was significantly altered under drought stress in tolerant hybrid ND 476, we observed that ubiquitin-fold modifier 1 (Ufm1) was up-regulated. Ufm1 is a ubiquitin-like modifier and conjugates to target proteins in cells [Citation47, Citation48]. In addition to being a recognition signal, ubiquitin regulates cellular processes directly by altering the biophysical properties of the substrate, suggesting that ubiquitination facilitates the unfolding process and improves the substrate degradation efficiency [Citation49], hence Ufm1 is up-regulated under drought stress, which enhances the rate of protein degradation. Moreover, Xia et al. [Citation50] found that the ubiquitin ligase was up-regulated in maize response to drought stress.
The response of maize to drought stress is a complex process involving DNA repair process, hydrolysis process, signal transduction, intracellular environment stability, and transcription and translation, etc. [Citation51]. In particular, both genetic analyses and the rapid accumulation of small ubiquitin-like modifiers (SUMO) conjugates in response to various adverse environmental conditions suggest that SUMOylation plays a key role in the stress responses [Citation52]. SUMO-conjugating enzyme (SCE) mediates addition of SUMO group to various cell proteins, through process referred to as SUMOylation [Citation53]. SUMO, in conjunction with intracellular proteins, actively participate in several biological functions such as altering signal transduction in the cytoplasm, transcription regulation, DNA repair, as well as regulation of nuclear material importation and subnuclear compartmentalization [Citation52, Citation54]. Moreover, the ubiquitin-dependent proteolytic pathway degrades most proteins and is the major proteolytic mechanism in eukaryotic cells [Citation55]. Thus, here, the up-regulation of SCE1 implies that cells promote the proteins and enzymes involved in protein ubiquitination in order to protect themselves under drought stress [Citation35].
Other proteins for drought tolerance in ND 476
Among the dominating up-regulated DAPs in ND 476, we observed 17.5 kDa class II heat shock proteins (see TC_TD comparison, Supplemental Table S2). Heat shock proteins (HSPs) are generally activated in plants’ response to external stimuli emanating from a wide array of abiotic stressors such as water deficit, nutrient deficiency, oxidative stress, among others [Citation56–59]. Thus, HSPs are key members of the plant stress proteins involved in stress responses [Citation60]. Particularly, HSPs provide chaperone functions to stabilize newly synthesized proteins by maintaining correct folding, or by assisting the refolding of damaged proteins [Citation61]. Molecular chaperones facilitate the stabilization of other macromolecular structures under stress conditions [Citation62]. By stabilizing partially unfolded proteins, HSPs help the easy transportation of other proteins and cellular materials across membranes [Citation63, Citation64]. Additionally, HSPs help preserve other proteins in their functional confirmations, thereby offering plant protection against environmental stresses [Citation65]. A previous study has demonstrated that HSPs are greatly accumulated in alfalfa (Medicago sativa L.) leaves in response to salinity stress [Citation66].
In our study, we found that starch phosphorylase 1 (STP-1) was down-regulated. STP-1 is largely known for the phosphorolytic degradation of starch [Citation67]. Moreover, drought stress caused a marked reduction in the starch content of the kernel [Citation68], shortened the grain filling period, and increased the grain filling rate [Citation69]. Therefore, STP-1 was down-regulated to resist the decrease of starch content in kernel caused by drought stress, and reduced material consumption to protect the kernels from drought damage. In addition, Prasch et al. [Citation70] suggested that impaired starch breakdown was accompanied by decreased stomatal opening in Arabidopsis thaliana, resulting in improved drought tolerance of Arabidopsis plants.
Enhancement of DNA replication in ZX 978
In the present study, adenine phosphoribosyltransferase 2 (APRTase 2) and DNA primase were up-regulated in response to drought stress (see SC_SD comparison, Supplemental Table S3). APRTase is an enzyme encoded by the APRT gene [Citation71], and being part of the type I PRT enzyme family, it is involved in the nucleotide salvage pathway [Citation72]. APRTase provides a mechanism for the production of adenine [Citation73]. It was found that the increase of adenine in wheat significantly increased the levels of free proline, amino acid and soluble sugar, which increased the photosynthesis, water status and chlorophyll content under drought stress [Citation74]. Therefore, the up-regulation of APRTase 2 in ZX 978 is used to combat drought stress by regulating osmotic potential and enhancing photosynthesis.
DNA primase is an enzyme involved in the replication of DNA and is a type of RNA polymerase. DNA primase plays an essential role in controlling cell cycle progression in response to DNA damage [Citation75]. Faria et al. [Citation76], studying Medicago truncatula, suggested that desiccation-tolerant seedlings may apply distinct strategies to survive in drought, either by avoidance or further repair of the DNA damage. Hence, DNA primase is an up-regulated enzyme that mitigates the damage caused by drought by repairing damage in DNA replication.
Overlapping drought responsive proteins between ND 476 and ZX 978
Venn diagram (, Area IV) analysis showed that 18 DAPs were common between TD_TC and SD_SC. All the 18 DAPs () were down-regulated in tolerant hybrid ND 476 in response to drought treatment. Conversely, among these 18 common DAPs, two (salt stress-induced protein and flowering locus T protein) were up-regulated, whilst 16 (globulin-1 S allele, RAB17 protein, sucrose synthase 1, farnesyl pyrophosphate synthase1 and vicilin) were down-regulated in sensitive hybrid ZX 978 in response to drought treatment ().
Flowering locus T protein (FT), a member of the phosphatidylethanolamine-binding protein family, functions as a florigen or a component of florigen [Citation77]. Long-distance signal called florigen is produced in the leaf under inductive day length conditions, and is transported to the shoot apex where it triggers floral morphogenesis [Citation78]. Regulation of the transition process from vegetative to anthesis is vital in plants’ drought stress acclimation [Citation79, Citation80]. The drought-escape response allows plants to adaptively shorten their life cycle to make seeds before severe stress leads to death [Citation80]. Therefore, FT is up-regulated in ZX 978 to promote seeds generation to shorten the life cycle to achieve drought escape. Similarly, Su et al. [Citation81] found that the FT was up-regulated in Arabidopsis in response to drought stress. In addition, FT is down-regulated in ND 476 maybe to slow the long-distance transportation of the signal, thereby saving energy to resist drought.
Vicilin is used as a storage protein and commonly found in plants like pea or lentil [Citation82]. It has been suggested to be an allergen in allergic reactions to peas [Citation83]. Vicilin is synthesised as preprovicilin with subsequent removal of a signal peptide and a C-terminal peptide as well as post translational endo-proteolytic cleavage [Citation84]. Additionally, the down-regulation of vicilin in the two hybrid lines was consistent with the inhibition of starch biosynthesis under drought stress in plants. Similarly, globulin-1 S allele, a type of polymorphic Globulin-1 gene, encodes a storage globulin [Citation85] that is down-regulated in two hybrid lines.
In the present study, sucrose synthase 1 (SS-1) was significantly down-regulated in response to drought stress. It has been noted that premature formation of black layer following cold temperatures, drought or defoliation might all be explained on the basis of reduced sucrose availability [Citation13]. It was suggested that SS plays a key role in the regulation of carbon metabolism and, therefore, of nitrogen fixation under drought stress conditions [Citation86, Citation87]. Moreover, SS, which reversibly catalyzes sucrose synthesis and cleavage, plays pivotal roles in regulating sucrose flux [Citation88].
The maize abscisic acid-responsive RAB17 protein localizes to the nucleus and cytoplasm in maize cells, involved in plant responses to stress [Citation89]. Based on the similar domain arrangements of Rab17 and a nuclear localization signal (NLS) binding phosphoprotein, interaction of Rab17 with NLS peptides was found [Citation90]. Thus, Rab17 may play a role in nuclear protein transport [Citation91]. Moreover, binding of the NLS peptide to Rab17 is dependent upon phosphorylation by protein kinase CK2 [Citation92]. Under drought conditions, the reduction in RAB17 protein may be due to reduced protein transport to save energy against drought stress, similar to our previous findings where the tolerant maize genotype YE8112 plants had to reduce the synthesis of redundant proteins to help themselves save energy for drought stress endurance [Citation35]. Figueras et al. [Citation93] observed a protective effect of RABl7 protein in vegetative tissues under osmotic stress conditions.
GO annotation and functional classification of the drought responsive DAPs
We performed GO annotation to assign GO terms to the DAPs. Further, GO functional classification of the GO-term-assigned-DAPs into biological processes (BP), molecular functions (MF), and cellular component (CC) categories was carried out. For the tolerant hybrid line ND 476 specific DAPs, the most significantly enriched GO terms in the MF category were GO: 0008194 (UDP-glycosyltransferase activity), GO: 0035251 (UDP-glucosyltransferase activity), GO: 0016758 (transferase activity, transferring hexosyl groups), GO: 0046527 (glucosyltransferase activity) and GO: 0016757 (transferase activity, transferring glycosyl groups) (Supplemental Table S4).
The significantly enriched GO terms in the sensitive hybrid line ZX 978 (SC_SD) were GO: 0010466 (negative regulation of peptidase activity), GO: 0030162 (regulation of proteolysis), GO: 0043086 (negative regulation of catalytic activity), GO: 0045861 (negative regulation of proteolysis), GO: 0050790 (regulation of catalytic activity), GO: 0051336 (regulation of hydrolase activity), GO: 0051346 (negative regulation of hydrolase activity) and GO: 0052547 (regulation of peptidase activity) in the BP category (Supplemental Table S5); GO: 0004866 (endopeptidase inhibitor activity), GO: 0035251 (UDP-glucosyltransferase activity), GO: 0061134 (peptidase regulator activity), GO: 0061135 (endopeptidase regulator activity) and GO: 0030414 (peptidase inhibitor activity) in the MF functions (Supplemental Table S5).
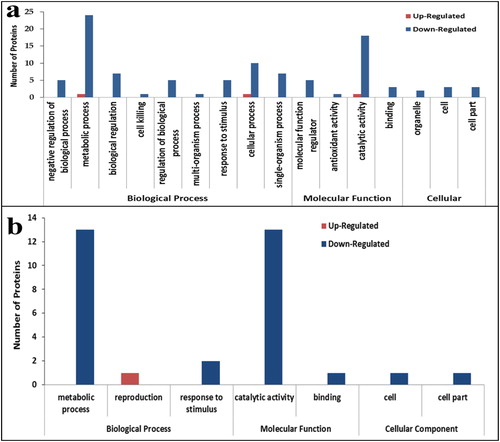
Additionally, the amount of DAPs in the two comparison combinations (TC_TD, SC_SD) annotated into each biological function is shown in . Among the tolerant hybrid line ND 476 (TC_TD) -specific DAPs, metabolic process was the most enriched BP function; and catalytic activity was most prominent in the MF category (). Among the sensitive hybrid line ZX 978 (SD_SC)-specific DAPs, metabolic process was the most common biological processes; whereas catalytic activity was prominent in the MF category ().
Figure 3. Gene ontology (GO) functional classification of differentially abundant proteins (DAPs) under drought conditions. Most significantly enriched GO terms in tolerant hybrid line ND 476 (a) and sensitive hybrid line ZX 978 (b). Note: The number above each bar graph shows the enrichment factor of each GO term. Red indicates up-regulated and blue indicates down-regulated.
KEGG pathway enrichment analysis of DAPs
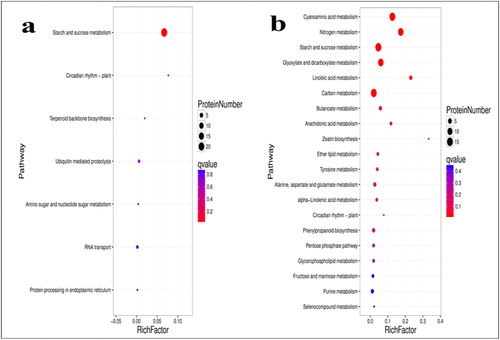
To further analyze the functional consequences of the drought-responsive DAPs, we mapped them to the Kyoto Encyclopedia of Genes and Genomes (KEGG, <seurld>https://www.genome.jp/kegg/</seurld>) database and the DAPs were assigned to various biological pathways. The top three enriched pathways in ND 476 were starch and sucrose metabolism (22 proteins), ubiquitin mediated proteolysis (2) and RNA transport (2) (). However, the composition of the enriched KEGG pathways in ZX 978 differed significantly, with cyanoamino acid metabolism (14 proteins), nitrogen metabolism (12), starch and sucrose metabolism (15), glyoxylate and dicarboxylate metabolism (12) and carbon metabolism (14) being the most enriched pathways ().
Figure 4. KEGG pathway enrichment analysis of the DAPs; TD_TC comparison (a); SD_SC comparison (b). Note: The bubble size represents the number of DAPs enriched in the corresponding pathway. The vertical coordinate shows KEGG pathway. The horizontal coordinate shows the enrichment factor (rich factor ≤ 1), and the enrichment factor indicates the number of differentially abundant proteins (DAPs) participating in a KEGG pathway as a proportion of proteins involved in the pathway in all identified proteins. The significance of the enrichment of the KEGG pathway is based on Student’s t-test, q < 0.05. The color gradient represents the size of the q value; the color is from blue to red, and the nearer red represents the smaller q value, the higher the significant level of enrichment of the corresponding KEGG pathway.
Most significantly enriched metabolic pathways in the two genotypes’ response to drought stress
Using the hypergeometric test, KEGG pathways that had a q value < 0.05 were considered to be significantly affected by drought stress. Consequently, we observed that ‘starch and sucrose metabolism’, ‘terpenoid backbone biosynthesis’ and ‘ubiquitin mediated proteolysis’ were the most significantly enriched pathways in drought-tolerant hybrid ND 476 (). For those pathways, sucrose synthase1 and starch phosphorylase1 were down-regulated (Supplemental Table S2). These results resonate well with our GO analysis results where sucrose synthase1 were most significantly enriched in the prominent GO terms (catalytic activity and metabolic process), suggesting their role as the major drought resistant signature in ND 476 hybrid. Moreover, the sucrose content increased and the starch content decreased concomitantly, indicating that drought facilitated the partitioning of photosynthates into sucrose [Citation94].
On the other hand, ‘cyanoamino acid metabolism’, ‘carbon metabolism’ and ‘nitrogen metabolism’ were the significantly enriched metabolic pathways in the sensitive hybrid ZX 978 (). Under drought conditions, the grain filling rate of maize is accelerated, but the time is short, which will lead to a decline in yield [Citation95]. Because of this phenomenon, maize under drought stress needs more complex molecular mechanisms to maintain its own stability.
Quantitative real-time PCR (qRT-PCR) analysis
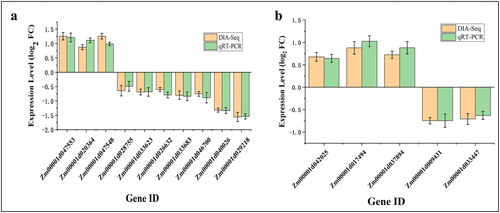
To confirm the accuracy of the DIA-seq data, we performed a verification experiment using quantitative real-time PCR (qRT-PCR). Fifteen genes were selected from different groups of DAPs (Supplemental Table S1). We found that the expression levels (log2 FC) (FC, fold change) of 15 genes measured by qRT-PCR were not significantly different from those measured by DIA-seq (; Supplemental Table S6). In addition, we conducted a correlation analysis of qRT-PCR date and DIA-seq date of 15 genes, showing that the correlation coefficient (R2) was 83.56% (Supplemental Figure S3). Thus, this confirmed the accuracy and reliability of our DIA-seq results.
Figure 5. Confirmation of DIA-seq results by quantitative real-time PCR (qRT-PCR). Quantitative RT-PCR analysis of the expression patterns of the maize kernel genes encoded by differentially abundant proteins (DAPs) from different comparisons: DAPs specific to TD_TC (a) and SD_SC (b). Note: The y-axis represents qRT-PCR (green) relative expression levels (log2-fold change) and log2-fold change of the DIA-seq data (orange). Negative values of expression level mean that the genes were down-regulated in response to drought stress. Maize gene GAPDH (accession no. X07156) was used as the internal reference. Error bars represent the SE (n = 3).
Comparative analysis of DIA and iTRAQ proteomics derived results of maize hybrid cultivars’ drought stress response at the grain-filling stage
More recently [Citation38], we performed comparative iTRAQ proteomics analysis of the same maize hybrids (ND476 and ZX978) responses to drought stress at the same (kernel filling) growth stage. Our comparative analysis of the current (DIA) study and our most recent proteomics study [Citation38] revealed some similarities and variations with regards to the detection capacity of the two proteomic analysis approaches and drought stress response evaluation in maize. Firstly, in terms of the detection capacity of the two strategies, the number of spectra (815,688) that were matched to known spectra using Mascot software (for iTRAQ proteomics) was close to the number of spectra (830,311) that was matched by Spectronaut software (for DIA proteomics). This may suggest that both tools can be effectively used for proteomics analysis with comparably similar success. However, a higher number of unique peptides (87,018) was identified by iTRAQ [Citation38] as compared to significantly fewer peptides (36,717) identified by DIA in this paper. Additionally, the iTRAQ approach detected a higher number of DAPs (1655), although it had fewer (9895) total proteins [Citation38]. Conversely, the DIA approach detected significantly fewer DAPs (826) from a significantly higher number (13,659) of total proteins (this study). Subsequently, the number of DAPs identified to be specifically accumulated in the tolerant genotype ND476 under drought stress conditions were significantly fewer (seven) by the DIA approach () as compared to twenty by the iTRAQ method (see also of Dong et al. [Citation38]). This observation may suggest DIA’s high data utilization and quantitative accuracy [Citation43]. However, both strategies can be employed in proteomics analysis with effectiveness. Further detailed comparisons of this nature will need to be performed to be able to draw up an authoritative conclusion.
With regards to the key drought responsive protein and metabolic networks identified from the two studies, our iTRAQ proteomics analysis results revealed that cellular redox homeostasis maintenance associated- and stress defense proteins were the predominant contributors of maize drought tolerance through their participation in purine metabolism, proteasome and plant hormone signal transduction metabolic pathways [Citation38]. However, our current DIA analysis results attribute maize drought tolerance to stress responsive chaperones, DNA replication related enzymes and seed storage (vicilin) proteins being involved in starch and sucrose metabolism, terpenoid backbone biosynthesis and carbon metabolism metabolic pathways. Although both studies similarly identify redox homeostasis maintenance and stress defense proteins (particularly peroxidases, oxidoreductases and chaperones) critical for maize drought tolerance, they showed varied metabolic pathways in which these proteins were involved. This variation could possibly be explained by the differences in the number of DAPs that were significantly enriched in different pathways. Considering that the DIA approach had fewer DAPs identified, as a result of its rigorous screening, it is possible that some proteins that were excluded from analysis could have obviously caused differences in the significant pathways enrichment of DAPs. Overall, both iTRAQ and DIA approaches are effective and vital tools for performing proteomics analysis of drought stress responses that can be used with confidence either singly or complementarily. Meanwhile, our findings derived from both previous [Citation38] and current studies add to the body of knowledge related to, and offer useful reference data for, maize drought stress tolerance breeding.
Conclusions
The DIA-based analysis of the maize filling-kernel proteomic responses of two contrasting hybrid cultivars exposed to a 12-day drought treatment at the grain filling stage identified a total of 826 DAPs. Among them, we screened out 7 DAPs specifically accumulated in the tolerant genotype ND 476 and 45 DAPs uniquely expressed in the sensitive genotype ZX 978. Our analysis revealed that the maize drought tolerance is related to some key proteins such as chaperones, DNA replication related enzymes, flowering locus T protein and vicilin, ABA-responsive RAB17 protein. These proteins chiefly participate in starch and sucrose metabolism, terpenoid backbone biosynthesis, cyanoamino acid metabolism and carbon metabolism pathways. Our findings provide useful reference data for maize drought stress tolerance during the grain filling stage, as well as offer some theoretical basis for maize drought resistance breeding.
Supplemental Material
Download PDF (229.2 KB)Supplemental Material
Download PDF (327.6 KB)Conflicts of interest
The authors declared no conflict of interest. Furthermore, the funders played no part in designing, data collection, analysis and interpretation of results, writing, editing or decision making process related to the publication of this study.
Additional information
Funding
References
- Campos H, Cooper M, Habben JE, et al. Improving drought tolerance in maize: a view from industry. Field Crops Res. 2004;90(1):19–34.
- International Service for Acquisition of Agri-biotech Applications (ISAAA). Biotechnology for the development of drought tolerant crops. [Internet]. 2008. [cited 2020-08-08]. Available from: http://www.isaaa.org/resources/publications/pocketk/32/default.asp
- Dai A. Increasing drought under global warming in observations and models. Nat Clim Change. 2013;3(1):52–58.
- Feller U, Vaseva II. Extreme climatic events: impacts of drought and high temperature on physiological processes in agronomically important plants. Front Environ Sci. 2014;2:39.
- Zhu JK. Abiotic stress signaling and responses in plants. Cell. 2016;167(2):313–324.
- FAO’s Director-General on How to Feed the World in 2050. Popul Dev Rev. 2009;35:837–839.
- Maazou ARS, Tu JL, Qiu J, et al. Breeding for drought tolerance in maize (Zea mays L.). Am J Plant Sci. 2016;7:1858–1870.
- Xu J, Yuan YB, Xu YB, et al. Identification of candidate genes for drought tolerance by whole-genome resequencing in maize. BMC Plant Biol. 2014;14:83–15.
- Zheng J, Fu JJ, Gou MY, et al. Genome-wide transcriptome analysis of two maize inbred lines under drought stress. Plant Mol Biol. 2010;72(4-5):407–421.
- Sah RP, Chakraborty M, Prasad K, et al. Impact of water deficit stress in maize: phenology and yield components. Sci Rep. 2020;10(1):1–5.
- Jurgens SK, Johnson RR, Boyer JS. Dry matter production and translocation in maize subjected to drought during grain fill. Agronj. 1978;70(4):678–682.
- Afuakwa JJ, Crookston RK. Using the kernel milk line to visually monitor grain maturity in maize. Crop Sci. 1984;24(4):687–691.
- Afuakwa JJ, Crookston RK, Jones RJ. Effect of temperature and sucrose availability on kernel black layer formation in maize. Crop Sci. 1984;24(2):285–288.
- Upadhyaya H, Sahoo L, Panda SK. Molecular physiology of osmotic stress in plants. In: Rout GR, Das AB, editors Molecular stress physiology of plants. Springer India; 2013. p. 179–192.
- Islam M, Begum MC, Kabir AH, et al. Molecular and biochemical mechanisms associated with differential responses to drought tolerance in wheat (Triticum aestivum L.). Plant Interact. 2015;10(1):195–201.
- Bhargava S, Sawant K. Drought stress adaptation: metabolic adjustment and regulation of gene expression. Plant Breed. 2013;132(1):21–32.
- Gill SS, Tuteja N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol Biochem. 2010;48(12):909–930.
- Jogaiah S, Govind SR, Tran LSP. Systems biology-based approaches toward understanding drought tolerance in food crops. Crit Rev Biotechnol. 2013;33(1):23–39.
- Oliver SN, Dennis ES, Dolferus R. ABA regulates apoplastic sugar transport and is a potential signal for cold-induced pollen sterility in rice. Plant Cell Physiol. 2007;48(9):1319–1330.
- Luan MD, Xu MY, Lu YM, et al. Expression of zma-miR169 miRNAs and their target ZmNF-YA genes in response to abiotic stress in maize leaves. Gene. 2015;555(2):178–185.
- Wang G, Zhu QG, Meng QW, et al. Transcript profiling during salt stress of young cotton (Gossypium hirsutum) seedlings via Solexa sequencing. Acta Physiol Plant. 2012;34(1):107–115.
- Jiang QY, Niu FJ, Sun XJ, et al. RNA-seq analysis of unintended effects in transgenic wheat overexpressing the transcription factor GmDREB1. The Crop Journal. 2017;5(3):207–218.
- Min HW, Chen CX, Wei SW, et al. Identification of drought tolerant mechanisms in maize seedlings based on transcriptome analysis of recombination inbred lines. Front Plant Sci. 2016;7:1080.
- Pour-Benab SM, Fabriki-Ourang S, Mehrabi AA. Expression of dehydrin and antioxidant genes and enzymatic antioxidant defense under drought stress in wild relatives of wheat. Biotechnol Biotechnol Equip. 2019;33(1):1063–1073.
- Guo YX, Zhang HH, Yuan YH, et al. Identification and characterization of NAC genes in response to abiotic stress conditions in Picea wilsonii using transcriptome sequencing. Biotechnol Biotechnol Equip. 2020;34(1):93–103.
- Jin H, Liu S, Zenda T, et al. Maize leaves drought-responsive genes revealed by comparative transcriptome of two cultivars during the filling stage. PLoS One. 2019;14(10):e0223786.
- Zenda T, Liu S, Wang X, et al. Key maize drought-responsive genes and pathways revealed by comparative transcriptome and physiological analyses of contrasting inbred lines. Int J Mol Sci. 2019;20(6):1268.
- Liu G, Zenda T, Liu S, et al. Comparative transcriptomic and physiological analyses of contrasting hybrid cultivars ND476 and ZX978 identify important differentially expressed genes and pathways regulating drought stress tolerance in maize. Genes Genomics. 2020;42(8):937–955.
- Cánovas FM, Dumas-Gaudot E, Recorbet G, et al. Plant proteome analysis. Proteomics. 2004;4(2):285–298.
- Zhao Q, Zhang H, Wang T, et al. Proteomics-based investigation of salt-responsive mechanisms in plant roots. J Proteomics. 2013; 82:230–253.
- Komatsu S, Hiraga S, Yanagawa Y. Proteomics techniques for the development of flood tolerant crops. J Proteome Res. 2012;11(1):68–78.
- Wu S, Ning F, Zhang QB, et al. Enhancing omics research of crop responses to drought under field conditions. Front Plant Sci. 2017; 8:174.
- Kosova K, Vitamvas P, Prasil IT, et al. Plant proteome changes under abiotic stress-contribution of proteomics studies to understanding plant stress response. J Proteomics. 2011;74:1301–1322.
- Yang LM, Jiang TB, Fountain JC, et al. Protein profiles reveal diverse responsive signaling pathways in kernels of two maize inbred lines with contrasting drought sensitivity. Int J Mol Sci. 2014;15(10):18892–18918.
- Zenda T, Liu S, Wang X, et al. Comparative proteomic and physiological analyses of two divergent maize inbred lines provide more insights into drought-stress tolerance mechanisms. Int J Mol Sci. 2018;19(10):3225.
- Liu S, Zenda T, Dong A, et al. Comparative proteomic and morpho-physiological analyses of maize wild-type Vp16 and mutant vp16 germinating seeds responses to PEG-induced drought stress. Int J Mol Sci. 2019;20(22):5586.
- Wang X, Zenda T, Liu S, et al. Comparative proteomics and physiological analyses reveal important maize filling-kernel drought-responsive genes and metabolic pathways. Int J Mol Sci. 2019;20(15):3743.
- Dong A, Yang Y, Liu S, et al. Comparative proteomics analysis of two maize hybrids revealed drought-stress tolerance mechanisms. Biotechnol Biotechnol Equip. 2020;34(1):763–780.
- Olfa B, Fethi BM, Beligh M, et al. Response to drought of two olive tree cultivars (cv Koroneki and Meski). Sci Hortic. 2008;116:1–393.
- Doerr A. DIA mass spectrometry. Nat Methods. 2015;12(1):35–35.
- Law KP, Lim YP. Recent advances in mass spectrometry: data independent analysis and hyper reaction monitoring. Expert Rev Proteomics. 2013;10(6):551–566.
- Chapman JD, Goodlett DR, Masselon CD. Multiplexed and data-independent tandem mass spectrometry for global proteome profiling. Mass Spectrom Rev. 2014;33(6):452–470.
- Bruderer R, Bernhardt OM, Gandhi T, et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol Cell Proteomics. 2015;14(5):1400–1410.
- Schgger H. Tricine-SDS-PAGE. Nat Protoc. 2006;1:16–22.
- Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology ConsortiumNat Genet. 2000;25(1):25–29.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–408.
- Natsume T, Tanida I, Iemura S, et al. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J. 2004;23(9):1977–1986.
- Sasakawa H, Sakata E, Yamaguchi Y, et al. Solution structure and dynamics of Ufm1, a ubiquitin-fold modifier 1. Biochem Biophys Res Commun. 2006;343(1):21–26.
- Hagai T, Levy Y. Ubiquitin not only serves as a tag but also assists degradation by inducing protein unfolding. Proc Natl Acad Sci USA. 2010;107(5):2001–2006.
- Xia ZL, Liu QJ, Wu JY, et al. ZmRFP1, the putative ortholog of SDIR1, encodes a RING-H2 E3 ubiquitin ligase and responds to drought stress in an ABA-dependent manner in maize. Gene. 2012;495(2):146–153.
- Farooq M, Wahid A, Kobayashi N, et al. Plant drought stress: effects, mechanisms and management. Agron Sustain Dev. 2009;29(1):185–212.
- Saracco SA, Miller MJ, Kurepa J, et al. Genetic analysis of SUMOylation in Arabidopsis: conjugation of SUMO1 and SUMO2 to nuclear proteins is essential. Plant Physiol. 2007;145(1):119–134.
- Nigam N, Singh A, Sahi C, et al. SUMO-conjugating enzyme (Sce) and FK506-binding protein (FKBP) encoding rice (Oryza sativa L.) genes: genome-wide analysis, expression studies and evidence for their involvement in abiotic stress response. Mol Genet Genomics. 2008;279(4):371–383.
- Mazur MJ, Van DB, Harrold A. Global SUMO proteome responses guide gene regulation, mRNA biogenesis, and plant stress responses. Front Plant Sci. 2012;3:215.
- Zhao Y, Wang Y, Yang H, et al. Quantitative proteomic analyses identify aba-related proteins and signal pathways in maize leaves under drought conditions. Front Plant Sci. 2016;7:1827–1823.
- Ritossa F. A new puffing pattern induced by temperature shock and DNP in drosophila. Experimental. 1962;18(12):571–573.
- Matz JM, Blake MJ, Tatelman HM, et al. Characterization and regulation of cold-induced heat shock protein expression in mouse brown adipose tissue. Am J Physiol. 1995;269(1 Pt 2):R38–47.
- Cao Y, Ohwatari N, Matsumoto T, et al. TGF-beta1 mediates 70-kDa heat shock protein induction due to ultraviolet irradiation in human skin fibroblasts. Pflugers Arch. 1999;438(3):239–244.
- Laplante AF, Moulin V, Auger FA, et al. Expression of heat shock proteins in mouse skin during wound healing. J Histochem Cytochem. 1998;46(11):1291–1301.
- Santoro MG. Heat shock factors and the control of the stress response. Biochem Pharmacol. 2000;59(1):55–63.
- Wong HR. Heat shock proteins. Facts, thoughts, and dreams. A. De Maio. Shock 11:1-12, 1999. Shock. 1999;12(4):323–325.
- Cuimei Z, Shangli S. Physiological and proteomic responses of contrasting alfalfa (Medicago sativa L.) varieties to PEG-induced osmotic stress. Front Plant Sci. 2018;9:242.
- Walter S, Buchner J. Molecular chaperones–cellular machines for protein folding. Angew Chem Int Ed. 2002;41(7):1098–1113.
- Júlio CB, Carlos HIR. Protein folding assisted by chaperones. Protein Peptide Lett. 2005;12:257–261.
- Wang W, Vinocur B, Shoseyov O, et al. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004;9(5):244–252.
- Ma QL, Kang JM, Long RC, et al. Proteomic analysis of salt and osmotic-drought stress in alfalfa seedlings. J Integr Agric. 2016;15(10):2266–2278.
- Rathore RS, Garg N, Garg S, et al. Starch phosphorylase: role in starch metabolism and biotechnological applications. Crit Rev Biotechnol. 2009;29(3):214–224.
- Ahmadi A, Baker DA. The effect of water stress on the activities of key regulatory enzymes of the sucrose to starch pathway in wheat. Plant Growth Regulation. 2001;35(1):81–91.
- Yang JC, Zhang JH, Wang ZQ, et al. Activities of starch hydrolytic enzymes and sucrose-phosphate synthase in the stems of rice subjected to water stress during grain filling. J Exp Bot. 2001;52(364):2169–2179.
- Prasch CM, Ott KV, Bauer H, et al. ß-amylase1 mutant arabidopsis plants show improved drought tolerance due to reduced starch breakdown in guard cells. J Exp Bot. 2015;66(19):6059–6067.
- Lee JB, Hite RK, Hamdan SM, et al. DNA primase acts as a molecular brake in DNA replication. Nature. 2006;439(7076):621–624.
- Cavanaugh NA, Kuchta RD. Initiation of new DNA strands by the herpes simplex virus-1 primase-helicase complex and either herpes DNA polymerase or human DNA polymerase alpha. J Biol Chem. 2009;284(3):1523–1532.
- Keck JL, Berger JM. Primus inter pares (first among equals). Nat Struct Biol. 2001;8(1):2–4.
- Gupta S, Agarwal VP, Gupta NK. Efficacy of putrescine and benzyladenine on photosynthesis and productivity in relation to drought tolerance in wheat (Triticum aestivum L.). Physiol Mol Biol Plants. 2012;18(4):331–336.
- Marini F, Pellicioli A, Paciotti V, et al. A role for DNA primase in coupling DNA replication to DNA damage response. EMBO J. 1997;16(3):639–650.
- Faria JMR, Buitink J, Van L, et al. Changes in DNA and microtubules during loss and re-establishment of desiccation tolerance in germinating Medicago truncatula seeds. J Exp Bot. 2005;56(418):2119–2130.
- Kim SJ, Hong SM, Yoo SJ, et al. Post-translational regulation of flowering locus t protein in Arabidopsis. Mol Plant. 2016;9(2):308–311.
- Notaguchi M, Daimon Y, Abe M, et al. Long-distance, graft-transmissible action of arabidopsis flowering locus t protein to promote flowering. Plant Cell Physiol. 2008;49(11):1645–1658.
- Yan YY, Shen LS, Chen Y, et al. A MYB-domain protein EFM mediates flowering responses to environmental cues in Arabidopsis. Dev Cell. 2014;30(4):437–448.
- Riboni M, Galbiati M, Tonelli C, et al. Gigantea enables drought escape response via abscisic acid-dependent activation of the florigens and suppressor of overexpression of constans1. Plant Physiol. 2013;162(3):1706–1719.
- Su Z, Ma X, Guo H, et al. Flower development under drought stress: morphological and transcriptomic analyses reveal acute responses and long-term acclimation in Arabidopsis. Plant Cell. 2013;25(10):3785–3807.
- Gunning BES, Steer MW. Plant cell biology structure and function. J Microsc. 1996;189:100–101.
- Sanchez-Monge R, Lopez-Torrejón G, Pascual CY, et al. Vicilin and convicilin are potential major allergens from pea. Clin Exp Allergy. 2004;34(11):1747–1753.
- Lycett GW, Delauney AJ, Gatehouse JA, et al. The vicilin gene family of pea (Pisum sativum L.): a complete cdna coding sequence for preprovicilin. Nucleic Acids Res. 1983;11(8):2367–2380.
- Belanger FC, Kriz AL. Molecular-basis for allelic polymorphism of the maize globulin-1 gene. Genetics. 1991;129(3):863–872.
- Amor Y, Haigler CH, Johnson S, et al. A membrane-associated form of sucrose synthase and its potential role in synthesis of cellulose and callose in plants. Proc Natl Acad Sci USA. 1995;92(20):9353–9357.
- González EM, Gordon AJ, James CL, et al. The role of sucrose synthase in the response of soybean nodules to drought. J Exp Bot. 1995;46(10):1515–1523.
- Zheng Y, Anderson S, Zhang Y, et al. The structure of sucrose synthase-1 from Arabidopsis thaliana and its functional implications. J Biol Chem. 2011;286(41):36108–36118.
- Jensen AB, Goday A, Figueras M, et al. Phosphorylation mediates the nuclear targeting of the maize rab17 protein. Plant J. 1998;13(5):691–697.
- Riera M, Figueras M, Lopez C, et al. Protein kinase ck2 modulates developmental functions of the abscisic acid responsive protein rab17 from maize. Proc Natl Acad Sci USA. 2004;101(26):9879–9884.
- Goday A, Jensen AB, Culiáñez-Macià FA, et al. The maize abscisic acid-responsive protein Rab17 is located in the nucleus and interacts with nuclear localization signals. Plant Cell. 1994;6(3):351–360.
- Vilardell J, Goday A, Freire MA, et al. Gene sequence, developmental expression, and protein phosphorylation of rab-17 in maize. Plant Mol Biol. 1990;14(3):423–432.
- Figueras M, Pujal J, Saleh A, et al. Maize rab17 overexpression in arabidopsis plants promotes osmotic stress tolerance. Ann Applied Biol. 2004;144(3):251–257.
- Yang CQ, Liu RX, Yang FQ, et al. Effects of drought on the sucrose metabolism of subtending leaves of cotton bolls at different fruiting branches and boll weight during flowering and bolling stages. Cotton Sci. 2014;26:452–458.
- Wardlaw IF. Interaction between drought and chronic high temperature during kernel filling in wheat in a controlled environment. Ann Bot. 2002;90(4):469–476.