
Abstract
Salix triandroides W. P. Fang, an indigenous small willow with high potential for heavy-metal soil remediation and levee protection, has been used for wetland ecological restoration for decades. However, studies on the evolution and genetic breeding of S. triandoides have been hindered by the lack of its genetic information. Here, we present the complete chloroplast genome of S. triandroides, which was assembled from Illumina sequencing data. The chloroplast genome of this species is circular, 155,683 bp in length and includes two inverted repeat regions (IRs) (27,490 bp) separated by a large single-copy (LSC) region (84,463 bp) and a small single-copy (SSC) region (16,240 bp). A total of 126 unique genes were identified, including 81 protein-coding genes, 37 tRNA genes and eight rRNA genes. Comparative analysis identified some hypervariable regions, with potential to be used as specific DNA barcodes or candidate markers for phylogenetic studies. Based on the sequences of the protein-coding genes, the phylogenetic analysis assigned 32 Salix species into two major clades and revealed that S. triandoides was a sister taxon to S. triandra. Our results provide a foundation for further molecular breeding of S. triandroides and insights into the evolution of Salix.
Supplemental data for this article is available online at https://doi.org/10.1080/13102818.2021.2023326 .
Introduction
Salix L. (often know as willows) is the largest genus of the family Salicaceae and has 450–520 species [Citation1–3]. Salix species are distributed mainly in the Northern Hemisphere [Citation2] and over half of the willow species (275) could be found in China [Citation4,Citation5]. Salix species have been used for ecological remediation and restoration in heavy metal-contaminated soil because these trees are highly productive and thrive on many nutrient-poor soils [Citation6–8]. Salix triandroides W. P. Fang, an indigenous small willow, has been used to remediate Cd-contaminated soils for decades [Citation9,Citation10]. Being highly adaptable in wet conditions, S. triandroides are often planted for wave reduction, levee protection and biodiversity conservation in wetlands [Citation11]. Previous studies on this species are few and mainly focus on effects of various stress factors (e.g. flooding and heavy-metal toxicity). So far few genomic studies have been documented, except for phylogenetic analysis with a few short gene sequences [Citation3].
The plant chloroplast genome contains a typical quadripartite construction, which contains two inverted repeat regions (IRa and IRb), each insulated by a large single-copy (LSC) at one end and by a small single-copy (SSC) at the other end. The chloroplast genome is a haploid and maternally inherited in angiosperms. It is a good complement to the nuclear genetic markers and has been widely employed for phylogenetic analysis and domestication studies of higher plants [Citation12–14]. The complete chloroplast genome sequences also show potential in understanding structural and functional evolutionary pathways [Citation12,Citation15]. In addition, chloroplast genetic engineering has been used for improving crops productivity and stress-resistance [Citation16]. In genus Salix, nearly 30 chloroplast genomes have been published, but the chloroplast genome of S. triandroides has not yet been reported.
In this study, we de novo sequenced and assembled a complete chloroplast genome of S. triandroides. In parallel, we assembled two chloroplast genomes of two other closely related species, S. brachista and S. dunnii, from published Illumina sequencing data [Citation17,Citation18]. Then we characterized their genomic structures and features including the gene content, repeated sequences and IR expansions/contractions, etc. We detected sequence divergence and phylogenetic relationship in Salix by comparative chloroplast genome analyses. We also predicted mutation hotspot regions in Salix, which would be helpful for developing potential molecular markers. This study will also provide a powerful tool to accelerate breeding and biotechnological studies for S. triandroides.
Materials and methods
Sample preparation, de novo sequencing and assembly
For chloroplast genome sequencing, we chose a mature and healthy S. triandroides tree from Yueyang Forestry and Ecology Garden (Yueyang City, Hunan Province, China). Total genomic DNA was extracted from fresh leaves following the CTAB-chloroform protocol [Citation19]. We constructed short-insert libraries with sizes of 500 bp for sequencing on the Illumina HiSeq X Ten platform under the PE 150 bp protocol. After quality assessment, reads were assembled into contigs using the program NOVOPlasty [Citation20]. A ribulose-1, 5-bisphosphate carboxylase/oxygenase (rbcL) gene sequence from S. triandra (GenBank accession no. MK722343) was used as the seed sequence, and complete chloroplast genome sequences of S. triandra and S. wilsonii (NC053549) were used as references to resolve the inverted repeat in the chloroplast genome of S. triandroides.
For the lack of available annotations of the chloroplast genome, we assembled the chloroplast genomes of another two Salix species (S. brachista and S. dunnii) which had been whole-genome sequenced [Citation17,Citation18]. These two chloroplast genomes were assembled with Illumina reads which were downloaded from the NCBI (National Center for Biotechnology Information) database (SRX9358501 and SRX4215130).
Genome annotation and sequence characterization
The assembled chloroplast genomes were annotated by a combination of GeSeq [Citation21] and CPGAVAS2 techniques [Citation22]. The different annotations of protein-coding sequences were determined using BLASTx. The tRNAs were scanned with tRNAscan-SE v2.0.3 [23]. Final chloroplast genome maps were drawn using OGDRAW program [Citation21]. The annotated chloroplast genomic sequence has been deposited in GenBank with an accession number: MW929213-MW929215.
We acquired chloroplast genome sequences of Salix species and two outgroup species in genus Populus (Salicaceae) from GenBank () to compare features among them and the chloroplast genome assembled in this study. We calculated the GC content for chloroplast genomes using an in-house perl script. The codon usage frequencies and the relative synonymous codon usage (RSCU) were analyzed for all protein-coding sequences using MEGA-X [Citation24]. In addition, we predicted RNA editing sites in protein-coding genes using the PREP suite with a cut-off value of 0.8 [Citation25]. To identify simple sequence repeats (SSRs), identified mono-, di-, tri-, tetra-, penta- and hexanucleotides with the minimum repeat number were set at 10, 5, 4, 3, 3 and 3, respectively, through the MIcroSAtellite (MISA) program. Redundant SSRs were manually checked and discarded. In addition, complex repeat sequences (i.e. forward, reverse, complement and palindromic) were identified using REPuter online software setting hamming distance ≤ 3 bp and the minimum repeat size ≥ 30 bp [Citation26].
Table 1. Summary of the chloroplast genomes of three Salix species.
Comparison of the chloroplast genomes and divergent hotspot identification
To estimate the conservation of chloroplast genes, we compared six Salix species with the annotation of S. triandroides as the reference. We used mVISTA online software to visualize rearrangements and inversions in sequences through the Shuffle-LAGAN program [Citation27]. Junction regions of IR-LSC and IR-SSC in the chloroplast genomes of these six Salix plants and Populus trichocarpa were compared by using IRscope [Citation28].
For the nucleotide diversity analysis, we downloaded the chloroplast genomes of 29 Salix species from NCBI (). The nucleotide diversity (Pi value) was calculated based on the sliding window analysis using the DnaSP v5.10 [Citation29]. The window size was set to 600 bp with a step size of 100 bp. After identifying hypervariable regions by sliding widows, we recalculated Pi values for the complete genic or intergenic sequences of these potential hotspot regions.
Phylogenetic analysis
For the phylogenetic analysis, 32 Salix species and two outgroup Populus species were recruited to construct the phylogenetic tree. The whole chloroplast genome sequences of the analyzed species were aligned using MAFFT v7.475 with the default settings [Citation30]. The best-fitting nucleotide substitution model was determined using the Akaike Information Criterion in the model-finder IQ-TREE [Citation31]. ML analysis was performed using IQ-TREE with the best model of GTR + F + R2 and 1000 bootstrap replicates.
Results
Organization of chloroplast genomes of Salix species
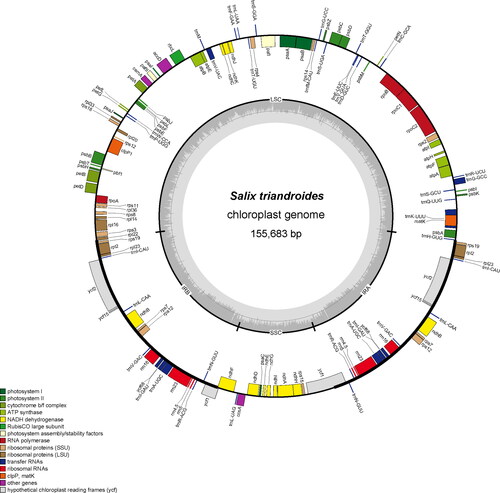
The chloroplast genomes of three Salix species (S. triandroides, S. brachista and S. dunnii) contain the typical quadripartite structures (; ), which include a large single copy (LSC) region, two inverted repeats (IR) and a small single copy (SSC) region. These three Salix chloroplast genomes ranged from 155,600 bp to 155,683 bp in length (), with IRs ranging from 27,429 to 27,490 bp, LSCs ranging from 84,422 to 84,497 bp and SSCs ranging from 16,240 to 16,292 bp. Each chloroplast genome encoded 37 transfer RNAs (tRNAs) and 8 ribosomal RNAs (rRNAs). The chloroplast genome of Salix dunnii included 82 protein-coding genes, while ycf1 around JSA (joint of SSC and IRa regions) was lacking in the other two species. In total, each chloroplast genome included 126 (ycf1 loss) or 127 (ycf1 present) genes. In addition, 17 intron-containing genes were detected in all genomes, and three of the intron-containing genes (rps12, pafI and clpP1) contained three exons (). The total GC contents of these three assembled chloroplast genomes were 36.7% on average ().
Figure 1. Circular map of the chloroplast genome of S. triandroides. Genes belonging to different functional groups are highlighted in different colours. Genes on the outside of the circle are transcribed counter-clockwise, while genes on the inside are transcribed clockwise. The darker grey in the inner circle corresponds to GC content, and the lighter grey corresponds to AT content.
To detect length variations, we compared the chloroplast genomes of nine Salix plants and nine Populus plants. There were higher genetic variabilities in the single copy regions (both LSC and SSC) than in the IR regions in Salix species. However, the IR regions diverged more than the SSC region in Populus species (). In addition, we compared the total GC content of nine Salix plants and nine Populus plants. The Salix species have similar total GC contents to Populus species (average in 36.7%) ().
Table 2. Relative synonymous codon usage (RSCU) comparison of three Salix plants.
Codon usage analysis and RNA editing in protein coding genes
In this study, we found that there were 23,879, 23,991 and 22,601 codons (including stop codons) in the chloroplast genomes of S. triandroides, S. brachista and S. dunnii, respectively. Twenty amino acids were translated from each genome. Leucine was the most abundant translated amino acid and corresponded to 2423 to 2545 codons, followed secondly by isoleucine corresponding to 1938 to 2076 codons.
Commonly, a RSCU (relative synonymous codon usage) larger than 1 means that the codon is a frequently used one, while a value less than 1 suggests a relatively less used codon [Citation32]. A RSCU at 1 indicates there is no preference for codon use [Citation32]. In this study, we found that there were 64 types of codons in the Salix chloroplast genome. RSCU showed high level of similarities in the three Salix chloroplast genomes (). Leucine and isoleucine were the most abundant amino acids and had a frequency of 10.6% to 10.7% and 8.6% to 8.7% in these genomes. In each genome, there were 30 codons with RSCU values larger than 1, and 32 codons with RSCU values less than 1. In addition, the RSCU values of AUG and UGG were equal to 1, which suggests that these two types of codons have no bias in expression. The codon usage bias in Salix was similar to those in Garcinia species [Citation33] and Theobroma species [Citation14], but different from Lilium species [Citation34].
There were 23 protein coding genes in the chloroplast genome of S. triandroides with a total of 58 RNA editing sites (). Both S. brachista and S. dunnii contained 60 RNA editing sites, and they were distributed on 23 and 25 coding genes (Supplemental Table S4 and S5). All sites served the function of changing the nucleobases from Cytosine to Uracil. Both ndhB and ndhD genes contained 11 RNA editing sites in these three chloroplast genomes.
Table 3. The variability of traditional chloroplast markers and the hypervariable markers.
Repeat sequences and SSR analysis
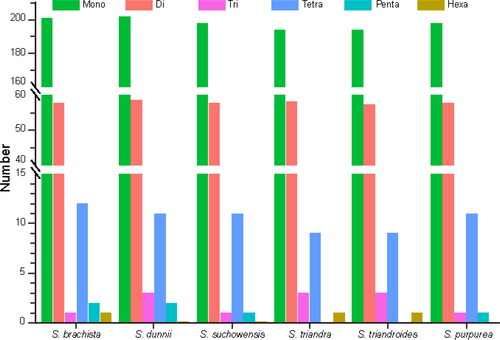
SSR molecular markers have been well-developed and frequently used in plant genetic analysis. In this study, MISA was used to analyze the SSRs in six Salix chloroplast genomes (). S. dunnii had the largest number of SSRs at 275, whereas S. triandroides had the least number at 261 (). The majority of these SSRs were mononucleotides (poly-A or poly-T mainly), with 194–202 members in these chloroplast genomes (). In each species, there were more than 50 dinucleotide repeats, about ten tetranucleotide repeats and at least one trinucleotide repeat. Pentanucleotide and hexanucleotide repeats were rarely found in some species. For most SSRs (more than 65%), the A/T content is much higher than that of G/C. It was consistent with the fact that the complete chloroplast genomes of Salix species are rich in A/T (> 63.2%).
Figure 2. Comparative analysis of simple sequence repeats (SSRs) in six chloroplast genomes.
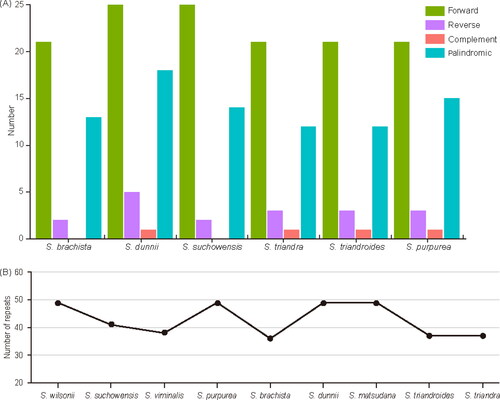
We also detected long complex repetitive sequences including forward repeats, reverse repeats, palindromic repeats and complement repeats in these six species. Most of the repeats were of 30–60 bp in length. The minimum number of repeats was found in S. brachista (36) whereas the maximum was found in S. dunnii (49) (). In addition, we studied the correlation between the repeat number and size of the genome for the nine species, and found that they were not related (). These observations strengthen the independence of repeats number from genome size.
Figure 3. (A) Identification and comparison of complex repetitive sequences in six chloroplast genomes. (B) Correlation of repeats with chloroplast genome size. Species were arranged based on genome size. The discontinuous pattern of the graph shows the random distribution of repeats independent of genome size.
Inverted repeats contraction and expansion
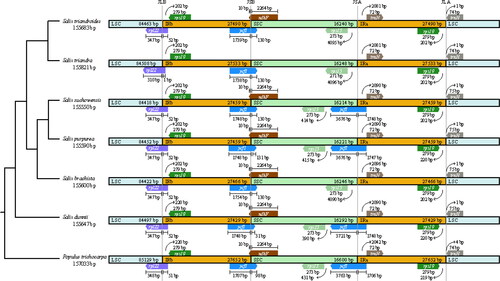
The inverted repeats contraction and expansion revealed variations at the junctions of any two of the regions (LSC/IRs/SSC). The comparative analysis indicated that the junction of LSC and IRb regions (JLB) and the junction of LSC and IRa regions (JLA) were conserved across species. In all species, rps19 is present completely in the LSC region and rpl22 starts in LSC regions and extends into the SSC region. A functional copy of ycf1 gene was observed at JSA in four species, whereas the truncated copy was found at the junction of IRb/SSC (JSB) in all species. At the junction of LSC and IRa (JLA), trnH exists in the LSC and rps19 exists in the IRa ().
Figure 4. Comparison of quadripartite junction sites in Salix chloroplast genomes. The start and end of each gene from the junctions is shown with arrows. JLB (IRb/LSC), JSA (SSC/IRa), JSB (IRb/SSC) and JLA (IRa/LSC) denote the junction sites between the quadripartite regions of the genome.
Sequence divergence and hotspots
The mVISTA plots display sequence similarities, collinearity patterns and genetic variations among the chloroplast genomes from six Salix species (). Our results indicated that the gene number, order and orientation were conserved in Salix. The sequences in the IR regions were more conserved than those in the single copy (LSC and SSC) regions. This may be due to the conservative characteristics of rRNA genes, which were all distributed in IR regions. The sequences in the non-coding regions were more divergent than those in the coding regions. The most divergent non-coding regions among these chloroplast genomes were trnK-psbK, ndhC-trnM and psbE-petL.
Figure 5. Sequence comparison of five chloroplast genomes in mVISTA using S. triandroides as a reference. Arrows indicate the annotated genes and their transcriptional direction. The vertical scale represents the percent identity.

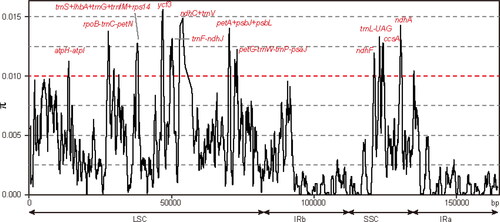
Additionally, we downloaded another 29 Salix chloroplast sequences from NCBI () and measured the nucleotide diversity by DNAsp to identify the mutation hotspot regions in the whole Salix chloroplast genomes (). Nucleotide sequences less than 600 bp long had diversity values varying from 0 to 0.01559. Highly diverged regions were shown in . We extracted the whole sequences of highly diverged regions and calculated their Pi values (). The region ndhC + trnV has the highest Pi values (Pi = 0.01295) followed by the other four spacer regions (Pi > 0.01) including trnS-lhbA-trnG-trnfM-rps14, trnF-ndhJ, rpoB-trnC-petN and atpH-atpI (). All these features were in the LSC region.
Figure 6. Sliding-window analysis of the whole chloroplast genomes of 32 Salix species. Window length: 600 bp; step size: 100 bp. X-axis: position of the midpoint of a window. Y-axis: nucleotide diversity of each window.
Phylogenetic analysis
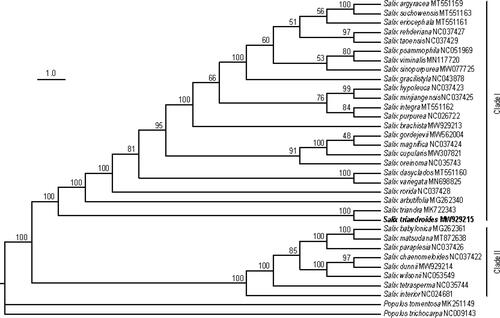
To study the phylogenetic position of S. triandroides, we constructed a maximum likelihood (ML) phylogenetic tree by using the amino acid sequences of protein-coding genes of 32 Salix species (29 downloaded from NCBI database and three assembled in this study) with two Populus species as outgroups (). Salix plants were clearly subdivided into two well-supported clades (boot-strap values = 100%), consistent with previous studies [Citation35,Citation36]. The phylogenetic analysis strongly supported that S. triandroides was closely related to S. triandra, and they were assigned to Clade I. This is congruent with previous research [Citation3]. This analysis substantially increases our understanding of the evolutionary relationship among Salix species.
Figure 7. Phylogenetic tree constructed using the maximum likelihood (ML) method based on the whole chloroplast genomes from 32 Salix species and two Populus species. The numbers above the branches represent the ML bootstrap values.
Discussion
In terms of species number, Salix is the largest genus in Salicaceae [Citation37]. Due to its wide geographic distribution, strong adaptability and fast growth rates, most Salix species are economically valuable. Some of the Salix species have been cultivated as raw materials for a wide range of wood products, as well as for paper and pulp, rope making and others [Citation38]. Due to the great demand of wood, genetic engineering within the chloroplast genome has been used as an effective way to improve plant productivity [Citation16]. Chloroplast genome resources can facilitate genetic engineering in Salix species. Here, we obtained the complete chloroplast genome of S. triandroides using Illumina sequencing data.
The structure of the chloroplast genome of S. triandroides was conservative and displayed a classical quadripartite structure. The gene content, gene order and gene orientation in S. triandroides were conserved. Some length differences in the chloroplast genomes were observed among strains of both Salix and Populus species (). In Salix, the length of LSC regions accounted for the size difference in the chloroplast genome, consistent with similar studies for Gossypium genus [Citation39] and Chrysanthemum boreale strains [Citation40]. In contrast, in Populus, the IR regions rather than the SSC regions contributed more to the variations in lengths of chloroplast genomes, which agree with a study on duckweed species (Lemnoideae) [Citation41]. This indicated that variations in chloroplast size may be lineage-specific in Salicaceae.
Even though universal DNA barcodes have been used for phylogenetic analysis at various taxonomic levels, they often discriminate the closely related species poorly for the lack of variations and lower divergence [Citation42]. In Salix, traditional chloroplast markers () lack variability, the employment of which may lead to unsuccessful species identification [Citation36,Citation43,Citation44]. Chloroplast genome sequences provide an efficient tool for taxonomic studies and provide valuable genetic information for subsequent analysis. The mutation events are unevenly distributed within the chloroplast genome and are concentrated in certain regions, called ‘hotspot’ regions [Citation42]. In this study, we identified some hotspot regions (; ) for 32 Salix species. These hypervariable regions could be developed to be specific DNA barcodes for further studies.
In Salix, the occurrence of repeats (SSR and LDR) was more prevalent in the non-coding regions (intergenic regions and introns) than in the coding regions, which is similar to the reports in Chrysanthemum boreale [Citation40], Artemisia selengensis [Citation45] and Aster tataricus [Citation46]. Weng et al. inferred that the distribution patterns of repeat sequences in plastid genomes are associated with genome rearrangements and nucleotide substitution rates [Citation47]. Therefore, these repeats could be used to develop genetic markers not only for phylogenetic studies, but also for detecting the diversity and differentiation of Salix and other Salicaceae species.
Phylogenetic analysis of willows has been reported for more than two decades. Azuma et al. (2000) firstly assigned 23 Salix species, based on rbcL genes, into two major clades [Citation35], which was supported by many subsequent studies [Citation36,Citation43,Citation44]. However, all these phylogenetic analyses were conducted based on less than three markers and relationships within species were not well resolved. A similar result has been found in subgenus Salix s.l. [Citation3]. Based on the whole genome assembly, Zhang et al. [Citation48] and He et al. [Citation18] constructed phylogenetic trees with high support values by using thousands of single-copy orthologous genes. However, these studies analyzed only three and five Salix species. In the present study, 32 Salix species were also subdivided into two major clades and the relationships among species were well resolved (). To the best of our knowledge, this is the first phylogenetic tree generated for so many Salix species based on the whole chloroplast genome sequences. The low support values of some nodes may be due to the high frequency of interspecific hybridization within this genus [Citation49]. Nevertheless, two major clades were supported with high bootstrap values.
To summarize, we have presented a de novo assembled chloroplast genome sequence of S. triandroides. We detected some hypervariable regions of Salix and they could be developed to potential DNA barcodes. Further phylogenetic studies based on chloroplast genome sequences with more species are necessary to shed light on the phylogenetic relationships among Salix plants.
Conclusions
Salix triandroides is an important small willow for levee protection and ecological restoration of wetland. In this study, we reported a complete chloroplast genome of S. triandroides which is 155,683 bp in length, including a pair of inverted repeat regions (27,490 bp), a large single-copy region (84,463 bp) and a small single-copy region (16,240 bp). We annotated a total of 126 unique genes in this circular chloroplast genome. We further predicted some hypervariable regions in this chloroplast genome for candidate marker development. Phylogenetic analysis assigned Salix species into two major clades and revealed that S. triandoides was a sister to S. triandra. Our research will provide a foundation for molecular breeding of S. triandroides and represent new insights into the evolution of Salix.
Supplemental Material
Download MS Excel (22.5 KB)Acknowledgement
We are sincerely grateful to Peizhen Zhang, Weirong Qian, Zhongnan Wang and Jianming Xiong (Yueyang Forestry and Ecology Garden) for plants cultivation.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The raw genomic Illumina data have been deposited in the NCBI Sequence Read Archive under accession numbers PRJNA735582. The chloroplast genomes of the S. brachista, S. dunnii and S. triandroides were submitted to GenBank under accession numbers: MW929213-MW929215.
Supplemental data
Supplemental data are deposited in Figshare (10.6084/m9.figshare.17143661.v1).
Additional information
Funding
Related Research Data
References
- Fang ZF, Zhao SD, Skvortsov AK. Salicaceae. In: Wu Z, Raven PH, editors. Flora of China. Vol. 4. St. Louis (MO): Missouri Botanical Garden Press; 1999. p. 139–274.
- Argus GW. Salix. In: Flora of North America Editorial Committee, editors. Vol. 7: Magnoliophyta: Salicaceae to Brassicaceae. New York: Oxford University Press; 2010. p. 23–51.
- Wu J, Nyman T, Wang D, et al. Phylogeny of Salix subgenus Salix s.l. (Salicaceae): delimitation, biogeography, and reticulate evolution. BMC Evol Biol. 2015;15:31.
- Gullberg U. Towards making willows pilot species for coppicing production. For Chron. 1993;69(6):721–726.
- Lindegaard KN, Barker J. Breeding willows for biomass. Asp Appl Biol. 1997;49:155–162.
- Dos Santos Utmazian MN, Wieshammer G, Vega R, et al. Hydroponic screening for metal resistance and accumulation of cadmium and zinc in twenty clones of willows and poplars. Environ Pollut. 2007;148(1):155–165.
- Kuzovkina YA, Volk TA. The characterization of willow (Salix L.) varieties for use in ecological engineering applications: co-ordination of structure, function and autecology. Ecol Eng. 2009;35(8):1178–1189.
- Marmiroli M, Pietrini F, Maestri E, et al. Growth, physiological and molecular traits in salicaceae trees investigated for phytoremediation of heavy metals and organics. Tree Physiol. 2011;31(12):1319–1334.
- Li Y, Li X, Zhang C, et al. Change trend of Salix ligneous plants in the dongting lake wetlands and its reasons. Wetl Sci. 2014;12:646–649. (in Chinese).
- Yao X, Ma F, Li Y, et al. Effect of water cadmium concentration and water level on the growth performance of Salix triandroides cuttings. Environ Sci Pollut Res Int. 2018;25(8):8002–8011.
- Ding X, Zou J, Li Y, et al. Acclimation of Salix triandroides cuttings to incomplete submergence is reduced by low light. Aquat Ecol. 2017;51(2):321–330.
- Jansen RK, Cai Z, Raubeson LA, et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci U S A. 2007;104(49):19369–19374.
- Wu Z, Gui S, Quan Z, et al. A precise chloroplast genome of Nelumbo nucifera (Nelumbonaceae) evaluated with Sanger, Illumina MiSeq, and PacBio RS II sequencing platforms: insight into the plastid evolution of basal eudicots. BMC Plant Biol. 2014;14:289.
- AbdullahWaseem S, Mirza B, et al. Comparative analyses of chloroplast genomes of Theobroma cacao and Theobroma grandiflorum. Biologia. 2020;75:761–771.
- Moore MJ, Soltis PS, Bell CD, et al. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc Natl Acad Sci U S A. 2010;107(10):4623–4628.
- Devine AL, Daniell H. Chloropalst genetic engineering. In: Moller S, editor. Plastids. London: Blackwell Publisher; 2004. p. 283–320.
- Chen J, Huang Y, Brachi B, et al. Genome-wide analysis of cushion willow provides insights into alpine plant divergence in a biodiversity hotspot. Nat Commun. 2019;10(1):5230.
- He L, Jia K, Zhang R, et al. Chromosome-scale assembly of the genome of Salix dunnii reveals a male-heterogametic sex determination system on chromosome 7. Mol Ecol Resour. 2021;21(6):1966–1982.
- Doyle JJ, Doyle JL. A rapid DNA isolation procedure from small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–15.
- Dierckxsens N, Mardulyn P, Smits G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017;45(4):e18.
- Tillich M, Lehwark P, Pellizzer T, et al. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017;45(W1):W6–W11.
- Shi L, Chen H, Jiang M, et al. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019;47(W1):W65–W73.
- Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33:686–689.
- Kumar S, Stecher G, Li M, et al. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549.
- Mower JP. The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009;37:253–259.
- Kurtz S, Choudhuri JV, Ohlebusch E, et al. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29(22):4633–4642.
- Brudno M, Do CB, Cooper GM, et al. Lagan and multi-Lagan: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003;13(4):721–731.
- Amiryousefi A, Hyvönen J, Poczai P. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 2018;34(17):3030–3031.
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–1452.
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780.
- Nguyen LT, Schmidt HA, von Haeseler A, et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–274.
- Oresic M, Shalloway D. Specific correlations between relative synonymous codon usage and protein secondary structure. J Mol Biol. 1998;281(1):31–48. 1998
- Wang Y, Zhao B, Lu Z, et al. The complete chloroplast genome provides insight into the polymorphism and adaptive evolution of Garcinia paucinervis. Biotechnol Biotec Eq. 2021;35(1):377–391.
- Liu H, Yu Y, Deng Y, et al. The chloroplast genome of Lilium henrici: genome structure and comparative analysis. Molecules. 2018;23(6):1276.
- Azuma T, Kajita T, Yokoyama J, et al. Phylogenetic relationships of Salix (Salicaceae) based on rbcL sequence data. Am J Bot. 2000;87(1):67–75.
- Lauron-Moreau A, Pitre FE, Argus GW, et al. Phylogenetic relationships of American willows (Salix L., Salicaceae). PLoS One. 2015;10(9):e0138963.
- Argus GW. Salix (Salicaceae) distribution maps and a synopsis of their classification in North America, North of Mexico. Harv Pap Bot. 2007;12(2):335–368.2.0.CO;2]
- Sharma KR, Kilemwab AM, Singh NB, et al. Variability in wood properties of promising willow clones. Int J Lignoc Product. 2014;1:82–92.
- Chen Z, Grover CE, Li P, et al. Molecular evolution of the plastid genome during diversification of the cotton genus. Mol Phylogenet Evol. 2017;112:268–276.
- Tyagi S, Jung J-A, Kim JS, et al. A comparative analysis of the complete chloroplast genomes of three Chrysanthemum boreale strains. PeerJ. 2020;8:e9448.
- Ding Y, Fang Y, Guo L, et al. Phylogenic study of lemnoideae (duckweeds) through complete chloroplast genomes for eight accessions. PeerJ. 2017;5:e4186.
- Dong W, Liu J, Yu J, et al. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS One. 2012;7(4):e35071.
- Chen J, Sun H, Wen J, et al. Molecular phylogeny of Salix L. (Salicaceae) inferred from three chloroplast datasets and its systematic implications. Taxon. 2010;59(1):29–37.
- Hardig TM, Anttila CK, Brunsfeld SJ. A phylogenetic analysis of Salix (Salicaceae) based on matK and ribosomal DNA sequence data. J Bot. 2010;2010:1–12.
- Meng D, Xiaomei Z, Wenzhen K, et al. Detecting useful genetic markers and reconstructing the phylogeny of an important medicinal resource plant, Artemisia selengensis, based on chloroplast genomics. PLoS One. 2019;14(2):e0211340.
- Shen X, Guo S, Yin Y, et al. Complete chloroplast genome sequence and phylogenetic analysis of aster tataricus. Molecules. 2018;23(10):2426.
- Weng M, Blazier JC, Govindu M, et al. Reconstruction of the ancestral plastid genome in geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol Biol Evol. 2014;31(3):645–659.
- Zhang J, Yuan H, Li Y, et al. Genome sequencing and phylogenetic analysis of allotetraploid Salix matsudana Koidz. Hortic Res. 2020;7(1):201.
- Hörandl E, Florineth F, Hadacek F. Weiden in österreich und angrenzenden gebieten [Willows in Austria and adjacent regions]. 2nd ed. Vienna (Austria): University of Agriculture; 2012. p. 164.