
Abstract
Publicly available genome-wide data, sequenced from 2730 ancient human samples were analyzed for genetic predisposition to malignancy. The temporal and spatial incidence of risk variants for cancer diseases in ancient genomes was recorded, allowing for estimates of their frequencies in ancient human communities. We identified 55 risk alleles associated with oncological conditions in the screened ancient samples. For further analysis, we selected three variants conclusively deemed to entail pathogenic effect, the VHL gene’s rs28940298, the TP53 gene’s rs78378222 and the BRCA2 gene’s rs11571833. The contemporary population frequency of these three variants is lower, or similar, to their estimated frequency in ancient human communities, indicating that they might have been subject to negative selection. This is also suggested by their temporal dynamics during the last 10000 years, which show an overall temporal decrease in population frequency. The oldest samples in which these three variants were established testify their ancient origin, including the presence of TP53 gene’s rs78378222 variant in 50000 BP old Neanderthal and Denisovan genomes. Our results demonstrate the presence of cancer-causing mutations in ancient human communities and suggest that they differed in frequency among ancient populations as they do among contemporary ones. Data on germline mutations in tumour suppressor genes in ancient human genomes are scarce and their historic prevalence gives insights into the evolution of predisposition to cancer disease and hence helps advance paleogenomic medicine.
Introduction
There has been a rise in the prevalence of oncological diseases in modern times but the reasons behind this rise remain unclear [Citation1]. Detrimental factors associated with modern lifestyle, the prolongation of human lifespan in recent decades, and novel screening methods could all be contributing to this trend. Analyses of human remains from ancient communities, facilitated by the achievements of contemporary oncology research, could give insights into the evolution and epidemiology of cancer diseases. The rarity of preserved soft tumour tissues, and difficulties in the analysis of neoplastic lesions in mummies [Citation2], partially explains why data on cancer prevalence in ancient human communities are scarce. Short lifespan and the absence of carcinogenic substances in the environment such as pollution, food additives and tobacco, might also be factors explaining this scarcity of data. Presumptive cancer cases based on visible pathologic bone lesions have been demonstrated in ancient Egyptian populations [Citation3, Citation4]. Analysis of skeletal remains from necropolises in the vicinity of Rome from the Imperial Age (first century BC – third century AD) establish a neoplasm typical of osteosarcoma [Citation5], and a rare case of bone metastasis of prostate carcinoma in an elderly man [Citation6]. Merczi et al. (2014) detect three cases of probable skeletal metastatic carcinoma in Hungary from the Roman period (1st-fifth century AD) [Citation7].
The effect of mutations predisposing to cancer is currently mostly based on genetic studies on contemporary populations. However, exceptionally preserved neoplastic cells in the mummy of Ferrante I of Aragon, King of Naples and leading figure of the Italian Renaissance (1431–1494), indicate the presence of an infiltrating epithelial malignant tumour, probably in the colon [Citation8]. The subsequent molecular analyses establish mutations in codon 12 in the K-RAS oncogene, a main hotspot for mutations causing colon cancer. Another study on eighteenth century mummies establishes a missense mutation in the APC gene, mutations in which are known to be common and strongly associated with the development of colorectal carcinomas [Citation9]. These studies indicate that genetic predisposition to cancer already existed in the pre-industrialization era. Genomic methods are just beginning to be utilized for analyses of disease occurrence in ancient DNA (aDNA). Such efforts are however complicated by the difficulty in characterizing the relationship between genotype and tumour phenotype in ancient samples. Also, DNA extracted from ancient bone material is highly fragmented, which does not allow complete reading of the information and provides only limited opportunities for analyzing germline mutations. Insight can be gained however by virtue of the ever increasing data of ancient whole-genome data, coupled with accumulating knowledge about the clinical significance of mutations predisposing to hereditary types of cancer in contemporary human populations.
The aim of this study was to analyze publicly available ancient genome-wide data for the prevalence of risk variants in genes predisposing to cancer.
Materials and methods
This study is based on analysis of 2730 publicly available ancient genome-wide data with information for up to 1.23 million positions in the genome in hg19 coordinates [Citation10]. The allocation to geographic regions of the aDNA samples is given in .
Table 1. Allocation to geographic region of the ancient DNA samples analyzed.
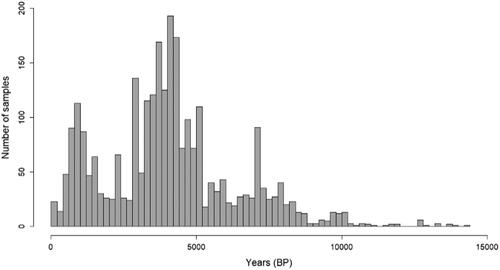
The age distribution from 100 BP to 15000 BP of the analyzed ancient samples is given in .
Figure 1. Age distribution of all aDNA samples examined (100–15000 BP).
Of all the genomic positions in the aDNA for which data were available (up to 1.23 million), initially we filtered out variants listed in the publicly available DisGeNet database [Citation11] of genes and variants associated with human diseases (of the 369554 variants listed in DisGeNet, there was information for 32643 in the ancient genome data samples). A list of 167 (Supplementary material S1) genes associated with oncologic conditions was compiled of genes used in screening tests of five renowned genomic laboratories, i.e. Genologica Medica (Spain), Sema4 (United States), Baylor Genetics (United States), Invitae (United States) and Genomic Diagnostic Laboratory, Children’s Hospital of Philadelphia (United States). The ancient genome-wide data contain information about 479 variants in the 167 genes associated with oncological conditions (Supplementary material S1). Based on large-scale case-control studies, WGS analyses, meta-analyses and other relevant studies, the DisGeNet database identifies 55 of these variants to be risk alleles for oncological conditions (Supplementary material S2).
Results
The estimated frequency of these 55 risk variants in ancient populations is consistently lower than their global frequency in contemporary populations (). Genetic factors of marked importance for diseases, including cancer are, however, rare variants, as the functional effect of rare variants is usually larger than that of common variants [Citation12]. Therefore for further analyses we selected three variants with low frequency in contemporary populations (MAF ≤ 0.01): the VHL gene’s rs28940298, the TP53 gene’s rs78378222 and the BRCA2 gene’s rs11571833. These three variants are represented with black triangles in and are highlighted with bold text in Table S2 (Supplementary material), and their contemporary frequencies are lower or similar to their estimated frequencies in ancient populations. The temporal dynamics of these variants during the last 10000 years indicate an overall temporal decrease in population frequency ().
Figure 2. Estimated population frequencies of the 55 variants identified to be risk factors for oncologic diseases in ancient vs. contemporary populations. Points above the identity line (x = y) are variants estimated to have higher frequencies in contemporary compared to ancient populations. The three variants (rs28940298 in VHL, rs11571833 in BRCA2 and rs7837822 in TP53) are represented with black triangles.
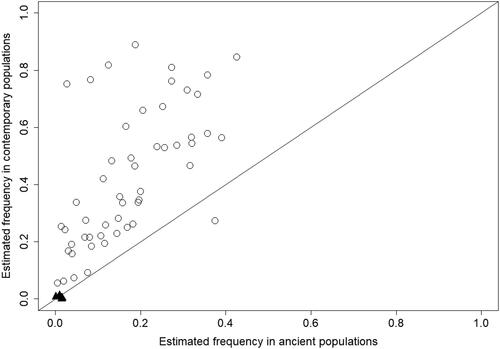
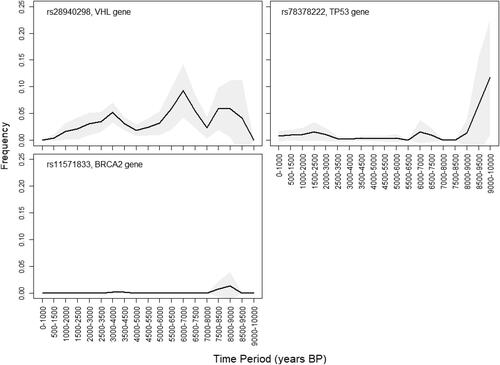
Figure 3. Temporal dynamics (1000–10000 BP) of the three examined variants established in ancient samples. The frequency trajectories are plotted using bins of 1000 years and sliding windows of 500 years. Uncertainty of the frequency estimation is indicated by a grey coloured area, representing the normal approximation of the 95% binomial proportion CI.
Among the ancient genome-wide samples analyzed in this study, there are two from Neanderthal individuals (from the Vindija Cave in Croatia and from the Altai region in Russia), and from a Denisovan individual from Siberia, all dated from around 50000 BP. Intriguingly, all these three archaic human individuals harboured one of the three mutations, the TP53 gene’s rs78378222.
The mutation rs28940298 C > T in the VHL gene is evaluated as likely pathogenic by VarSome. It is established in homozygous state (TT) in a total of 40 ancient genomes, 23 of which are from Europe, the rest being from different parts in Asia, but also in a 1000-year-old sample from the Caribbean ().
Figure 4. Geographical location of the aDNA samples in which the three variants were established. The samples are allocated in panels corresponding to the time period to which they have been dated (0–3000 BP, 3000–5000 BP, and 5000–52000 BP).
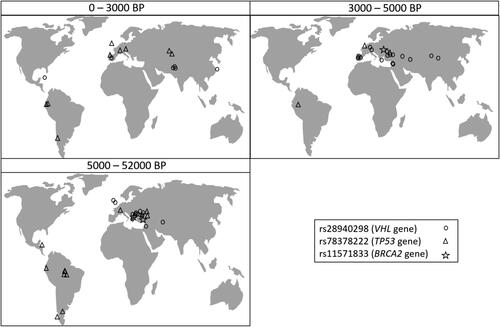
In contemporary human populations, a search for the rs28940298 C > T variant in 251358 genomes finds it 53 times, the overall population frequency being much lower (MAF = 0.00015) than that estimated in ancient human communities (MAF = 0.015). Among contemporary populations, its frequency varies from 0.0007 in South Asian populations, 0.0003 in European (Non-Finnish) populations, to as low as 0.00006 in African populations, and the mutation is not detected at all in Latin populations. The five oldest samples in which this mutation was established (around 7800–8700 BP) are all from the Balkan region and Anatolia, indicating a possible region of origin of this mutation. Its temporal dynamics suggest, despite fluctuations and three peaks (at around 9000 BP, 7000 and 9000 BP), an apparent gradual frequency decline through time (). Examining the population frequencies of all disease associated variants from DisGeNet that are also found in the ancient genome-wide data, this mutation is among 0.0006% with largest relative drop in frequency between the overall ancient and contemporary estimates. Disease associated variants are expected to be affected by negative selection, and this result suggests that the rs28940298 mutation has been subject to strong negative selection.
The TP53 gene’s rs78378222 variant is found in 19 ancient human genomes (). In contemporary populations, its frequency is highest in European (MAF = 0.014) and Latin American (MAF = 0.013) populations, corresponding to its incidence in ancient human communities. The presence of this variant in 50000-old Neanderthal genomes and a Denisovan genome from Central Asia is an indication of their considerable age, as well as of their possible place of origin.
The mutation rs11571833 A > T in the BRCA2 (K3326* mutation) is a stop-gain variant. It is established in only three aDNA samples, from Anatolia (8000 − 8500 BP), South Ural (3500 − 4000 BP) and Caucasus (13000–13500 BP) (MAF = 0.001). In contemporary human populations, the frequency of the T allele is also very low (MAF = 0.007), being highest in European populations.
Discussion
For this paleogenomic study, publicly available genome-wide data were examined for the incidence of SNP variants associated with cancer. Human populations differ in frequency of SNP variants and these differences have been the basis for studying differentiation and migration among human populations [Citation13]. Genetic variants also play a role in adaptation to extreme environmental conditions such as cold and altitude [Citation14], malaria resistance [Citation15], lactose [Citation16] and arsenic [Citation17] tolerance and other environmental factors. Some variants, on the other hand, are the result of spontaneous or induced mutagenesis, and lead to disease and reduced lifespan.
We initially hypothesized that oncological diseases were less prevalent in ancient human communities, as potentially mutagenic factors, e.g. toxic chemicals in the environment or in the food, tobacco smoking, radiation, etc., were not as widespread. Also, we hypothesized that mutations in tumour-suppressor genes might have been expressed to a lesser extent in ancient communities, as only a small portion of individuals reached advanced age for the clinical effect of pathogenic mutations to manifest itself.
The VHL rs28940298 mutation was initially identified in the contemporary Chuvash people of Russia where it is endemic and 1 in 20 individuals are heterozygous carriers [Citation18]. It has been established in other populations, e.g. Indian [Citation19], Russian and Ukrainian [Citation20], in patients of Asian and Western European ancestry [Citation21]. A study by Liu et al. (2004), based on the worldwide distribution of the mutation and haplotype analysis of 8 polymorphic markers around this gene concludes that this mutation was spread from a single founder 14000 to 62000 years ago [Citation22]. Even though we establish extensive worldwide distribution of this variant from ancient samples, the hypothesis of spread from a single founder cannot be dismissed due to the antiquity of the mutation and subsequent human migrations. Negative selection, and attenuation of endemic and other environmental factors, could explain the gradual frequency decrease of this variant through time.
The normal functioning of the VHL gene can be disrupted by a multitude of mutations leading to Von Hippel-Lindau disease predisposition [Citation23]. The molecular pathogenesis is dictated by the loss of function of the VHL protein, which binds and degrades transcription of hypoxia-inducible factor α (HIF-α), an oxygen-dependent pathway. Protein from mutant VHL gene cannot recognize HIF-α, so HIF-α accumulates affecting the expression of a group of downstream genes, including erythropoietin (EPO), vascular endothelial growth factor (VEGF) and transforming growth factor (TGF-223 α), to promote oncogenesis [Citation24]. This tumour-suppressor gene is often associated with renal cell carcinoma (RCC), pancreatic cyst or tumour (PCT), central nervous system haemangioblastoma (CHB), retinal angiomas (RA) and phaeochromocytoma (PHEO). A number of studies have shown that the rs28940298 (c.598C > T) mutation in the VHL gene in homozygote state causes Chuvash polycythaemia, an autosomal recessive form of erythrocytosis [Citation18, Citation22, Citation25]. This mutation results in a substitution of arginine for tryptophan at codon 200 (p.Arg200Trp). The mutant protein causes HIF-α levels to increase, promoting erythropoiesis and resulting in polycythaemia. Chuvash polycythaemia is characterized by increased red blood cell mass and haemoglobin levels, increased mass of solid organs [Citation26], and is associated with arterial and venous thrombosis, major bleeding episodes, cerebral vascular events and premature mortality [Citation27].
TP53 is considered a key tumour suppressor gene which acts as a transcription factor and induces the expression of genes involved in cell cycle regulation and genes involved in the intrinsic (PUMA, BAX) and extrinsic (TNFRSF10B, FAS) pathways of apoptosis [Citation28]. Studies of TP53 gene have mostly focussed on mutations in exons [Citation29], while the UTR regions have been largely ignored [Citation30]. A GWAS from 2011 identifies association of the rs78378222 mutation with prostate cancer, glioma and colorectal adenoma [Citation31], while a meta-analysis from 2016 suggests that this mutation is a potent risk factor for overall cancer [Citation32].
Pathogenic mutations in the BRCA2 gene predispose to hereditary breast and ovarian cancer (HBOC). However, the germ-line stop-gain mutation K3326* (rs11571833) is also associated with risk of lung cancer [Citation33, Citation34], cancers of the upper-aero-digestive tract [Citation35] and with the risk of urinary tract cancers [Citation36]. The oldest instance the BRCA2 gene rs11571833 A > T mutation was established in a sample from the Caucasus region from 13000 to 13500 BP, an approximate estimate of its minimum age. It was established only two more times in the analyzed samples, in a 8000–8500-old sample from Anatolia and in a 3500–4000-old sample from Southern Ural. The scarcity of established cases of this variant in ancient samples prevents us from making any reliable inferences on its historical prevalence. Yet, all ancient samples in which it was detected are from Western Asia, whereas its contemporary distribution is global, which might indicate worldwide spread from this region. Also, compared to the estimated ancient frequencies (0.0003), the contemporary frequencies are higher, i.e. the global estimate being 0.0066 and regionally ranging from as high as 0.011 in Finnish down to 0.0012 in African populations.
The BRCA2 gene encodes the breast cancer type two susceptibility protein (BRCA2), an essential protein in genome maintenance, homologous recombination (HR) and replication fork protection. One role of BRCA2, common to DNA break repair, DNA crosslink repair, and replication fork protection, is delivery of RAD51 to sites where it is needed [Citation37]. The BRCA2 gene rs11571833 A > T mutation is a germline nonsense mutation entailing stop codon K3326X and loss of the terminal 93 amino acids of the protein chain. It is located in the C-terminus of BRCA2, which also contains a RAD51 binding domain [Citation38]. This mutation eliminates Thr3387 of the BRCA2 protein obstructing the localization of BRCA2 to the nucleus and the release of Rad51. Recent studies show that the key role of BRCA2 in the repair and recovery from stalled replication forks involves exon 27, which harbours the K3326X mutation [Citation39].
Geographic bias is inherent to all aDNA studies and it has inevitably also affected our results about the incidence and prevalence of cancer associated variants in ancient human communities. Our results demonstrate the presence of such mutations and suggest that they differed in frequency among ancient communities as they do among contemporary populations. Key to understanding disease aetiology is identifying the processes that cause the genetic disease prevalence among human populations to differ. Disease-causing variants are studied intensively so that screening programs can be established in populations where they are found with relatively high frequency [Citation40].
Conclusions
For the present study, publicly available genome-wide data from ancient DNA samples were analyzed for the incidence of variants associated with cancer diseases. Of the established 55 risk alleles for oncological conditions, we selected three variants conclusively deemed to entail pathogenic effect, VHL rs28940298, BRCA2 rs11571833 and TP53 rs78378222. The contemporary population frequency of these three variants is lower, or similar to their estimated frequency in ancient human communities, indicating that they might have been subject to negative selection. This is also suggested by the temporal dynamics of these variants during the last 10000 years that show an overall temporal decrease in population frequency. The oldest samples in which these three variants were established testify their ancient origin, especially intriguing being the presence of TP53 rs78378222 variant in 50000 BP Neanderthal and Denisovan genomes. Data on germline mutations in cancer-causing genes in ancient human genomes are scarce and their historic prevalence might give insight into the evolution of cancer disease and help advance modern medicine.
Supplemental Material
Download Zip (292.5 KB)Data availability
Genome-wide data used for this study are publicly available from Allen Ancient DNA Resource 2021, version 50.0, https://reich.hms.harvard.edu/allen-ancient-dna-resource-aadr-downloadable-genotypes-present-day-and-ancient-dna-data. Datasets used for the analyses are given in the Supplementary material S1 and S2. Any additional data used to support the findings of this study are available from the corresponding author upon request.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249.
- Fornaciari G. Histology of ancient soft tissue tumors: a review. Int J Paleopathol. 2018;21:64–76.
- Giuffra V, Ciranni R, Fornaciari G. I TUMORI MALIGNI NELL'ANTICO EGITTO E IN NUBIA’. Egitto e Vicino Oriente. 2004;27:81–93.
- Strouhal E. Tumors in the remains of ancient Egyptians. Am J Phys Anthropol. 1976;45(3 pt. 2):613–620.
- Minozzi S, Catalano P, Caldarini C, et al. Palaeopathology of human remains from the roman imperial age. Pathobiology. 2012;79(5):268–283.
- Minozzi S, Lunardini A, Caldarini C, et al. Metastatic prostate carcinoma from imperial Rome (1st to 2nd centuries AD). Pathobiology. 2018;85(5–6):289–299.
- Merczi M, Marcsik A, Bernert Z, et al. Skeletal metastatic carcinomas from the roman period (1st to 5th century AD) in Hungary. Pathobiology. 2014;81(2):100–111.
- Marchetti A, Pellegrini S, Bevilacqua G, et al. K-RAS mutation in the tumour of ferrante I of Aragon, king of Naples. Lancet (London, England). 1996;347(9010):1272–1nil.
- Feldman M, Hershkovitz I, Sklan EH, et al. Detection of a tumor suppressor gene variant predisposing to colorectal cancer in an 18th century Hungarian mummy. PLoS One. 2016;11(2):e0147217.
- Allen Ancient DNA Resource. 2021. version 50.0. [Available from: https://reich.hms.harvard.edu/allen-ancient-dna-resource-aadr-downloadable-genotypes-present-day-and-ancient-dna-data.
- Piñero J, Bravo À, Queralt-Rosinach N, et al. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017;45(D1):D833–D839.
- Gibson G. Rare and common variants: twenty arguments. Nat Rev Genet. 2012;13(2):135–145.
- Deguilloux M-F, Leahy R, Pemonge M-H, et al. European neolithization and ancient DNA: an assessment. Evol Anthropol. 2012;21(1):24–37.
- Ilardo M, Nielsen R. Human adaptation to extreme environmental conditions. Curr Opin Genet Dev. 2018;53:77–82.
- Kariuki SN, Williams TN. Human genetics and malaria resistance. Hum Genet. 2020;139(6–7):801–811.
- Anguita-Ruiz A, Aguilera CM, Gil Á. Genetics of lactose intolerance: an updated review and online interactive world maps of phenotype and genotype frequencies. Nutrients. 2020;12(9):2689.
- Apata M, Pfeifer SP. Recent population genomic insights into the genetic basis of arsenic tolerance in humans: the difficulties of identifying positively selected loci in strongly bottlenecked populations. Heredity (Edinb). 2020;124(2):253–262.
- Ang SO, Chen H, Gordeuk VR, et al. Endemic polycythemia in Russia: mutation in the VHL gene. Blood Cells Mol Dis. 2002;28(1):57–62.
- Mallik N, Sharma P, Kaur Hira J, et al. Genetic basis of unexplained erythrocytosis in Indian patients. Eur J Haematol. 2019;103(2):124–130.
- Pastore YD, Jelinek J, Ang S, et al. Mutations in the VHL gene in sporadic apparently congenital polycythemia. Blood. 2003;101(4):1591–1595.
- Pastore Y, Jedlickova K, Guan Y, et al. Mutations of von Hippel-Lindau tumor-suppressor gene and congenital polycythemia. Am J Hum Genet. 2003;73(2):412–419.
- Liu E, Percy MJ, Amos CI, et al. The worldwide distribution of the VHL 598C > T mutation indicates a single founding event. Blood. 2004;103(5):1937–1940.
- Nordstrom-O'Brien M, van der Luijt RB, van Rooijen E, et al. Genetic analysis of von Hippel-Lindau disease. Hum Mutat. 2010;31(5):521–537.
- Na X, Wu G, Ryan CK, et al. Overproduction of vascular endothelial growth factor related to von Hippel-Lindau tumor suppressor gene mutations and hypoxia-inducible factor-1 alpha expression in renal cell carcinomas. J Urol. 2003;170(2 Pt 1):588–592.
- Lenglet M, Robriquet F, Schwarz K, et al. Identification of a new VHL exon and complex splicing alterations in familial erythrocytosis or von Hippel-Lindau disease. Blood. 2018;132(5):469–483.
- Yoon D, Okhotin DV, Kim B, et al. Increased size of solid organs in patients with Chuvash polycythemia and in mice with altered expression of HIF-1alpha and HIF-2alpha. J Mol Med (Berl). 2010;88(5):523–530.
- Gordeuk VR, Prchal JT. Vascular complications in Chuvash polycythemia. Seminars in Thrombosis and Hemostasis. 2006;32(3):289–294.
- Carmela C, Erhan A, Laura W, et al. Non-genotoxic MDM2 inhibition selectively induces a pro-apoptotic p53 gene signature in chronic lymphocytic leukemia cells. Haematologica. 2019;104(12):2429–2442.
- Voropaeva EN, Orlov YL, Pospelova TI, et al. The rs78378222 prevalence and the copy loss of the protective allele A in the tumor tissue of diffuse large B-cell lymphoma. PeerJ. 2020;8:e10335.
- Li Y, Gordon MW, Xu-Monette ZY, et al. Single nucleotide variation in the TP53 3’ untranslated region in diffuse large B-cell lymphoma treated with rituximab-CHOP: a report from the international DLBCL Rituximab-CHOP consortium program. Blood. 2013;121(22):4529–4540.
- Stacey SN, Sulem P, Jonasdottir A, Swedish Low-risk Colorectal Cancer Study Group, et al. A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat Genet. 2011;43(11):1098–1103.
- Wang Y, Wu XS, He J, et al. A novel TP53 variant (rs78378222 A > C) in the polyadenylation signal is associated with increased cancer susceptibility: evidence from a meta-analysis. Oncotarget. 2016;7(22):32854–32865.
- McKay JD, Hung RJ, Han Y, SpiroMeta Consortium, et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet. 2017;49(7):1126–1132.
- Rafnar T, Sigurjonsdottir GR, Stacey SN, et al. Association of BRCA2 K3326* with small cell lung cancer and squamous cell cancer of the skin. J Natl Cancer Inst. 2018;110(9):967–974.
- Delahaye-Sourdeix M, Anantharaman D, Timofeeva MN, et al. A rare truncating BRCA2 variant and genetic susceptibility to upper aerodigestive tract cancer. JNCI. 2015;107(5):djv037.
- Ge Y, Wang Y, Shao W, et al. Rare variants in BRCA2 and CHEK2 are associated with the risk of urinary tract cancers. Sci Rep. 2016;6(1):33542.
- Paul MW, Sidhu A, Liang Y, et al. Role of BRCA2 DNA-binding and C-terminal domain in its mobility and conformation in DNA repair. eLife. 2021;10:e67926.
- Baughan S, Tainsky MA. K3326X and other C-terminal BRCA2 variants implicated in hereditary cancer syndromes: a review. Cancers (Basel). 2021;13(3):447.
- Kolinjivadi AM, Sannino V, de Antoni A, et al. Moonlighting at replication forks – a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 2017;591(8):1083–1100.
- Guo XE, Ngo B, Modrek AS, et al. Targeting tumor suppressor networks for cancer therapeutics. Curr Drug Targets. 2014;15(1):2–16.