
Abstract
The G1/S transition is crucial for regulating the initiation of cell division and is highly conserved across eukaryotes. This phase of the cell cycle involves intricate transcriptional controls that are essential for understanding cell proliferation dynamics. The G1/S transition integrates diverse intracellular and extracellular signals, including growth factors and cell sizes. Saccharomyces cerevisiae, an important model organism, plays an integral role in unveiling the regulatory mechanisms of the G1/S transition. Recent studies on the G1/S transition in both yeast and vertebrates have demonstrated a direct correlation between dysregulation of the G1/S transition and the development of cancer. This review explores the G1/S transition regulatory network and its association with cancer formation, providing a theoretical foundation for future research in fundamental cell cycle dynamics and cancer.
Introduction
Cell proliferation is a strictly regulated process involving multiple decision points that control the initiation of cell division [Citation1]. Notably, the transition from the G1 to the S phase is irreversible. In Saccharomyces cerevisiae, this crucial step is called the “START” point, whereas in vertebrates, it is referred to as the “RESTRICTION” point [Citation2]. The G1/S transition plays a pivotal role in regulating this cellular process. Dysfunctions in the G1/S transition can lead to uncontrolled cell proliferation, a significant contributing factor to the development of cancer in vertebrates. The retinoblastoma (Rb) gene, which regulates the G1/S transition, was the first tumor suppressor identified [Citation3]. The Rb gene is not only mutated in rare cancers, such as those affecting the eye, but also in a broader array of cancer types [Citation3]. Moreover, the Rb protein is functionally inactivated in nearly all human cancers, indicating its crucial role in maintaining cellular homeostasis [Citation3, Citation4]. The inactivation of genes related to the G1/S transition, an important mechanism in the formation of cancer, is particularly associated with mutations in the suppressor genes p53 and p21 [Citation5–9]. Consequently, research on the G1/S transition regulatory network has become a focal point in cell cycle studies, providing an important theoretical foundation for understanding the mechanisms of cancer.
Cyclin-dependent kinase 1 (Cdk1) plays a critical role in controlling the G1/S transition in yeast [Citation10]. Cdk1 also serves as the central component in cell cycle regulation in eukaryotes, and its regulatory mechanisms are well conserved across several species [Citation11–15]. The regulation of Cdk1 activity primarily involves the interaction between cyclin molecules at different phases of the cycle, the phosphorylation of Cdk1 kinases, and the association with inhibitory proteins, specifically the Cdk1 kinase inhibitor CKI [Citation16]. Cdk1 kinase activity depends on its binding to cyclin molecules that are selectively expressed during the cell cycle [Citation17]. This binding enables Cdk1 to phosphorylate distinct substrates at specific times and locations, thereby facilitating various regulatory functions within the cell cycle and ensuring its progression. In S. cerevisiae, Cdk1 and its structural subunit Cks1 exhibit consistent expression patterns throughout the cell cycle, indicating that the activity of Cdk1 is contingent upon the sequential production of cyclins. The impact of the structural subunit Cks1 on CDK activity is negligible, but it does augment the interaction between Cdk1 and the substrate [Citation18]. In S. cerevisiae, Cak1 functions upstream of Cdk1, where it phosphorylates threonine 169 of Cdk1 [Citation19]. This phosphorylation event is essential for the subsequent binding of cyclin [Citation19]. Another mechanism of Cdk1 activity involves the negative control of the inhibitory protein CKI. The spatial binding of substrate is hindered by the interaction between CKI and Cdk1, resulting in the inhibition of substrate phosphorylation by Cdk1. The occurrence and progression of cancer are facilitated by uncontrolled cell proliferation resulting from CKI mutations [Citation20]. Understanding Cdk1 function is essential for unraveling the intricate mechanisms that regulate the cell cycle in eukaryotes. For the discovery of Cdk1 and its dependence on cyclin, Paul Nurse and Leland H. Hartwell won the Nobel Prize in Physiology or Medicine in 2001 [Citation21].
As research progresses, an increasing number of novel genes linked to cell cycle regulation have been identified. To date, hundreds of genes associated with the regulation of G1/S transition have been discovered in S. cerevisiae. Cip1, a recently identified p21 analog in yeast, functions as a Cln3-CDK to suppress the START phase of cell-cycle [Citation22]. However, the precise regulatory mechanism governing its activity requires further study. Relevant research suggests that histone acetyltransferase Gcn5 and histone deacetylase Rpd3 act as transcriptional regulators of the G1/S phase in S. cerevisiae [Citation23]. In eukaryotes, Gcn5 is a supplementary activator of transcription, whereas Rpd3 controls transcription by modulating chromatin remodeling [Citation23]. These latest developments provide new perspectives on the study of G1/S transition. This review will explore the evolution of the G1/S transition regulatory network and its relationship to cancer development, aiming to provide a basis for understanding the mechanisms underlying cancer occurrence.
Classic regulatory network of the cell cycle G1/S transition
The G1/S transition is primarily regulated by two transcription factors: the MBF (MCB-binding factor) and the SBF (SCB-binding factor) [Citation24]. Both heterodimeric, each is made up of a DNA-binding subunit, Swi4 and Mbp1, respectively, and a common regulatory subunit, Swi6 [Citation25]. SBF regulates the expression of CLN1/2 and cell morphology, whereas MBF modulates the expression of CLB5/6 and DNA replication-related genes in the S phase [Citation26]. Despite their abilities to regulate different genes, SBF and MBF share significant functional overlap [Citation27, Citation28], highlighted by the lethal phenotype observed in swi4swi6 and swi4mbp1 double mutants, which emphasizes the redundancy as well as the significance of this transcriptional pathway [Citation29, Citation30].
Transcriptional activation in the G1/S phase is regulated by cyclin-dependent protein kinases and cyclin activity in the G1 phase—specifically, the Cdk1 and G1 cyclins Cln1-3 in S. cerevisiae (). The binding of SBF to the promoter of its target gene occurs during the early G1 phase, but the binding of the transcriptional repressor Whi5 delays transcriptional activation until the late G1 phase [Citation31, Citation32]. The Whi3 protein binds to the CLN3 mRNA in the endoplasmic reticulum (ER) membrane during the early G1 phase, preventing the synthesis of the Cln3 protein [Citation33]. Due to the interaction between Whi3 and the Cln3-Cdk1 complex, and between Cln3 and molecular chaperones Ssa1-4, the Cln3 complex is blocked from entering the nucleus [Citation33]. In the late G1 phase, the chaperone protein Ydj1 binds to Ssa1-4, which allows the Cln3-Cdk1 complex to be released from the ER, accumulate in the nucleus, and subsequently prompt Whi5 to dissociate from SBF [Citation34, Citation35], initiating transcription. Ultimately, the Cln1/2-Cdk1 complex accumulates and acts on SBF, Whi5, and MBF to increase transcriptional activity of the whole genome, generating a positive feedback loop, which enforces G1/S transition consensus and cellular activity [Citation36–38].
Figure 1. Transcriptional regulation of G1/S transition genes in yeast.
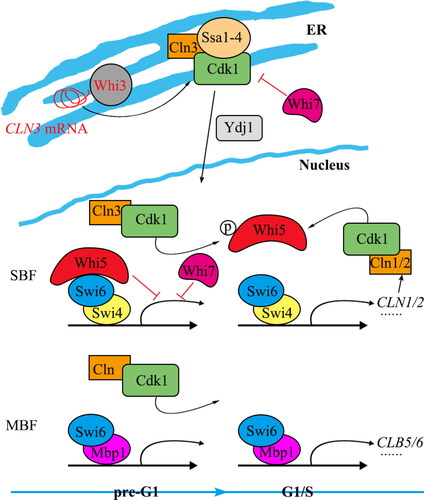
Whi7/Srl3 was initially discovered as a multicopy suppressor of the rad53 mutant. Whi7/Srl3 belongs to a family of proteins that feature a G1/S transition factor binding (GTB) motif. This family also includes Whi5 and the transcriptional repressor Nrm1. The GTB motifs in Whi5 and Nrm1 mediate the transcriptional repression of SBF and MBF, respectively [Citation39]. However, a recent study has linked Whi7 with the regulation of the “START” point in the cell cycle. Specifically, Whi7/Srl3 has been implicated in controlling the activity of the Cln3-Cdk1 complex in the ER membrane [Citation40] (). Whi7 is critical for retaining Cln3 within the ER membrane; however, its function is impeded by phosphorylation dependent on CDK [Citation40].
Distinct from its role in localizing Cln3 to the ER, Whi7 also regulates the “START” point independently. In the early G1 phase, Whi7 interacts with and inhibits Cln3-Cdk1 activity. Consequently, this inhibition prevents the phosphorylation of Whi5 by Cln3-Cdk1 [Citation40]. Notably, Whi7, serving as a functional counterpart to Whi5, is a liable protein within the G1/S transcriptional program. Its stability is influenced by Cdk1 phosphorylation, and it is subjected to degradation by the ubiquitin ligase SCF Grr1. The coordinated action of Whi5 and Whi7 underscores the reliance of yeast cells on multiple transcriptional repressors to control the initiation of the cell cycle via the “START” point [Citation41]. In summary, Whi7 exerts a dual inhibitory effect on transcription during the G1/S phase, playing a pivotal role in the transition from the G1 to the S phase.
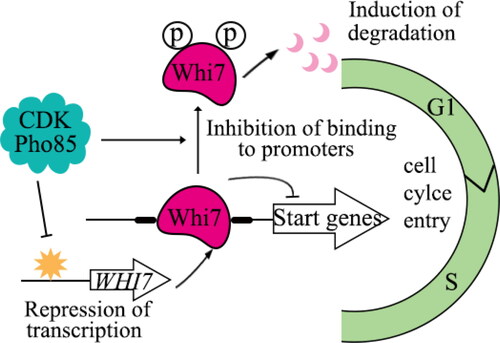
Furthermore, a recent study has shown that Pho85, a multifunctional CDK, transmits signals to the cell under favorable environmental conditions [Citation42]. It is particularly involved in the regulation of the cell cycle, especially in promoting the G1/S transition in the “START” phase. Pho85 acts as a key regulator of cell cycle initiation by targeting the “START” repressor Whi7. This repressor is deactivated through the phosphorylation of Whi7 by Pho85, a process that significantly contributes to the activation of START [Citation42]. Mechanistically, Pho85 modulates Whi7 by regulating both the transcription of the WHI7 gene and the stability of the Whi7 protein. The ability of Whi7 to bind with promoters is further limited by its phosphorylation by Pho85. In addition, while Whi5 is typically the primary START repressor of normal cycling cells, the absence of Pho85 shifts this role to Whi7, leading to G1 arrest. Overall, these findings highlight a novel mechanism by which Pho85 facilitates the initiation of START by controlling the Whi7 repressor at multiple levels. Therefore, Whi7 serves as a crucial link between the response to adverse conditions and the regulation of the cell cycle () [Citation42].
Figure 2. Pho85 promotes the G1/S transition through regulating Whi7.
Subsequent studies have furthered these initial findings and suggested various cellular strategies for synchronizing the activation of transcriptional programs with the growth of cells. These strategies include the dilution of Whi5 as a consequence of cellular growth [Citation43], the role played by Ydj1 in balancing cell cycle entry and cell growth [Citation44], and the dynamic interaction between a fixed number of SBF binding sites on DNA and an increasing number of Cln3 proteins induced by cell growth [Citation45].
Discovery of a new regulator of the G1/S transition
All organisms have developed regulatory mechanisms to optimize their fitness in adapting to changing environmental conditions. For single-celled organisms such as S. cerevisiae that cannot create homeostatic conditions, this adaptability is crucial to their survival. Cellular growth, division, and developmental processes in S. cerevisiae are significantly influenced by different carbon and nitrogen sources [Citation46]. Towards the end of the G1 phase, yeast cells commence division in a process known as the “START” point [Citation47]. To initiate the “START” phase of cell cycle, cells must reach a specific size, which regulates the size distribution within an asynchronous population [Citation47]. Dynamic adjustments of the size threshold occur upon changes in nutritional availability to improve competitive fitness [Citation47]. However, the molecular mechanisms by which nutritional availability influences cell growth and division remain unclear.
Approximately 200 genes are activated during the initiation of the G1/S transcriptional program at “START” [Citation1]. This complex program is responsible for the production of proteins that are crucial for spindle replication, DNA replication, and yeast budding [Citation1]. This program is regulated by two transcription factor complexes: MBF (MluI cell cycle box binding factor) and SBF (Swi4/6 cell cycle box binding factor) [Citation24]. The MBF and SBF complexes contain the DNA-binding proteins Mbp1 and Swi4, respectively, and both complexes bind to the Swi6 regulatory subunit [Citation48, Citation49]. The Whi5 transcriptional repressor can inhibit the transcriptional activity of SBF before the critical cell size is reached [Citation50]. At the initiation of the “START” point, the G1 phase cyclin Cln binds to the protein kinase Cdc28, which phosphorylates SBF and Whi5, and this phosphorylation event disrupts the SBF-Whi5 interaction [Citation35]. CLN3 gene expression is independent of transcriptional control by SBF and is believed to trigger a positive feedback loop in the upstream G1 phase [Citation51].
Major yeast nutrient signaling pathways, such as the glucose-activated protein kinase A (PKA) pathway, that mediate the link between cell size threshold and division initiation have been identified [Citation52]. The PKA pathway inhibits the expression of CLN1 and other G1/S transcripts, potentially increasing cell size thresholds in nutrient-rich environments [Citation52]. Conversely, nutrient-poor environments lead to increased expression of G1/S phase transcription factors, which activate “START” when cells are smaller [Citation53]. Nsr1, a microprotein localized in the nucleus, interacts with SWI4 and SWI6, which together constitute the SBF complex, restarting the G1/S transition machinery in nutrient-poor conditions [Citation54].
Much of our understanding of the proteins involved in regulating the SBF-MBF complex comes from studies in yeast. Our research group studied Cdk1-interacting protein 1 (Cip1), a novel repressor of the cell cycle [Citation13, Citation22] (). The Cip1 protein is involved in the G1/S transition and specifically inhibits G1-Cdk1. In addition, its expression level and phosphorylation exhibit cellular periodicity [Citation14]. An increase in Cln3-Cdk1 activity within the nucleus leads to the phosphorylation of the transcriptional repressor Whi5 and its subsequent dissociation from SBF. This results in the activation of SBF-mediated G1/S transition [Citation55]. CIP1 also limits cell proliferation and impedes S phase progression when overexpressed in yeast [Citation13, Citation15]. Conversely, deletion of the CIP1 gene promotes the G1/S transition [Citation13].
Figure 3. Regulation of Cip1 activity during the G1 phase [Citation22, Citation57].
![Figure 3. Regulation of Cip1 activity during the G1 phase [Citation22, Citation57].](/cms/asset/c744faa2-277f-48c2-bb07-23d3b64aa75a/tbeq_a_2362842_f0003_c.jpg)
During the G1/S transition, CLN2 is expressed significantly earlier in cln3Δcip1Δ mutant yeast cells than in cln3Δ mutant cells [Citation22]. Additionally, increased CIP1 expression inhibits cell budding (a marker of G1/S transition) and cell proliferation in cln3Δ mutants. These findings suggest that Cip1 regulates the G1/S transition via a mechanism that is independent of Cln3-Cdk1 [Citation22]. Our research supports this theory by demonstrating that Cip1 interacts with Ccr4 and Caf120, components of the Ccr4-NOT complex [Citation22] (). The CCR4 gene encodes cytoplasmic deadenylate, which negatively impacts the half-life of WHI5 mRNA. Additionally, Ccr4 impacts the timing of cyclin gene expression in the G1 phase by regulating the stability of WHI5 mRNA [Citation56]. Research has demonstrated that Cip1 functions as a dual repressor, suppressing the Cln3-Cdk1 complex and Ccr4 [Citation22]. This maintains the activity of Whi5, which stops SBF from transcribing G1/S genes. Notably, the Ccr4-NOT complex interacts with the tumor suppressor gene p21, which is a homolog of Cip1 in humans. This raises the possibility of p21 regulating the human cell cycle in a similar manner.
In the regulation of G1/S transition, the histone deacetylase complex plays a crucial role in the promoter regions. In higher eukaryotes, the DREAM complexes, comprising E2F4, DP, RBL, and MuvB, are responsible for recruiting the highly conserved histone deacetylating enzyme 1 (HDAC1). This recruitment results in targeted histone deacetylation, leading to the suppression of genes that promote the S phase [Citation58]. Histone acetylation typically promotes chromatin opening and active transcription, whereas deacetylation leads to chromatin closing and transcriptional repression [Citation59]. The histone deacetylase Rpd3 regulates transcription through chromatin remodeling, and the histone acetyltransferase Gcn5 acts as a coactivator. Both Rpd3 and Gcn5 are essential for the regulation of G1/S transition [Citation23, Citation60]. In addition, the coactivator Rep2 recruits Gcn5 in an Mlu1-binding factor (MBF)-dependent manner. Gcn5-mediated acetylation of the MBF promoter facilitates transcription during the G1/S phase and is crucial for accurate and timely gene expression. The acetylation of histone residues H3K9 and H3K18 on the MBF promoter by Gcn5 during the G1/S transition is essential for maintaining precise transcription timing [Citation23].
In human cells, the suppression of G1/S transcriptional control during the G1 phase requires Rb to recruit HDAC1 to the G1/S target promoter [Citation61]. The G1/S transition also depends on an interaction between Rpd3 (a homolog of HDAC1) and the transcriptional repressor Whi5 [Citation62]. Future research should examine the effect of histone acetyltransferases (HAT) and HDAC recruitment on the histone acetylation status of G1/S target promoters during the cell cycle and their impact on G1/S transition control. Additionally, distinct regulators, namely Msa1 and Msa2, play key roles in managing G1/S transition in yeast cells [Citation63]. These regulators are components of the yeast counterpart of the DREAM complex and are crucial for the initial stages of transcriptional regulation, promoting G1 arrest and the transition to a quiescent state [Citation63].
Research on G1/S transition of yeasts provides new insight into the molecular mechanisms of cancer development
Regulation of transcription during the G/S transition is critical for the progression of the cell cycle. DRTFl/E2F is a transcription factor implicated in the coordination of the cell cycle machinery with the transcription apparatus. As the central hub for signals that either promote or restrict growth, it serves as a crucial target within cells for molecules that disrupt the normal control of the cell cycle, such as proteins produced by oncogenic viruses [Citation64]. Previous research has shown that proteins belonging to two separate families, DP and E2F, interact in a combined manner in DRTFl/E2F as DP/E2F heterodimers [Citation64, Citation65]. The activity of E2F and DP proteins, regulated by the cell cycle, is influenced by phosphorylation levels mediated by CDKs (cyclin-dependent kinases). Both proteins have proto-oncogenic activity and regulate apoptosis. Consequently, current evidence suggests that the function of DRTF1/E2F is pivotal in controlling cell cycle progression [Citation64].
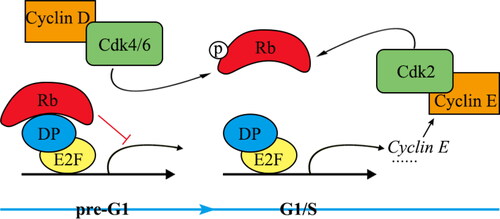
In vertebrates, gene expression is regulated at the G1/S transition through two distinct events [Citation66]. Gene regulation mediated by DP-E2F is similar to that mediated by SBF/MBF in yeast (). The Rb gene, the first tumor suppressor gene discovered, regulates the G1/S transition through the DP-E2F pathway. The cell cycle inhibitor Rb, similar to the yeast Whi5, undergoes dilution during cell development in the G1 phase, thereby facilitating the initiation of G1/S transition [Citation67]. Notably, Rb functions as the mammalian equivalent of Whi5 by inhibiting the primary transcription factor E2F, which is crucial for the mammalian cell cycle. However, it is important to note that their amino acid sequences exhibit significant differences [Citation68]. Cyclin D-Cdk4/6 phosphorylation controls Rb during the G1 phase. As a result of phosphorylation, the Rb protein dissociates from DP-E2F, leading to the initiation of the G1/S transition [Citation69]. Additionally, expression of the cyclin E gene is mediated by DP-E2F. Cyclin E protein forms a complex with Cdk4/6 to phosphorylate Rb. Phosphorylation of Rb leads to enhanced transcriptional activity of DP-E2F throughout the genome [Citation70]. Mutations in the Rb gene result in the dysfunction of DP-E2F, causing uncontrolled cell proliferation that culminates in the development of cancer [Citation58].
Figure 4. Transcriptional regulation of G1/S transition genes in invertebrates.
During the G1 phase, the concentration of Rb decreases through a newly identified pathway. Research findings indicate that this reduction in Rb content is due to the regulated degradation of proteins via the E3 ubiquitin ligase UBR5. In cells with UBR5 mutations, Rb levels are elevated in the early G1 phase, leading to a slower G1/S transition and increased sensitivity to Cdk4/6 inhibition. This discovery suggests that inhibiting UBR5 could enhance the effectiveness of cancer therapies based on Cdk4/6 inhibitors [Citation71]. Plakophilin 3 (PKP3), a component of desmosomes, binds to phosphorylated RB in the cytoplasm, thereby facilitating the activation of E2F1 and promoting cell cycle progression [Citation72]. These data reveal a mechanism by which PKP3 promotes cell growth and functions as an oncogene.
The E2F family of transcription factors possesses numerous regulatory activities, which have been studied since 1980 [Citation73, Citation74]. Currently, eight members, E2F1–8, have been identified in this family, among these, E2F1 has been extensively studied, whereas E2F4 is typically the predominant E2F activity in cells [Citation74]. E2F4 acts as a transcriptional repressor associated with cell cycle arrest, a function that depends on its interaction with Rb family members [Citation75]. E2F4 engages in functional interactions with chromatin regulators that are associated with gene activation. Specifically, in cells with mutations in the Rb family, the absence of E2F4 leads to reduced histone acetylation at the promoters of E2F target sites and cell cycle genes [Citation76]. The Rb-E2F complex is essential for G1/S transition, and the five conventional E2F transcription factors, E2F1-5, can modulate the function of Rb proteins through direct interaction. The E2F transcription factors are divided into two major categories based on their activity at cell cycle gene promoters: activators (E2F1, E2F2, and E2F3a) and canonical repressors (E2F3b, E2F4, and E2F5) [Citation65]. E2F repressors interact with Rb family proteins to suppress the transcription of cell cycle genes involved in G1/S transition, whereas E2F activators facilitate the initiation of cell cycle by enhancing the expression of related genes.
According to the published data, E2F1-3 and E2F4-7 are transcriptional activators and repressors, respectively. E2F8 is a newly identified E2F family member [Citation74], and little is known about its function. It possesses a distinct repeating DNA-binding domain that sets it apart from E2F1-6. This unique characteristic allows E2F8 to regulate gene expression in a manner that is independent of dimer formation [Citation74]. The overexpression of E2F8 promotes the turnover of the G1/S phase by activating E2F1 and cyclin D1 [Citation3]. The E2F family has long been a target for the development of antitumor therapies. However, the molecular mechanisms of E2F remain unknown, particularly with regard to the new member E2F8. Recently, E2F8 has been implicated in the regulation of esophageal squamous cell carcinoma (ESCC) growth and the modulation of cell cycle [Citation77]. These findings suggest that targeting E2F8 could help treat ESCC. E2F8 is correlated with a delay in the progression of the cell cycle, particularly the S phase. E2F8 encodes a transcription factor belonging to a family containing other members known to regulate the expression of genes crucial for cell cycle progression. E2F8 regulates nuclear division by promoting the G1/S transition at the appropriate time.
It is possible that E2F8 regulates the cell cycle in ESCC through its influence on the CCND1/p21 pathway. The involvement of CCND1 as an oncogene in various cancers is attributed to its regulatory role in facilitating the G1/S transition. Additionally, p21 serves as a significant effector in multiple pathways associated with tumor suppression. E2F8 can initiate or inhibit the activity of CCND1 and p21 to influence cell proliferation. Additionally, the expression of E2F8 enhances ESCC cell proliferation in vivo [Citation78]. Previous studies have demonstrated that E2F8 is important in different types of cancer. For example, a study conducted by Sun et al. revealed that E2F8 influences the progression of the cell cycle to promote papillary thyroid cancer [Citation79]. Related research has revealed that E2F8 is a potential therapeutic target in hepatocellular cancer [Citation80]. These findings indicate that E2F8 exhibits oncogenic properties in many cancer types, although further work is required to elucidate the intricate processes that facilitate cell cycle regulation by E2F8. In summary, E2F8 has been successfully identified as an oncogene that mediates cellular proliferation both in vitro through the regulation of CCND1 and p21 and in vivo.
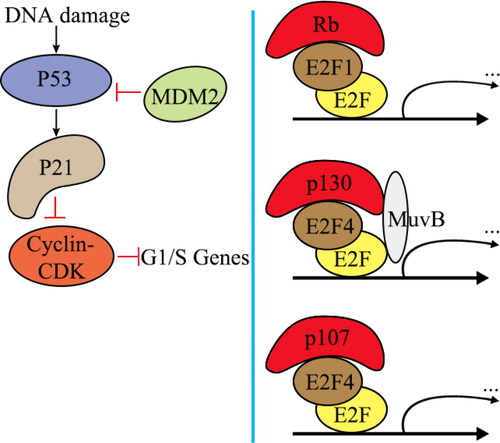
The Rb proteins p130 (encoded by RBL2) and p107 (encoded by RBL1) form the DREAM complex with the E2F4 repressor and multivulva class B (MuvB) [Citation81] (). E2F4 binds to the E2F site in the promoters of G1/S genes. DREAM and Rb synergistically inhibit G1/S on the transcriptional level. This inhibition, which occurs when p53 is activated, is partially dependent on the phosphorylation of p130 and Rb by cyclin-CDK [Citation82–84]. The downregulation of cell cycle genes mediated by p53 occurs in both human and mouse cells upon DNA damage [Citation85]. When MDM2, a p53 repressor, is inhibited or DNA damage occurs, p53 activation results in enhanced expression of the CDK inhibitor p21 and reduced expression of the cyclin E-CDK2 complex [Citation86]. Suppression of G1/S transcriptional control, which is mediated by both Rb and p103-DREAM, is restored due to decreased CDK2 activity. When Rb and p130 are absent, p107-E2F4 can effectively bind to and inhibit the transcription of G1/S genes, causing cell cycle arrest [Citation83]. The inactivation of G1/S transcriptional control is an important mechanism by which cancer can form, and it can be caused by mutations in p53 and p21 [Citation5, Citation6].
Figure 5. Inhibition of cell cycle gene expression by DREAM and Rb following p53 activation.
G1/S transition versus cancer treatment
Substantial advancements have been made in our understanding of the regulation of G1/S transition in the past few years, forming the foundation of numerous cancer studies. The control of G1/S transition, which represents the intersection of intricate signaling networks, is a main factor in abnormal cell proliferation in cancer. Furthermore, several of its constituent proteins exhibit significant evolutionary conservation, making them good targets for diverse tumor types in clinical settings.
Cell cycle disruption is a characteristic feature observed in all types of cancer that results in the unrestrained proliferation of cells. CDKs are important for the regulation of cell cycle and are frequently overexpressed or altered in cancerous cells. This characteristic makes CDKs promising targets for therapeutic intervention as CDK inhibitors. The first generation of CDK inhibitors is non-selective and highly toxic [Citation87, Citation88]. The second generation of CDK inhibitors, particularly CDK4/6 inhibitors (CDK4/6i), are highly specific and less toxic for CDK4 and CDK6 [Citation89]. Upon oncogenic signaling and growth factor stimulation, cyclin D1 promotes G1/S transition by activating CDK4/6. CDK4/6i is a small oral drug that prevents Rb phosphorylation and releases inactive Rb from its association with E2F1, which arrests the cell cycle between the G1 and S phases [Citation90]. Three CDK4/6i drugs for treating metastatic hormone receptor-positive breast cancer have been approved by the United States Food and Drug Administration (palbociclib, ribociclib, and abemaciclib) [Citation91].
More recently, a novel compound known as G1T38 has emerged as another highly selective and efficacious inhibitor of CDK4/6. G1T38 confers significant antiproliferative effects in CDK4/6-expressing cells and malignancies and is particularly effective against CDK4/cyclin D1, CDK9/cyclin T, and CDK6/cyclin D3 [Citation92]. Compared to CDK2/cyclin A and CDK2/cyclin E, G1T38 is more than 1,000 times more selective towards CDK4/cyclin D1. Overall, G1T38 is a new, selective highly effective, and orally administered small-molecule CDK4/6 inhibitor that significantly inhibits tumor growth in CDK4/6-dependent malignancies without producing severe neutropenia. Consequently, G1T38 may be the best therapeutic agent to take orally on a regular basis to combat cancer.
Given the intricate and unpredictable nature of cancer development and progression, further studies of CDK4/6i in different malignancies are required. An important area of focus is the optimization of CDK4/6i efficacy and further mitigation of adverse effects [Citation93]. Targeting individual proteins or pathways may not result in sustained control of cancer because tumor cells employ redundant mechanisms and continuously generate mutations that can lead to acquired resistance against treatments with a single mode of action. Gaining a deeper understanding of the resistance mechanisms of CDK4/6i inhibitors is crucial for developing better approaches to mitigate drug resistance. Drugs targeting multiple pathways are one approach, but there is a risk of increased toxicity. Additional clinical trials are needed to enhance the efficacy of CDK4/6 inhibitors and other combinations of pharmaceutical products in patients with breast cancer and other malignancies. Beginning treatment at an early stage of the disease may yield the most favorable results.
p21 is important for cellular responses to both internal and external stimuli by controlling the G1/S transition. Interestingly, low levels of p21 can stimulate cancer cell proliferation by facilitating the formation and activation of the cyclin D-Cdk4 complex and Cdk6 [Citation94], suggesting that p21 functions as either an activator or inhibitor of the cell cycle depending on its abundance in the cell. Owing to this unique feature, p21 serves as a biomarker and therapeutic target. The potential of p21 inhibitors as therapeutic targets is significant, as they improve tumor cell survival by promoting cell migration, inhibiting apoptosis and conferring drug resistance [Citation95].
Phosphorylation of various kinases at different sites can lead to the cytoplasmic displacement of p21, which impairs its function as well as the therapeutic response of patients to chemotherapies. Therefore, changes in p21 subcellular localization can be used as a biomarker for targeted therapeutics or prognostic evaluation [Citation96]. The cytoplasmic p21 protein is activated by several kinases, and phosphorylation at specific residues is associated with distinct outcomes. For example, phosphorylation of p21 at Ser-146 and Thr-145 by Akt increases cell viability and promotes resistance against the chemotherapeutic drug paclitaxel in glioblastoma cells [Citation94]. In breast cancer cells, cytoplasmic p21 has also been linked to a negative reaction to tamoxifen in MCF7 [Citation97]. Heregulin beta1 inhibits tamoxifen-induced apoptosis by activating the MAPK and PI3K/Akt pathways, thereby enhancing the cytoplasmic p21 expression. The cellular localization of p21 influences the effectiveness of tamoxifen treatment [Citation97]. In summary, the cytoplasmic localization of p21 has the potential to be a dependable biomarker and a potential target for therapeutic intervention.
The development of drugs targeting HDACs has been an area of active research in recent years. Studies have shown that HDAC6 expression may serve as a diagnostic tool for cancer [Citation98]. HDAC6 inhibitors, such as SAHA, tubacin, and tubastatin A, hyperacetylate HDAC6 substrates and disrupt tumor cell function [Citation99]. CAY10603 (N-[4-[3-[[7-(hydroxyamino)-7-oxoheptyl] carbamoyl]-1,2-oxazol-5-yl] phenyl] carbamate) was recently identified as a novel HDAC6 selective inhibitor that inhibits the catalytic site of HDAC6 [Citation100]. Low doses of CAY arrest the G1/S transition and suppress the proliferation of some tumor cells [Citation101]. Although the introduction of these compounds has provided new therapeutic avenues, the questions of clinical safety, efficacy, and resistance to the agents must still be addressed.
Future perspectives
Interrupted G1/S transition is frequently observed in higher animals during the development of cancer. Deregulation of gene expression in yeast cells is also associated with genomic instability. Furthermore, external environmental cues may impede cell cycle progression and cause cell cycle arrest. The stringent regulation of the transition from the G1 to the S phase ensures the proper progression of the cell cycle. Further studies are necessary to investigate the newly identified regulatory mechanism of the Cip1 protein in the G1/S transition. It is important to decipher the mechanisms that are involved in the degradation of Cip1 and the precise role of Cip1 phosphorylation during stress. A series of unknown questions, such as whether Cip1 directly regulates the G1/S transition and whether there are other new proteins involved in its regulation, remain to be answered. Exploring new regulatory mechanisms for the G1/S transition using the model organism S. cerevisiae will provide new insights for studying the development of various diseases in humans caused by cell cycle disorders.
Authors’ contributions
C.Z. and P.L. contributed equally to this work. Y.Q. and J.Z. conceptualized and organized the manuscript. Z.H. and T.L. reviewed and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
The authors are grateful for the support provided by the Central Laboratory of the Affiliated Hospital of Hebei University for this work. The authors would like to thank A&L Scientific Editing (www.alpublish.com) for its linguistic assistance during the preparation of this manuscript.
Disclosure statement
The authors declare that they have no competing interests.
Data availability statement
The data that supports the findings of this study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Bertoli C, Skotheim JM, de Bruin RAM. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol. 2013;14(8):518–528. doi: 10.1038/nrm3629.
- Johnson A, Skotheim JM. Start and the restriction point. Curr Opin Cell Biol. 2013;25(6):717–723. doi: 10.1016/j.ceb.2013.07.010.
- Roufayel R, Mezher R, Storey KB. The role of retinoblastoma protein in cell cycle regulation: an updated review. Curr Mol Med. 2021;21(8):620–629. doi: 10.2174/1566524020666210104113003.
- Delston RB, Harbour JW. Rb at the interface between cell cycle and apoptotic decisions. Curr Mol Med. 2006;6(7):713–718. doi: 10.2174/1566524010606070713.
- Engeland K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018;25(1):114–132. doi: 10.1038/cdd.2017.172.
- Barr AR, Cooper S, Heldt FS, et al. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat Commun. 2017;8(1):14728. doi: 10.1038/ncomms14728.
- Engeland K. Cell cycle regulation: p53-p21-Rb signaling. Cell Death Differ. 2022;29(5):946–960. doi: 10.1038/s41418-022-00988-z.
- Li Y, Li S, Shi X, et al. KLF12 promotes the proliferation of breast cancer cells by reducing the transcription of p21 in a p53-dependent and p53-independent manner. Cell Death Dis. 2023;14(5):313. doi: 10.1038/s41419-023-05824-x.
- Wu JY, Chien YC, Tsai IC, et al. Capsanthin induces G1/S phase arrest, erlotinib-sensitivity and inhibits tumor progression by suppressing EZH2-mediated epigenetically silencing of p21 in triple-negative breast cancer cells. Aging (Albany NY). 2021;13(9):12514–12525. doi: 10.18632/aging.202925.
- Hartwell LH, Culotti J, Pringle JR, et al. Genetic control of the cell division cycle in yeast. Science. 1974;183(4120):46–51. doi: 10.1126/science.183.4120.46.
- Nurse P, Thuriaux P, Nasmyth K. Genetic control of the cell division cycle in the fission yeast Schizosaccharomyces pombe. Mol Gen Genet. 1976;146(2):167–178. doi: 10.1007/BF00268085.
- Evans T, Rosenthal ET, Youngblom J, et al. Cyclin: a protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell. 1983;33(2):389–396. doi: 10.1016/0092-8674(83)90420-8.
- Ren P, Malik A, Zeng F. Identification of YPL014W (Cip1) as a novel negative regulator of cyclin-dependent kinase in Saccharomyces cerevisiae. Genes Cells. 2016;21(6):543–552. doi: 10.1111/gtc.12361.
- Zhang Z, Ren P, Vashisht AA, et al. Cdk1-interacting protein Cip1 is regulated by the S transition in response to genotoxic stress. Genes Cells. 2017;22(10):850–860. doi: 10.1111/gtc.12518.
- Chang Y-L, Tseng S-F, Huang Y-C, et al. Yeast Cip1 is activated by environmental stress to inhibit Cdk1-G1 cyclins via Mcm1 and Msn2/4. Nat Commun. 2017;8(1):56. doi: 10.1038/s41467-017-00080-y.
- Al-Rawi A, Kaye E, Korolchuk S, et al. Cyclin a and Cks1 promote kinase consensus switching to non-proline-directed CDK1 phosphorylation. Cell Rep. 2023;42(3):112139. doi: 10.1016/j.celrep.2023.112139.
- Enserink JM, Chymkowitch P. Cell cycle-dependent transcription: the cyclin dependent kinase Cdk1 is a direct regulator of basal transcription machineries. Int J Mol Sci. 2022;23(3):1293. doi: 10.3390/ijms23031293.
- Lanker S, Valdivieso MH, Wittenberg C. Rapid degradation of the G1 cyclin Cln2 induced by CDK-dependent phosphorylation. Science. 1996;271(5255):1597–1601. doi: 10.1126/science.271.5255.1597.
- Ross KE, Kaldis P, Solomon MJ. Activating phosphorylation of the Saccharomyces cerevisiae cyclin-dependent kinase, cdc28p, precedes cyclin binding. Mol Biol Cell. 2000;11(5):1597–1609. doi: 10.1371/journal.pgen.1002851.
- Kaldis P, Pitluk ZW, Bany IA, et al. Localization and regulation of the CDK-activating kinase (Cak1p) from budding yeast. J Cell Sci. 1998;111(24):3585–3596. doi: 10.1242/jcs.111.24.3585.
- Kurreck J, Stein CA. Molecular medicine: an introduction. Weinheim: Wiley-VCH Verlag GmbH and Co. KGaA; 2016. p. 9.
- Li P, Liu X, Hao Z, et al. Dual repressive function by Cip1, a budding yeast analog of p21, in cell-cycle START regulation. Front Microbiol. 2020;11:1623. doi: 10.3389/fmicb.2020.01623.
- Kishkevich A, Cooke SL, Harris MRA, et al. Gcn5 and Rpd3 have a limited role in the regulation of cell cycle transcripts during the G1 and S phases in Saccharomyces cerevisiae. Sci Rep. 2019;9(1):10686. doi: 10.1038/s41598-019-47170-z.
- Stephan OOH. Interactions, structural aspects and evolutionary perspectives of the yeast ‘START’-regulatory network. FEMS Yeast Res. 2022;22(1):foab064. doi: 10.1093/femsyr/foab064.
- Black L, Tollis S, Fu G, et al. G1/S transcription factors assemble in increasing numbers of discrete clusters through G1 phase. J Cell Biol. 2020;219(9):e202003041. doi: 10.1083/jcb.202003041.
- Simon I, Barnett J, Hannett N, et al. Serial regulation of transcriptional regulators in the yeast cell cycle. Cell. 2001;106(6):697–708. doi: 10.1016/S0092-8674(01)00494-9.
- Tollis S, Singh J, Palou R, et al. The microprotein Nrs1 rewires the G1/S transcriptional machinery during nitrogen limitation in budding yeast. PLoS Biol. 2022;20(3):e3001548. doi: 10.1371/journal.pbio.3001548.
- Sheu Y, Kawaguchi RK, Gillis J, et al. Prevalent and dynamic binding of the cell cycletransition kinase Rad53 to gene promoters. eLife. 2022;11:e84320. doi: 10.7554/eLife.84320.
- Bean JM, Siggia ED, Cross FR. High functional overlap between MluI cell-cycle box binding factor and Swi4/6 cell-cycle box binding factor in the G1/S transcriptional program in Saccharomyces cerevisiae. Genetics. 2005;171(1):49–61. doi: 10.1534/genetics.105.044560.
- De Bruin RAM, Kalashnikova TI, Chahwan C, et al. Constraining G1-specific transcription to late G1 phase: the MBF-associated corepressor Nrm1 acts via negative feedback. Mol Cell. 2006;23(4):483–496. doi: 10.1016/j.molcel.2006.06.025.
- Dirick L, Böhm T, Nasmyth K. Roles and regulation of Cln-Cdc28 kinases at the start of the cell cycle of Saccharomyces cerevisiae. Embo J. 1995;14(19):4803–4813. doi: 10.1002/j.1460-2075.1995.tb00162.x.
- Stuart D, Wittenberg C. CLN3, not positive feedback, determines the timing of CLN2 transcription in cycling cells. Genes Dev. 1995;9(22):2780–2794. doi: 10.1101/gad.9.22.2780.
- Wang H, Garí E, Vergés E, et al. Recruitment of Cdc28 by Whi3 restricts nuclear accumulation of the G1 cyclin-Cdk complex to late G1. Embo J. 2004;23(1):180–190. doi: 10.1038/sj.emboj.7600022.
- Palumbo P, Vanoni M, Cusimano V, et al. Whi5 phosphorylation embedded in the G1/S network dynamically controls critical cell size and cell fate. Nat Commun. 2016;7(1):11372. doi: 10.1038/ncomms11372.
- Wagner MV, Smolka MB, de Bruin RA, et al. Whi5 regulation by site specific CDK-phosphorylation in Saccharomyces cerevisiae. PLoS One. 2009;4(1):e4300. doi: 10.1371/journal.pone.0004300.
- Charvin G, Oikonomou C, Siggia ED, et al. Origin of irreversibility of cell cycle start in budding yeast. PLoS Biol. 2010;8(1):e1000284. doi: 10.1371/journal.pbio.1000284.
- Eser U, Falleur-Fettig M, Johnson A, et al. Commitment to a cellular transition precedes genome-wide transcriptional change. Mol Cell. 2011;43(4):515–527. doi: 10.1016/j.molcel.2011.06.024.
- Skotheim JM, Di Talia S, Siggia ED, et al. Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature. 2008;454(7202):291–296. doi: 10.1038/nature07118.
- Travesa A, Kalashnikova TI, de Bruin RAM, et al. Repression of G1/S transcription is mediated via interaction of the GTB motifs of Nrm1 and Whi5 with Swi6. Mol Cell Biol. 2013;33(8):1476–1486. doi: 10.1128/MCB.01333-12.
- Yahya G, Parisi E, Flores A, et al. A Whi7-anchored loop controls the G1 cdk-cyclin complex at start. Mol Cell. 2014;53(1):115–126. doi: 10.1016/j.molcel.2013.11.015.
- Gomar-Alba M, Méndez E, Quilis I, et al. Whi7 is an unstable cell-cycle repressor of the start transcriptional program. Nat Commun. 2017;8(1):329. doi: 10.1038/s41467-017-00374-1.
- Ros-Carrero C, Spiridon-Bodi M, Igual JC, et al. The CDK Pho85 inhibits Whi7 start repressor to promote cell cycle entry in budding yeast. EMBO Rep. 2024;25(2):745–769. doi: 10.1038/s44319-023-00049-7.
- Schmoller KM, Turner JJ, Kõivomägi M, et al. Dilution of the cell cycle inhibitor Whi5 controls budding-yeast cell size. Nature. 2015;526(7572):268–272. doi: 10.1038/nature14908.
- Ferrezuelo F, Colomina N, Palmisano A, et al. The critical size is set at a single-cell level by growth rate to attain homeostasis and adaptation. Nat Commun. 2012;3(1):1012. doi: 10.1038/ncomms2015.
- Wang H, Carey LB, Cai Y, et al. Recruitment of Cln3 cyclin to promoters controls cell cycle entry via histone deacetylase and other targets. PLoS Biol. 2009;7(9):e1000189. doi: 10.1371/journal.pbio.1000189.
- Broach JR. Nutritional control of growth and development in yeast. Genetics. 2012;192(1):73–105. doi: 10.1534/genetics.111.135731.
- Jorgensen P, Tyers M. How cells coordinate growth and division. Curr Biol. 2004;14(23):R1014–R1027. doi: 10.1016/j.cub.2004.11.027.
- Lowndes NF, Johnson AL, Breeden L, et al. Swi6 protein is required for transcription of the periodically expressed DNA synthesis genes in budding yeast. Nature. 1992;357(6378):505–508. doi: 10.1038/357505a0.
- Primig M, Sockanathan S, Auer H, et al. Anatomy of a transcription factor important for the start of the cell cycle in Saccharomyces cerevisiae. Nature. 1992;358(6387):593–597. doi: 10.1038/358593a0.
- Costanzo M, Nishikawa JL, Tang X, et al. CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell. 2004;117(7):899–913. doi: 10.1016/j.cell.2004.05.024.
- McInerny CJ, Partridge JF, Mikesell GE, et al. A novel Mcm1-dependent element in the SWI4, CLN3, CDC6, and CDC47 promoters activates M/G1-specific transcription. Genes Dev. 1997;11(10):1277–1288. doi: 10.1101/gad.11.10.1277.
- Flick K, Chapman-Shimshoni D, Stuart D, et al. Regulation of cell size by glucose is exerted via repression of the CLN1 promoter. Mol Cell Biol. 1998;18(5):2492–2501. doi: 10.1128/MCB.18.5.2492.
- Dorsey S, Tollis S, Cheng J, et al. G1/S transcription factor copy number is a growth-dependent determinant of cell cycle commitment in yeast. Cell Syst. 2018;6(5):539–554.e11. doi: 10.1016/j.cels.2018.04.012.
- Tollis S, Singh J, Thattikota Y, et al. Nsr1, a nitrogen source-regulated microprotein, confers an alternative mechanism of G1/S transcriptional activation in budding yeast. bioRxiv. 2020. doi: 10.1101/2020.04.20.033787.
- Hendler A, Medina EM, Buchler NE, et al. The evolution of a G1/S transcriptional network in yeasts. Curr Genet. 2018;64(1):81–86. doi: 10.1007/s00294-017-0726-3.
- Manukyan A, Zhang J, Thippeswamy U, et al. Ccr4 alters cell size in yeast by modulating the timing of CLN1 and CLN2 expression. Genetics. 2008;179(1):345–357. doi: 10.1534/genetics.108.086744.
- Li P, Hao Z, Zeng F. Tumor suppressor stars in yeast G1/S transition. Curr Genet. 2021;67(2):207–212. doi: 10.1007/s00294-020-01126-3.
- Miles S, Breeden L. A common strategy for initiating the transition from proliferation to quiescence. Curr Genet. 2017;63(2):179–186. doi: 10.1007/s00294-016-0640-0.
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005.
- Montanari A, Leo M, De Luca V, et al. Gcn5 histone acetyltransferase is present in the mitoplasts. Biol Open. 2019;8(2):bio041244. doi: 10.1242/bio.041244.
- Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92(4):463–473. doi: 10.1016/s0092-8674(00)80940-x.
- Huang D, Kaluarachchi S, van Dyk D, et al. Dual regulation by pairs of cyclin-dependent protein kinases and histone deacetylases controls G1 transcription in budding yeast. PLoS Biol. 2009;7(9):e1000188. doi: 10.1371/journal.pbio.1000188.
- Miles S, Croxford MW, Abeysinghe AP, et al. Msa1 and Msa2 modulate G1-specific transcription to promote G1 arrest and the transition to quiescence in budding yeast. PLoS Genet. 2016;12(6):e1006088. doi: 10.1371/journal.pgen.1006088.
- Lam EW, La Thangue NB. DP and E2F proteins: coordinating transcription with cell cycle progression. Curr Opin Cell Biol. 1994;6(6):859–866. doi: 10.1016/0955-0674(94)90057-4.
- Slansky JE, Farnham PJ. Introduction to the E2F family: protein structure and gene regulation. Curr Top Microbiol Immunol. 1996;208:1–30. doi: 10.1007/978-3-642-79910-5_1.
- Fischer M, Schade AE, Branigan TB, et al. Coordinating gene expression during the cell cycle. Trends Biochem. Sci. 2022;47(12):1009–1022. doi: 10.1016/j.tibs.2022.06.007.
- Zatulovskiy E, Zhang S, Berenson DF, et al. Cell growth dilutes the cell cycle inhibitor Rb to trigger cell division. Science. 2020;369(6502):466–471. doi: 10.1126/science.aaz6213.
- Medina EM, Turner JJ, Gordân R, et al. Punctuated evolution and transitional hybrid network in an ancestral cell cycle of fungi. eLife. 2016;5:e09492. doi: 10.7554/eLife.09492.
- Fischer M, Grossmann P, Padi M, et al. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016;44(13):6070–6086. doi: 10.1093/nar/gkw523.
- Cooper K. Rb, whi it’s not just for metazoans anymore. Oncogene. 2006;25(38):5228–5232. doi: 10.1038/sj.onc.1209630.
- Zhang S, Valenzuela LF, Zatulovskiy E, et al. The G1/S transition is promoted by Rb degradation via the E3 ligase UBR5. bioRxiv. 2023. doi: 10.1101/2023.10.03.560768.
- Müller L, Keil R, Hatzfeld M. Plakophilin 3 facilitates G1/S phase transition and enhances proliferation by capturing RB protein in the cytoplasm and promoting EGFR signaling. Cell Rep. 2023;42(1):112031. doi: 10.1016/j.celrep.2023.112031.
- Wang H, Wang X, Xu L, et al. Integrated analysis of the E2F transcription factors across cancer types. Oncol Rep. 2020;43(4):1133–1146. doi: 10.3892/or.2020.7504.
- Kassab A, Gupta I, Moustafa AA. Role of E2F transcription factor in oral cancer: recent insight and advancements. Semin Cancer Biol. 2023;92:28–41. doi: 10.1016/j.semcancer.2023.03.004.
- Nakajima R, Zhao L, Zhou Y, et al. Deregulated E2F activity as a cancer-cell specific therapeutic tool. Genes (Basel). 2023;14(2):393. doi: 10.3390/genes14020393.
- Hsu J, Arand J, Chaikovsky A, et al. E2F4 regulates transcriptional activation in mouse embryonic stem cells independently of the RB family. Nat Commun. 2019;10(1):2939. doi: 10.1038/s41467-019-10901-x.
- Chang H, Song J, Wu J, et al. E2F transcription factor 8 promotes cell proliferation via CCND1/p21 in esophageal squamous cell carcinoma. Onco Targets Ther. 2018;11:8165–8173. doi: 10.2147/OTT.S180938.
- Li J, Wang H, Cao F, et al. A bioinformatics analysis for diagnostic roles of the E2F family in esophageal cancer. J Gastrointest Oncol. 2022;13(5):2115–2131. doi: 10.21037/jgo-22-855.
- Sun J, Shi R, Zhao S, et al. E2F8, a direct target of miR-144, promotes papillary thyroid cancer progression via regulating cell cycle. J Exp Clin Cancer Res. 2017;36(1):40. doi: 10.1186/s13046-017-0504-6.
- Lee DY, Chun JN, Cho M, et al. Emerging role of E2F8 in human cancer. Biochim Biophys Acta Mol Basis Dis. 2023;1869(6):166745. doi: 10.1016/j.bbadis.2023.166745.
- Schade AE, Fischer M, DeCaprio JA. RB, p130 and p107 differentially repress G1/S and G2/M genes after p53 activation. Nucleic Acids Res. 2019;47(21):11197–11208. doi: 10.1093/nar/gkz961.
- Uxa S, Bernhart SH, Mages CFS, et al. DREAM and RB cooperate to induce gene repression and cell-cycle arrest in response to p53 activation. Nucleic Acids Res. 2019;47(17):9087–9103. doi: 10.1093/nar/gkz635.
- Schade AE, Oser MG, Nicholson HE, et al. Cyclin D-CDK4 relieves cooperative repression of proliferation and cell cycle gene expression by DREAM and RB. Oncogene. 2019;38(25):4962–4976. doi: 10.1038/s41388-019-0767-9.
- Mages CF, Wintsche A, Bernhart SH, et al. The DREAM complex through its subunit Lin37 cooperates with Rb to initiate quiescence. eLife. 2017;6:e26876. doi: 10.7554/eLife.26876.
- Fischer M. Conservation and divergence of the p53 gene regulatory network between mice and humans. Oncogene. 2019;38(21):4095–4109. doi: 10.1038/s41388-019-0706-9.
- Hafner A, Bulyk ML, Jambhekar A, et al. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20(4):199–210. doi: 10.1038/s41580-019-0110-x.
- Liu M, Liu H, Chen J. Mechanisms of the CDK4/6 inhibitor palbociclib (PD 0332991) and its future application in cancer treatment. Oncol Rep. 2018;39(3):901–911. doi: 10.3892/or.2018.6221.
- Vijayaraghavan S, Moulder S, Keyomarsi K, et al. Inhibiting CDK in cancer therapy: current evidence and future directions. Target Oncol. 2018;13(1):21–38. doi: 10.1007/s11523-017-0541-2.
- Turner NC, Ro J, André F, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373(3):209–219. doi: 10.1056/NEJMoa1505270.
- Vilgelm AE, Saleh N, Shattuck-Brandt R, et al. MDM2 antagonists overcome intrinsic resistance to CDK4/6 inhibition by inducing p21. Sci Transl Med. 2019;11(505):eaav7171. doi: 10.1126/scitranslmed.aav7171.
- Cejuela M, Gil-Torralvo A, Castilla MÁ, et al. Abemaciclib, palbociclib, and ribociclib in real-world data: a direct comparison of first-line treatment for endocrine-receptor-positive metastatic breast cancer. Int J Mol Sci. 2023;24(10):8488. doi: 10.3390/ijms24108488.
- Bisi JE, Sorrentino JA, Jordan JL, et al. Preclinical development of G1 T38: a novel, potent and selective inhibitor of cyclin dependent kinases 4/6 for use as an oral antineoplastic in patients with Cdk4/6 sensitive tumors. Oncotarget. 2017;8(26):42343–42358. doi: 10.18632/oncotarget.
- Teh JLF, Cheng PF, Purwin TJ, et al. In vivo E2F reporting reveals efficacious schedules of MEK1/2-CDK4/6 targeting and mTOR-S6 resistance mechanisms. Cancer Discov. 2018;8(5):568–581. doi: 10.1158/2159-8290.CD-17-0699.
- Kreis NN, Louwen F, Yuan J. The multifaceted p21 (Cip1/Waf1/CDKN1A) in cell differentiation, migration and cancer therapy. Cancers (Basel). 2019;11(9):11–1220. doi: 10.3390/cancers11091220.
- Liu H, Liu K, Dong Z. The role of p21-activated kinases in cancer and beyond: where are we heading? Front Cell Dev Biol. 2021;9:641381. doi: 10.3389/fcell.2021.641381.
- Shamloo B, Usluer S. p21 in cancer research. Cancers (Basel). 2019;11(8):1178. doi: 10.3390/cancers11081178.
- Pérez-Tenorio G, Berglund F, Merca AE, et al. Cytoplasmic p21WAF1/CIP1 correlates with Akt activation and poor response to tamoxifen in breast cancer. Int J Oncol. 2006;28(5):1031–1042. doi: 10.3892/ijo.28.5.1031.
- Zhang Z, Cao Y, Zhao W, et al. HDAC6 serves as a biomarker for the prognosis of patients with renal cell carcinoma. Cancer Biomark. 2017;19(2):169–175. doi: 10.3233/CBM-160298.
- Zhang X, Ma Q, Wu H, et al. A review of progress in histone deacetylase 6 inhibitors research: structural specificity and functional diversity. J Med Chem. 2021;64(3):1362–1391. doi: 10.1021/acs.jmedchem.0c01782.
- Sixto-López Y, Bello M, Rodríguez-Fonseca RA, et al. Searching the conformational complexity and binding properties of HDAC6 through docking and molecular dynamic simulations. J Biomol Struct Dyn. 2017;35(13):2794–2814. doi: 10.1080/07391102.2016.1231084.
- Kukushkin AN, Svetlikova SB. Histone deacetylase HDAC6 inhibitor CAY10603 blocks G1/S of the cell cycle and promotes senescence of murine fibroblasts. Cell Tiss Biol. 2019;13(4):268–275. doi: 10.1134/S1990519X19040047.