
Abstract
Background
Numerous studies suggest a progressive accumulation of post-translationally modified peptides within amyloid fibrils, including isoaspartate (isoD) modifications. Here, we generated and characterised novel monoclonal antibodies targeting isoD-modified transthyretin (TTR). The antibodies were used to investigate the presence of isoD-modified TTR in deposits from transthyretin amyloidosis patients and to mediate antibody-dependent phagocytosis of TTR fibrils.
Methods
Monoclonal antibodies were generated by immunisation of mice using an isoD-modified peptide and subsequent hybridoma generation. The antibodies were characterised in terms of affinity and specificity to isoD-modified TTR using surface plasmon resonance, transmission electron microscopy and immunohistochemical staining of human cardiac tissue. The potential to elicit antibody-dependent phagocytosis of TTR fibrils was assessed using THP-1 cells.
Results
We developed two mouse monoclonal antibodies, 2F2 and 4D4, with high nanomolar affinity for isoD-modified TTR and strong selectivity over the unmodified epitope. Both antibodies show presence of isoD-modified TTR in human cardiac tissue, but not in freshly purified recombinant TTR, suggesting isoD modification only present in aged fibrillar deposits. Likewise, the antibodies only facilitated phagocytosis of TTR fibrils and not TTR monomers by THP-1 cells.
Conclusions
These antibodies label aged, non-native TTR deposits, leaving native TTR unattended and thereby potentially enabling new therapeutic approaches.
Introduction
Systemic amyloidoses represent a group of rare diseases that are often underdiagnosed due to their multifaceted symptomatic appearance. In these multisystem diseases, misfolded fibrillar proteins are deposited extracellularly in various organs, resulting in a successive loss of their function and integrity [Citation1].
Transthyretin amyloidosis (ATTR) results from the gradual accumulation of the plasma protein transthyretin (TTR) in tissues, leading to heart failure, neuropathy, and death. There are two forms of ATTR amyloidosis, wild-type (ATTRwt), often underdiagnosed in older individuals, and hereditary ATTR amyloidosis (ATTRv), caused by TTR gene mutations. Over 130 mutations exist, with varying onset and symptoms; some primarily affect nerves, while others impact the heart or present mixed symptoms [Citation2]. The most widespread mutation is V30M, associated with neuropathy [Citation3], followed by V122I, primarily affecting the heart [Citation4].
TTR, a 55 kDa homotetrameric protein primarily synthesised in the liver, transports thyroxine and holo-retinol binding protein in the cerebrospinal fluid and plasma [Citation5]. Factors like low pH, elevated temperature or TTR gene mutations increase instability, prompting faster dissociation and monomer release, initiating amyloid fibril formation [Citation6]. Recently, Schmidt et al. solved the first cryo-EM structure of a human ATTRv-V30M fibril, revealing a significant unfolding of native TTR confirmation and offering insights into potential protein-protein interaction sites within the fibril [Citation7].
Therapeutic approaches for treating ATTRv include liver transplantation [Citation8], gene silencing by siRNAs [Citation9,Citation10] or antisense oligonucleotides [Citation11], in vivo gene-editing via CRISPR-Cas9 [Citation12] and stabilising the TTR complex to prevent misfolding/unfolding, aggregation, and tissue deposition. While these strategies slow amyloid progression, additional methods are needed to clear existing deposits. Passive immunotherapy applying monoclonal antibodies (mAbs) targeting abnormal TTR structures might represent such a promising approach. To our knowledge, all mAbs developed so far target a cryptic epitope on the surface of amyloidogenic TTR in ATTRwt and ATTRv. The mAb PRX004 has advanced to phase 2 trials [Citation13,Citation14], while NI301A recently completed phase 1 for ATTR cardiomyopathy [Citation15,Citation16].
Recent advancements in treating amyloidosis, such as the successful completion of phase 3 trials for the mAb donanemab in Alzheimer’s disease (AD), suggest potential implications for ATTR therapy [Citation17,Citation18]. While donanemab’s targeting of the pyroglutamate form of Aβ underscores tailored therapeutic approaches, knowledge of pathogenic PTMs of TTR in amyloidosis remains limited [Citation19,Citation20].
Based on the comprehensive analysis of PTMs in AD [Citation21–23], some of these might be also deduced for TTR in ATTRwt and ATTRv amyloidosis. One such modification is isoaspartate (isoAsp, isoD), which is formed from L-aspartyl or L-asparaginyl precursors [Citation24]. Isomerisation to L-isoaspartate or L-aspartate occurs spontaneously and has been shown to determine the half-life of a protein and to alter its structure by introducing an additional methylene group into the amide backbone [Citation25–27] with potential impact on protein solubility, conformation, and function.
By means of two mAbs, 2F2 and 4D4, we here provide evidence for isoD formation in TTR at position 38 and the presence of isoD-TTR in amyloid deposits. The antibodies bind to amyloid deposits of human pathological tissue but not native TTR protein. Likewise, they only facilitate cellular uptake of isoD-modified TTR fibrils into THP-1 cells via phagocytosis. Thus, we propose these antibodies could have further therapeutic applications by labelling TTR protein deposits for clearance in patients.
Materials and methods
Preparation of recombinant TTRwt and variants
The TTRwt encoding sequence was PCR amplified from TTR cDNA in pGEM-T-vector (Biozol) into pQE30 (Qiagen) (SI Table 1). The genetic information for a cleavage site of the tobacco etch virus protease was inserted. The mutant TTR expression vectors were generated using appropriate primers and site-directed mutagenesis.
TTR proteins were expressed in Escherichia coli and purified, essentially as described elsewhere [Citation28]. Briefly, after transformation of E. coli M15, cells were cultured in Luria-Bertani-medium/ampicillin/kanamycin at 37 °C. IPTG was added at a final concentration of 40 µM (TTR-V122I) or 400 µM (TTRwt, TTR-V30M). The culture was incubated at 30 °C overnight (TTRwt), at 37 °C for 4 h (TTR-V122I) or at 21 °C overnight (TTR-V30M). The harvested cells were resuspended in 50 mM Tris, 300 mM NaCl, 5 mM imidazole, 2 mM dithioerythritol, protease inhibitor, pH 7.6, followed by sonification for 15 min. The supernatant was subjected to nickel-nitrilotriacetic acid-agarose (GE Healthcare Life Sciences). After washing with 50 mM Tris, 300 mM NaCl, 20 mM imidazole, pH 7.6, elution of His-tagged protein was obtained by using 50 mM Tris, 100 mM NaCl, 250 mM imidazole. The elution fractions were dialysed overnight against 50 mM Tris, 100 mM NaCl, 1 mM dithioerythritol, pH 7.6. The dialysed protein was cleaved overnight at 13 °C using TEV protease (enzyme:substrate 1:1.16) in 10 mM Tris, 100 mM NaCl, 5% (w/v) glycerol, 0.5 mM EDTA, pH 8. The cleaved protein was centrifuged and collected supernatant was subjected to Ni2+-NTA (HisTrap HP, GE Healthcare Life Sciences).
Amyloid fibril formation monitored by thioflavin T (ThT) binding
To investigate the time course of amyloid fibril formation, a ThT assay was performed as described previously [Citation28]. Recombinant TTRwt, TTR-V30M or TTR-V122I (200 µM) were incubated with 20 µM ThT in aggregation buffer (50 mM sodium acetate pH 4.0) in a 96-well plate using a CLARIOstar plate reader (BMG Labtech). The excitation and emission wavelengths were 440 nm and 490 nm. Signals were recorded at 37 °C under continuous shaking (300 rpm). Aggregate-solutions were transferred to 1.5 ml tubes after 72 h and then incubated continuously at 37 °C for several months.
Extraction of amyloid fibrils from tissue
The extraction of fibrils from human tissue was based on the water extraction protocol from Annamalai et al. [Citation29]. In brief, ∼2 g of frozen human heart tissue was thawed, diced into small pieces and washed for five times with 20 mM Tris, 138 mM NaCl, 2 mM CaCl2, 0.1% (w/v) NaN3, pH 8. The tissue was then incubated in 1 ml collagenase solution (Sigma-Aldrich) overnight at 37 °C under continuous shaking. After centrifugation for 30 min at 3100 xg and 4 °C, the pellet was homogenised in 3 ml 20 mM Tris, 140 mM NaCl, 10 mM EDTA, 0.1% (w/v) NaN3, pH 8 using a glass potter pestle and centrifuged for 5 min at 3100 xg and 4 °C. This homogenisation step was repeated nine times. For amyloid fibril extraction, the remaining pellet was resuspended in 3 ml ice-cold water and centrifuged for 5 min at 3100 xg and 4 °C, collecting the supernatant. This extraction process was repeated six times. The use of human heart tissue was approved by Heidelberg University’s ethical committees (123/2006).
Antibody reactivity against in vitro amyloid and extracted TTR fibrils
For dot blots, 0.5 µg of recombinant TTR protein (TTRwt, TTR-V30M, TTR-V122I) and ATTR fibrils or 0.5 µg of either freshly solved Aβ(1-42) or aggregated Aβ(1-42) with confirmed isoD-content were spotted on nitrocellulose membranes. Membranes were blocked for 1 h in blocking solution (5% (w/v) milk powder in TBS + 0.05% (v/v) Tween-20 (Carl Roth)). Antibodies 2F2, 4D4 or polyclonal total-TTR antibody (Biozol) were diluted to 1 µg/ml in blocking solution and incubated overnight at 4 °C. Alkaline phosphatase-conjugated, anti-mouse or rabbit IgG antibodies (abcam) in combination with chromogenic substrates BCIP and NBT (Carl Roth) were used subsequently for detection.
Preparation of the immunogen, antibody generation and hybridoma selection
A peptide consisting of amino acids 35-43 of human TTR, containing an isoD residue at position 38 instead of aspartate and an additional artificial C-terminal amino acid for coupling (Ac-KAA(isoD)DTWEPC-amide), was synthesised by Peptide&Elephants (Berlin, Germany). The peptide was covalently linked to keyhole limpet haemocyanin via the C-terminal cysteine residue. For generation of mAbs, 8-week-old female BALB/c mice were used. Animal experiments were approved by the local authorities (Landesverwaltungsamt Sachsen-Anhalt, Department of Consumer Protection and Veterinary Affairs, Halle (Saale), Saxony-Anhalt, Germany, 42502-3-851). Mice were immunised subcutaneously with a 1:1 emulsion of KLH-conjugated TTR peptide and Freund’s complete adjuvant (VWR), followed by boosts with another 95 µg incomplete Freund’s adjuvant 44 and 73 days later and a final immunogen boost every day starting 4 days prior to fusion. Antibody titre was confirmed by enzyme-linked immunosorbent assay (ELISA). Homogenised spleen cells were fused to SP2/0 myeloma cells using the PEG method [Citation30]. Cells were immediately plated on prepared feeder cell plates in HAT medium and screened after 10 days. Hybridomas were selected upon specific binding to isoD38-TTR(35-43), but not to D38-TTR(35-43), using an indirect ELISA and surface plasmon resonance (SPR). Stable antibody-producing hybridomas have been selected and subsequently cloned by limited dilution and an additional single-cell-separation to ensure the monoclonality of the hybridomas.
Antibody expression and purification
Hybridoma cell clones were cultured in T175 flasks. The supernatant was collected, sterile filtered and mixed with binding buffer (40 mM Na2HPO4, 150 mM NaCl, pH 7.0) at an equal volume and subjected to a pre-equilibrated Protein G Sepharose Fast Flow column (GE Healthcare Life Sciences). Elution was done by using 40 mM Na2HPO4 and 2 M KSCN at pH 7.0. Collected fractions were dialysed against PBS at 4 °C overnight.
Antibody characterisation by SPR analysis
Antibody binding kinetics to D38/isoD38-TTR(35-43)-PEG-Biotin peptides were analysed using SPR on a BIAcore 3000 device at 25 °C. The surface of a CM5 sensor chip (Cytiva) was modified with a goat anti-mouse antibody by activation of mixing 0.1 M N-hydroxysuccinimide (NHS) with 0.4 M N-ethyl-N′-(dimethylaminopropyl) carbodiimide hydrochloride (EDC) 1:1. EDC/NHS was applied to the sensor chip with a flow rate of 20 μl/min for 7 min. Goat anti-mouse IgG was diluted to 50 μg/ml in 10 mM sodium acetate, pH 5.5, and injected for 2 × 4 min contact time with a flow rate of 30 μl/min. Deactivation was done by applying 1 M ethanolamine pH 8.5 with a flow rate of 10 µl/min for 2 × 7 min contact time. About 20,000 RU of the goat anti-mouse antibody were immobilised onto the chip surface. 2F2 and 4D4 were captured using antibody dilutions of 25 µg/ml in HBS-EP buffer and a flow rate of 2 µl/min to achieve a level of about 2,000 RU for both antibodies on the chip surface. For 4D4 antibody, multi cycle kinetic analysis was carried out by applying 10 nM to 100 µM of isoD38-TTR(35-43) and 1 µM to 200 µM of D38-TTR(35-43), respectively. For 2F2 antibody, five consecutive injections of 2 nM to 512 nM of isoD38-TTR(35-43) and 512 nM to 131 µM D38-TTR(35-43) were applied.
The sensorgrams were evaluated using the BIAevaluation software 4.1.1. For the 4D4 antibody the sensorgrams were corrected by double-referencing using blank surface (anti-mouse antibody) and buffer-only injections and the dissociation constant was calculated using the steady-state binding model. For the 2F2 antibody, the sensorgrams were evaluated using the single cycle kinetic model.
Antibody binding kinetics to isoD38-TTR(28-50)-PEG-Biotin peptide were analysed by SPR using a Bruker SPR-24 Pro device at 25 °C. For this purpose, onto an IgG-Capture Sensor (Bruker) about 4,000 RU of 2F2 and 4D4 were immobilised using antibody dilutions of 10 µg/ml in HBS-EP buffer and a flow rate of 5 µl/min. For both antibodies, a single injection cycle kinetic analysis was carried out by applying 0.5 nM to 50 nM of isoD38-TTR(28-50).
Histology and imaging
Fresh frozen cardiac tissue with confirmed genetic diagnoses of three different forms of hereditary ATTR cardiac amyloidosis and one amyloid light chain (AL) amyloidosis were provided by Amyloidosis Centre Heidelberg, Germany. FFPE tissue of one sporadic ATTR case was provided by the Department of Nephropathology of University Hospital of Erlangen, Germany. The use of FFPE tissue rest material was approved by the Ethics Committee of the Friedrich-Alexander-University of Erlangen-Nürnberg for the archive of the Department of Nephropathology (Re.-No.4415) waiving the need of informed consent due to its retrospective nature. FFPE tissue of normal control heart was commercially obtained from Morphisto (Offenbach am Main, Germany).
Paraffin-embedded sections were deparaffinized in xylene and rehydrated in graded alcohols. Both paraffin-embedded and slide-mounted cryosections (10 µm) were treated with 1% (v/v) hydrogen peroxide (Sigma-Aldrich) in methanol (frozen sections) or PBS (FFPE tissue) to block endogenous peroxidase activity at RT for 10 min. After additional blocking using 5% (v/v) normal goat serum for 30 min, slides were incubated with primary antibody in a moist chamber at 4 °C overnight. The secondary antibody (anti-mouse or anti-rabbit HRP-linked antibody; Invitrogen) was applied at a 1:1,000 dilution in TBS containing 2% (w/v) bovine serum albumin (BSA, Sigma-Aldrich) at RT for 1 h. Staining was performed using diaminobenzidine.
For Congo red staining, slides were immersed in freshly filtered Congo red staining solution (1.2% (w/v) Congo red (Carl Roth) in absolute ethanol with 5% KOH). For thioflavin S staining, sections were incubated in 1% (w/v) thioflavin S solution (Sigma-Aldrich). Fluorescence was observed using a Keyence BZ-X800 fluorescence microscope. Apple-green birefringence of Congo red was obtained using a polariser and analyser with a Leica M205C microscope.
Transmission electron microscopy and immunogold labelling
A sample (5 µl) from extracted human ATTR-V30M fibrils were directly incubated on a formvar carbon-coated copper grid (Plano) for 10 min and washed three times with distilled water. Staining was obtained with 2% (v/v) uranyl acetate (SERVA Electrophoresis GmbH) for 5 min.
For immunogold labelling, the fibril samples were fixed on the grids using 2% (w/v) paraformaldehyde (Merck) in PBS buffer for 20 min followed by washing with distilled water. The sections were incubated for 30 min at RT with blocking solution (1% (w/v) BSA containing 0.1% Tween-20 in PBS). After blocking, the grids were incubated with 4 µg/ml of primary antibody: mAbs 2F2 and 4D4, polyclonal total-TTR antibody (Biozol), and appropriate isotype controls (BD Biosciences). After additional washing with blocking solution the grids were incubated with secondary antibodies coupled to colloidal gold: anti-rabbit IgG 5 nm gold (goat polyclonal, Sigma-Aldrich) and anti-mouse IgG 20 nm gold (goat polyclonal, Abcam), diluted 1:50 in blocking solution for 90 min. After three washing steps with distilled water, the grids were treated with contrasting solution (2% (v/v) uranyl acetate) for 5 min.
Fibrils were imaged with a LEO EM 912 Omega TEM (Zeiss) at 80 kV and digital micrographs were obtained with a dual speed 2K-on-axis CCD camera based YAG scintillator (TRS-Tröndle).
Phagocytosis assay
A sample containing heart extracted human ATTR-V30M fibrils or 1 mg/ml native recombinant TTRwt was amine-coupled with pHrodo Red dye (Thermo Fisher) at RT for 1 h, applying a protein:dye ratio of 10:1. In case of for native TTRwt, excess pHrodo Red dye was removed by diafiltration using a 5 kDa molecular weight cut-off (Merck). For amyloid fibrils, three centrifugation steps at 17,000 xg for 10 min followed by resuspension in PBS removed excess free dye label.
For stimulation, 2x105 human THP-1 monocytes were cultured in cell culture media (RPMI, 10% low IgG FBS) and differentiated using 200 nM phorbol 12-myristate 13-acetate (PMA, Sigma-Aldrich) for 3 days. After this initial stimulus, PMA-containing media was replaced by fresh RPMI + 10% low IgG FBS for further 2 days.
A 20 µg/ml aliquot of pHrodo-labelled TTR (either monomer TTRwt or extracted human TTR-V30M fibril) was separately pre-incubated with 40 µg/ml antibody in cell culture media at 37 °C for 1 h. After addition of 2x105 PMA-treated THP-1 cells the tissue culture was incubated for 3 h. Antigen-antibody-complexes were removed by washing the cells three times with PBS.
The extent of pHrodo-labelled TTR uptake was quantified by flow cytometric analysis using a FACSCalibur (Becton Dickinson). Forward and side scatter light was used to identify cell populations and to measure size and granularity of the cells. Red-fluorescent cells were recorded at 585 nm after excitation at 560 nm. Remaining cells were transferred to glass chamber slides and imaged by bright field and fluorescence microscopy using a Keyence BZ-X800.
Results
Generation and biophysical characterisation of isoD-specific TTR mAbs
Based on the solved structure of a TTR fibril from a patient suffering from ATTR-V30M [Citation7], we searched for potential isoD residues with exposure to the fibril surface (). Schmidt et al. state that the two fragments of the solved TTR fibril structure can possibly be linked with the missing residues by a flexible region (). This highly flexible and unstructured region contains an aspartate residue at position 38, which is prone for isoD formation, as already shown in various studies [Citation25–29].
Figure 1. TTR fibril and localisation of the isoaspartate epitope. (A) Native TTRwt structure with asterisk-indicated location of D38 for each monomer (PDB 4ACU) (B) Ribbon diagram of four layers of the TTR-V30M fibril (PDB 6SDZ). Rainbow colour from N- (blue) to C-terminus (red). (C) Cα-trace of TTR residues Pro11-Lys35 (orange) and Gly57-Thr123 (blue). Residues Ala36-His56 (red) are a schematic representation of the possible connection between the two fragments, with the asterisk marking estimated position of Asp38. (D) Top: The amino acid sequence of human TTR highlighting the location of the epitope for the developed isoD38-TTR mAbs. Bottom: Synthetic peptide sequence of TTR(35-43) with additional cysteine residue (lower case), linked to keyhole limpet haemocyanin as immunogen.
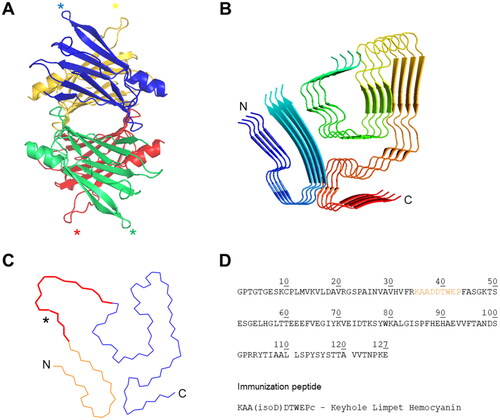
For immunisation of mice, we selected the peptide TTR(35-43), containing an isoD residue at position 38 (). The generated hybridoma clones were selected according to strong reactivity towards isoD38-TTR(35-43)-peptide and little/no reactivity towards D38-TTR(35-43)-peptide (data not shown). The isotype of the two resulting murine mAbs 2F2 and 4D4 were determined as IgG3/κ and IgG2a/κ, respectively. Sequences of variable regions were determined and revealed unique CDR composition.
The isoD specificity of 2F2 and 4D4 was further explored using kinetic analysis by SPR measurements (SI Figures 1 and 2). The dissociation constants for the binding of 2F2 and 4D4 to either isoD38-TTR(35-43), D38-TTR(35-43) or isoD38-TTR(28-50) are shown in SI Table 2. Both TTR mAbs showed high affinity for isoD38-TTR(35-43), as suggested by the KD-values of 1.55 nM (2F2) and 124 nM (4D4). In comparison, the dissociation constants for the non-modified peptide D38-TTR(35-43) are 233-fold (719 nM, 4D4) and 464-fold (29 µM, 2F2) higher compared to isoD38-TTR(35-43).
For further characterisation of the new mAbs, we analysed their ability to detect isoD-residues in different TTR species by dot blot analysis ( and SI Figure 3) and ELISA (SI Figure 4). Recombinantly expressed TTRwt, TTR-V30M and TTR-V122I were not identified by 2F2 and 4D4, indicating absence of isoD residues at position 38. Instead, both antibodies recognised aggregated, one-year old samples of the recombinant TTR-derived fibrils. Furthermore, for the first time, we observed isoD38-TTR-species in extracted fibrils of human cardiac tissue of patients with pathological ATTR-V30M, detected by 2F2 and 4D4 antibodies. In contrast, the total-TTR pAb did not discriminate between recombinant TTR variants and aggregated, aged TTR fibrils either in vitro produced or extracted from human tissue. Both TTR mAbs showed no reactivity against other isoD-containing amyloid fibrils (isoD7-Aβ) or their monomeric peptides.
Figure 2. Specificity of the TTR mAbs 2F2 and 4D4, characterised by dot blot analysis. The two mAbs 2F2 and 4D4 specific for isoD38-modified TTR and a polyclonal total TTR antibody were compared. The mAbs bind to aged amyloid aggregates of recombinant TTR protein and extracted fibrils. No binding was detected with native recombinant TTR and aggregated Aβ1-42 (verified isoD content). Detection was carried out based on alkaline phosphatase activity. The data support high specificity of the isoD-specific mAbs for aged, modified TTR.

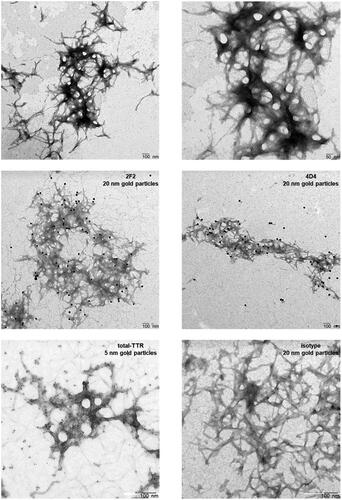
The binding of 2F2 and 4D4 to TTR fibrils was further analysed using transmission electron microscopy with subsequent immunogold labelling. TEM analyses revealed typical TTR fibrils in cluster networks (, upper panel). The isoD-TTR mAbs 2F2 and 4D4 bind to TTR-V30M fibrils as visualised by immunogold staining (, middle panel). In contrast, the total-TTR pAb detected the entire TTR fibril structure (5 nm gold particle). No fibril labelling was observed using a non-sense primary antibody (, lower panel). These findings corroborate the previous analyses and strongly support the specific binding of isoD38-TTR mAbs to ATTR-fibrils from human tissue.
Figure 3. Visualisation of extracted human ATTR-V30M fibrils, immunogold-labelled using isoD38-TTR mAbs 2F2 and 4D4. Transmission electron microscopy reveal the typical long-branched structure of amyloid TTR fibrils (upper panel). 2F2 and 4D4 showing strong labelling of human ATTR-V30M fibrils, indicated by 20 nm gold particles (middle panel). Immunogold labelling with an antibody binding to total-TTR and a non-sense isotype control is shown in comparison (lower panel).
Application of mAbs 2F2 and 4D4 in immunohistochemistry
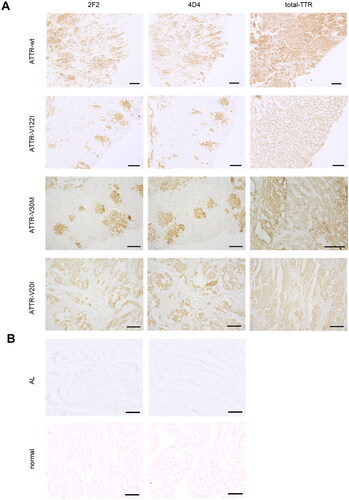
We next examined whether the isoD38-TTR mAbs 2F2 and 4D4 could specifically immunolabel TTR amyloid deposits in paraffin-embedded or frozen sections from cardiac tissue of patients with sporadic and hereditary ATTR (). 2F2 and 4D4 immunoreactivities showed a very similar pattern. The antibodies detected deposits in the extracellular space between cardiomyocytes. These areas were also stained using the total-TTR antibody, which also displayed reactivity towards non-amyloid tissue. There were no specific immunoreactivities detected using the 2F2 and 4D4 antibody with cardiac tissue derived from healthy persons nor with heart tissue from cardiomyopathy patients suffering from AL amyloidosis.
Figure 4. Immunolabeling of cardiac tissue from patients with hereditary and sporadic ATTR amyloidosis. (A) 2F2 and 4D4 antibodies specifically label amyloid TTR deposits in various ATTR amyloidosis cases (wild-type, V30M, V122I, V20I) in contrast to staining of the polyclonal total-TTR antibody. (B) No 2F2 or 4D4 immunolabeling was observed in cardiac tissue from cardiomyopathy patients with AL amyloidosis (AL) or healthy individuals (normal). Scale bar = 200 µm.
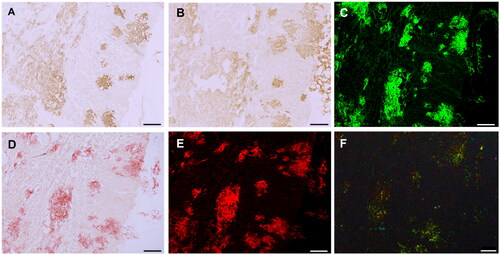
To confirm the specific staining of amyloid deposits by 2F2 and 4D4, frozen sections of cardiac tissue from ATTR-V30M were stained with thioflavin S and Congo red (). These areas showed high consistency with positive areas of isoD38-TTR mAbs 2F2 and 4D4 (). Less intense 2F2 and 4D4 immunoreactivity was also found in regions not staining for amyloid, and thus may represent staining of pre-amyloid TTR deposits.
Figure 5. Amyloid-specific staining of cardiac TTR deposits. Sections of cardiac tissue with ATTR-V30M amyloidosis were stained with isoD38-TTR mAbs 2F2 (A) and 4D4 (B). Consecutive sections stained with thioflavin S (C) and Congo red (D) reveal amyloid deposits, confirmed by Congo red fluorescence (E) and Congo red polarisation (F). Scale bar = 250 µm.
2F2 and 4D4 promote phagocytosis of TTR fibrils
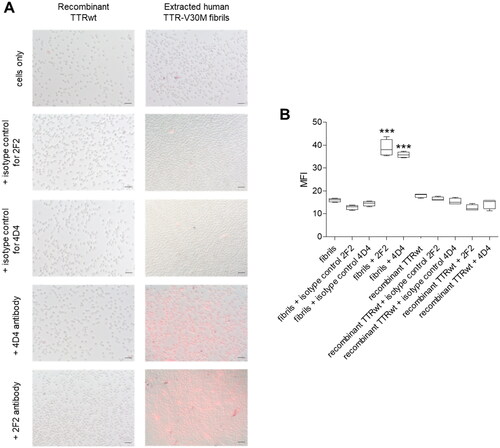
In order to analyse whether 2F2 and 4D4 are capable to induce Fc-mediated phagocytosis of TTR fibrils, we established a cellular phagocytosis assay using human macrophage-like THP-1 cells in vitro. The assay is based on covalent labelling of extracted human ATTR-V30M fibrils or, as a control, recombinant monomeric TTRwt to the pH-sensitive dye pHrodo-red. THP-1 cells were added after treatment of the pHrodo-labelled TTR with 2F2 and 4D4 or isotype control, respectively. The cellular uptake was visualised by bright field and fluorescence microscopy () and measured by flow cytometry analysis and displayed as mean fluorescence intensity (MFI, ). Low fluorescence was observed with either TTRwt monomer treated with isoD38-TTR-specific antibodies or the isotype controls. In contrast, fluorescence was significantly increased after treatment of ATTR-V30M fibrils with 2F2 or 4D4 antibody, indicating phagocytosis of TTR fibrils mediated by these antibodies. These data strongly suggest that the formation of isoD38 during ageing and deposition of TTR is a viable anchor point for therapeutic antibodies.
Figure 6. 2F2/4D4 mAb-dependent phagocytosis. Recombinant TTRwt proteins or extracted human ATTR-V30M fibrils were covalently labelled with the pH-sensitive fluorescent dye pHrodo Red, which shows enhanced fluorescence upon exposure to lowered pH in endocytic vesicles. (A) Overlays of bright field and fluorescence images of THP-1 cells reveal enhanced uptake of aggregated pHrodo-labelled ATTR-V30M fibrils in presence of isoD38-TTR-antibodies 2F2 and 4D4. Scale = 50 µm. (B) Engulfment of pHrodo-labelled fibrils was measured by relative mean fluorescence intensity (MFI) of THP-1 cells. Antibody-dependent phagocytosis was specific for pHrodo-ATTR-V30M-fibrils that were treated with 2F2 or 4D4 antibody, but both isotype control antibodies failed to induce phagocytosis. 2F2 and 4D4 had no effect on the uptake of native recombinant TTRwt. Data were represented by a boxplot with 1.5 IQR. Statistical analysis was done by One-way ANOVA, Tukey post-hoc test, *** p < 0.001.
Discussion
Transthyretin amyloidosis is a rare disease characterised by fibrillar TTR deposits in tissues, impacting organ function [Citation31]. The clinical picture of ATTRwt and ATTRv comes along with varying organ involvement and prognosis, leading to frequent misdiagnosis or underdiagnosis. Michalon et al. recently showed hidden ATTR involvement in 16% of heart failure cases [Citation15], highlighting its underdiagnosed nature [Citation32]. Diagnosing ATTR remains challenging, and current therapies offer limited improvement in patients’ quality of life. Thus, selective immunotherapeutics could enhance both diagnosis and treatment efficacy.
In this regard, PTMs of amyloid peptides might represent attractive anchor points for monoclonal antibodies. As prominent example, in the Aβ peptide contributing to amyloid pathology in AD, numerous PTMs have been described [Citation33]. Among these, the most prominent modification represents the pyroglutamate modification of the N-terminus [Citation34,Citation35]. Several studies have shown that a specific passive vaccination with antibodies against such modifications leads to reduction of brain amyloid load [Citation36,Citation37]. Just recently this was successfully shown for donanemab in clinical trial phase 3, which targets a pyroglutamate-modified Aβ (N3pG-Aβ). Moreover, we were able to show in a preclinical study a therapeutic effect with an isoD7-specific Aβ antibody [Citation38].
So far, there is a lack of data about the occurrence and proportion of isoD modifications in ATTR. However, Yang et al. have identified an isoD modification at position 38 of the transthyretin protein in plasma samples from individuals suffering from either mild cognitive impairment or AD [Citation39]. This could indicate a possible involvement of isoD in other amyloid diseases. Furthermore, D38 is located in a loop region and does not appear to be involved in any fixed rigid structure of the fibril, as seen from the human TTR fibril structure solved by Schmidt et al. [Citation7].
Based on these findings, we developed two monoclonal antibodies 2F2 and 4D4 that recognise an isoD modification at position 38 of TTR. Due to a lack of isoD within the native, physiological form of TTR, the developed antibodies were intended to specifically react with aged TTR protein as found in deposited fibrils.
Both antibodies show nanomolar affinities to the isoD38 peptide but differ considerably in their dissociation rate and thus in their general binding. The specificity to isoD38-containing TTR-species could be confirmed by dot blot analysis. Here it was shown that recombinant TTRwt, TTR-V30M and TTR-V122I did not contain any recognisable isoD38-containing residues. In contrast, when the same species were used in aggregation studies and aged for several months, isoD38 modifications were detected using the mAbs 2F2 and 4D4. These results were substantiated by immunogold labelling and TEM analysis of extracted ATTR-V30M fibrils from human cardiac tissue and immunohistochemical staining of cardiac tissue from patients with confirmed ATTRwt and ATTRv amyloidosis. All these analyses substantiate the presence of isoD amyloid deposits within pathological tissue and the specificity of the mAbs 2F2 and 4D4 to detect modified TTR leaving native TTR unattended.
In order to assess a potential therapeutic use of the antibodies in future, we utilised an in vitro phagocytosis assay based on extracted fibrils from ATTR deposits within the myocardium. Similar assays have been used previously, applying antibodies against other amyloidogenic proteins [Citation40–42]. It is noteworthy that despite the use of a human cell line with human Fcγ receptors, phagocytosis of the murine antibodies was ensured. Temming et al. have already shown that the murine subtypes IgG2a (4D4) and IgG3 (2F2) are recognised by the human FcγRIa with an affinity of 3.6 and 9.9 nM, respectively [Citation43]. From a clinical perspective, the potential success of 2F2 and 4D4 as therapeutics might hinge on their inability to bind with physiological TTR. This characteristic is crucial, as binding to native TTR, which is abundant in plasma, could negatively affect pharmacokinetics and hinder effective interaction with deposited ATTR in tissue (SI Figure 5). Hence, the results of this study imply that targeting of isoD38-TTR might generate a new tailored approach of immunotherapy for ATTR.
Current therapeutic approaches for treating ATTR amyloidosis aim to slow or prevent amyloid formation. TTR stabilisers like tafamidis and repurposed anti-inflammatory drug diflunisal, along with TTR gene silencers patisiran, vutrisiran, and inotersen, have significantly slowed or halted disease progression [Citation9–11, Citation44]. The isoD38-TTR mAbs 2F2 and 4D4 might complement these treatments by removing pre-existing amyloid, potentially offering functional improvements such as reducing cardiac muscle stiffness and enhancing heart contractility and elasticity.
Antibody-mediated amyloid removal is a crucial treatment approach for amyloidosis, currently in advanced clinical development with proof-of-mechanism in patients. In AD, antibodies like gantenerumab, aducanumab, and lecanemab have notably reduced Aβ plaque burden [Citation45–47]. Donanemab, another antibody, has passed phase 3 clinical trials successfully, targeting a pyroglutamate variant of Aβ unlike other mentioned antibodies [Citation17]. In Parkinson’s disease, advances have been seen with the α-synuclein aggregate-selective antibodies cinpanemab and PRX002/RG793 [Citation48,Citation49]. Treatment with dezamizumab, a humanised mAb that targets the plasma protein SAP which has been identified in all types of amyloid deposits, was associated with signs of organ response such as a decrease in liver stiffness, which was highly consistent with macrophage-dependent phagocytic clearance of amyloid demonstrated in animal models [Citation50].
Previously developed mAbs for the treatment of ATTR like PRX004 and NI006 are structure-selective antibodies whereas the focus of our studies relied on the detection of an age-related PTM. Reducing non-native TTR in pre-existing amyloid deposits, along with inhibiting TTR nucleation and targeting fibrils for phagocytic clearance, may block further TTR aggregation. Thus, isoD38-TTR-specific antibodies which target TTR fibrils rather than physiological TTR may play a crucial role in removing amyloid deposits and thereby potentially be the starting point for therapeutic developments.
| Abbreviations | ||
| Aβ | = | Amyloid-β |
| AD | = | Alzheimer’s disease |
| AL | = | light chain amyloidosis |
| ATTR | = | transthyretin amyloidosis |
| ATTRwt | = | wild-type transthyretin amyloidosis |
| ATTRv | = | hereditary transthyretin amyloidosis |
| BCIP | = | 5-Bromo-4-chloro-3-indolyl phosphate |
| BSA | = | bovine serum albumin |
| cryo-EM | = | cryogenic electron microscopy |
| DTT | = | dithiothreitol |
| EDC | = | N-ethyl-N′-(dimethylaminopropyl) carbodiimide hydrochloride |
| EDTA | = | ethylenediaminetetraacetic acid |
| E. coli | = | Escherichia coli |
| ELISA | = | enzyme-linked immunosorbent assay |
| FBS | = | fetal bovine serum |
| FFPE | = | formalin-fixed paraffin-embedded |
| HAT | = | hypoxanthine-aminopterin-thymidine |
| HBS-EP | = | HEPES-buffered saline with EDTA and polysorbate |
| HRP | = | horse-radish peroxidase |
| IgG | = | Immunoglobulin G |
| IPTG | = | isopropyl β-D-1-thiogalactopyranoside |
| isoAsp or isoD | = | isoaspartate |
| KLH | = | keyhole limpet hemocyanin |
| mAb | = | monoclonal antibody |
| NBT | = | nitro blue tetrazolium |
| NHS | = | N-hydroxysuccinimide |
| pAb | = | polyclonal antibody |
| PBS | = | phosphate-buffered saline |
| PCR | = | polymerase chain reaction |
| PEG | = | polyethylene glycol |
| PMA | = | phorbol 12-myristate 13-acetate |
| PTM | = | post-translational modification |
| RT | = | room temperature |
| siRNA | = | small interfering ribonucleic acid |
| SPR | = | surface plasmon resonance |
| TBS | = | Tris-buffered saline |
| TEM | = | transmission electron microscopy |
| TEV | = | tobacco etch virus |
| ThT | = | thioflavin T |
| TTR | = | transthyretin |
| TTRwt | = | wild-type transthyretin |
Supplemental Material
Download MS Word (1.4 MB)Acknowledgements
The authors thank Dr. Anke Piechotta, Dr. Kathrin Gnoth and Dr. Stefanie Geißler for their critical comments on the experiments. The authors further acknowledge the assistance of Victoria Gröger, Anja Künemund, and Edith Jansig for animal husbandry and antibody injection.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Nevone A, Merlini G, Nuvolone M. Treating protein misfolding diseases: therapeutic successes against systemic amyloidoses. Front Pharmacol. 2020;11:1024. doi:10.3389/fphar.2020.01024.
- Thomas VE, Smith J, Benson MD, et al. Amyloidosis: diagnosis and new therapies for a misunderstood and misdiagnosed disease. Neurodegener Dis Manag. 2019;9(6):289–299. doi:10.2217/nmt-2019-0020.
- Suhr OB, Lindqvist P, Olofsson BO, et al. Myocardial hypertrophy and function are related to age at onset in familial amyloidotic polyneuropathy. Amyloid. 2006;13(3):154–159. doi:10.1080/13506120600876849.
- Adams D, Ando Y, Beirão JM, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2021;268(6):2109–2122. doi:10.1007/s00415-019-09688-0.
- Buxbaum JN, Reixach N. Transthyretin: the servant of many masters. Cell Mol Life Sci. 2009;66(19):3095–3101. doi:10.1007/s00018-009-0109-0.
- Hurshman AR, White JT, Powers ET, et al. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry. 2004;43(23):7365–7381. doi:10.1021/bi049621l.
- Schmidt M, Wiese S, Adak V, et al. Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis. Nat Commun. 2019;10(1):5008. doi:10.1038/s41467-019-13038-z.
- Holmgren G, Steen L, Suhr O, et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet. 1993;341(8853):1113–1116. doi:10.1016/0140-6736(93)93127-m.
- Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. doi:10.1056/NEJMoa1716153.
- Adams D, Tournev IL, Taylor MS, et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid. 2023;30(1):1–9. doi:10.1080/13506129.2022.2091985.
- Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31. doi:10.1056/NEJMoa1716793.
- Gillmore JD, Maitland ML, Lebwohl D. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med. 2021;385(18):1722–1723. doi:10.1056/NEJMc2114592.
- Higaki JN, Chakrabartty A, Galant NJ, et al. Novel conformation-specific monoclonal antibodies against amyloidogenic forms of transthyretin. Amyloid. 2016;23(2):86–97. doi:10.3109/13506129.2016.1148025.
- Fontana M, Buchholtz K, Engelmann MDM, et al. NNC6019–0001, a humanized monoclonal antibody, in patients with transthyretin amyloid cardiomyopathy (ATTR-CM): rationale and study design of a phase 2, randomized, placebo-controlled trial. Eur Heart J. 2022;43(2):ehac544.1767. doi:10.1093/eurheartj/ehac544.1767.
- Michalon A, Hagenbuch A, Huy C, et al. A human antibody selective for transthyretin amyloid removes cardiac amyloid through phagocytic immune cells. Nat Commun, 12(1):3142. doi:10.1038/s41467-021-23274-x.
- Garcia-Pavia P, Aus Dem Siepen F, Donal E, et al. Phase 1 trial of antibody NI006 for depletion of cardiac transthyretin amyloid. N Engl J Med. 2023;389(3):239–250. doi:10.1056/NEJMoa2303765.
- Mintun MA, Wessels AM, Sims JR. Donanemab in early alzheimer’s disease. N Engl J Med. 2021;385(7):667–1704. doi:10.1056/NEJMoa2100708.
- Sims JR, Zimmer JA, Evans CD, et al. Donanemab in early symptomatic alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA. 2023;330(6):512–527. doi:10.1001/jama.2023.13239.
- Zhang Q, Kelly JW. Cys10 mixed disulfides make transthyretin more amyloidogenic under mildly acidic conditions. Biochemistry. 2003;42(29):8756–8761. doi:10.1021/bi030077a.
- Gales L, Saraiva MJ, Damas AM. Structural basis for the protective role of sulfite against transthyretin amyloid formation. Biochim Biophys Acta - Proteins Proteomics. 2007;1774(1):59–64. doi:10.1016/j.bbapap.2006.10.015.
- Roher AE, Kokjohn TA, Clarke SG, et al. APP/Aβ structural diversity and alzheimer’s disease pathogenesis. Neurochem Int. 2017;110:1–13. doi:10.1016/j.neuint.2017.08.007.
- Park J, Lai MKP, Arumugam TV, et al. O-GlcNAcylation as a therapeutic target for alzheimer’s disease. Neuromolecular Med. 2020;22(2):171–193. doi:10.1007/s12017-019-08584-0.
- Wang J, Mukherjee S, Zubarev RA. Isoaspartate and neurodegeneration. Aging (Albany NY). 2022;14(22):8882–8883. doi:10.18632/aging.204420.
- Shimizu T, Watanabe A, Ogawara M, et al. Isoaspartate formation and neurodegeneration in alzheimer’s disease. Arch Biochem Biophys. 2000;381(2):225–234. doi:10.1006/abbi.2000.1955.
- Robinson NE, Robinson AB. Molecular clocks. Proc Natl Acad Sci U S A. 2001;98(3):944–949. doi:10.1073/pnas.98.3.944.
- Aswad DW, Paranandi MV, Schurter BT. Isoaspartate in peptides and proteins: formation, significance, and analysis. J Pharm Biomed Anal. 2000;21(6):1129–1136. doi:10.1016/s0731-7085(99)00230-7.
- Geiger T, Clarke S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J Biol Chem. 1987;262(2):785–794. doi:10.1016/s0021-9258(19)75855-4.
- Matsubara K, Mizuguchi M, Kawano K. Expression of a synthetic gene encoding human transthyretin in Escherichia coli. Protein Expr Purif. 2003;30(1):55–61. doi:10.1016/s1046-5928(03)00069-x.
- Annamalai K, Gührs KH, Koehler R, et al. Polymorphism of amyloid fibrils in vivo. Angew Chemie - Int Ed. 2016;55(15):4822-4825. doi:10.1002/anie.201511524.
- Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–497. doi:10.1038/256495a0.
- Gonzalez-Duarte A, Ulloa-Aguirre A. A brief journey through protein misfolding in transthyretin amyloidosis (ATTR amyloidosis). Int J Mol Sci. 2021;22(23):13158. doi:10.3390/ijms222313158.
- Rozenbaum MH, Large S, Bhambri R, et al. Impact of delayed diagnosis and misdiagnosis for patients with transthyretin amyloid cardiomyopathy (ATTR-CM): a targeted literature review. Cardiol Ther. 2021;10(1):141–159. doi:10.1007/s40119-021-00219-5.
- Kummer MP, Heneka MT. Truncated and modified amyloid-beta species. Alzheimer’s Res. Ther. 2014;6(3):28. doi:10.1186/alzrt258.
- Bayer TA. Pyroglutamate Aβ Cascade as drug target in alzheimer’s disease. Mol Psychiatry. 2021;27(4):1880–1885. doi:10.1038/s41380-021-01409-2.
- Schilling S, Zeitschel U, Hoffmann T, et al. Glutaminyl cyclase inhibition attenuates pyroglutamate abeta and alzheimer’s disease-like pathology. Nat Med. 2008;14(10):1106–1111. doi:10.1038/nm.1872.
- Frost JL, Liu B, Kleinschmidt M, et al. Passive immunization against pyroglutamate-3 amyloid-β reduces plaque burden in alzheimer-like transgenic mice: a pilot study. Neurodegener Dis. 2012;10(1-4):265–270. doi:10.1159/000335913.
- Bakrania P, Hall G, Bouter Y, et al. Discovery of a novel pseudo β-hairpin structure of N-truncated amyloid-β for use as a vaccine against alzheimer’s disease. Mol Psychiatry. 2021;27(2):840–848. doi:10.1038/s41380-021-01385-7.
- Gnoth K, Piechotta A, Kleinschmidt M, et al. Targeting isoaspartate-modified Aβ rescues behavioral deficits in transgenic mice with Alzheimer’s disease-like pathology. Alzheimer’s Res Ther. 2020;12(1):149. doi:10.1186/s13195-020-00719-x.
- Yang H, Lyutvinskiy Y, Soininen H, et al. Alzheimer’s disease and mild cognitive impairment are associated with elevated levels of isoaspartyl residues in blood plasma proteins. J Alzheimer’s Dis. 2011;27(1):113–118. doi:10.3233/JAD-2011-110626.
- Wall JS, Kennel SJ, Williams A, et al. AL amyloid imaging and therapy with a monoclonal antibody to a cryptic epitope on amyloid fibrils. PLoS One. 2012;7(12):e52686. doi:10.1371/journal.pone.0052686.
- Hrncic R, Wall J, Wolfenbarger DA, et al. Antibody-mediated resolution of light chain-associated amyloid deposits. Am J Pathol. 2000;157(4):1239–1246. doi:10.1016/S0002-9440(10)64639-1.
- Hallé M, Tribout-Jover P, Lanteigne AM, et al. Methods to monitor monocytes-mediated amyloid-beta uptake and phagocytosis in the context of adjuvanted immunotherapies. J Immunol Methods. 2015;424:64–79. doi:10.1016/j.jim.2015.05.002.
- Temming AR, Bentlage AEH, de Taeye SW, et al. Cross-reactivity of mouse IgG subclasses to human Fc gamma receptors: antibody deglycosylation only eliminates IgG2b binding. Mol Immunol. 2020;127:79–86. doi:10.1016/j.molimm.2020.08.015.
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016. doi:10.1056/NEJMoa1805689.
- Bateman RJ, Cummings J, Schobel S, et al. Gantenerumab: an anti-amyloid monoclonal antibody with potential disease-modifying effects in early Alzheimer’s disease. Alzheimer’s Res. Ther. 2022;14(1):178. doi:10.1186/s13195-022-01110-8.
- Budd Haeberlein S, Aisen PS, Barkhof F, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J Prev Alzheimer’s Dis. 2022;9(2):197–210. doi:10.14283/jpad.2022.30.
- Van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early alzheimer’s disease. N Engl J Med. 2023;388(1):9–21. doi:10.1056/nejmc2301380.
- Lang AE, Siderowf AD, Macklin EA, et al. Trial of cinpanemab in early parkinson’s disease. N Engl J Med. 2022;387(5):408–420. doi:10.1056/NEJMoa2203395.
- Jankovic J, Goodman I, Safirstein B, et al. Results from a phase 1b multiple ascending-dose study of PRX002/RG7935, an anti-alpha-synuclein monoclonal antibody, in patients with Parkinson’s disease. Parkinsonism Relat Disord. 2018;46:e25. doi:10.1016/j.parkreldis.2017.11.083.
- Richards D, Millns H, Cookson L, et al. An observational, non-interventional study for the follow-up of patients with amyloidosis who received miridesap followed by dezamizumab in a phase 1 study. Orphanet J Rare Dis. 2022;17(1):259. doi:10.1186/s13023-022-02405-7.