
ABSTRACT
Background
Acute lung injury (ALI) is a severe and often fatal pulmonary disease. Current treatments for ALI and acute respiratory distress syndrome (ARDS) are limited. Natural product metabolites have shown promise as therapeutic alternatives. However, the effects of Licochalcone B (LCB) on ALI are largely unknown.
Methods
We investigated the effects of LCB on lipopolysaccharide-challenged mice and human pulmonary microvascular endothelial cells. Cell viability, apoptosis, and ROS production were assessed. Lung tissue histopathology and oxidative stress and inflammation markers were evaluated. Protein expression levels were measured.
Results
LCB had no cytotoxic effects on cells and increased cell viability. It reduced apoptosis and ROS levels in cells. In mice with ALI, LCB decreased lung tissue weight and improved oxidative stress and inflammation markers. It also enhanced expression levels of Nrf2, HO-1, and NQO1 while reducing Keap1.
Conclusion
LCB protects against LPS-induced acute lung injury in cells and mice. The Keap1/Nrf2 pathway may be involved in its protective effects. LCB shows potential as a strategy to alleviate ALI caused by LPS.
1. Introduction
Acute lung injury (ALI) and its most severe form, acute respiratory distress syndrome (ARDS), are serious pulmonary diseases typically caused by factors such as infection, trauma, smoke inhalation, and chemical poisoning. These conditions are significant causes of mortality in critically ill patients who have undergone multiple blood transfusions, experienced shock, sepsis, or ischemia-reperfusion [Citation1,Citation2]. Currently, there are no effective treatments for ALI and ARDS. Even with the best supportive care and lung-protective ventilation strategies, the 60-day mortality rate remains around 25% [Citation3,Citation4]. In ALI, persistent and uncontrolled inflammation results in increased permeability of pulmonary blood vessels and alveoli, infiltration of polymorphonuclear neutrophils, and subsequent pulmonary edema and lung tissue damage, which in turn impairs respiratory function [Citation5]. Oxidative stress and cell apoptosis have been implicated in the pathogenesis of ALI. Clinical observations have found that Gram-negative bacterial infections play a crucial role in the development and progression of Acute Lung Injury (ALI). Lipopolysaccharide (LPS) is an endotoxin found in the outer membrane of Gram-negative bacteria, which can stimulate alveolar epithelial cells, alveolar cells, airway epithelial cells, and alveolar immune cells to produce large amounts of Reactive Oxygen Species (ROS), leading to acute inflammatory responses [Citation6].
The Keap1/Nrf2 signaling pathway, a renowned protective mechanism and a vital antioxidative pathway, contributes to cellular safeguarding against the detrimental effects of oxidative stress by maintaining balance between the oxidizing and antioxidizing systems [Citation7]. Under physiological conditions, Keap1 acts as an inhibitor of Nrf2. In the presence of various activating factors, such as electrophilic compounds, hydrogen peroxide, and ultraviolet (UV) irradiation, among others, Keap1 releases its inhibitory hold on Nrf2 [Citation8]. Consequently, activated Nrf2 mediates diverse enzyme classes in shielding against oxidative stress. Notably, phase II detoxification enzymes, encompassing NAD(P)H:quinone oxidase (NQO-1) and heme oxygenase-1 (HO-1), are instrumental in defending cells from oxidative damage [Citation9].
The lung protection due to the supression of Keap1 and up-regulation of Nrf2 and its downstream detoxification enzymes has been confirmed in many experimental models of ALI and sepsis [Citation10–12]. Thus, suppression of the inflammatory responses and/or induction of Nrf2 may be a potential strategy to alleviate the progression of ALI caused by LPS.
Licochalcone B (LCB) is a chalcone compound derived from the licorice genus (Leguminosae) and can be found in the roots and stems of the Glycyrrhiza inflate. Previous investigations have explored LCB's anti-inflammatory, antioxidative, and antitumor properties, finding that LCB exerts anti-inflammatory effects by inhibiting the phosphorylation of NF-κB p65, disrupting NEK7-NLRP3 interactions, and activating the Nrf2 pathway, among other mechanisms [Citation13–15]. Nevertheless, its impact on ALI remains largely unexplored. In this study, the effects and underlying mechanisms of LCB on lipopolysaccharide-challenged mice and human pulmonary microvascular endothelial cells (HPMECs) were examined.
2. Materials and methods
2.1 Reagents
LCB (purity ≥ 98%) and Dexamethasone (DEX, purity > 99.6%) were purchased from Sichuan Victory Biological Technology Co., Ltd. LPS was purchased from Sinopharm Chemical Reagent Co., Ltd. SOD (E-BC-K020-M), MPO (E-BC-K074-M) and NO (E-BC-K035-M) assay kit were purchased from Elabscience. MDA (A003-1-1), GSH-Px (A005-1-2), CAT (A007-1-1) were obtained from Njjcbio. TNF-α ELISA Kit (E-EL-M3063), IL-6 ELISA Kit (E-EL-M0044c) and IL-1β ELISA Kit (E-EL-M0037c) were purchased from Elabscience. Antibodies against the following targets were took from Proteintech: Nrf2 (16396-1-AP), NQO1 (11451-1-AP), HO-1 (1070-1-AP) and KEAP1 (10503-1-AP). GAPDH (AF7021) was obtained from Affinity. Horseradish peroxidase (HRP)-labeled goat anti-rabbit IgG secondary antibody (70-GAR0072) was obtained from Multi sciences. Cell Counting Kit-8 (C0038), Calcein/PI Cell Viability/Cytotoxicity assay kit (C2015M), Reactive Oxygen Species assay kit (S0033S), Annexin V-FITC apoptosis detection kit (C1062S) were purchased from Beyotime Biotechnology Co., Ltd.
2.2 Cell culture and treatment
The Human Pulmonary Microvascular Endothelial Cell (HPMEC) line was purchased from the Shanghai Institute of Biochemistry and Cell Biology. HPMECs were cultured at 37 °C. All cells were cultured in the endothelial cell medium (ECM) with 2% fetal bovine serum (FBS) for nutritional support and 1% penicillin-streptomycin for antibacterial effects in vitro.
2.3 Measurement of cell viability
Cells were plated at a density of 1 × 10^4 cells/ml onto 96-well plates for 24 h. Cell viability was assessed using the cell counting kit-8 (CCK-8) assay at 24 and 48 h post-treatment. To minimize experimental errors, each concentration group was repeated six times. The optical density was measured at a wavelength of 450 nm using a microplate reader (Thermo Fisher Scientific, USA).
2.4 Reactive oxygen species (ROS) measurements
To obtain further evidence for the protective effect of LCB against LPS induced oxidative stress, alterations of intracellular ROS levels were determined. The production of intracellular ROS was measured using 2’,7'-dichlorofluorescin diacetate (DCFH-DA; Invitrogen, Carlsbad, CA, USA) by fluorescence microscope. Briefly, HPMECs were treated with LPS 1 µM alone for 48 h or LCB (400 ng/mL) for 1 h first and then co-treated with LPS 1 µM for 48 h. After harvest of HPMECs, the cells were incubated with DCFH-DA solution (10 µM) in the dark at 37˚C for 30 min, washed with PBS (pH 7.4), and analyzed within 30 min using a fluorescence microscope (The excitation wavelength was 488 nm, and the emission wavelength was 525 nm). For the in vivo experiment section, portions of the collected lung tissue were prepared into cell suspensions, and the subsequent analyzes were conducted using the same methods.
2.5 Calcein-AM/PI staining
Following a 48-hour seeding period, HPMEC cells were harvested, rinsed with phosphate-buffered saline (PBS), and resuspended in a medium supplemented with 2% fetal bovine serum (FBS) to attain a density of 1 × 105 cells/mL. The cells were then cultivated in a 24-well plate at 200 µL/well for an additional 24 h. After the incubation with LPS/LPS + LCB, cellular fluorescence was assessed using calcein-AM/PI staining as per the manufacturer's guidelines. Each sample was enriched with 0.5 µL (1 µM) of calcein-AM and 0.5 µL (1 µM) of PI, followed by a 30-minute incubation in darkness at 37°C. Subsequently, cells were inspected under a fluorescence microscope, employing sterile water as the control.
2.6 Measurement of apoptosis
The Annexin V-FITC/PI apoptosis detection kit was employed to quantitatively determine the apoptosis rate of HPMECs. After 48 h of seeding, HPMECs were harvested, washed with PBS, and resuspended in ECM medium. Following 48 h of treatment with LPS/LPS + LCB, the procedure was conducted according to the provided instructions. After the cells were resuspended, 195 µL Annexin V-FITC conjugate solution and 5 µL (2.5 µg/mL) of Annexin-V-FITC and 10 µL (50 µg/mL) of PI in the cell suspension were added to each sample, followed by a 15-minute incubation in darkness at room temperature (25°C). Cells were then analyzed using a flow cytometer (BD FACSCanto™ II) with sterile water serving as the control.
2.7 Animals and treatment
Specific pathogen-free (SPF) grade C57BL/6 male mice, aged 6–8 weeks and weighing 20–22 g, were acquired from Gempharmatech Co., Ltd. (Certificate No. SCXK2020-034). Mice were housed under a laboratory temperature of 22 ± 1°C and relative humidity of 55 ± 5%, with a 12-hour light/dark cycle, and provided food and water ad libitum. All animal procedures were approved by the Animal Care and Use Committee of Chengdu University of Traditional Chinese Medicine(2023-04)and conducted in accordance with the National Institutes of Health (NIH) Guidelines for the Care and Use of Laboratory Animals. Following a 7-day acclimatization period, mice were randomly allocated to six groups (eight per group): (I) Normal saline (Control group), (II) LPS (5 mg/kg) group, (III) Dexamethasone (2 mg/kg) + LPS (DEX group), (IV) LCB 10 mg/kg + LPS (LCB-L group), (V) LCB 20 mg/kg + LPS (LCB-M group), and (VI) LCB 40 mg/kg + LPS (LCB-H group). Intraperitoneal (i.p.) injections of LCB were administered, with LCB diluted in DMSO to various concentrations [Citation14,Citation16]. In the Control and LPS groups, mice received 0.1% DMSO-containing normal saline via i.p. once daily for seven consecutive days. In the DEX, LCB-L, LCB-M, and LCB-H groups, mice received dexamethasone or different LCB doses via i.p. The LPS-induced lung injury mouse model was established following previously published protocols. Successful construction of the acute lung injury (ALI) mouse model was confirmed upon observing symptoms such as dull coat color, diminished mental state, reduced food intake, rapid breathing, cyanosis around the nasal and lip areas, and sluggish movement in the mice [Citation17]. Excluding the Control group, mice in other groups underwent non-invasive tracheal instillation of LPS to induce lung injury, 120 min after the final prophylactic administration. Six hours post-LPS treatment, mice were anesthetized with 1% pentobarbital, and humanely euthanized. Lung tissues and bronchoalveolar lavage fluid were subsequently collected.
2.8 Lung wet-to-dry weight ratio
Following the mice's euthanasia, a section of each specimen's right lung was promptly excised and any surface moisture was absorbed using blotting paper to determine its wet weight. Subsequently, the tissue was desiccated in a drying oven at 80°C for 24 h, and its dry weight was ascertained. The wet-to-dry (W/D) weight ratio was employed to evaluate the extent of pulmonary edema.
2.9 Bronchoalveolar lavage
Following 6 h post-final LPS exposure, the mouse's left lung was lavaged three times with 0.5 ml of pre-chilled sterile PBS for 3 min each, achieving a recovery rate of over 80%. After combining the lavages, the bronchoalveolar lavage fluid (BALF) was centrifuged at 4°C for 10 min at 12,000 rpm. The collected BALF was stored at – 80°C for subsequent measurement of oxidative stress markers.
2.10 Histological study
Immediately after euthanizing the rats, lung tissue was collected for histopathological examination. The lung specimens were fixed with 4% paraformaldehyde at room temperature for 24 h. Following deparaffinization and dehydration, the lung tissue was sectioned into 5 mm slices. These lung sections were stained with hematoxylin and eosin (H&E) to assess histopathological alterations. The pathological features of the lung slices were evaluated from various aspects, including the infiltration or aggregation of neutrophils within the airspace or vascular walls, alveolar congestion, hemorrhaging, and the formation of thickened alveolar walls or hyaline membranes. Each criterion was assessed on a five-tiered scale of damage, ranging from 0 (minimal damage) to 4 (maximal damage). Two experienced pathologists, working independently and blinded to each other's assessments, graded the pathological characteristics of the lung slices based on the aforementioned index.
2.11 Determination of oxidative stress and inflammatory factors
During the oxidative stress process, antioxidant enzymes, such as catalase (CAT), superoxide dismutase (SOD), and glutathione peroxidase (GSH-Px), provide a protective function in maintaining cellular oxidative balance. In contrast, myeloperoxidase (MPO) and excessive nitric oxide (NO) may exacerbate oxidative damage. Conversely, malondialdehyde (MDA) serves as a biomarker for monitoring the extent of cellular oxidative damage and oxidative stress levels. Maintaining a balance among these biomolecules is crucial in regulating the oxidative stress process. In the process of ALI, interleukin-6 (IL-6), interleukin-1β (IL-1β), and tumor necrosis factor-alpha (TNF-α) serve as crucial inflammatory biomarkers. These cytokines play a significant role in mediating inflammation and tissue damage, contributing to the pathophysiology of ALI. Consequently, we employed corresponding commercial assay kits and followed the manufacturer’s instructions to assess the levels of these biomolecules in BALF.
2.12 Western blotting assay
The lung tissue samples were harvested, immediately frozen in liquid nitrogen for at least 12 h. Take out appropriate quantity of lung tissue and add radio-immunoprecipitation assay (RIPA) Buffer, cocktail, phenylmethylsulphonyl fluoride (PMSF) and protein phosphatase inhibitor respectively. After mechanically grinding the tissue, the protein concentration was determined according to the instructions of the BCA protein assay kit. The protein samples were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDSPAGE). Subsequently, proteins were transferred to polyvinylidene difluoride (PVDF) membranes and incubated with 5% skim milk for 60 min and then incubated overnight at 4°C with primary antibodies including Nrf2, HO-1,NQO1 and KEAP1. The membrane was washed 3 times, and then incubated with the secondary antibody for 1 h at room temperature. After incubation, the membrane was put in an electrochemiluminescence (ECL) colors solution and developed with a gel imager.
2.13 Immunohistochemistry
Lung specimens were fixed, embedded in paraffin, and sectioned at 4 μm thickness. Following deparaffinization and rehydration, sections underwent antigen retrieval via sodium citrate buffer for 5 min at 100°C. Post-incubation with 5% normal goat serum for 20 min, the sections were exposed to a polyclonal nitrotyrosine primary antibody overnight at 4°C, succeeded by the application of a goat anti-rabbit immunoglobulin horseradish peroxidase-conjugated secondary antibody for 1 h at ambient temperature. Nitrotyrosine presence was denoted by the brown peroxidase reaction product and captured using light micrographs.
2.14 Statistical analysis
Data are presented as mean ± SD and assessed using SPSS software (Version 26.0, Chicago, USA). One-way analysis of variance facilitated multiple comparisons, while non-normally distributed data were analyzed via the Kruskal-Wallis one-way analysis of variance. A P-value < 0.05 was deemed statistically significant.
3. Results
3.1 Effects of LCB on cell viability, apoptosis and ROS production
Following 24-hour and 48-hour incubation periods, the potential cytotoxicity of LCB was assessed using the CCK8 assay. After 24 h of treatment with LCB at concentrations of 0.2-0.7 μM, cell viability exhibited a significant increase, and after 48 h of treatment, the LCB concentration range that increased cell viability expanded to 0.1-0.7 μM. This indicates that LCB did not exhibit cytotoxic effects on HPMECs within this range. Moreover, cell viability reached its peak at 400 nM, and thus, LCB concentrations of 400 nM were selected for subsequent experiments (A).
Figure 1. Effects of LCB on cell viability, apoptosis and ROS production. (A) Effect of LCB on the viability of HPMECs. (B) Effect of 1 μg/mL LPS + LCB on the viability of HPMECs. (C) Live/dead cell ratio measured with Calcein-AM/PI staining at 24 and 48 h. (D) Apoptosis rate measured with Annexin V-FITC/PI staining. (E) ROS production evaluated by DCFH-DA. Data are expressed as means ± SD and analyzed by one-way ANOVA. HMPECs = 2 batches of cells, repeat 3 multiple wells for each bacth of cells. * P < 0.05, ** P < 0.01, *** P < 0.001 vs control group. + P < 0.05, ++ P < 0.01, +++ P < 0.001 vs LPS group.
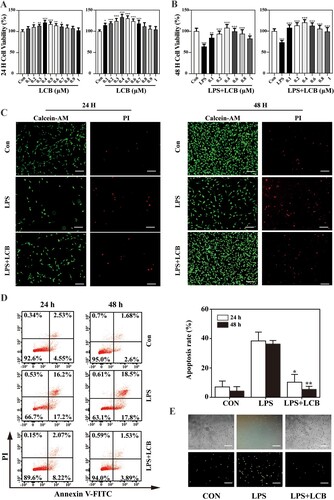
B shows that the cell viability of HPMECs induced by 1 μg/mL LPS was decreased significantly compared to the control group. Treatment with LCB and the addition of LPS resulted in dose-dependent increases in cell viability compared to the cells treated with 1 μg/mL LPS alone.
Subsequently, we analyzed the live/dead cell ratio using Calcein-AM/PI staining (C). Following a 24-hour treatment, the control group exhibited a relatively low proportion of red cells, while the LPS group displayed a red cell ratio of approximately 62%. In contrast, the LCB group presented a significantly lower proportion of red fluorescent cells compared to the LPS group. This observation suggests that a 24-hour LCB treatment initially exhibited a restorative effect on HPMECs under oxidative damage. After a 48-hour treatment, the number of green cells in the LCB group markedly increased, while the LPS group sustained a higher proportion of red cells relative to the control group. Consequently, the results of the Calcein-AM/PI staining analysis were consistent with the findings obtained from the CCK-8 assay.
To evaluate the effect of LCB on apoptosis, flow cytometry analysis was conducted using Annexin V-FITC/PI staining. The apoptosis rates in the control group at 24 and 48 h were 7.08% and 4.28%, respectively. In the LPS group, the apoptosis rates rose significantly to 41.4% at 24 h and 36.3% at 48 h. The LCB group exhibited apoptosis rates of 10.29% and 5.42% at 24 and 48 h respectively (D). These results indicate that LCB can reduce LPS-induced apoptosis in HPMECs.
The impact of LCB on ROS production was evaluated by staining cells with the ROS indicator DCFH-DA. When LCB was incubated with oxidatively damaged HPMECs for 24 h, the green fluorescence intensity was noticeably reduced, indicating that LCB addition can decrease ROS levels. In the LPS group, the green fluorescence intensity did not decline even after 48 h, suggesting that it is challenging for oxidatively damaged cells to autonomously restore normal activity. Therefore, LCB can repair HPMECs under oxidative damage. (E).
3.2 Effects of LCB on LPS-induced dysfunction, oxidative stress and inflammation in mice with ALI
LPS instillation induced severe histopathological alterations in the lungs, characterized by increased alveolar wall thickness, leukocyte infiltration, and pulmonary edema. Treatment with dexamethasone notably ameliorated these pathological manifestations (A). The W/D weight ratio of lung tissue serves as an indicator of pulmonary edema severity. LPS-induced elevation of the W/D weight ratio was significantly mitigated by dexamethasone. LCB also reduced the W/D weight ratio in a dose-dependent manner (B). Consequently, LCB can alleviate LPS-induced pulmonary edema in mice. The activation of oxidative stress is recognized as a pivotal event in LPS-induced ALI [Citation18]. Consequently, we evaluated the levels of reactive oxygen species (ROS) in lung tissues, and the results demonstrated that LPS instillation resulted in ROS accumulation (C). Oxidative stress markers in bronchoalveolar lavage fluid (BALF) were presented in A – F, which revealed a depletion of superoxide dismutase (SOD) and increased levels of myeloperoxidase (MPO), catalase (CAT), glutathione peroxidase (GSH-Px), nitric oxide (NO), and malondialdehyde (MDA). Following LCB treatment, the alterations in oxidative stress markers were mitigated. Nonetheless, no significant difference was observed between the DEX group and LCB-H group concerning ROS levels. Consequently, LCB can ameliorate the levels of LPS-induced oxidative stress markers.
Figure 2. Effects of LCB on LPS-induced dysfunction and ROS in mice with ALI. (A) H&E stanning of pulmonary sections. (B) A semi-quantitative histopathological score of lung injury and W/D weight ratio of lung tissues. (C) ROS levels of lung tissues analyzed by a flow cytometer. LCB-L: LCB 10 mg/kg + LPS; LCB-M: LCB 20 mg/kg + LPS; LCB-L: LCB 40 mg/kg + LPS. Data are presented as means ± SD for eight replicates and analyzed using a one-way ANOVA. * P < 0.05, ** P < 0.01, *** P < 0.001 vs control group. + P < 0.05, ++ P < 0.01, +++ P < 0.001 vs LPS group.
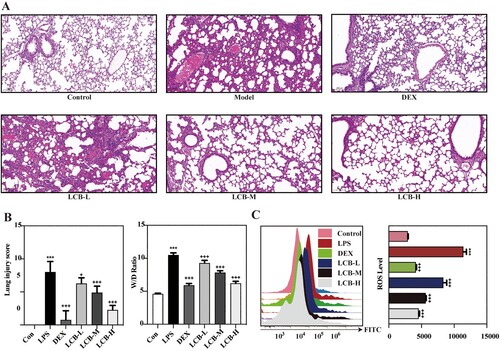
Figure 3. Effects of LCB on LPS-induced oxidative stress markers and inflammatory factors in mice with ALI. (A) Myeloperoxidase (MPO). (B) Catalase (CAT). (C) Superoxide dismutase. (D) Glutathione peroxidase (GSH-Px). (E) Nitric oxide (NO). (F) Malondialdehyde (MDA). (G) Interleukin-6 (IL-6). (H) Interleukin-1β (IL-1β). (I) Tumor necrosis factor-alpha (TNF-α). Data are presented as means ± SD for eight replicates and analyzed using a one-way ANOVA. * P < 0.05, ** P < 0.01, *** P < 0.001 vs control group. + P < 0.05, ++ P < 0.01, +++ P < 0.001 vs LPS group.
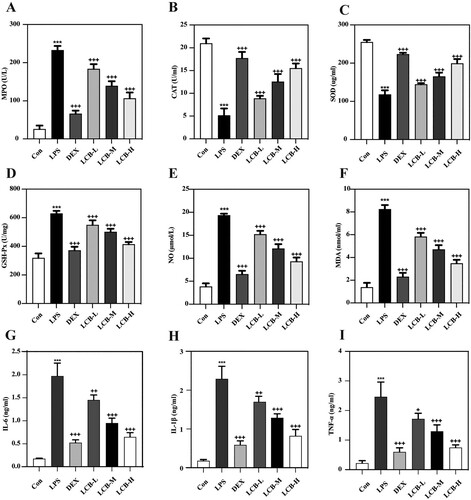
In BALF, the pro-inflammatory cytokines IL-6 (G), IL-1β (H), and TNF-α (I) exhibited a significant increase following LPS induction. Treatment with LCB reduced these inflammatory markers in a concentration-dependent manner. Moreover, the inhibitory effects of high-dose LCB on these cytokines show no significant difference when compared with DEX. Collectively, the data above indicated that LCB possessed an effective anti-inflammatory role in LPS-induced ALI.
3.3 Effects of LCB on keap1/nrf2 signaling pathways
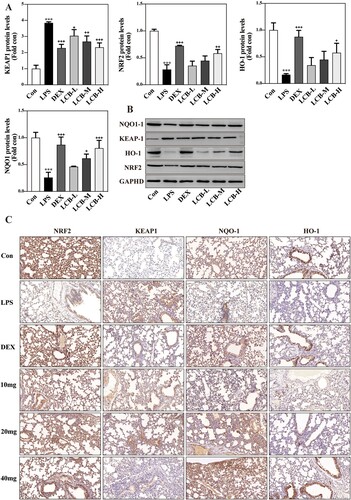
To investigate the effect of LCB on the Keap1/Nrf2 signaling pathway in LPS-induced lung injury, the expression levels of Nrf2, Keap1, HO-1, and NQO1 were analyzed using western blot and immunohistochemistry. As illustrated in A,B, the expression of Nrf2, NQO1, and HO-1 decreased significantly, while the expression of Keap1 increased after LPS instillation. LCB exhibited a concentration-dependent enhancement of Nrf2, NQO1, and HO-1 expression, and a reduction in Keap1 expression. Furthermore, there was no significant difference between the LCB-H group and the dexamethasone group in the expression of all proteins. Subsequently, we performed immunohistochemical analysis for the aforementioned four proteins, and the results were consistent with those obtained from the western blotting (C). These findings suggest that LCB may exert its protective effects against LPS-induced lung injury through the Keap1/Nrf2 signaling pathway.
Figure 4. Effects of LCB on Keap1/Nrf2/HO-1 signaling pathways. (A) The expression of KEAP1, NRF2, HO-1 and NQO1 was measured by western blotting in the lung tissues. (B) Immunohistochemical staining of KEAP1, NRF2, HO-1 and NQO1 in the lung tissues. Data are presented as means ± SD for eight replicates and analyzed using a one-way ANOVA. * P < 0.05, ** P < 0.01, *** P < 0.001 vs control group. + P < 0.05, ++ P < 0.01, +++ P < 0.001 vs LPS group.
4. Discussion
Although great progress has been made in understanding the pathogenesis of ALI, it still is a heavy burden for the public due to its high morbidity and mortality in critically ill patients due to limited treatments in clinical practice such as supportive treatment, nutritional support, mechanical ventilation, etiological treatment and comprehensive treatment to maintain fluid, electrolyte, acid and alkali balance [Citation19]. Therefore, it is of great value and significance to find a new drug or treatment for ALI. In recent years, more and more researchers have applied complementary and alternative medicine in various refractory diseases, and traditional Chinese medicine originating from herbal medicines has also been excavated many bioactive components that play a certain role in ALI. Among them, licorice has been used in the Asia-Pacific region for nearly two thousand years. Traditional Chinese medicine theory believes that it has the effect of “supplementing qi, detoxifying and clearing heat”, and believes that such an effect is in line with the pathogenesis of ALI. As a bioactive ingredient in licorice, LCB has been confirmed to have great values in various human such as cancer [Citation20–22], stroke [Citation14], and neurodegenerative disease [Citation23]. However, whether LCB has therapeutic potential for ALI is unclear.
This study elucidated the protective effect of LCB on LPS-induced ALI both in vitro and in vivo. Owing to the distinct metabolic processes, varying concentrations of LCB were employed in the in vivo and in vitro experiments. We treated HMPECs with different concentration gradients of LCB and found that LCB had no apparent cytotoxicity. On the contrary, after 24 h of treatment, LCB at various concentrations could improve cell viability to a certain extent. LPS-treated HMPECs showed a significant decrease in activity, accompanied by ROS production and apoptosis. LCB pretreatment significantly reversed this process, demonstrating in vitro that LCB is related to alleviating oxidative stress and LPS-induced cell death.
To further verify the antioxidant and reparative effects of LCB on LPS-induced lung injury in vivo, we established an LPS-induced lung injury model and evaluated it from multiple perspectives. During the ALI process, the overactivation of alveolar macrophages and infiltration of neutrophils lead to the excessive release of oxidative stress products. For example, NO will be overproduced under the induction of iNOS, directly or indirectly damaging alveolar epithelium and microvascular epithelium, leading to increased permeability. This further exacerbates the activation of macrophages and neutrophil infiltration, forming an uncontrolled and self-amplifying pulmonary inflammation.
Pulmonary edema is a characteristic manifestation of localized inflammation and injury in the lungs [Citation24]. Therefore, we utilized the wet-to-dry (W/D) ratio to quantify pulmonary edema, and found that LCB significantly reduced the elevation of the W/D ratio induced by LPS, suggesting that LCB has a protective effect against LPS-induced lung injury. This finding was validated at the pathological level, as LCB significantly ameliorated the typical pathological alterations caused by LPS-induced lung injury. Subsequently, to explore the potential protective mechanism of LCB against LPS-induced lung injury, we measured various antioxidants and peroxidation products involved in oxidative stress processes.
Superoxide dismutase (SOD) is a well-established antioxidant enzyme responsible for converting superoxide anions (O2·-) into hydrogen peroxide (H2O2), thereby alleviating cellular damage caused by oxygen free radicals [Citation25]. Glutathione peroxidase (GSH-Px) is an antioxidant enzyme that catalyzes the conversion of reduced glutathione (GSH) to oxidized glutathione (GSSG). Both catalase (CAT) and glutathione peroxidases (GPxs) reduce toxic H2O2 to water and oxygen, preventing the formation of highly toxic hydroxyl free radicals and protecting cells from oxidative damage [Citation26]. Malondialdehyde (MDA) is a lipid peroxidation product commonly utilized as a biomarker for evaluating oxidative stress and lipid peroxidation damage. MDA formation can induce cross-linking and aggregation of crucial macromolecules, such as proteins and nucleic acids, exerting cytotoxic effects [Citation27]. Nitric oxide (NO) is a biologically active molecule involved in various physiological processes, including vasodilation, neurotransmitter release, and immune regulation. However, excessive NO production can react with other free radicals to generate highly reactive peroxynitrite (ONOO-), leading to cell damage and oxidative stress [Citation28,Citation29]. Myeloperoxidase (MPO) is a peroxidase enzyme found in leukocytes that facilitates reactions between chloride, hydrogen peroxide, and other substrates, producing highly oxidative chlorinated free radicals (such as hypochlorous acid). Although MPO plays a crucial role in immune defense, the reactive oxygen species it generates can induce oxidative stress and tissue damage [Citation30]. LCB exhibited antioxidative and protective effects in LPS-challenged mice, as evidenced by ameliorated LPS-induced histological changes in lung tissues, increased SOD activity, reduced ROS, MPO, CAT, GSH-Px, NO, and MDA levels following LCB pretreatment.
Continuous oxidative stress has been found to contribute to inflammation and is associated with LPS-induced ALI. LPS can trigger inflammatory cell infiltration and the generation of inflammatory mediators, including TNF-α, IL-1β, IL-6, INOS, COX-2, and later HMGB-1, all of which play crucial roles in inflammation-related diseases [Citation31]. LCB has been reported to significantly inhibit LPS-induced phosphorylation at serine 276 and transcriptional activation of NF-κB. Moreover, LCB effectively inhibited LPS-induced activation of PKA, which is necessary for the phosphorylation of NF-κB p65 at serine 276. As a result, licochalcone B significantly reduced LPS-induced production of NO, TNFα, and MCP-1 [Citation13]. In this study, we examined the expression levels of TNF-α, IL-1β, and IL-6 in BALF and discovered that they were suppressed by LCB in LPS-induced ALI, highlighting the anti-inflammatory effect of LCB. Additionally, pretreatment with LCB substantially ameliorated LPS-induced histological alterations in lung tissues and decreased inflammatory cell infiltration.
Activation of NF-κB, MAPK, and PI3K-AKT pathways by LPS can lead to the production of iNOS and pro-inflammatory cytokine [Citation32]. These pathways play a key role in regulating Nrf2-dependent transcription [Citation33]. Therefore, to further understand the mechanism of LCB’s anti-oxidative properties, Keap1/Nrf2 signaling pathway, an important anti-oxidative pathway that regulates the balance between the oxidation system and anti-oxidation system, was investigated after LCB treatment.
Under the normal physiological state, Nrf2 and Keap1 are located in the cytoplasm. However, when oxidative stress is activated, protein kinases will be activated to induce the phosphorylation of Nrf2, which further disassociates with Keap1 and is transferred into the nucleus . The entered Nrf2 combines with the antioxidant reaction element (ARE) to initiate the transcription of anti-oxidation proteins, such as heme oxygenase-1 (HO-1) and NQO1 [Citation34,Citation35]. Previous studies have reported that in various models of ALI, the Keap1/Nrf2 signaling pathway is inhibited [Citation36–38]. Similarly, our study found that LPS induced an increase in Keap1 and a downregulation of its downstream targets Nrf2, HO-1, and NQO1, explaining the decline in antioxidative capacity caused by LPS. The downregulation of inflammatory markers induced by LCB is likely a result of its anti-oxidative effects, which can be attributed to the activation of key transcription factors in the Keap1/Nrf2 signaling pathway and their association with multiple inflammation-related pathways. LCB pretreatment reversed the inhibition of the Keap1/Nrf2 signaling pathway in a concentration-dependent manner, reducing Keap1 levels and elevating Nrf2, HO-1, and NQO1 levels.
Taken together, these findings suggested that oxidative stress plays a significant role in LPS-induced ALI. Accordingly, alleviating oxidative stress and inflammation could have a protective effect against LPS-induced lung injury. Specifically, LCB pre-treatment inhibited oxidative stress, enhanced the antioxidant capacity, alleviated inflammation, and ultimately reversed LPS-induced damage. The underlying mechanism may be related to the activation of the Keap1/Nrf2 axis. Further experimentation using Keap1/Nrf2 pathway inhibitors, such as ML385 and brusatol, or Nrf2 knockout mice, can provide evidence that the effect of LCB on LPS-induced inflammation or tissue injury is Keap1/Nrf2 pathway-dependent. Collectively, this study provides new insights into the therapeutic potential of LCB and suggests that it may be a promising candidate for prevention of LPS-induced respiratory diseases.
5. Conclusion
Our study demonstrated that LCB possesses the potential to protect against LPS-induced ALI in both cells and mice. The Keap1/Nrf2 pathway could be a potential target of LCB's protective effects. These findings suggest that LCB could be a viable therapeutic approach to combat ALI.
Ethics statement
All animal procedures were approved by the Animal Care and Use Committee of Chengdu University of Traditional Chinese Medicine (2023-04).
Acknowledgements
Ju Huang contributed to the preparation of the figures and tables, and writing of the manuscript. Yu Zhu, Songtao Li and Nianzhi Chen contributed to data collection and preparation of Figures and Tables. Huangyu Jiang, Jingwen Liu, Niang Dan, Hang Xiao and Qiao Zheng reviewed the manuscript. Jianyuan Tang and Xiangrui Meng contributed to the establishment of the structure of this manuscript and acted as the main supervisor. All authors read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its supplementary materials.
Additional information
Funding
References
- Vlaar AP, Juffermans NP. Transfusion-related acute lung injury: a clinical review. Lancet. 2013;382:984–94. doi:10.1016/S0140-6736(12)62197-7
- Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202:145–56. doi:10.1002/path.1491
- Aoyama H, Uchida K, Aoyama K, et al. Assessment of therapeutic interventions and lung protective ventilation in patients With moderate to severe acute respiratory distress syndrome: A systematic review and network meta-analysis. JAMA Netw Open. 2019;2:1–16. doi:10.1001/jamanetworkopen.2019.8116
- Wang CY, Calfee CS, Paul DW, et al. One-year mortality and predictors of death among hospital survivors of acute respiratory distress syndrome. Intensive Care Med. 2014;40:388–96. doi:10.1007/s00134-013-3186-3
- Meyer NJ, Gattinoni L, Calfee CS. Acute respiratory distress syndrome. Lancet Lond Engl. 2021;398:622–37. doi:10.1016/S0140-6736(21)00439-6
- Standiford Theodore J., Ward Peter A. Therapeutic targeting of acute lung injury and acute respiratory distress syndrome. Transl Res J Lab Clin Med. 2016;167:183–191. doi:10.1016/j.trsl.2015.04.015
- Cho H-Y, Reddy SP, Kleeberger SR. Nrf2 defends the lung from oxidative stress. Antioxid Redox Signal. 2006;8:76–87. doi:10.1089/ars.2006.8.76
- Alfieri A, Srivastava S, Siow RCM, et al. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood-brain barrier disruption and neurological deficits in stroke. Free Radic Biol Med. 2013;65:1012–22. doi:10.1016/j.freeradbiomed.2013.08.190
- Lin Y, Kuang Y, Li K, et al. Nrf2 activators from glycyrrhiza inflata and their hepatoprotective activities against CCl4-induced liver injury in mice. Bioorg Med Chem. 2017;25:5522–30. doi:10.1016/j.bmc.2017.08.018
- Sarady-Andrews JK, Liu F, Gallo D, et al. Biliverdin administration protects against endotoxin-induced acute lung injury in rats. Am J Physiol Lung Cell Mol Physiol. 2005;289:L1131–L1137. doi:10.1152/ajplung.00458.2004
- Ryter SW, Choi AMK. Heme oxygenase-1/carbon monoxide: novel therapeutic strategies in critical care medicine. Curr Drug Targets. 2010;11:1485–94. doi:10.2174/1389450111009011485
- An L, Liu C-T, Yu M-J, et al. Heme oxygenase-1 system, inflammation and ventilator-induced lung injury. Eur J Pharmacol. 2012;677:1–4. doi:10.1016/j.ejphar.2011.12.010
- Furusawa J, Funakoshi-Tago M, Mashino T, et al. Glycyrrhiza inflata-derived chalcones, licochalcone A, licochalcone B and licochalcone D, inhibit phosphorylation of NF-kappaB p65 in LPS signaling pathway. Int Immunopharmacol. 2009;9:499–507. doi:10.1016/j.intimp.2009.01.031
- Zhou B, Wang H, Zhang B, et al. Licochalcone B attenuates neuronal injury through anti-oxidant effect and enhancement of Nrf2 pathway in MCAO rat model of stroke. Int Immunopharmacol. 2021;100:1–12. doi:10.1016/j.intimp.2021.108073
- Li Q, Feng H, Wang H, et al. Licochalcone B specifically inhibits the NLRP3 inflammasome by disrupting NEK7-NLRP3 interaction. EMBO Rep. 2022;23:1–18. doi:10.15252/embr.202153499
- Wang J, Wang C-Y. Integrated miRNA and mRNA omics reveal the anti-cancerous mechanism of licochalcone B on human hepatoma cell HepG2. Food Chem Toxicol Int J Publ Br Ind Biol Res Assoc. 2021;150:1–9. doi:10.1016/j.fct.2021.112096
- Huang B, Liu J, Ju C, et al. Licochalcone A prevents the loss of dopaminergic neurons by inhibiting microglial activation in lipopolysaccharide (LPS)-induced Parkinson’s disease models. Int J Mol Sci. 2017;18:2043–2052. doi:10.3390/ijms18102043
- Xie X, Sun S, Zhong W, et al. Zingerone attenuates lipopolysaccharide-induced acute lung injury in mice. Int Immunopharmacol. 2014;19:103–9. doi:10.1016/j.intimp.2013.12.028
- Butt Y, Kurdowska A, Allen TC. Acute lung injury: A clinical and molecular review. Arch Pathol Lab Med. 2016;140:345–50. doi:10.5858/arpa.2015-0519-RA
- Sadek K, Abouzed T, Nasr S, et al. Licochalcone B ameliorates liver cancer via targeting of apoptotic genes, DNA repair systems, and cell cycle control. Iran J Pharm Res IJPR. 2020;19:372–86. doi:10.22037/ijpr.2020.1101292
- Huang Z, Jin G. Licochalcone B induced apoptosis and autophagy in osteosarcoma tumor cells via the inactivation of PI3 K/AKT/mTOR pathway. Biol Pharm Bull. 2022;45:730–7. doi:10.1248/bpb.b21-00991
- Song M, Yoon G, Choi J-S, et al. Janus kinase 2 inhibition by Licochalcone B suppresses esophageal squamous cell carcinoma growth. Phytother Res PTR. 2020;34:2032–43. doi:10.1002/ptr.6661
- Cao Y, Xu W, Huang Y, et al. Licochalcone B, a chalcone derivative from Glycyrrhiza inflata, as a multifunctional agent for the treatment of Alzheimer’s disease. Nat Prod Res. 2020;34:736–9. doi:10.1080/14786419.2018.1496429
- Su Z-Q, Mo Z-Z, Liao J-B, et al. Usnic acid protects LPS-induced acute lung injury in mice through attenuating inflammatory responses and oxidative stress. Int Immunopharmacol. 2014;22:371–8. doi:10.1016/j.intimp.2014.06.043
- Halliwell B. Commentary for “Oxygen free radicals and iron in relation to biology and medicine: some problems and concepts”. Arch Biochem Biophys. 2022;718:109151–109153. doi:10.1016/j.abb.2022.109151
- Mirault ME, Tremblay A, Furling D, et al. Transgenic glutathione peroxidase mouse models for neuroprotection studies. Ann N Y Acad Sci. 1994;738:104–15. doi:10.1111/j.1749-6632.1994.tb21795.x
- Li J, Lu K, Sun F, et al. Panaxydol attenuates ferroptosis against LPS-induced acute lung injury in mice by Keap1-Nrf2/HO-1 pathway. J Transl Med. 2021;19:96–121. doi:10.1186/s12967-021-02745-1
- Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33(829–837):837a–837d. doi:10.1093/eurheartj/ehr304
- Radi R. Oxygen radicals, nitric oxide, and peroxynitrite: redox pathways in molecular medicine. Proc Natl Acad Sci U S A. 2018;115:5839–48. doi:10.1073/pnas.1804932115
- Eiserich JP, Baldus S, Brennan M-L, et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science. 2002;296:2391–4. doi:10.1126/science.1106830
- Yang H, Lv H, Li H, et al. Oridonin protects LPS-induced acute lung injury by modulating Nrf2-mediated oxidative stress and Nrf2-independent NLRP3 and NF-κB pathways. Cell Commun Signal. 2019;17:62–78. doi:10.1186/s12964-019-0366-y
- Wang Z-F, Liu J, Yang Y-A, et al. A review: The anti-inflammatory, anticancer and antibacterial properties of four kinds of licorice flavonoids isolated from licorice. Curr Med Chem. 2020;27:1997–2011. doi:10.2174/0929867325666181001104550
- Grahame Hardie D. AMP-activated protein kinase: a key regulator of energy balance with many roles in human disease. J Intern Med. 2014;276:543–59. doi:10.1111/joim.12268
- Yao Y, Wang H, Xu F, et al. Insoluble-bound polyphenols of adlay seed ameliorate H2O2-induced oxidative stress in HepG2 cells via Nrf2 signalling. Food Chem. 2020;325:126865–9. doi:10.1016/j.foodchem.2020.126865
- Kim JY, Kim DY, Son H, et al. Protease-activated receptor-2 activates NQO-1 via Nrf2 stabilization in keratinocytes. J Dermatol Sci. 2014;74:48–55. doi:10.1016/j.jdermsci.2013.11.010
- Qing R, Huang Z, Tang Y, et al. Cordycepin alleviates lipopolysaccharide-induced acute lung injury via Nrf2/HO-1 pathway. Int Immunopharmacol. 2018;60:18–25. doi:10.1016/j.intimp.2018.04.032
- Qiu Y-L, Cheng X-N, Bai F, et al. Aucubin protects against lipopolysaccharide-induced acute pulmonary injury through regulating Nrf2 and AMPK pathways. Biomed Pharmacother Biomedecine Pharmacother. 2018;106:192–9. doi:10.1016/j.biopha.2018.05.070
- Cc C, Sy C, Cc C, et al. Djulis (Chenopodium formosanum) and its bioactive compounds protect human lung epithelial A549 cells from oxidative injury induced by particulate matter via Nrf2 signaling pathway. Mol Basel Switz. Molecules. 2021;27:253–281. doi:10.3390/molecules27010253