
ABSTRACT
Introduction
Huntington’s Disease (HD) is a genetic neurodegenerative disease for which there is currently no disease-modifying treatment. One of several underlying mechanisms proposed to be involved in HD pathogenesis is inflammation; there is now accumulating evidence that the immune system may play an integral role in disease pathology and progression. As such, modulation of the immune system could be a potential therapeutic target for HD.
Areas covered
To date, the number of trials targeting immune aspects of HD has been limited. However, targeting it, may have great advantages over other therapeutic areas, given that many drugs already exist that have actions in this system coupled to the fact that inflammation can be measured both peripherally and, to some extent, centrally using CSF and PET imaging. In this review, we look at evidence that the immune system and the newly emerging area of the microbiome are altered in HD patients, and then present and discuss clinical trials that have targeted different parts of the immune system.
Expert opinion
We then conclude by discussing how this field might develop going forward, focusing on the role of imaging and other biomarkers to monitor central immune activation and response to novel treatments in HD.
1. Introduction
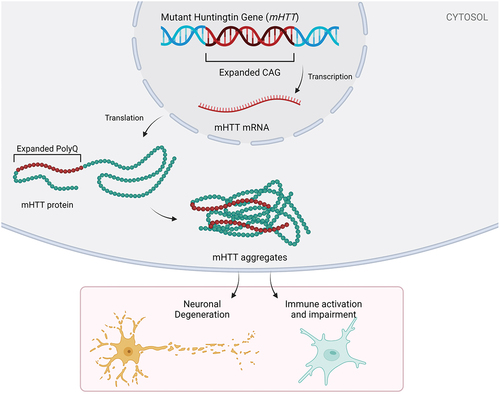
Huntington’s disease (HD) is an autosomal dominant, neurodegenerative disorder caused by a trinucleotide expansion in the huntingtin gene (HTT) [Citation1]. Exon 1 of HTT contains a repetitive sequence of cytosine, adenine, and guanine (CAG) repeats that encodes a poly-glutamine (polyQ) tract in the huntingtin protein (HTT). Healthy individuals have less than 36 CAG repeats, whereas mutant HTT gene carriers have over 36 (often 40 or more) repeats [Citation2]. Due to the high penetrance observed over 39 repeats, most studies categorize individuals with >39 CAG repeats as premanifest or manifest, depending on whether they have displayed symptoms and/or signs of HD. A new HD integrated staging system, HD-ISS, has been proposed that adds better granularity to this classification, which is likely to impact on clinical trials going forward [Citation3]. Pathological CAG repeats result in an expanded polyQ tract and subsequent misfolding of the HTT protein (). The mutant form of the HTT protein (mHTT) forms soluble fragments and aggregates that can be neurotoxic, with apparent neuronal selectivity, e.g. the medium spiny neurons in the striatum are particularly vulnerable to degeneration [Citation4]. This degeneration results in brain atrophy in HD gene carriers that is estimated to begin 15 years before predicted disease onset [Citation5], which is currently diagnosed in the clinic based on the emergence of motor features, usually accompanied by additional cognitive and neuropsychiatric abnormalities. Several mechanisms have been implicated in HD pathology, primarily around proteostasis, but there is also evidence that neuroinflammation may play a role, as seen in other chronic neurodegenerative disorders of the central nervous system (CNS). However, the exact contribution of the inflammatory process in HD is unknown. In the following review, we have summarized recent literature describing the role of neuroinflammation and peripheral immune activation in HD pathology and progression. We did this using initial PubMed search terms (‘Huntington’s Disease,’ ‘neuroinflammation,’ ‘central immune system,’ and ‘peripheral immune system’) which informed preliminary reading, after which relevant articles were selected, described in detail, and cited in this review.
Figure 1. HD pathology and its potential contribution to neuronal degeneration and an impaired immune system. Huntington’s disease is characterized by an expanded CAG repeat (often >39 CAG repeats) on the huntingtin gene. Pathological CAG repeats result in an expanded polyQ tract, subsequent misfolding of the mHTT protein and mHTT aggregation. Neuronal degeneration and impaired immune system are the downstream effects of this CAG repeat gene mutation.
2. Central immune system in HD
2.1. Microglia in HD
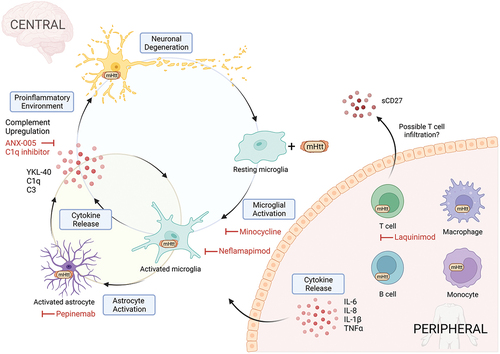
Microglia are the primary resident immune cells of the brain [Citation6]. They make up 5–10% of all cells in the CNS and are responsible for the elimination of microbes, dead cells, protein aggregates, and other cellular debris [Citation7,Citation8]. Microglia patrol the CNS in a resting state and then become activated when encountering a pathogen or changes in the microenvironment. When activated, they communicate to their surroundings via cytokine release, the nature of which varies depending on the insult. Previously, microglia were classified using an M1/M2 terminology, reflecting the peripheral macrophage nomenclature, where M1 broadly dictated the pro-inflammatory phenotype and M2 anti-inflammatory. Morphologically, microglia are often referred to as ramified (or resting), or amoeboid (or active). However, over the past decade, a better understanding of the complexity of microglial expression patterns has led to a new classification of microglial subtypes as a broad activation spectrum [Citation9,Citation10]. The gradient of phenotypes is based upon transcriptional, proteomic, and epigenetic signatures, encompassing homeostatic, activated, dysfunctional, and disease-associated microglia (‘DAM’), and in the context of neurodegeneration ‘MGnD’ (microglia neurodegenerative phenotype) [Citation11]. HTT is highly expressed in microglia. However, in HD, it is not clear whether microglial dysfunction and neuroinflammation contribute to disease pathogenesis – e.g. through impaired phagocytosis of mHTT – or occurs downstream of protein aggregation, though it is likely that the two feed forward to promote inflammation ().
Figure 2. A simplified diagram of the immune system in HD. In the central immune system, the presence of mHTT is thought to invoke an inflammatory response in microglia resulting in secretion of pro-inflammatory cytokines. Pro-inflammatory cytokines promote neurodegeneration directly in neurons or indirectly through astrocyte activation which in turn amplifies and sustains the pro-inflammatory response in microglia. In the peripheral immune system, mHTT inclusions are present in both innate and adaptive immune cells resulting in a ‘primed’ inflammatory state and increased cytokine release. Limited studies show possible T-cell infiltration to the brain and activation through elevated sCD27 in the CSF. Clinical trial drug interventions targeting these immune changes are in red.
2.1.1. Preclinical studies
In vitro studies of microglia have attempted to tackle this question of cause or effect. Overexpression of mHTT in the mouse microglial cell line BV2 increases the expression of several pro-inflammatory genes including Il6, Tnfa, even in the absence of pro-inflammatory stimuli [Citation12]. HD-derived induced pluripotent stem cell (iPSC)-microglia show increased secretion of pro-inflammatory cytokines (without inflammatory stimuli) that correlates with increased CAG repeat length, suggesting a direct role of mHTT expression in microglial function. Higher neuronal cell death is also observed in co-cultures with mHtt-microglia vs wildtype (WT) microglia [Citation13].
Various degrees of microgliosis have been found in the brains of HD mouse models. Several studies of R6/2 mice aged between 4-weeks [Citation14] and 8–12 weeks [Citation15,Citation16] have reported increased Iba1-positive cells in the striatum, though this has not been found in all studies [Citation17]. Microglial activity in HD mice models is also altered; increased phagocytosis in R6/2 mice [Citation18], as well as increased microglial soma size with extended short processes are seen in both R6/2 and YAC128 mice, indicating a more activated rather than resting state [Citation16,Citation19]. Suppression of Galectin-3 in R6/2 mice reduces microglial-mediated inflammation [Citation20]. Microglia isolated from the HD knock-in Q140 mice also demonstrate a typical pro-inflammatory response but are partially impaired in repressing proinflammatory gene expression with repeated inflammatory stimulation both in vitro and in vivo [Citation21].
An interesting model for testing microglia-specific effects was undertaken in BACHD mice (with full-length human mHTT), using a Cre depletion of mHTT specifically in microglia. While the BACHD mouse showed several HD-associated phenotypes and microglial activation, this was not rescued by microglia-specific correction of the mHTT gene, leading the authors to conclude that mHTT expression in microglia does not contribute to disease process [Citation22]. In another study, microglia depletion in R6/2 mice using colony-stimulating factor 1 receptor inhibition (CSF1Ri) resulted in a reduction of striatal atrophy, astrogliosis, and mHTT accumulation – suggesting that microglia may have a positive role in slowing down disease progression in HD. Thus, the work to date would suggest that the microglial effects seen in HD models are not cell autonomous but rather are secondary to mHTT expression/pathology in other cell types or the extracellular space.
2.1.2. Human studies
2.1.2.1. Postmortem
In human postmortem tissue, histological studies have shown that reactive microglia are found in the earliest stages of HD (Vonsattel et al. [Citation23] scoring method stage 1), then accumulate and increase in density with increasing disease severity and neuronal loss (stage 2/3) before declining in the more advanced stages (stage 5) [Citation24]. Reactive microglia are widespread across multiple brain regions including the striatum [Citation16,Citation24,Citation25], midcingulate cortex [Citation26], internal capsule [Citation27], and cortex [Citation24]. In addition, the microglial density in the midcingulate has been found to positively correlate with 1C2-positive mHTT aggregate burden [Citation26]. Functionally, microglia also appear to be more phagocytic in HD postmortem brains than controls [Citation28].
This reaction may change, or be perpetuated, following intracerebral interventions in HD such as the direct injection of cells or miRNAs designed to repair or slow down disease pathology, respectively. In this respect, there is one striking postmortem study in an HD patient who received a bilateral allogeneic fetal striatal transplant many years before death. In this case, densely packed, proinflammatory microglia were found surrounding and within the graft site [Citation29]. In addition, CD4 and CD8 T cells were found forming clusters within the graft. It was suggested that the immune infiltration may have contributed to the poor graft survival. It is unclear whether this would be seen in other cases of direct brain surgery in HD designed to deliver a therapeutic agent but does highlight that the microglia in HD may be primed to react more vigorously in the face of such interventions.
2.1.3. PET imaging
Positron emission tomography (PET) imaging offers a unique insight into the pathophysiology of the living brain and provides information that one cannot get from postmortem tissues, biofluids, or structural magnetic resonance imaging (MRI) and thus is invaluable when it comes to clinical trials targeting inflammation in HD. The current gold standard for measuring microglia-associated neuroinflammation is through translocator protein (TSPO) PET imaging. TSPO is regarded as a biomarker of active neuroinflammation due to its upregulation by activated microglia, and partially by other CNS cells such as activated astrocytes [Citation30]. Therefore, TSPO binding signal may not be exclusively attributed to activated microglia nor does it distinguish different subtypes of microglia (as described above). However, this field is rapidly evolving.
2.1.3.1. 1st generation TSPO tracers
[11C]PK11195 has been the most used PET tracer in HD. In premanifest patients, microglial activation in the striatum has been consistently reported [Citation31–35]. The postcentral gyrus, globus pallidus, thalamus, and hypothalamus have also been reported to have elevated [11C]PK11195 binding in premanifest subjects [Citation35], but these findings are somewhat inconsistent [Citation31–33]. [11C]PK11195 has been key in demonstrating the link between neuroinflammation and neurodegeneration in living HD patients, but its low binding affinity and high signal-to-noise ratio have led to the development of better second and third-generation TSPO ligands.
2.1.3.2. 2nd and 3rd generation TSPO tracers
The 2nd generation tracer [11C]PBR28 has been evaluated in HD patients. In a small study of eight HD gene carriers (one premanifest, seven manifest), they found increased binding in the putamen and pallidum, but not in the caudate of HD gene carriers compared to controls [Citation36]. [11C]PBR28 was also used as a supplementary measure in the LEGATO-HD trial (discussed in the peripheral immune system section) to test the efficacy of Laquinimod on microglial activation with a 1 year follow-up [Citation37]. The TSPO sub study (HD n = 15, Con n = 21) found no significant change in [11C]PBR28 distribution volume ratios post treatment in the caudate and putamen in either those treated with Laquinimod (N = 10) or those treated with placebo (N = 5) [Citation38]. In both studies, microglial activation significantly correlated with disease severity measures and could differentiate HD gene carriers from controls [Citation36,Citation37]. Similar findings have now been found with the 3rd generation tracer [11C]ER176 in which binding is increased in the putamen and pallidum in manifest patients, which is associated with disease burden (CAP (CAG-Age-Product) score) and striatal atrophy [Citation38].
However, it is important to note that the later generations of TSPO PET ligands are affected by a single nucleotide polymorphism (SNP) in the TSPO gene, which results in subsets of the patient population being either a high, low, or mixed affinity binder. Low-affinity binders are often excluded from studies because of a low signal to noise.
2.1.3.3. Preclinical PET
Microglial PET imaging has also been undertaken in mouse models of HD. One such study, using TSPO PET, examined the tracer [18 F]PBR06 in the HD mouse models BACHD and R6/2 [Citation39]. A positive signal was detected in the striatum, cortex, and hippocampus in early and mid-stage BACHD mice and in R6/2 mice at a late disease stage, which was further supported by immunostaining/blotting of Iba-1 and TSPO.
In addition, this study evaluated the use of PET in response to treatment with LM11A-31. LM11A-31 is a small-molecule p75NTR ligand that decreases microglial activation and has previously been found to improve dendritic spine density as well as ameliorate cognitive and motor impairments in both BACHD and R6/2 mice [Citation40,Citation41]. LM11A-31 has also been tested in a phase 2a trial for Alzheimer’s disease, making it an appealing target for HD clinical trials. [18 F]PBR06 was able to detect decreased microglial activation in both mouse models treated with LM11A-31, which was further validated with significant reduction of IBA1 and TSPO in immunostaining and western blots. All of the findings show the utility of [18 F]PBR06 to measure microglia, as was supported by the correlation of its signal to postmortem findings. As such, it may be a useful tool for clinical trials.
2.1.3.4. Alternative neuroinflammation radiotracers
TSPO radiotracers currently dominate PET studies, but the research field is moving away from TSPO to alternative targets of neuroinflammation. Promising alternative radiotracer targets include COX-1, COX-2, and colony-stimulating factor 1 receptor (CSF1R) for microglia, monoamine oxidase B (MAO-B) for measuring astrogliosis, and purinergic P2X7 receptor (P2X7R) ligands as a pan glial marker expressed primarily on microglia, but also oligodendrocytes and astrocytes [Citation42]. Radiotracers for these targets have been tested in humans but have yet to be fully validated across human neurodegenerative diseases [Citation43].
2.1.4. Clinical trials
2.1.4.1. DOMINO study – Minocycline
Minocycline is a semisynthetic tetracycline-derived antibiotic currently used systemically to treat a range of infections [Citation44]. Although minocycline has been in clinical use since 1971, its use increased in the neurodegeneration field during the 2000´s as it was shown to cross the blood–brain barrier and in animal studies to inhibit microglial activation [Citation45,Citation46] as well as reduce the phagocytic activity of microglia and macrophages, thus reducing the apoptosis of neurons [Citation47]. Preclinically, minocycline has been shown to have various effects across different HD animal models both efficacious [Citation45,Citation46,Citation48] and detrimental [Citation49–51]. A phase 2/3 trial of 114 HD participants was run by the Huntington Study group from April 2006 to November 2008 (NCT00277355) to determine the safety and efficacy of minocycline [Citation52].
Minocycline was well tolerated, with only five adverse events reported leading to withdrawal from the trial. The primary endpoint was not achieved, as the change in mean decline in TFC suggested futility, which was further supported by the secondary outcome measures (Unified Huntington’s Disease Rating Scale 1999 (UHDRS’99) total score) [Citation52]. As this trial did not reach its prespecified primary outcome, further investigation of this drug in HD was not felt warranted.
2.1.4.2. Neflamapimod
Neflamapimod (VX-745) is an inhibitor of p38 mitogen activated protein kinase alpha (p38 MAPK) and initially designed as a modulator of inflammatory diseases such as rheumatoid arthritis by Vertex Pharmaceuticals [Citation53]. Neflamapimod has been shown to inhibit IL-1β and TNF-α release in peripheral blood mononuclear cells (PBMC) [Citation54] and centrally shifts microglial activation from a pro-inflammatory to a phagocytic state in animal models, a mechanism that would be expected to have beneficial effects in HD [Citation55].
In 2019, 15 HD participants were enrolled in a study to evaluate the safety, tolerability, and effect of VX-745 (NCT03980938). The trial was terminated early in October 2020 due to COVID-19 delays, and additional results from another study involving patients with a different neurodegenerative disorder (NCT04001517) suggested a higher dose may be more beneficial. Only one participant completed both placebo and drug periods. The data is yet to be published.
There is currently no indication that Neflamapimod will continue to be trialed in HD.
2.1.5. Summary
Activated microglia have been shown to be present at higher levels in the HD brain compared to controls, both at postmortem and throughout the disease process using PET imaging. TSPO PET imaging has revealed that this begins in the prodromal stage and increases into the manifest phase of the disease, increasing with disease severity. Despite these findings being validated in transgenic HD mouse models with evidence for dysfunctional microglia, their role in disease pathogenesis is still unresolved. Preclinical and human observational studies have strongly suggested involvement of microglia in the disease process, but clinical trials have not supported this contention to date. Importantly, neither PET imaging nor biofluid markers were available at the time of the DOMINO trial (not to be confused with the Domino-HD study that looked at lifestyle interventions in HD) that could confirm target engagement. Future work looking at the role of microglia in HD is needed, which tries to build on the preclinical studies showing that these cells have some secondary role in the disease process.
2.2. Astrocytes in HD
Although not by definition an immune cell, astrocytes are important glial cells in the CNS thought to have a significant influence on the immune response, especially via their crosstalk with microglia [Citation56]. Astrocytes are derived from neural progenitor cells (NPCs) and are integral to many processes in the CNS including neurotransmitter recycling, ionic balance, modulation of synaptogenesis and synaptic transmission, and maintenance of the blood–brain barrier (BBB) [Citation57].
2.2.1. Preclinical studies
Glial fibrillary acidic protein (GFAP)-positive astrocytes are increased in the striatum of zQ175 mice compared to wild-type and are typically found in clusters and in areas of low HTT aggregate load [Citation58]. mHtt aggregation in astrocytes has been linked to impaired glutamate uptake and the resulting neuronal excitotoxicity [Citation59]. For example, a study investigating exon-1 and full-length mHTT expression on astrocytes in mice models found exon-1 astrocytes (R6/2 model) suffer substantially more transcriptional dysregulation (4526 differentially expressed genes) compared to full-length mHTT astrocytes (zQ175 model) (800 differentially expressed genes) [Citation60]. This finding complements an additional study showing that primary astrocytes from R6/2 mice exhibit prolonged NF-κB activation and increased neuronal toxicity compared to WT astrocytes [Citation61]. Furthermore, and unlike microglia, it appears that astrocytes can be primary drivers of the disease process; transgenic mice that selectively overexpress mHtt in astrocytes display HD phenotypes such as motor dysfunction and reduced lifespan [Citation62], and astrocyte-specific deletion of mHtt in a BACHD mouse model improved their motor and psychiatric-like phenotypes [Citation63].
Further support for the primary role of mHTT-expressing astrocytes in the disease process has come from the work of Steve Goldman and his team. Immunodeficient mice engrafted with mHTT-expressing human glia developed an HD-related phenotype with motor features and hyperexcitable striatal neurons. Conversely, engraftment of normal human glial cells in R6/2 HD mouse models ameliorated disease phenotypes through restoring potassium homeostasis, slowing disease progression and extending survival [Citation64].
2.2.2. Human Studies
GFAP positive reactive astrocytes have been found to increase with HD severity and inversely correlate with striatal neuron loss [Citation23,Citation65]. Previously, no visible GFAP-reactive astrocytes were found in Grade 0 HD brains [Citation23,Citation65]. However, a more recent study found significant grade-dependent increases in GFAP immunoreactivity that were first seen in the striatum of Grade 0 brains (n = 2) [Citation59]. In addition, wild-type huntingtin has been found to aggregate in HD astrocytes despite these cells having lower levels of HTT RNA than control astrocytes [Citation66].
Astrocyte gene expression also changes with disease severity. For striatal astrocyte–enriched genes around 20% were altered at grade 1, rising to ~50% at grades 2 and 3. Marked astrocyte reactivity occurs at stage 3, but additional astrocyte genes are altered at grade 1 [Citation67]. Single nucleus RNA sequencing (snRNAseq) analysis in tandem with neuroimaging found astrocyte-specific genes positively correlated with cortical cell loss [Citation68]. More recently, a genome-wide analysis study using cortical mRNA from HD patients found GFAP to score highest in differential gene expression [Citation69]. A snRNAseq study has shown gene expression in astrocytes significantly differed between HD and controls and interestingly, the gene expression patterns could distinguish several different astrocyte ’states’ [Citation66].
Overall, there is clear evidence that astrocytes are central to the disease process in HD and have some cell autonomous role in driving disease progression. How much of this is due to inflammation per se is less clear.
2.2.3. Clinical trials
2.2.3.1. SIGNAL-HD – Pepinemab
Pepinemab is a humanized Ig4 monoclonal antibody that actively blocks the protein semaphorin 4D (SEMA4D). Plexin receptor activation (via SEMA4D) leads to activation of glial cells, inhibition of glial migration and replacement, and disruption of blood–brain barrier integrity [Citation69–71]. In the transgenic HD mouse model YAC128, anti-SEMA4D treatment resulted in a reduction in atrophy in the striatum, cortex, and corpus callosum and improved cognitive and behavioral deficits. Motor features, however, did not show any improvement [Citation72].
Following a phase 1/2a study of 36 HD patients, a larger scale phase 2 safety study was conducted in 265 HD gene carriers (CAG repeats ≥36) defined as early manifest (EM, n = 179) or late prodromal (LP, n = 86) with completion in 2020 (NCT02481674). Pepinemab was generally well tolerated, and there was a high completion rate of participants in the trial (13/265 study discontinuations). However, the primary endpoints (one-touch stockings of Cambridge (OTS), paced tapping (PTAP) test, and clinical global impression of change (CGIC)) did not reach statistical significance when compared with placebo. Interestingly, MRI (n = 264) results found a 26% reduction in caudate atrophy and FDG-PET (n = 76) measured an increase in metabolic activity in the early manifest group but not in the late prodromal group relative to placebo. Although the primary clinical endpoints were not met, the decrease in caudate atrophy coupled with an increase in metabolic activity as seen on FDG-PET in early manifest subjects warrants further investigation in this particular cohort [Citation73].
As of April 2023, Vaccinex is exploring partnering and financing opportunities for a phase 3 study of this drug in HD.
2.2.4. Summary
Evidence suggests that astrocyte dysfunction influences HD pathology in a significant way. Postmortem studies suggest an increase in GFAP positive reactive astrocytes and transcriptomic analysis indicates abnormal astrocyte gene expression in HD. Immunodeficient mice engrafted with mHTT-expressing human glia developed an HD phenotype and engraftment of normal human glial cells in R6/2 HD mouse models ameliorated disease phenotypes, highlighting a significant primary role for astrocytes in HD pathology. Pepinemab, a SEMA4D inhibitor, was trialed in a phase 2 clinical trial in HD and had mixed results. The primary clinic-based outcomes did not show significant improvement, but secondary MRI and FDG PET measures had positive effects in terms of a reduction in atrophy and increase in brain metabolism. Based on the secondary measures, Pepinemab may be continued into phase 3 trials (financing dependent). Future studies using PET ligands for astrocytes (in development) could also answer critical questions on astrocyte activation in HD human brains throughout disease progression and response to therapeutics.
3. Peripheral immune system in HD
In the last few decades, the peripheral immune system has been more closely interrogated in many neurodegenerative diseases. The peripheral immune system is comprised of innate (macrophages, monocytes, neutrophils, and natural killer (NK) cells) and adaptive (T- and B-lymphocytes) immune cells. Immune cells are all predominantly found in the blood and the periphery but also have the capacity to cross the BBB into the CSF and brain, initiating local activity that is thought to be deeply intertwined with the glial immune activation in many neurodegenerative diseases. The BBB is also dysfunctional in HD. Human iPSC BBB models of HD have found intrinsic abnormalities in angiogenesis and barrier properties [Citation74,Citation75]. Similar vascular and BBB changes are found preclinically in R6/2 mice models and clinically with MRI and postmortem studies in HD patients) [Citation76]. BBB impairment in HD may thus be key to allowing pathogenic peripheral immune cells and cytokines to enter the CNS.
3.1. Preclinical studies
Peripheral immune cells have been investigated in several transgenic HD mouse models, including R6/2, HdhQ150 knock-in, YAC128, and zQ175 mice. Each transgenic mouse model has suggested that there is activation of the peripheral immune system [Citation77,Citation78], though this varies as to what is seen and the regional specificity of the affected cells. Macrophages from R6/2 mice have shown increased phagocytosis and YAC128 mouse peritoneal macrophages produce higher levels of IL-6 in comparison to controls when stimulated [Citation77]. Additionally, the number of activated T cells is increased in both the spleen and peritoneal cavity of zQ175 and R6/2 mice [Citation78].
A novel immune deficient mouse strain, YACNSG, has recently been developed using the YAC128 mouse line [Citation79]. YACNSG showed significant striatal atrophy compared to wild-type mice although this does not reach the degree of atrophy seen in typical the YAC128 model. When YACNSG is engrafted with the human immune system, striatal atrophy increases to the extent seen in the YAC128 strain, which suggests a role for the systemic immune system in striatal degeneration in HD. This might make YACNSG a good model to study the efficacy of human anti-inflammatory agents in HD [Citation80].
3.2. Human studies
Immune cells express HTT and several subtypes have also been found to express high levels of mHTT, including myeloid cells, which have been the most studied in patients with HD. Monocytes isolated from HD patient blood not only express mHTT but also produce significantly higher levels of IL-6 [Citation81,Citation82] and TNFα [Citation81,Citation82] than cells from control individuals when stimulated with IFNg and LPS in vitro, suggesting they exist in a more ‘primed’ or pro-inflammatory state. The increased pro-inflammatory cytokine production is proposed to be directly induced by HTT expression, and this has been validated by the finding that cytokine production is rescued back to WT level when HTT expression is reduced in HD monocytes/macrophages treated with anti-HTT small interfering RNA (siRNA) [Citation82]. Further exploration has revealed that this pathway is mediated by mHTT increasing NFkB translocation [Citation82]. While NK cells have not been directly studied in HD, one paper describes the enhanced recognition of HTT-overexpressing tumor cells by NK cells, suggesting that NK cells may be more activated in HD as well [Citation80]. Other innate immune cells have not been studied in HD, suggesting a gap in the field to better elucidate the peripheral immune system changes through disease progression.
In HD, B cells and T cells also express high levels of mHTT and this expression level increases with disease stage [Citation83]. However, the contribution of the adaptive system to HD is less well elucidated, and there is no consensus as to whether there is any functional consequence of mHTT on the lymphocyte populations. One study showed that the increased mHTT expression in T cells does not appear to affect their proliferative capacity, cytokine production, or transcriptional profile [Citation84]. This suggests that the NFkB-mediated increased cytokine production in monocytes and macrophages with high mHTT expression may be cell-type or myeloid specific. However, another study showed sCD27 (Soluble CD27 receptor), a highly specific biomarker of activated T cell inflammation [Citation85], to be increased in the CSF of both premanifest and manifest patients compared to controls [Citation86]. Additionally, the frequency of intrathecal T helper 17.1 cells is significantly increased in the CSF of premanifest patients compared to manifest [Citation87]. This could suggest early involvement of Th17.1 cells in the disease process but would need further exploration. B cells have not been studied functionally in HD.
Infiltration of immune cells into the brain has also not been well studied. One postmortem study that used qPCR to examine the neuroinflammatory environment in the brain stated that T cells were not significantly enriched, based upon the expression of CD247; however, this is likely due to the lack of sensitivity of the method to detect changes in small cell populations in bulk RNA [Citation88].
3.3. Clinical trials
3.3.1. LEGATO-HD – Laquinimod
Developed by TEVA Pharmaceuticals, Laquinimod is an oral immunomodulator that has been shown to have favorable efficacy and good tolerance in patients. The mechanism of Laquinimod has yet to be fully understood, but it is thought that it primarily affects cells of the peripheral innate immune system as well as cells within the CNS [Citation89]. In YAC128 and R6/2 HD mice, motor function improved with Laquinimod treatment [Citation90,Citation91].
In 2014, TEVA initiated a phase 2 trial of LEGATO-HD, testing the safety and efficacy of Laquinimod in 352 HD patients (NCT02215616). Overall, Laquinimod was generally safe and well tolerated in HD patients [Citation92]. However, LEGATO-HD did not meet its primary clinical endpoint, as no significant improvement was detected in the Unified Huntington’s Disease Rating Scale-Total Motor Score (UHDRS-TMS). Additionally, TSPO PET imaging found no differences in microglial activation between treatment and placebo [Citation37]. A treatment period of more than 12 months may have been needed to see any clinical effects, as the trial only ran for a year [Citation92]. However, a positive secondary outcome was seen in LEGATO-HD in terms of a reduction in caudate atrophy; however, as the trial did not meet its primary endpoint, TEVA pharmaceuticals have decided not to pursue Laquinimod treatment in HD [Citation92].
3.4. Inflammation and the gut microbiome
The role of the gut microbiome in inflammation is currently a very active area of research, including in the study of neurodegenerative diseases. In HD, many patients experience gastrointestinal symptoms and issues, as well as weight-loss [Citation93,Citation94]. This, coupled with the fact that mHTT is ubiquitously expressed, has raised questions as to whether the microbiome is normal in HD and if not, does this have consequences for the patient, given that microbes in the gut can directly secrete bacteria proteins or release cytokines that can then diffuse into the peripheral circulation and potentially even cross into the CNS [Citation95].
The gut microbiome has been investigated in a number of transgenic HD mouse models (R6/1 [Citation96–101], R6/2 [Citation102], and BACHD [Citation103]). These studies have found differences in microbial and fungal composition compared to controls, which coincided with gut dysbiosis and weight-loss. Altered microbial composition observations have also been replicated in HD patients [Citation104]. Despite these changes, one intervention trial using probiotics failed to ameliorate gut dysbiosis, cognitive or mood symptoms in HD patients [Citation105], though notably this was a small study.
Investigating the relationship between the gut and cytokines in the plasma of HD patients found that two genus populations of bacteria, Intestimonias and Biphila, correlated positively with cytokine IL-4 and negatively with IL-6 levels, respectively [Citation106]. This demonstrates a potential correlation between changes in inflammation in the systemic circulation and the gut microbiome in HD.
In summary, the gut microbiome seems to be altered in HD but exactly what this means in terms of inflammation and disease pathogenesis is unknown.
3.5. Summary
Although relatively few studies have been done on the peripheral immune system in HD, there does appear some evidence for it being affected due to the presence of mHTT in peripheral immune cells. The high rate of turnover of these cells may reduce any impact they have on disease pathogenesis. However, an altered BBB in HD may contribute to active immune cell infiltration, which could also have some of its origins in the microbiome, but further research is needed on this. A clinical trial using Laquinimod in HD, a drug targeting both peripheral and central immune systems, showed no major efficacy. TSPO PET imaging revealed that Laquinimod did not affect microglial activation. Possible reasons for this include Laquinimod being given at too low a dosage; being unable to effectively cross the BBB in HD or simply that it is unable to reduce microglial activation.
4. Cytokines in HD
Cytokines are a broad category of small proteins that act as pro- or anti-inflammatory signals to immune cells. They are produced by a broad number of innate and adaptive immune cells in the periphery but are also released in the central nervous system by glial cells. They can be measured peripherally in the blood or centrally through CSF or postmortem brain samples.
4.1. Preclinical studies (therapeutic targets and biomarker identification)
Increased levels of cytokines have been measured across multiple transgenic HD animal models (see ). Plasma or serum studies have been conducted in five transgenic mouse models (R6/2 [Citation41,Citation77,Citation78,Citation81,Citation107–109], YAC128 [Citation81,Citation90], BACHD [Citation110], CAG140 [Citation111], HdhQ150 [Citation81], N171-82Q [Citation111]). The cytokines interleukin IL-6, IL-10, IL-12, and TNF-α are commonly found to be elevated in most of the HD mouse models in postmortem brain tissue and through blood samples. The YAC128 model is reported to be the closest in mimicking HD human immune system dysfunction, with blood profiles most closely reflecting those seen in patients [Citation77,Citation81]. Cytokine levels in the plasma have also been used as a biomarker of neuroinflammation in animal models. However, in a study investigating Kynurenine 3-monooxygenase (KMO) deletion, normalization of pro-inflammatory cytokines did not translate into behavioral phenotype improvements in R6/2 mice [Citation112]. Therefore, cytokine levels alone in preclinical models may not be important in disease expression/pathogenesis.
Table 1. Cytokine levels in different transgenic HD animal models.
Two of the cytokines discussed above have been considered as potential therapeutic targets, leading to testing in animal models.
4.1.1. IL-6 neutralizing antibody
An IL-6 neutralizing antibody administered to R6/2 mice reduced weight loss at late stages and partially rescued motor deficits on the rotarod performance when compared with the control IgG treatment [Citation113]. Following these findings, an R6/2 mice model lacking IL-6 was recently created to test the potentially beneficial effects of reducing IL-6. However, the lack of IL-6 exacerbated R6/2 associated behavioral phenotypes, calling into question the therapeutic effects of targeting this cytokine [Citation114].
4.2. TNF-α inhibition
TNF-α increases the excitatory and decreases the inhibitory synaptic strength of medium spiny neurons in the YAC128 HD mouse model. Genetically or pharmacologically blocking TNF-α restored synaptic strength to normal [Citation115]. Treatment with XPro1595, a selective soluble inhibitor of TNF-α, increased neuronal density, decreased mHtt aggregates, and improved motor function in R6/2 mice [Citation108]. In contrast, improvements have not been reported with other anti-TNF-α drugs. R6/2 mice treated with etanercept, a drug that inhibits TNF-α, dampened plasma levels of TNF-α, IL-1β, and IL-6; however, the level of local TNF-α and IL-6 expression in the striatum was not affected. Etanercept partially reduced brain atrophy but failed to improve HD-related functional and cognitive deficits in R6/2 mice [Citation109]. Similarly, striatal injections of adeno-associated virus (AAV)-vectors expressing dominant-negative-TNF-α (AAV-DN-TNF) into YAC128 mice did not modify motor function, neuronal degeneration, or neuroinflammation [Citation116].
4.3. Human studies
4.3.1. Postmortem
Cytokine gene expression has been examined in the human striatum, cortex, and cerebellum using quantitative real time-PCR (qRT-PCR) of postmortem tissues [Citation81,Citation88]. In the striatum, mRNA expression of the pro-inflammatory cytokines IL-6 [Citation81,Citation88], IL-8 [Citation81], TNF-α [Citation81] and CCL2 [Citation88] and anti-inflammatory cytokine IL-10 [Citation88] are upregulated.
4.3.2. Plasma and CSF
lists other studies that have looked at inflammatory markers in HD in the CSF and plasma divided by disease stage: premanifest and manifest. This table highlights the lack of consensus in the field on the contribution of cytokines to disease process, so below we have summarized a few key findings that relate to preclinical research as well as highlighting the use of cytokines in biofluids as potential biomarkers for disease.
Table 2. Plasma and CSF protein levels in premanifest and manifest HD patients.
4.3.2.1. IL-6
A meta-analysis of the studies examining plasma IL-6 levels in HD pooled data from nine studies, together involving 469 HD gene mutation carriers and 209 healthy controls. Overall, it was found that plasma IL-6 levels are significantly elevated in HD in the premanifest stage and then increased further with disease advancement [Citation121]. While the majority of studies support the claim that IL-6 increases in manifest disease [Citation81,Citation107,Citation122–125], in premanifest cohorts, IL-6 levels are much more varied [Citation5,Citation35,Citation81,Citation107,Citation123–126] possibly reflecting differences between pre-manifest and manifest disease, but also perhaps due to the wide range of estimated phenoconversion times of individuals in this cohort. IL-6 blood levels have been found to positively correlate with CSF levels, suggesting that blood IL-6 could be used as a biomarker of disease state if variability were lower [Citation81].
4.3.2.2. TNF-α
Studies investigating patient-derived monocytes found that LPS stimulation led to higher production of TNF- α by HD monocytes compared to controls [Citation35,Citation82]. However, there is little supporting evidence for changes in TNF-α levels from studies examining CSF or plasma of human patients (see ).
4.3.2.3. YKL-40
Another promising marker that has appeared more recently in the HD literature is YKL-40 (studied in mice with the murine homologue Chitinase-3-like I (Chi3l1)). YKL-40 has received attention due to its early detection in neurodegenerative diseases [Citation127]. YKL-40 transcription in astrocytes is induced by cytokine release from macrophages, which triggers morphological changes and enhances astrocytic migration [Citation128]. CSF YKL-40 trends toward, or is significantly increased, in manifest HD participants compared to controls [Citation86,Citation129,Citation130]. Differences in CSF YKL-40 have also been found in a study looking at a premanifest cohort, in which CSF YKL-40 was found to be elevated in far-from-onset premanifest individuals compared to controls [Citation5]. While a potential biomarker for the disease, plasma YKL-40 has not been studied in HD.
4.4. Clinical trials
Despite multiple cytokine studies being done in preclinical models of HD, a lack of efficacy in animal models has prevented them from being trialed in patients.
4.5. Summary
Elevated cytokine levels in human and animal HD models suggest there is nonspecific immune dysfunction peripherally and centrally in HD. Premanifest and prodromal patients are the prime targets for therapeutic treatment. While some cytokine levels in the CSF or plasma in manifest patients are reliably elevated compared to controls, levels in premanifest patients are not consistent and therefore cannot currently be used as a reliable metric of disease progression or therapeutic efficacy in clinical trials. Few findings from preclinical studies have been replicated in humans, highlighting the barriers to clinical translation for cytokine research in HD. Cytokine inhibitors are an attractive therapeutic, given that many are already in clinical use for other conditions, but poor pharmacokinetic properties and systemic side effects with the limited preclinical data currently do not support their translation to clinical trials in this instance.
5. Complement system in HD
The complement system is integral to a functioning innate and adaptive immune response, first named for its ability to ‘complement’ the antibacterial properties of antibodies in the heat-labile fraction of serum [Citation131]. There are three complement pathways: classical, lectin, and alternative, each with different activation pathways and responses. Activation of complement pathways culminates in the formation of major effector molecules such as proinflammatory anaphylatoxins, membrane attack complexes (MAC) that directly lyse cell membranes, and opsonins that promote phagocytosis. A few studies have shown dysregulation of the complement system in HD, but initiation of a phase 2 trial investigating the C1q inhibitor ANX005 in HD patients has renewed interest in the topic.
5.1. Preclinical studies
R6/2 mice crossed with complement C3 deficient mice exhibited no alteration in multiple behavioral assays, weight, or survival [Citation132]. However, the authors also found a translational discrepancy, in that R6/2 mice do not mirror the large-scale complement upregulation seen in HD patients, and thus alternate models were needed. In light of this, further work was done, and a postmortem study using 3-month-old zQ175 mice found that the levels of C1q and C3 were elevated in the dorsolateral striatum, which correlated with the earliest observation of corticostriatal synapse loss [Citation28].
5.2. Human studies
5.2.1. Postmortem
The level of complement mRNA in the HD striatum is two- to fivefold higher than that seen in controls [Citation27]. In another study, complement proteins 3 (C3), C4a, and C4b were among the top 30 increased genes in the striatum of HD subjects [Citation133]. Higher levels of both C3 and iC3b have been found in the globus pallidus of HD patients compared to controls, but more interestingly in two rare samples of caudate nucleus from premanifest HD patients, it was found that levels of C3 and CR3 (microglia-specific complement receptor 3) transcripts were elevated relative to controls [Citation28].
5.2.2. Plasma and CSF
Complement proteins in the CSF and plasma have been investigated in HD but are limited to a handful of contradictory studies (). C1QC had a trend for increased expression with disease progression (control < HD-early < HD-mid) in one study [Citation134] but was not significantly different from controls in another study [Citation135]. In CSF samples from HD Clarity (a longitudinal observational HD cohort study), levels of C3 and iC3b were significantly increased in HD patients with early manifest HD, but plasma samples from the same patients did not detect any change, suggesting that complement changes are localized to the CNS. iC3b was also increased in premanifest patients, making it the only complement marker in the CSF to be detected prior to symptom onset [Citation28].
5.3. Clinical trials
5.3.1. ANX005
Annexon Biosciences is undertaking trials of ANX005, a monoclonal antibody, in Guillain-Barré Syndrome (GBS) (phase 3), Amyotrophic Lateral Sclerosis (ALS) (phase 2) and Huntington’s disease (phase 2). ANX005 is a humanized anti-C1q antibody. C1q initiates activation of the classical complement cascade, blocking it, blocks downstream activation of active components C3 and C5, while leaving the protective functions of other complement pathways intact [Citation136]. It is thought that abnormal C1q accumulation drives complement activation, initiating chronic neuroinflammation and resulting in synaptic loss in HD [Citation137]. Preclinically, dosing, frequency, and tolerability of the C1q inhibitor was established in non-human primate and rat models of other neurodegenerative disorders using ANX-M1, the murine equivalent of ANX005 [Citation138]. These studies confirmed the functional inhibition of the classical complement cascade and protected against disease pathology in mouse models of GBS and AD [Citation138]. In zQ175 HD mice treated with ANX-M1, a 60% reduction in C3 deposition was seen in the dorsolateral striatum accompanied by a 70% increase in corticostriatal synapses compared to IgG controls. Interestingly, ANX-M1 had no effect on baseline complement deposition or corticostriatal synapse numbers in WT mice [Citation28].
A Phase 2 study was conducted in 2020 at multiple US centers to assess the safety and tolerability of ANX005 (NCT04514367). Twenty-eight gene carrier HD adults were treated with ANX005 intravenously biweekly for 6 months and followed up for 3 months post treatment. The primary objective was achieved with the treatment being generally well tolerated with no serious infections or deaths reported during the study. A measurement of neuronal loss, neurofilament light chain level, was no different in the trial in the CSF nor plasma compared to historical controls. Of the 28 patients, 5 discontinued treatment, 3 due to autoimmune-like side effects that resolved after the termination of treatment. In the CSF and plasma, complete blocking of C1q activity and the classical complement pathway was observed in the six-month treatment period, which was maintained into the 3-month follow-up period, illustrating target engagement of ANX005.
Overall, the secondary objectives, cUHDRS and TFC, saw a trend toward improvement that did not reach significance. The study highlighted a heterogeneous response in the patients, so subgroup analysis was also performed and patients with higher baseline C4a/C4 levels (n = 12) exhibited greater clinical improvement. Patients with C4/C4a levels above the median at baseline saw increases in their cUHDRS scores after 6 months relative to those with a ratio below the median (75% vs. 36%) [Citation139].
In conclusion ANX005 was generally well tolerated, maintained full target engagement, and showed clinical improvement in a subgroup of HD patients. Annexon Biosciences are now aiming to initiate a phase 3 trial of the ANX005 in HD in 2024.
5.4. Summary
A handful of studies have examined the role of the complement system in HD. R6/2 mice are deficient in C3 and therefore using other HD mice models such as zQ175 should be done when studying the complement system in vivo preclinically. iC3b in CSF samples in premanifest subjects suggests there may be early involvement of the complement system in HD. The most convincing proof of principle for complement involvement in HD pathogenesis comes from the early clinical trial findings using the C1q complement inhibitor, ANX005. The phase 3 clinical trial, which is set to begin in 2024, will shed further light on this both mechanistically and as a therapeutic target. This will be the only immunotherapy currently being trialed in HD.
6. Expert Opinion
6.1. What is the evidence that there are immune abnormalities in animal models of HD?
There is evidence of immune abnormalities in many transgenic models of HD, including abnormalities in microglia, cytokines, complement, peripheral immune cells, and astrocytes. Animal models are important to test a proof of concept, but all of these models have major limitations that makes translating their findings to the clinic difficult.
6.2. Have drugs been shown to reduce inflammation in HD and slow disease down in preclinical models of HD?
There have been preclinical studies investigating the effects of cytokine inhibitors with some degree of success, but they are not without concerns given their broad range of actions and thus the development of more sensitive cytokine assays as well as better, more specific cytokine inhibitors are needed.
6.3. What is the evidence that there are immune abnormalities in patients with HD?
There is evidence of immune abnormalities in patients with HD, including abnormal microglia activity, cytokine production, complement system activation, peripheral immune cell proportions and activation state, and astrocyte function. TSPO PET studies have been integral in showing a CNS immune response starting in the premanifest stage of the condition, while traditional blood profiling has shed light on the peripheral immune changes. However, the role these immune abnormalities play in disease pathogenesis and progression is unclear and clinical trials targeting the immune system will provide the only robust answer.
6.4. Have drugs been shown to reduce inflammation in HD and slow disease down in HD gene carriers?
Past immune therapies trialed in HD are borrowed from trials using them in other neurodegenerative diseases. A few preclinical studies investigating drug effects in HD models have though been conducted before trialing in HD patients. The lack of clinical trial success to date suggests there is a need for more rigorous preclinical testing or drugs directed at better characterized immune targets known to be altered in HD pathology. Furthermore, the new classification of HD stages using the HD-ISS may also help inform decisions as to the optimal group for any such trials, which to date have concentrated only on manifest patients, when the disease may not be amenable to disease modification through immune modulation.
Laquinimod and Pepinemab both showed reductions in caudate atrophy, but this did not translate to any significant clinical improvement. There are several possible reasons for this, including targeting the right disease stage for a long enough time. Currently, there are two new potential candidates targeting different aspects of the immune system in HD; Annexon’s ANX005 is planned for a phase 3 trial in 2024 and Pepinemab may be continued into phase 3 trial (financing dependent). Results from these trials would be critical in offering proof of concept that the immune system plays a direct role in disease progression.
6.5. What next?
It is clear that more work is needed to better define the state of the immune system in HD as a function of disease stage. There is a need for more sensitive biofluid assays and better imaging, especially PET ligands, that target pro/anti-inflammatory subtypes of microglia/astrocytes. This knowledge would also help answer the question as to when one should optimally target disease progression in HD using this class of drugs. The ability to understand disease progression and track treatment response through specific and sensitive techniques would greatly augment drug candidate success.
Once this is achieved, the next question is whether to target the immune system systemically or specifically within the CNS. Considering the evidence for both central and peripheral immune involvement, both strategies are valid; however, the interplay between the systems is not sufficiently understood to be confident which would have a stronger effect. Targeting the immune system systemically has the advantage that we already have a plethora of licensed, well-tolerated drugs on the market that could be repurposed, as is being done in PD, for example, with Azathioprine [Citation140]. However, such approaches are limited by the potential systemic side effects that may accompany their use, especially if required for a sustained period. Targeting local neuroinflammation in the brain via microglia or astrocytes directly may prove difficult due to the issues of delivery to the brain, and more specifically to the right region. This may become especially relevant when thinking about the role of inflammation after local CNS interventions such as implanted cell or injected gene therapies. Nevertheless, the continued interest in the field of neuroinflammation in neurodegenerative diseases offers hope that immune specific drugs could be available in the coming years that could positively benefit HD patients.
6.6. General discussion
In this review, we have tried to answer the question ‘is the immune system dysregulated in HD?’ with considerable evidence to show that it is. However, the most crucial question we have not been able to answer is ‘can targeting the immune system slow down the progression of Huntington’s disease?’ The only way to be able to answer this question is through intervention studies. Microglia have been shown to be activated in prodromal HD patients, which worsens with disease progression and could act as a good initial target, but currently there is no drug known that selectively targets microglia, which has been tested in mice or humans.
Research into immune abnormalities in HD is not just valuable in finding potential therapeutic targets but could also be applied to other aspects of HD therapies. For example, mHTT lowering drugs currently being trialed, such as tominersen, elicit an immune response. This highlights a possible need for immunosuppression during dosing to reduce negative reactions. The same is true for the emerging injected therapies in HD such as the uniQure gene therapy study, AMT-130, with its AAV-delivered miRNA. Understanding the HD patients’ immune system may also help stratify patients into those likely to be the optimal trial cohort – for example, with ANX005, patients with a higher complement activation showed higher improved clinical measures than those with lower complement.
Finally, there has also been work looking at whether one can recruit the immune system to help clear mHTT using vaccines, though this success would be contingent on a functional immune system in vaccinated recipients. Thus, there is much that needs to be done in this field of the immune system in HD and only with time and more trials will we be able to determine whether it is a feasible target to halt or slow disease progression in HD using this therapeutic approach.
Article highlights
Inflammation is seen in Huntington’s disease although the extent varies with disease stage;
The role of inflammation in HD is still unknown and trials to date targeting this pathway have not shown positive results;
The largely negative trial results may be due to limitations of preclinical models of HD; thus, targets found to be successful in animal models are not translatable when tested in humans;
New targets such as the compliment system have been identified which are now the subject of clinical trials;
Imaging for microglia and brain inflammation is improving and may prove invaluable in future trials;
The role of the microbiome in HD and its influence on inflammation both peripherally and centrally is an emerging area of research.
This box summarizes key points contained in the article.
Declaration of Interests
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Tabrizi SJ, Flower MD, Ross CA, et al. Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat Rev Neurol. 2020;16(10):529–546. Available from: https://www.nature.com/articles/s41582-020-0389-4.
- Migliore S, Jankovic J, Squitieri F Genetic counseling in Huntington’s disease: potential new challenges on horizon? Front Neurol. 2019;10:453.
- Tabrizi SJ, Schobel S, Gantman EC, et al. A biological classification of Huntington’s disease: the integrated staging system. Lancet Neurol. 2022;21(7):632–644. Available from: http://www.thelancet.com/article/S147444222200120X/fulltext.
- Li S, Li XJ Multiple pathways contribute to the pathogenesis of Huntington disease. Mol Neurodegener. 2006;1(1):1–10. Available from: https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/1750-1326-1-19
- Scahill RI, Zeun P, Osborne-Crowley K, et al. Biological and clinical characteristics of gene carriers far from predicted onset in the Huntington’s disease Young Adult Study (HD-YAS): a cross-sectional analysis. Lancet Neurol. 2020;19(6):502–512. Available from: http://www.thelancet.com/article/S1474442220301435/fulltext.
- Li Q, Barres BA Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2017;18(4):225–242.
- Frost JL, Schafer DP Microglia: architects of the developing nervous system. Trends Cell Biol. 2016;26(8):587. Available from: /pmc/articles/PMC4961529/
- Colonna M, Butovsky O Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35(1):441. Available from: /pmc/articles/PMC8167938/
- Jurga AM, Paleczna M, Kuter KZ Overview of general and discriminating markers of differential microglia phenotypes. Front Cell Neurosci. 2020;14:544457.
- Ransohoff RM A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19(8):987–991. Available from: https://www.nature.com/articles/nn.4338
- Paolicelli RC, Sierra A, Stevens B, et al. Microglia states and nomenclature: a field at its crossroads. Neuron. 2022;110(21):3458. Available from: /pmc/articles/PMC9999291/.
- Crotti A, Benner C, Kerman BE, et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci. 2014;17(4):513. Available from: /pmc/articles/PMC4113004/
- Stöberl N, Donaldson J, Binda CS, et al. Mutant huntingtin confers cell-autonomous phenotypes on Huntington’s disease iPSC-derived microglia. Sci Rep. 2023;13(1):1–15. Available from: https://www.nature.com/articles/s41598-023-46852-z
- Paldino E, D’Angelo V, Sancesario G, et al. Pyroptotic cell death in the R6/2 mouse model of Huntington’s disease: new insight on the inflammasome. Cell Death Discov. 2020;6:6. Available from: https://pubmed.ncbi.nlm.nih.gov/32821438/
- Simmons DA, Casale M, Alcon B, et al. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia. 2007;55(10):1074–1084. Available from: https://pubmed.ncbi.nlm.nih.gov/17551926/
- Palazuelos J, Aguado T, Pazos MR, et al. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington’s disease excitotoxicity. Brain. 2009;132(11):3152–3164.
- Godavarthi SK, Narender D, Mishra A, et al. Induction of chemokines, MCP-1, and KC in the mutant huntingtin expressing neuronal cells because of proteasomal dysfunction. J Neurochem. 2009;108(3):787–795. Available from: https://pubmed.ncbi.nlm.nih.gov/19187096/
- Savage JC, St-Pierre MK, Carrier M, et al. Microglial physiological properties and interactions with synapses are altered at presymptomatic stages in a mouse model of Huntington’s disease pathology. J Neuroinflammation. 2020;17(1):1–18. Available from: https://jneuroinflammation.biomedcentral.com/articles/10.1186/s12974-020-01782-9
- Franciosi S, Ryu JK, Shim Y, et al. Age-dependent neurovascular abnormalities and altered microglial morphology in the YAC128 mouse model of Huntington disease. Neurobiol Dis. 2012;45(1):438–449. Available from: https://pubmed.ncbi.nlm.nih.gov/21946335/
- Siew JJ, Chen HM, Chen HY, et al. Galectin-3 is required for the microglia-mediated brain inflammation in a model of Huntington’s disease. Nat Commun. 2019;10(1):1–18. Available from: https://www.nature.com/articles/s41467-019-11441-0
- Steinberg N, Galleguillos D, Zaidi A, et al. Naïve Huntington’s disease microglia mount a normal response to inflammatory stimuli but display a partially impaired development of innate immune tolerance that can be counteracted by ganglioside GM1. J Neuroinflammation. 2023;20(1):1–20. Available from: https://jneuroinflammation.biomedcentral.com/articles/10.1186/s12974-023-02963-y
- Petkau TL, Hill A, Connolly C, et al. Mutant huntingtin expression in microglia is neither required nor sufficient to cause the Huntington’s disease-like phenotype in BACHD mice. Hum Mol Genet. 2019;28(10):1661–1670.
- Vonsattel J-P, Myers RH, Stevens TJ, et al. Neuropathological Classification of Huntingtonʼs Disease. J Neuropathol Exp Neurol. 1985;44(6):559–577.
- Sapp E, Kegel KB, Aronin N, et al. Early and progressive accumulation of reactive Microglia in the Huntington disease brain. J Neuropathol Exp Neurol. 2001;60(2):161–172.
- Dowie MJ, Grimsey NL, Hoffman T, et al. Cannabinoid receptor CB2 is expressed on vascular cells, but not astroglial cells in the post-mortem human Huntington’s disease brain. J Chem Neuroanat. 2014;59–60:62–71.
- Palpagama T, Mills AR, Ferguson MW, et al. Microglial and astrocytic responses in the Human Midcingulate cortex in Huntington’s disease. Ann Neurol. 2023;94(5):895–910. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/ana.26753
- Singhrao SK, Neal JW, Morgan BP, et al. Increased complement biosynthesis by microglia and complement activation on neurons in huntington’s disease. Exp Neurol. 1999;159(2):362–376. doi: 10.1006/exnr.1999.7170.
- Wilton DK, Mastro K, Heller MD, et al. Microglia and complement mediate early corticostriatal synapse loss and cognitive dysfunction in Huntington’s disease. Nat Med. 2023.
- Maxan A, Mason S, Saint-Pierre M, et al. Outcome of cell suspension allografts in a patient with Huntington’s disease. Ann Neurol. 2018;84(6):950. Available from: /pmc/articles/PMC6587549/
- Lavisse S, Guillermier M, Hérard AS, et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci. 2012;32(32):10809–10818. Available from: https://www.jneurosci.org/content/32/32/10809
- Pavese N, Gerhard A, Tai YF, et al. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66(11):1638–1643. Available from: https://pubmed.ncbi.nlm.nih.gov/16769933/
- Tai YF, Pavese N, Gerhard A, et al. Imaging microglial activation in Huntington’s disease. Brain Res Bull. 2007;72(2–3):148–151. Available from: https://pubmed.ncbi.nlm.nih.gov/17352938/
- Politis M, Pavese N, Tai YF, et al. Hypothalamic involvement in Huntington’s disease: an in vivo PET study. Brain. 2008;131(11):2860–2869.
- Politis M, Pavese N, Tai YF, et al. Microglial activation in regions related to cognitive function predicts disease onset in Huntington’s disease: a multimodal imaging study. Hum Brain Mapp. 2011;32(2):258–270. Available from: https://pubmed.ncbi.nlm.nih.gov/21229614/
- Politis M, Lahiri N, Niccolini F, et al. Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol Dis. 2015;83:115–121. Available from: https://pubmed.ncbi.nlm.nih.gov/26297319/
- Lois C, González I, Izquierdo-García D, et al. Neuroinflammation in Huntington’s Disease: new Insights with 11 C-PBR28 PET/MRI. ACS Chem Neurosci. 2018;9(11):2563–2571. Available from: https://pubmed.ncbi.nlm.nih.gov/29719953/
- Roussakis AA, Gennaro M, Gordon MF, et al. A PET-CT study on neuroinflammation in Huntington’s disease patients participating in a randomized trial with laquinimod. Brain Commun. 2023 5;5(2). doi: 10.1093/braincomms/fcad084
- Rocha NP, Charron O, Latham LB, et al. Microglia activation in basal ganglia is a late event in huntington disease pathophysiology. Neurol Neuroimmunol Neuroinflamm. 2021 8;8(3). Available from: https://pubmed.ncbi.nlm.nih.gov/33795375/
- Simmons DA, James ML, Belichenko NP, et al. TSPO–PET imaging using [18F]PBR06 is a potential translatable biomarker for treatment response in Huntington’s disease: preclinical evidence with the p75NTR ligand LM11A-31. Hum Mol Genet. 2018;27(16):2893. Available from: /pmc/articles/PMC6077813/
- Simmons DA, Belichenko NP, Ford EC, et al. A small molecule p75NTR ligand normalizes signalling and reduces Huntington’s disease phenotypes in R6/2 and BACHD mice. Hum Mol Genet. 2016;25(22):4920. Available from: /pmc/articles/PMC5418739/
- Simmons DA, Mills BD, Butler RR, et al. Neuroimaging, urinary, and plasma biomarkers of treatment response in huntington’s disease: preclinical evidence with the p75NTR Ligand LM11A-31. Neurotherapeutics. 2021;18(2):1039–1063. Available from: https://pubmed.ncbi.nlm.nih.gov/33786806/
- Meyer JH, Cervenka S, Kim MJ, et al. Neuroinflammation in psychiatric disorders: PET imaging and promising new targets. Lancet Psychiatry. 2020;7(12):1064. Available from: /pmc/articles/PMC7893630/
- Kreisl WC, Kim MJ, Coughlin JM, et al. PET imaging of neuroinflammation in neurological disorders. Lancet Neurol. 2020;19(11):940. Available from: /pmc/articles/PMC7912433/
- Garrido-Mesa N, Zarzuelo A, Gálvez J Minocycline: far beyond an antibiotic. Br J Pharmacol. 2013;169(2):337. Available from: /pmc/articles/PMC3651660/
- Chen M, Ona VO, Li M, et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. 2000;6(7):797–801. Available from: https://pubmed.ncbi.nlm.nih.gov/10888929/
- Wang X, Zhu S, Drozda M, et al. Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington’s disease. Proc Natl Acad Sci U S A. 2003;100(18):10483–10487. Available from: https://pubmed.ncbi.nlm.nih.gov/12930891/
- Blecharz-Lang KG, Patsouris V, Nieminen-Kelhä M, et al. Minocycline attenuates microglia/macrophage phagocytic activity and inhibits SAH-induced neuronal cell death and inflammation. Neurocrit Care. 2022;37(2):410. Available from: /pmc/articles/PMC9519684/
- Stack EC, Smith KM, Ryu H, et al. Combination therapy using minocycline and coenzyme Q10 in R6/2 transgenic Huntington’s disease mice. Biochim Biophys Acta. 2006;1762(3):373–380. Available from: https://pubmed.ncbi.nlm.nih.gov/16364609/
- Diguet E, Rouland R, Tison F Minocycline is not beneficial in a phenotypic mouse model of Huntington’s disease. Ann Neurol. 2003;54(6):841–842. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/ana.10818
- Diguet E, Fernagut PO, Wei X, et al. Deleterious effects of minocycline in animal models of Parkinson’s disease and Huntington’s disease. Eur J Neurosci. 2004;19(12):3266–3276. Available from: https://pubmed.ncbi.nlm.nih.gov/15217383/
- Smith DL, Woodman B, Mahal A, et al. Minocycline and doxycycline are not beneficial in a model of Huntington’s disease. Ann Neurol. 2003;54(2):186–196. Available from: https://pubmed.ncbi.nlm.nih.gov/12891671/
- Thsgd I A futility study of minocycline in huntington’s disease. Mov Disord. 2010;25(13):2219. Available from: /pmc/articles/PMC8801051/
- Shi Y, Mader M Brain penetrant kinase inhibitors: learning from kinase neuroscience discovery. Bioorg Med Chem Lett. 2018;28(11):1981–1991.
- Madkour MM, Anbar HS, El-Gamal MI Current status and future prospects of p38α/MAPK14 kinase and its inhibitors. Eur J Med Chem. 2021;213:113216.
- Alam JJ Selective brain-targeted antagonism of p38 MAPKα reduces hippocampal IL-1β levels and improves Morris water maze performance in aged rats. J Alzheimers Dis. 2015;48(1):219. Available from: /pmc/articles/PMC4923728/
- Freeman MR Specification and morphogenesis of astrocytes. Science. 2010;330(6005):774–778. Available from: https://pubmed.ncbi.nlm.nih.gov/21051628/
- Lee HG, Wheeler MA, Quintana FJ Function and therapeutic value of astrocytes in neurological diseases. Nat Rev Drug Discov. 2022;21(5):339–358. Available from: https://www.nature.com/articles/s41573-022-00390-x
- Brown TG, Thayer MN, VanTreeck JG, et al. Striatal spatial heterogeneity, clustering, and white matter association of GFAP+ astrocytes in a mouse model of Huntington’s disease. Front Cell Neurosci. 2023;17:1094503. Available from: /pmc/articles/PMC10175581/
- Faideau M, Kim J, Cormier K, et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington’s disease subjects. Hum Mol Genet. 2010;19(15):3053–3067. Available from: https://pubmed.ncbi.nlm.nih.gov/20494921/
- Benraiss A, Mariani JN, Osipovitch M, et al. Cell-intrinsic glial pathology is conserved across human and murine models of Huntington’s disease. Cell Rep. 2021;36(1):109308. Available from: https://pubmed.ncbi.nlm.nih.gov/34233199/
- Hsiao HY, Chen YC, Chen HM, et al. A critical role of astrocyte-mediated nuclear factor-κB-dependent inflammation in Huntington’s disease. Hum Mol Genet. 2013;22(9):1826–1842. Available from: https://pubmed.ncbi.nlm.nih.gov/23372043/
- Bradford J, Shin JY, Roberts M, et al. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci U S A. 2009;106(52):22480. Available from: /pmc/articles/PMC2799722/
- Wood TE, Barry J, Yang Z, et al. Mutant huntingtin reduction in astrocytes slows disease progression in the BACHD conditional Huntington’s disease mouse model. Hum Mol Genet. 2019;28(3):487–500. Available from: https://pubmed.ncbi.nlm.nih.gov/30312396/
- Benraiss A, Wang S, Herrlinger S, et al. Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat Commun. 2016;7(1). doi: 10.1038/ncomms11758.
- Myers RH, Vonsattel JP, Paskevich PA, et al. Decreased neuronal and increased oligodendroglial densities in Huntington’s disease caudate nucleus. J Neuropathol Exp Neurol. 1991;50(6):729–742.
- Al-Dalahmah O, Sosunov AA, Shaik A, et al. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol Commun. 2020;8(1). doi: 10.1186/s40478-020-0880-6.
- Diaz-Castro B, Gangwani MR, Yu X, et al. Astrocyte molecular signatures in Huntington’s disease. Sci Transl Med. 2019;11(514):8546. Available from: https://www.science.org/doi/10.1126/scitranslmed.aaw8546
- Estevez-Fraga C, Altmann A, Parker CS, et al. Genetic topography and cortical cell loss in Huntington’s disease link development and neurodegeneration. Brain. 2023;146(11):4532–4546.
- Smith ES, Jonason A, Reilly C, et al. SEMA4D compromises blood-brain barrier, activates microglia, and inhibits remyelination in neurodegenerative disease. Neurobiol Dis. 2015;73:254–268. Available from: https://pubmed.ncbi.nlm.nih.gov/25461192/
- Basile JR, Gavard J, Gutkind JS Plexin-B1 utilizes RhoA and Rho kinase to promote the integrin-dependent activation of Akt and ERK and endothelial cell motility. J Biol Chem. 2007;282(48):34888–34895. Available from: https://pubmed.ncbi.nlm.nih.gov/17855350/
- Okuno T, Nakatsuji Y, Moriya M, et al. Roles of Sema4D-plexin-B1 interactions in the central nervous system for pathogenesis of experimental autoimmune encephalomyelitis. J Immunol. 2010;184(3):1499–1506. Available from: https://pubmed.ncbi.nlm.nih.gov/20038643/
- Southwell AL, Franciosi S, Villanueva EB, et al. Anti-semaphorin 4D immunotherapy ameliorates neuropathology and some cognitive impairment in the YAC128 mouse model of Huntington disease. Neurobiol Dis. 2015;76:46–56.
- Feigin A, Evans EE, Fisher TL, et al. Pepinemab antibody blockade of SEMA4D in early Huntington’s disease: a randomized, placebo-controlled, phase 2 trial. Nat Med. 2022;28(10):2183–2193. Available from: https://www.nature.com/articles/s41591-022-01919-8
- Lim RG, Quan C, Reyes-Ortiz AM, et al. Huntington’s disease iPSC-derived brain microvascular endothelial cells reveal WNT-mediated angiogenic and blood-brain barrier deficits. Cell Rep. 2017;19(7):1365–1377. Available from: https://pubmed.ncbi.nlm.nih.gov/28514657/
- Vignone D, Gonzalez Paz O, Fini I, et al. Modelling the human blood-brain barrier in Huntington disease. Int J Mol Sci. 202223;23(14):7813. Available from: https://pubmed.ncbi.nlm.nih.gov/35887162/
- Drouin-Ouellet J, Sawiak SJ, Cisbani G, et al. Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: potential implications for its pathophysiology. Ann Neurol. 2015;78(2):160–177. Available from: https://pubmed.ncbi.nlm.nih.gov/25866151/
- Träger U, Andre R, Magnusson-Lind A, et al. Characterisation of immune cell function in fragment and full-length Huntington’s disease mouse models. Neurobiol Dis. 2015;73:388–398. Available from: https://pubmed.ncbi.nlm.nih.gov/25447230/
- Pido-Lopez J, Andre R, Benjamin AC, et al. In vivo neutralization of the protagonist role of macrophages during the chronic inflammatory stage of Huntington’s disease. Sci Rep. 2018 8;8(1). Available from: https://pubmed.ncbi.nlm.nih.gov/30061661/
- Dahlenburg H, Cameron D, Yang S, et al. A novel Huntington’s disease mouse model to assess the role of neuroinflammation on disease progression and to develop human cell therapies. Stem Cells Transl Med. 2021;10(7):1033. Available from: /pmc/articles/PMC8235129/
- Lu Z, Bi J, Sun Y, et al. Polyglutamine protein in tumor cells sensitizes tumor cells to natural killer cell cytolysis. Mol Immunol. 2020;124:153–160.
- Björkqvist M, Wild EJ, Thiele J, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med. 2008;205(8):1869. Available from: /pmc/articles/ PMC2525598/
- Träger U, Andre R, Lahiri N, et al. HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFκB pathway dysregulation. Brain. 2014;137(3):819–833. Available from: https://pubmed.ncbi.nlm.nih.gov/24459107/
- Weiss A, Träger U, Wild EJ, et al. Mutant huntingtin fragmentation in immune cells tracks Huntington’s disease progression. J Clin Invest. 2012;122(10):3731–3736. Available from: https://pubmed.ncbi.nlm.nih.gov/22996692/
- Miller JRC, Träger U, Andre R, et al. Mutant Huntingtin does not affect the intrinsic phenotype of human huntington’s disease t lymphocytes. PLoS One. 2015:10. Available from: /pmc/articles/PMC4631523/
- Komori M, Blake A, Greenwood M, et al. Cerebrospinal fluid markers reveal intrathecal inflammation in progressive multiple sclerosis. Ann Neurol. 2015;78(1):3. Available from: /pmc/articles/PMC5568079/
- Niemelä V, Burman J, Blennow K, et al. Cerebrospinal fluid sCD27 levels indicate active T cell-mediated inflammation in premanifest Huntington’s disease. PLoS One. 2018:13. Available from: https://pubmed.ncbi.nlm.nih.gov/29474427/
- von Essen MR, Hellem MNN, Vinther-Jensen T, et al. Early intrathecal T helper 17.1 cell activity in Huntington disease. Ann Neurol. 2020;87(2):246–255. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/ana.25647
- Silvestroni A, Faull RLM, Strand AD, et al. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. Neuroreport. 2009;20(12):1098–1103. Available from: https://journals.lww.com/neuroreport/fulltext/2009/08050/distinct_neuroinflammatory_profile_in_post_mortem.12.aspx
- Kaye J, Piryatinsky V, Birnberg T, et al. Laquinimod arrests experimental autoimmune encephalomyelitis by activating the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2016:113:E6145–E6152. Available from: https://www.pnas.org/doi/abs/10.1073/pnas.1607843113
- Garcia-Miralles M, Hong X, Tan LJ, et al. Laquinimod rescues striatal, cortical and white matter pathology and results in modest behavioural improvements in the YAC128 model of Huntington disease. Sci Rep. 2016;6(1):6. Available from: https://pubmed.ncbi.nlm.nih.gov/27528441/
- Ellrichmann G, Blusch A, Fatoba O, et al. Laquinimod treatment in the R6/2 mouse model. Sci Rep. 2017;7(1). doi: 10.1038/s41598-017-04990-1.
- Reilmann R, Anderson KE, Feigin A, et al. Safety and efficacy of laquinimod for Huntington’s disease (LEGATO-HD): a multicentre, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Neurol. 2024;23(3):243–255. Available from: http://www.thelancet.com/article/S1474442223004544/fulltext
- Robbins AO, Ho AK, Barker RA Weight changes in Huntington’s disease. Eur J Neurol. 2006:13. Available from: https://pubmed.ncbi.nlm.nih.gov/16879284/.
- Van der Burg JMM, Winqvist A, Aziz NA, et al. Gastrointestinal dysfunction contributes to weight loss in Huntington’s disease mice. Neurobiol Dis. 2011;44(1):1–8. Available from: https://pubmed.ncbi.nlm.nih.gov/21624468/
- Han Y, Wang B, Gao H, et al. Vagus nerve and underlying impact on the gut microbiota-brain axis in behavior and neurodegenerative diseases. J Inflamm Res. 2022;15:6213. Available from: /pmc/articles/PMC9656367/
- Gubert C, Kong G, Costello C, et al. Dietary fibre confers therapeutic effects in a preclinical model of Huntington’s disease. Brain Behav Immun. 2024;116:404–418. Available from: https://pubmed.ncbi.nlm.nih.gov/38142919/
- Kong G, Cao K-A L, Hannan AJ Alterations in the gut fungal community in a mouse model of Huntington’s disease. Microbiol Spectr. 2022:10. Available from: https://pubmed.ncbi.nlm.nih.gov/35262396/.
- Gubert C, Love CJ, Kodikara S, et al. Gene-environment-gut interactions in Huntington’s disease mice are associated with environmental modulation of the gut microbiome. iScience. 2022;25(1):103687. Available from: /pmc/articles/PMC8760441/
- Kong G, Cao KAL, Judd LM, et al. Microbiome profiling reveals gut dysbiosis in a transgenic mouse model of Huntington’s disease. Neurobiol Dis. 2020:135. Available from: https://pubmed.ncbi.nlm.nih.gov/30194046/
- Kong G, Ellul S, Narayana VK, et al. An integrated metagenomics and metabolomics approach implicates the microbiota-gut-brain axis in the pathogenesis of Huntington’s disease. Neurobiol Dis. 2021:148. Available from: https://pubmed.ncbi.nlm.nih.gov/33249136/
- Gubert C, Choo JM, Love CJ, et al. Faecal microbiota transplant ameliorates gut dysbiosis and cognitive deficits in Huntington’s disease mice. Brain Commun. 2022:4. Available from: https://pubmed.ncbi.nlm.nih.gov/36035436/
- Stan TL, Soylu-Kucharz R, Burleigh S, et al. Increased intestinal permeability and gut dysbiosis in the R6/2 mouse model of Huntington’s disease. Sci Rep. 2020;10(1):1–9. Available from: https://www.nature.com/articles/s41598-020-75229-9
- Radulescu CI, Garcia-Miralles M, Sidik H, et al. Manipulation of microbiota reveals altered callosal myelination and white matter plasticity in a model of Huntington disease. Neurobiol Dis. 2019;127:65–75. Available from: https://pubmed.ncbi.nlm.nih.gov/30802499/
- Wasser CI, Mercieca EC, Kong G, et al. Gut dysbiosis in Huntington’s disease: associations among gut microbiota, cognitive performance and clinical outcomes. Brain Commun. 2020 2;2(2). Available from: https://pubmed.ncbi.nlm.nih.gov/33005892/
- Wasser CI, Mercieca EC, Kong G, et al. A randomized controlled trial of probiotics targeting gut dysbiosis in Huntington’s disease. J Huntingtons Dis. 2023;12(1):43–55. Available from: https://pubmed.ncbi.nlm.nih.gov/37005888/.
- Du G, Dong W, Yang Q, et al. Altered gut microbiota related to inflammatory responses in patients with Huntington’s disease. Front Immunol. 2021:11. Available from: https://pubmed.ncbi.nlm.nih.gov/33679692/
- Chang KH, Wu YR, Chen YC, et al. Plasma inflammatory biomarkers for Huntington’s disease patients and mouse model. Brain Behav Immun. 2015;44:121–127. Available from: https://pubmed.ncbi.nlm.nih.gov/25266150/
- Hsiao HY, Chiu FL, Chen CM, et al. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum Mol Genet. 2014;23(16):4328–4344. Available from: https://pubmed.ncbi.nlm.nih.gov/24698979/.
- Pido-Lopez J, Tanudjojo B, Farag S, et al. Inhibition of tumour necrosis factor alpha in the R6/2 mouse model of Huntington’s disease by etanercept treatment. Sci Rep. 2019;9(1). doi: 10.1038/s41598-019-43627-3.
- Valadão PAC, da S OB, Joviano-Santos JV, et al. Inflammatory changes in peripheral organs in the BACHD murine model of Huntington’s disease. Life Sci. 2019;232:116653.
- Corey-Bloom J, Aikin AM, Gutierrez AM, et al. Beneficial effects of glatiramer acetate in Huntington’s disease mouse models: evidence for BDNF-elevating and immunomodulatory mechanisms. Brain Res. 2017;1673:102–110. Available from: https://pubmed.ncbi.nlm.nih.gov/28823953/
- Bondulich MK, Fan Y, Song Y, et al. Ablation of kynurenine 3-monooxygenase rescues plasma inflammatory cytokine levels in the R6/2 mouse model of Huntington’s disease. Sci Rep. 2021;11(1):1–18. Available from: https://www.nature.com/articles/s41598-021-84858-7.
- Bouchard J, Truong J, Bouchard K, et al. Cannabinoid receptor 2 signaling in peripheral immune cells modulates disease onset and severity in mouse models of Huntington’s disease. J Neurosci. 2012;32(50):18259–18268. Available from: https://pubmed.ncbi.nlm.nih.gov/23238740/.
- Wertz MH, Pineda SS, Lee H, et al. Interleukin-6 deficiency exacerbates Huntington’s disease model phenotypes. Mol Neurodegener. 2020;15(1):15. Available from: https://pubmed.ncbi.nlm.nih.gov/32448329/.
- Chambon J, Komal P, Lewitus GM, et al. Early TNF-dependent regulation of excitatory and inhibitory synapses on striatal direct pathway medium spiny neurons in the YAC128 mouse model of Huntington’s disease. J Neurosci. 2023;43(4):672. Available from: /pmc/articles/PMC9888503/
- Alto LT, Chen X, Ruhn KA, et al. AAV-dominant negative tumor necrosis factor (DN-TNF) gene transfer to the striatum does not rescue medium spiny neurons in the YAC128 mouse model of Huntington’s disease. PLoS One. 2014;9(5):e96544. Available from: https://pubmed.ncbi.nlm.nih.gov/24824433/.
- Du G, Dong W, Yang Q, et al. Altered gut microbiota related to inflammatory responses in patients with Huntington’s disease. Front Immunol. 2021;11:603594.
- Wild E, Magnusson A, Lahiri Swales N, et al. Abnormal peripheral chemokine profile in Huntington’s disease. PLOS Curr. 2011:3. Available from: /pmc/articles/PMC3082446/
- Silajdžić E, Rezeli M, Végvári Á, et al. A critical evaluation of inflammatory markers in Huntington’s disease plasma. J Huntingtons Dis. 2013;2(1):125–134. doi: 10.3233/JHD-130049.
- You H, Wu T, Du G, et al. Evaluation of blood glial fibrillary acidic protein as a potential marker in Huntington’s disease. Front Neurol. 2021:12. Available from: /pmc/articles/PMC8639701/
- Eide S, Misztal M, Feng ZP Interleukin-6 as a marker of Huntington’s disease progression: systematic review and meta-analysis. Brain Behav Immun Health. 2023;30:100635.
- Rodrigues FB, Byrne LM, McColgan P, et al. Cerebrospinal fluid inflammatory biomarkers reflect clinical severity in Huntington’s disease. PLoS One. 2016:11. Available from: /pmc/articles/PMC5033331/
- Dalrymple A, Wild EJ, Joubert R, et al. Proteomic profiling of plasma in Huntington’s disease reveals neuroinflammatory activation and biomarker candidates. J Proteome Res. 2007;6(7):2833–2840. Available from: https://pubs.acs.org/doi/abs/10.1021/pr0700753.
- Corey-Bloom J, Fischer RS, Kim A, et al. Levels of interleukin-6 in saliva, but not plasma, correlate with clinical metrics in Huntington’s disease patients and healthy control subjects. Int J Mol Sci. 2020;21(17):6363. Available from: https://www.mdpi.com/1422-0067/21/17/6363/htm
- Sánchez-López F, Tasset I, Agüera E, et al. Oxidative stress and inflammation biomarkers in the blood of patients with Huntington’s disease. Neurol Res. 2012;34(7):721–724. Available from: https://www.tandfonline.com/doi/abs/10.1179/1743132812Y.0000000073.
- Wang R, Ross CA, Cai H, et al. Metabolic and hormonal signatures in pre-manifest and manifest Huntington’s disease patients. Front Physiol. 2014 5;5. Available from: https://pubmed.ncbi.nlm.nih.gov/25002850/
- Baldacci F, Lista S, Cavedo E, et al. Diagnostic function of the neuroinflammatory biomarker YKL-40 in Alzheimer’s disease and other neurodegenerative diseases. Expert Rev Proteomics. 2017;14(4):285–299. Available from: https://pubmed.ncbi.nlm.nih.gov/28281838/
- Bonneh-Barkay D, Bissel SJ, Kofler J, et al. Astrocyte and macrophage regulation of YKL-40 expression and cellular response in neuroinflammation. Brain Pathol. 2012;22(4):530–546. Available from: https://pubmed.ncbi.nlm.nih.gov/22074331/
- Vinther-Jensen T, Börnsen L, Budtz-Jørgensen E, et al. Selected CSF biomarkers indicate no evidence of early neuroinflammation in Huntington disease. Neurol Neuroimmunol Neuroinflamm. 2016;3(6). doi: 10.1212/NXI.0000000000000287.
- Vinther-Jensen T, Budtz-Jørgensen E, Simonsen AH, et al. YKL-40 in cerebrospinal fluid in Huntington’s disease–a role in pathology or a nonspecific response to inflammation? Parkinsonism Relat Disord. 2014;20(11):1301–1303. Available from: https://pubmed.ncbi.nlm.nih.gov/25219973/
- Dunkelberger JR, Song WC Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20(1):34–50. Available from: https://www.nature.com/articles/cr2009139
- Larkin PB, Muchowski PJ Genetic deficiency of complement component 3 does not alter disease progression in a mouse model of Huntington’s disease. J Huntingtons Dis. 2012;1(1):107. Available from: /pmc/articles/PMC3478099/
- Hodges A, Strand AD, Aragaki AK, et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet. 2006;15(6):965–977.
- Fang Q, Strand A, Law W, et al. Brain-specific proteins decline in the cerebrospinal fluid of humans with Huntington disease. Mol Cell Proteomics. 2009;8(3):451. Available from: /pmc/articles/PMC2649809/.
- Caron NS, Haqqani AS, Sandhu A, et al. Cerebrospinal fluid biomarkers for assessing Huntington disease onset and severity. Brain Commun. 2022;4. doi: 10.1093/braincomms/fcac309.
- Sivadasan A, Bril V Therapies in Autoimmune Peripheral Neuropathies beyond Intravenous Immunoglobulin, Plasma Exchange and Corticosteroids: an Analytical Review. Transfus Med Rev. 2022;36(4):220–229.
- Zhang W, Chen Y, Pei H C1q and central nervous system disorders. Front Immunol. 2023;14:1145649.
- Lansita JA, Mease KM, Qiu H, et al. Nonclinical development of ANX005: a humanized anti-c1q antibody for treatment of autoimmune and neurodegenerative diseases. Int J Toxicol. 2017;36(6):449–462. doi: 10.1177/1091581817740873.
- Beers B, Bredlau A-L, Bhattacharyya A, et al. J08 A phase 2 open label study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of intravenous ANX005 in patients with, or at risk of, manifest Huntington’s disease (HD). J Neurol Neurosurg Psychiatry. 2022;93(A96–A97). Available from: https://jnnp.bmj.com/content/93/Suppl_1/A96.3.
- Greenland JC, Cutting E, Kadyan S, et al. Azathioprine immunosuppression and disease modification in Parkinson’s disease (AZA-PD): a randomised double-blind placebo-controlled phase II trial protocol. BMJ Open. 2020;10:e040527. Available from: https://pubmed.ncbi.nlm.nih.gov/33234645/