
ABSTRACT
Background
Considering the rise of new SARS-CoV-2 variants that have reduced the efficacy of COVID-19 vaccines, the development of new antiviral medications for the disease has become increasingly necessary. In this study, ASC10, a novel antiviral prodrug, was studied in a phase 1 trial in healthy Chinese participants.
Research design and methods
Part 1 involved 60 participants, receiving 50–800 mg ASC10 or placebo twice daily for 5.5 days. Part 2, with 12 participants, explored ASC10 dosing in the fed/fasting states.
Results
ASC10-A, the main pharmacologically active metabolite, rapidly appeared in plasma (Tmax: 1.00–2.00 h) and decreased (t1/2: 1.10–3.04 h) without accumulation. The Cmax and area under the plasma concentration – time curve (AUC) of ASC10-A increased dose-dependently (50–800 mg BID) over 5.5 days, with no accumulation. The Tmax was slightly delayed in the fed state; however, the Cmax and AUC were similar between the fed and fasting states. Adverse events (AEs) were comparable (ASC10/placebo, 66.7%) and mostly mild (95%).
Conclusion
ASC10 was demonstrated to be safe and well tolerated and exhibited dose-proportional exposure and minimal food effects.
Clinical trial registration
www.clinicaltrials.gov identifier is NCT05523141.
GRAPHICAL ABSTRACT

1. Introduction
The ongoing coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), remains a global health burden. As of 12 December 2022, there were 645,084,824 confirmed cases of COVID-19, including 6,633,118 deaths, reported to the World Health Organization (WHO) [Citation1]. Owing to viral adaptation in the human host, several variants, including Alpha, Delta, and Omicron, have been described for SARS-CoV-2. The Delta variant was the dominant variant from May 2021 and caused the majority of severe COVID-19 cases [Citation2]. However, the Omicron variant, first identified in South Africa in November 2021, rapidly displaced the Delta variant and is currently the most predominant SARS-CoV-2 variant. The emergence of a novel SARS-CoV-2 variant has diminished the effectiveness of COVID-19 vaccines, consequently exacerbating the persistence of the ongoing pandemic [Citation2–4]. Additionally, the latest variant, BA.2.75.2, even exhibits extensive escape from neutralizing antibodies [Citation5]. Although three antiviral drugs (remdesivir, nirmatrelvir/ritonavir, and molnupiravir) have been approved or issued for Emergency Use Authorization (EUA) for the treatment of COVID-19, new antiviral drugs for the disease are still required.
ASC10 is a double prodrug of the active antiviral ribonucleoside analog ASC10-A (also known as β-d-N4-hydroxycytidine [NHC]) that exhibits broad-spectrum antiviral activity against various viruses, including SARS-CoV-2, respiratory syncytial virus (RSV), and influenza viruses [Citation6]. Following oral administration, ASC10 is rapidly metabolized into a single prodrug, molnupiravir, and then further rapidly metabolized into the active moiety ASC10-A in the intestine, liver, and blood. ASC10 can be directly metabolized to form ASC10-A. The main component in the plasma is the antiviral nucleoside analog ASC10-A, which is converted into the active triphosphate metabolite ASC10-A-TP in target tissue cells. ASC10-A-TP binds to RNA-dependent RNA polymerase (RdRp), serves as a substrate for RNA synthesis, and induces large-scale mutations in viral RNA, consequently affecting viral replication and exerting antiviral effects. In vivo studies in a murine SARS-CoV-2 model demonstrated that ASC10 could eliminate SARS-CoV-2 in the lungs and protect mice from virus-induced weight loss, the development of clinical symptoms, and death.
ASC10 displayed solubility in acetonitrile (33–50 mg/ml), limited solubility in methanol (10–33 mg/ml), and near insolubility in water (<0.1 mg/ml). In contrast, molnupiravir demonstrated solubility in these solvents at 9.0, >100, and 39.7 mg/ml, respectively. The pKa values for ASC10 were 2.07 and 10.21, whereas for Molnupiravir they were 2.2, 10.2, and 12.0. The measured Log P of ASC10 was 0.44 while Log D of molnupiravir was 0.46 [Citation7]. Stability assessments conducted over 12 months at 30°C ± 2°C/65%±5% relative humidity (RH) and accelerated studies over 6 months at 40°C ± 2°C/75%±5% RH attested to the stability of ASC10. The permeability of ASC10 in Caco-2 cells (human colorectal adenocarcinoma cells) was found to be 3.2-fold higher than that of molnupiravir. This enhanced permeability could be attributed to the increased lipophilicity of ASC10. To further explore its pharmacokinetics (PK), preclinical studies have been conducted to examine the single-dose administration of ASC10 and molnupiravir in rats, dogs, and crab-eating monkeys. After the oral administration of 100 mg/kg of ASC10 and molnupiravir, the average bioavailability was found to be 43.3% and 15.1%, respectively. Notably, the average exposure to the active metabolite ASC10-A following oral administration was 2.1-fold higher than that of molnupiravir. Similarly, when rats were administered 200 mg/kg of ASC10 and molnupiravir orally, the average bioavailability values were observed to be 76.9% and 48.7%, respectively. The average exposure to ASC10-A after oral administration was 1.2-fold higher than that after oral administration of molnupiravir. In comparison, dogs exhibit a higher permeability within the gastrointestinal tract, which makes them more suitable for investigating drug absorption. After oral administration of 100 mg/kg ASC10 and molnupiravir to dogs, the average exposure and bioavailability were similar. Notably, the absorption and bioavailability in monkeys are expected to provide a more accurate representation of outcomes in humans. These findings suggest that the dual prodrug ASC10 May result in increased drug exposure and bioavailability in patients compared with molnupiravir. Consequently, ASC10 has significant potential to offer improved therapeutic effects against COVID-19, warranting further investigation through clinical trials. Furthermore, given that molnupiravir had not yet been approved for COVID-19 treatment in China at that juncture, there was a critical need to develop a therapeutic agent exhibiting exceptional traits, including heightened bioavailability, potent antiviral efficacy, and enhanced resistance against viral resistance.
Here, we report the results of a first-in-human, phase 1, randomized, double-blind, placebo-controlled study of ASC10 in healthy participants to determine the safety, tolerability, and pharmacokinetics (PK) of multiple ascending oral doses. Additionally, a randomized, open-label, crossover evaluation in the fed and fasting states was also conducted to assess the effect of food on the PK of ASC10.
2. Patients and methods
2.1. Study design and participants
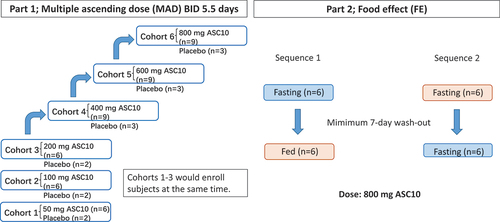
This study (trial registration: NCT05523141) was conducted at the First Affiliated Hospital, Zhejiang University School of Medicine, China; approved by the Ethics Committee of the First Affiliated Hospital, College of Medicine, Zhejiang University; and conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice. Written informed consent was obtained from each participant during screening prior to the initiation of any study-related procedures. This study consisted of two parts, as shown in . Part 1 (multiple ascending doses (MAD)) was a randomized, double-blind, placebo-controlled study that included six cohorts treated with various ASC10 doses (freely provided by Ascletis BioScience Co., Ltd.): 50, 100, 200, 400, 600, or 800 mg. The first three dose escalation cohorts comprised eight participants per cohort, while the other three cohorts comprised 12 participants per cohort. Eligible participants were randomized in a 3:1 ratio to receive either twice-daily (BID) doses of ASC10 or a placebo, using a computer-generated pseudorandom procedure at the time of the first dose administration. A placebo was chosen as the control treatment to assess whether any observed safety and tolerability effects were attributable to the treatment or simply reflected the study conditions. The participants received ASC10 or placebo BID for five consecutive days and received a single dose in the early morning of Day 6 to collect blood samples for the evaluation of steady-state PK parameters. Since the expected therapeutic dose of ASC10 is 800 mg BID [Citation5], the safety risk of doses lower than 200 mg was expected to be minimal; thus, participants from the first three cohorts (50, 100, and 200 mg) were enrolled simultaneously. For the remaining dose-escalation cohorts (400, 600, and 800 mg), the safety and tolerability data of the participants in the previous cohort up to 48 h post-dose were reviewed prior to dose escalation to ensure that it was safe to proceed with the planned protocol. Part 2 (food effect, FE) employed a randomized, open-label, crossover design. Twelve participants were enrolled and randomized in a 1:1 ratio to receive either 800 mg ASC10 in the fed state, followed by 800 mg ASC10 in the fasting state, or vice versa. The high-fat meal the participants received had a total caloric content ranging from 800 to 1000 kcal, with 150 kcal of protein, 250 kcal of carbohydrates, and a fat content of approximately 500–600 kcal. Randomization was performed, and the participants were monitored as described in Part 1. There was a 7-day washout period between the doses in the evaluation of FE. All participants were followed up for 7 days after dose administration (post-final dose for participants who received multiple doses) to assess safety and tolerability. The inclusion and exclusion criteria for the healthy participants were as follows: Key inclusion criteria: (1) male and female volunteers aged 18–45; (2) no planned pregnancy within a 6-month period, with a commitment to employ efficacious contraceptive measures throughout the study duration and for at least 3 months after the final administered dose; (3) maintenance of a favorable general health condition as evaluated by meticulous analysis of medical history, physical examination, vital signs, and supplementary screening evaluations; (4) a BMI of 19–26 kg/m2, with a minimal weight requirement of ≥50 kg for males and ≥45 kg for females. The key exclusion criteria were as follows: (1) pregnant and lactating women; (2) individuals with a history of fever within 14 days preceding the commencement of the study; (3) participants afflicted with acute or chronic diseases, encompassing but not limited to cardiovascular, gastrointestinal, liver, kidney, endocrine, and respiratory system diseases; and (4) individuals diagnosed with cancer.
Figure 1. Scheme of the study design. This study comprised two parts. Part 1 (multiple ascending dose) consisted of six cohorts (eight or 12 participants per cohort). Eligible participants were randomized in a 3:1 ratio to receive either twice-daily (BID) doses of 50 to 800 mg ASC10 or a placebo for 5.5 days and monitored for 7 days for safety. In Part 2 (food effect), 12 participants were randomized in a 1:1 ratio to receive either 800 mg ASC10 in the fed state followed by 800 mg in the fasting state, or vice versa, with a 7-day washout period between doses.
2.2. Outcome measures
The primary objective of this study was to determine the safety and tolerability of multiple ascending doses of ASC10 in healthy participants. The secondary objectives were to define the PK profiles of ASC10, molnupiravir, and ASC10-A (NHC) in plasma and urine following multiple doses of ASC10 and to assess the effect of food on the PK parameters of these compounds following a single dose in healthy participants. The safety and tolerability endpoints were determined through clinical laboratory evaluations, vital signs, electrocardiograms, physical examinations, and monitoring adverse events (AEs), which were monitored throughout the study through observing signs and symptoms, open questioning, and spontaneous reporting and were graded according to the Common Terminology Criteria for Adverse Events Version 5.0 (CTCAE V5.0). Participants who experienced more than one occurrence of the same AE were counted only once. The PK endpoints included plasma and urinary parameters. Following multiple doses, plasma and urine samples for PK analysis were collected up to 12 h after the first dose and up to 48 and 24 h after the final dose, respectively. For the FE study, plasma samples were collected up to 48 h post-dosing. PK samples were analyzed for ASC10, molnupiravir, and ASC10-A using a validated liquid chromatography with tandem mass spectrometry bioanalytical method [Citation6]. The validated ranges were 0.2–200 ng/mL in plasma and 0.5–500 ng/mL in urine for ASC10 and molnupiravir, and 10–10000 ng/mL in both plasma and urine for ASC10-A. The accuracy of the bioanalytical method was validated using incurred sample reanalysis, which revealed that all results had a relative percentage difference within the acceptable criteria of ± 20%.
2.3. Sample size and statistics
The sample size for this study was based on previous clinical experience, and sample size determination did not involve formal statistical hypotheses or tests. However, eight or 12 participants per cohort were considered sufficient for PK analysis following multiple doses. Twelve participants in the FE evaluation is the typical number employed for FE studies with a randomized crossover design. The PK parameters were calculated by non-compartmental methods using Phoenix WinNonlin, version 8.1. Concentration measurements below the limit of quantification (BLQ) were recorded as 0. The primary PK parameters included the maximum observed plasma concentration (Cmax), area under the plasma concentration – time curve (AUC), AUC from time zero (pre-dose) to the time of the last measurable concentration (AUC0-t), AUC from time zero (pre-dose) extrapolated to infinity (AUC0-∞), time of maximum plasma concentration (Tmax), terminal plasma elimination half-life (t1/2), apparent volume of distribution (Vd/F), apparent clearance (CL/F), and mean residence time (MRT). AUC0-t and AUC0-∞ were calculated using a Linear-Up Log-Down rule method. Tmax and Cmax were based on actual measured values. In the MAD study, the accumulation ratio after repeated dosing (Rac) was analyzed. PK parameters such as Tmax,ss, Cmax,ss, Cmin,ss, Cav,ss, AUC0-t,ss, AUC0-τ, drug fluctuation (DF), and range were calculated after the drug concentration reached steady state. A comparative analysis was performed between fed and fasting doses of ASC10, ASC10-A, and molnupiravir. A Wilcoxon-signed rank test was adopted for pairing data to analyze the difference between fasting and fed Tmax. Other primary PK parameters (AUC0–24, AUC0-t, AUC0-∞, and Cmax) were analyzed using a mixed linear model following natural logarithm transformation for multivariate analysis of variance and further analyzed using a t-test to test for differences. The confidence interval method was adopted to calculate the geometric least squares mean ratios (fed/fasting) and the corresponding 95% confidence intervals for the primary fasting and fed state PK parameters. The incidence of AEs and adverse reactions was calculated by listing their frequency and number according to the system and calculating the percentage.
3. Results
3.1. Participant demographics and disposition
In Part 1, of the MAD study, 234 participants were screened, and 60 healthy participants were enrolled. In Part 2, of the FE study, 41 participants were screened, and 12 healthy participants were enrolled. All enrolled participants completed the protocol-specific study procedures and assessments. The baseline demographics of the participants are presented in . The participants were aged 19–44 years, with a mean BMI of 19–25 kg/m2. There were no other notable differences in the participant demographics between the cohorts.
Table 1. Demographic profile of enrolled participants.
3.2. Safety evaluation
3.2.1. Part 1 MAD study
In total, 84 AEs were reported in 40 participants (). Participants receiving ASC10 or placebo had an equal incidence of AEs (30/45 or 10/15, 66.7%). Furthermore, the incidence of study drug-related AEs was similar between the two groups (46.7% vs. 33.3%). No significant dose-related trends were observed; however, more AEs were reported in the higher-dose group. All AEs were mild (grade 1: 95.2%) or moderate (grade 2: 4.8%) in severity. The most common AE was hypertriglyceridemia (16.7%), but none of these instances were considered study drug-related. Study drug-related AEs comprised increased blood creatinine (11.6%), increased lipase (11.6%), decreased neutrophil count (9.3%), increased alanine aminotransferase (9.3%), increased aspartate aminotransferase (9.3%), sinus bradycardia (9.3%), increased total bile acids (9.3%), decreased white cell count (9.3%), hyperuricemia (7.0%), anemia (4.7%), increased amylase (2.3%), allergic dermatitis (2.3%), atrial escape rhythm (2.3%), and ventricular extrasystoles (2.3%).
Table 2. Summary of adverse events using preferred terms.
3.2.2. Part 2 FE study
Seventeen AEs were reported in 10 participants, 13 of which were considered study drug-related (). All AEs were mild (grade 1), except for one study drug-unrelated moderate AE of presyncope (grade 2). Study drug-related AEs included decreased neutrophil count (30.8%), decreased white cell count (23.1%), increased blood creatinine (15.4%), hyperuricemia (15.4%), increased lipase (7.7%), and increased total bile acids (7.7%).
No serious AEs were observed during this study, and none of the participants discontinued study drug administration. Moreover, none of the participants experienced grade 3 or higher AEs. There were no clinically significant findings or dose-related trends in the clinical laboratory evaluations, vital signs, and electrocardiographic findings. Overall, ASC10 was generally safe and well tolerated when administered at multiple doses of up to 800 mg BID for 5.5 days in healthy participants.
3.3. Pharmacokinetics evaluation
Concentrations of ASC10 in the plasma were generally BLQ, and ASC10 was only detected in the urine samples from the 800 mg BID cohort; thus, the PK parameters of ASC10 were not calculable. Therefore, only the PK profiles of ASC10-A and molnupiravir are presented in the following sections.
3.3.1. Part 1 MAD study
The main PK parameters of molnupiravir on Days 1 and 6 in each dose cohort after multiple ASC10 doses are summarized in . On Day 1, concentrations of molnupiravir were quantifiable in three participants from the 50 mg cohort at 0.5 h postdose, two participants from the 100 mg cohort at up to 1.5 h postdose, and five participants from the 200 mg cohort at up to 1.5 h postdose. Molnupiravir was detectable in all participants at dose levels ≥400 mg BID until 4 h postdose. The concentration of molnupiravir detected ranged from 0.200 ng/mL to 3.014 ng/mL. On Day 6, three participants in the 50 mg cohort, two participants in the 100 mg cohort, and all participants in the ≥200 mg BID dose cohorts had low quantifiable molnupiravir, with concentrations ranging from 0.200 ng/mL to 2.380 ng/mL.
Table 3. Pharmacokinetic parameters of molnupiravir (50–800 mg ASC10 twice daily for 5.5 days).
The main PK parameters of ASC10-A on Days 1 and 6 in each dose cohort after multiple ASC10 doses are summarized in . The mean plasma concentration – time curves of ASC10-A on Days 1 and 6 are shown in . Following oral administration of ASC10, ASC10-A appeared rapidly in plasma, with a median Tmax in all dose cohorts of 1.00–1.99 h postdose at both Days 1 and 6. For all dose levels, plasma concentrations decreased in a monophasic manner on Day 1, with a mean t1/2 of 1.10–1.19 h. Similarly, plasma concentrations declined in a monophasic manner on Day 6, with a mean t1/2 of 1.15–3.04 h. The main PK parameters (Cmax, AUC0–24, AUC0-t, and AUC0-∞) of each dose cohort after multiple doses of ASC10 were highly correlated with dose, exhibiting a linear PK profile within a range of 80.00–125.00%. The AUC parameters and Cmax increased in an approximately dose-dependent manner in the dose range of 50–800 mg. Generally, across all cohorts and days, the Cmax of ASC10-A was between 552- and 5842-fold higher than that of ASC10 (where measurable).
Figure 2. Arithmetic mean plasma concentrations of ASC10-A (50–800 mg ASC10 BID, multiple ascending doses) on Days 1 (a) and 6 (b). Error bars indicate standard errors of the mean (SD).
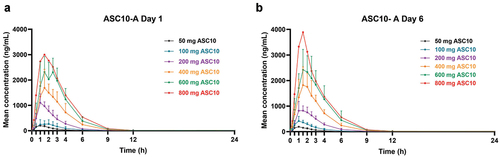
Table 4. Pharmacokinetic parameters of ASC10-A (50–800 mg ASC10 twice daily for 5.5 days).
Across all dosage levels, there were no indications of accumulation, as evident from the geometric mean accumulation ratios derived from the area under the plasma concentration–time curve during a dosing interval (AUCτ) and Cmax values. These ratios ranged between 0.94 and 1.06 for AUCτ and between 0.85 and 1.24 for Cmax.
After multiple oral doses of ASC10 BID for 5.5 days, up to 4.48% of the administered dose was excreted in urine as ASC10-A, as determined by the arithmetic mean percentage of the dose administered recovered in urine throughout the dosing interval (). The majority (generally >90%) of the total amount excreted was excreted within the first 8 h. Furthermore, a notable trend of increased recovery percentage with increasing dosage was observed. Across the dosage range of 200–800 mg BID, the amount of ASC10 excreted in urine during the dosing interval increased by approximately 12.6- and 10.6-fold on Days 1 and 6, respectively.
Table 5. Percentage of urine excretion for ASC10, molnupiravir, and ASC10-A on days 1 and 6 (50–800 mg ASC10 twice daily for 5.5 days).
3.3.2. Part 2 FE study
The main PK parameters of molnupiravir and ASC10-A after a single oral dose of 800 mg ASC10 under fed or fasting conditions are listed in . The corresponding mean plasma drug concentration – time curves of ASC10-A are presented in . Concentrations of molnupiravir were quantifiable between 0.25 and 4 h postdose for all participants under fed or fasting conditions, ranging between 0.27 and 6.17 ng/mL under fasting conditions and between 0.21 and 3.32 ng/mL under fed conditions. Following the administration of 800 mg ASC10 under fed conditions, the Tmax of ASC10-A was delayed, with a median of 3.99 h postdose versus 2.00 h postdose under fasting conditions. Generally, the slower absorption and delayed Tmax under fed conditions were reflected in lower Cmax values. The mean Cmax under fed conditions was approximately 90.7% of that under fasting conditions; however, exposure (assessed by AUClast and AUCinf) was similar for both fed and fasting conditions, demonstrating that the rate, but not the extent, of absorption was lower under fed conditions. The concentrations of ASC10-A declined in a monophasic manner in both fed and fasting conditions following Cmax and remained quantifiable until between 12 and 15 h post-dose under both conditions. The mean t1/2 was the same between fed and fasting conditions, with a value of 1.28 h. Hence, the administration of ASC10 with food was unlikely to have an effect on exposure.
Figure 3. Arithmetic mean plasma concentrations of ASC10-A (food effect, 800 mg ASC10). Error bars indicate standard errors of the mean (SD).
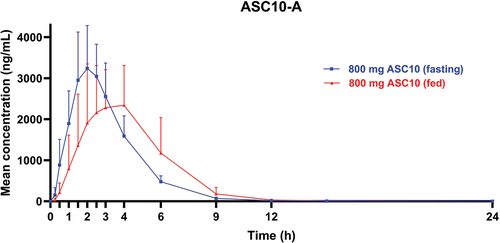
Table 6. Pharmacokinetic parameters of ASC10-A (food effect).
4. Discussion
ASC10 is a double prodrug of the active antiviral ribonucleoside analog ASC10-A (NHC) that is rapidly metabolized into the single prodrug molnupiravir, which can be rapidly converted into ASC10-A or directly metabolized into ASC10-A after oral dosing. Thus, in this study, ASC10 was hardly detected, and molnupiravir exposure was very low in the plasma after administration of doses between 50 and 800 mg. ASC10-A appeared shortly after oral administration and exhibited linear dose-proportional PK. However, no accumulation was observed. Although the Tmax of ASC10-A increased and the Cmax of ASC10-A was slightly lower under fed conditions, the overall exposure (AUC) to ASC10-A did not change, indicating that food had a minimal effect on ASC10-A. Because molnupiravir is a single prodrug and ASC10 is a double prodrug, it is important to compare the PK of ASC10-A (NHC) across studies. Comparing the ASC10-A (NHC) PK profiles in this study to those of the molnupiravir phase 1 study in the US revealed that the geometric mean AUCτ, 12 h, AUCinf, and Cmax of ASC10-A (NHC) on Days 1 and 6 were slightly higher in healthy Chinese participants following oral dosing with ASC10 than those in healthy American participants following oral dosing with molnupiravir [Citation8]. These differences appeared to be related to the differences in body weight, or BMI, between the two populations (approximately 60 kg for Chinese participants and 80 kg for American participants). However, the PK parameters were similar between Chinese participants after ASC10 oral dosing and Japanese participants after oral dosing with molnupiravir [Citation9]. This may be attributed to the Chinese and Japanese participants having similar BMIs (19–25 kg/m2 for the ASC10 phase 1 Chinese population and 18.5–24.9 kg/m2 for the molnupiravir phase 1 Japanese population). Following the oral administration of 400 and 800 mg ASC10 or molnupiravir, the geometric mean AUCτ, 12 h of ASC10-A (NHC) was 5748 and 11,154 ng·h/mL in the Chinese participants and 5240 and 12,100 ng·h/mL in the Japanese participants on Day 1, respectively, and 6065 and 11,922 ng·h/mL in the Chinese participants and 5410 and 12,700 ng·h/mL in the Japanese participants on Day 6, respectively.
A dosage of 800 mg BID of molnupiravir exhibits maximum antiviral activity (approximately 2.0 log10 copies/mL viral load change in SARS-CoV-2 RNA from the baseline) and is recommended as the efficacious dose [Citation10]. Considering the findings from this ASC10 phase 1 study, together with data from studies on molnupiravir in healthy American and Japanese participants [Citation11,Citation12], ASC10 should be administered at the same therapeutic dose as molnupiravir, which is 800 mg BID for 5.5 days.
ASC10 was well tolerated at doses of 50–800 mg BID for 5.5 days. The most frequently observed AE was hypertriglyceridemia, which was observed in seven (15.6%) and three (20%) participants receiving ASC10 and placebo, respectively. As more hypertriglyceridemia AEs were reported in participants in both the low-dose (50 and 100 mg ASC10) and placebo groups than the other groups, none were considered study drug-related. All participants were hospitalized at the study site for the first 5 days; thus, the hypertriglyceridemia AEs were considered transient phenomena due to the lack of exercise. All AEs were either mild or moderate. None of the participants discontinued or paused the dosing regimen during the study. Furthermore, no serious AEs were observed. Results from the clinical laboratory evaluations, vital signs, and electrocardiography did not reveal any significant findings or trends related to dosage. Together with the tolerability results, these results suggest that ASC10 is generally safe at the dose levels and durations tested in this study cohort.
The study has certain limitations. A majority of participants were of Han Chinese descent, and the patients recruited had to be aged between 18 and 45. Further assessment is needed to determine if race and age may affect PK and safety.
5. Conclusion
After multiple dosings of ASC10 for 5.5 days, both the Cmax and AUCτ, 12 h of the active metabolite ASC10-A (NHC) on Days 1 and 6 increased in an approximately dose-proportional manner across the dosage range of 50–800 mg ASC10 BID. Furthermore, there was no evidence of accumulation or FE. The AUCτ, 12 h, AUCinf, and Cmax of ASC10-A (NHC) on Days 1 and 6 in Chinese healthy participants following oral dosing with ASC10 were similar to those in healthy Japanese participants after oral dosing with molnupiravir. Based on this, the recommended therapeutic dosage of ASC10 should be the same as that of molnupiravir (800 mg BID for 5.5 days). Together with the favorable safety profile of ASC10, the results of this study support the continued investigation of ASC10 at a dosage regimen of 800 mg BID for 5.5 days in patients with SARS-CoV-2, RSV, or influenza virus infections.
List of abbreviations
| AEs | = | adverse events |
| AUC | = | area under the plasma concentration – time curve |
| AUC0-t | = | AUC from time zero (pre-dose) to the time of the last measurable concentration |
| AUC0-∞ | = | AUC from time zero (pre-dose) extrapolated to infinity |
| BID | = | twice-daily |
| BMI | = | body mass index |
| Cmax | = | maximum observed plasma concentration |
| CL/F | = | apparent clearance |
| COVID-19 | = | coronavirus disease 2019 |
| CTCAE V5.0 | = | Common Terminology Criteria for Adverse Events Version 5.0 |
| EUA | = | Emergency Use Authorization |
| FE | = | food effect |
| MAD | = | multiple ascending dose |
| MRT | = | mean residence time |
| NHC | = | β-d-N4-hydroxycytidine |
| PK | = | pharmacokinetics |
| Rac | = | accumulation ratio |
| RdRp | = | RNA-dependent RNA polymerase |
| RSV | = | respiratory syncytial virus |
| SARS-CoV-2 | = | severe acute respiratory syndrome coronavirus 2 |
| t1/2 | = | terminal plasma elimination half-life |
| Tmax | = | time of maximum plasma concentration |
| Vd/F | = | apparent volume of distribution |
| WHO | = | World Health Organization |
Declaration of interest
JJ Wu and Y Yan are employees and stockholder of Ascletis Pharma Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contribution
Y Qiu, as the corresponding author, was involved in designing the study, analyzing the data, and reviewing and approving the final version of the paper. JJ Wu, as the corresponding author, contributed to the study’s design, data analysis, and writing of most of the initial draft of the paper. J Liu and Q Zhao, the first authors, played a role in data generation and collection, data analysis, writing most of the original draft of the paper, and reviewing and approving the final version. Y Zhai, X Wu, J Kai, Je Ruan, M Wu, M Wu, Z Zhou, and Y Yan, as coauthors, contributed to data generation and collection for the study, and reviewed and approved the final version of the paper. Additionally, Y Yan participated in designing the study.
Data availability statement
The data that support the findings of this study are available from the corresponding author, Y Qiu and JJ Wu, upon reasonable request.
Additional information
Funding
References
- WHO COVID-19 dashboard [Internet]. 2024 [cited 2024 Jun 6]. Available from: https://covid19.who.int/
- Tian D, Sun Y, Zhou J, et al. The global epidemic of the SARS-CoV-2 delta variant, key spike mutations and immune escape. Front Immunol. 2021;12:751778. doi: 10.3389/fimmu.2021.751778
- Gupta RK, Topol EJ. COVID-19 vaccine breakthrough infections. Science. 2021;374(6575):1561–1562. doi: 10.1126/science.abl8487
- Li X. Omicron: Call for updated vaccines. J Med Virol. 2022;94(4):1261–1263. doi: 10.1002/jmv.27530
- Sheward DJ, Kim C, Fischbach J, et al. Omicron sublineage BA.2.75.2 exhibits extensive escape from neutralising antibodies. Lancet Infect Dis. 2022;22(11):1538–1540. doi: 10.1016/S1473-3099(22)00663-6
- Yoon JJ, Toots M, Lee S, et al. Orally efficacious broad-spectrum ribonucleoside analog inhibitor of influenza and respiratory syncytial viruses. Antimicrob Agents Chemother. 2018;62(8):e00766–18. doi: 10.1128/AAC.00766-18
- Product Assessment Report of Molnupiravir, European Medicines Agency, European Union. 2022 [cited 2022 Nov 1]. Available online https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&cad=rja&uact=8&ved=2ahUKEwj4lcP16rv5AhWchv0HHcLtDW0QFnoECAIQAQ&url=https%3A%2F%2Fwww.ema.europa.eu%2Fen%2Fdocuments%2Freferral%2Flagevrio-also-known-molnupiravir-mk-4482-covid-19-article-53-procedure-assessment-report_en.pdf&usg=AOvVaw3l1W4pGFCLhRRx3AAV3n7Q
- Drug label of LAGEVRIO-molnupiravir capsule, 10/2023 version, merck sharp & dohme LLC.
- Komarov T, Karnakova P, Archakova O, et al. Development and validation of a high-performance liquid chromatography with tandemmass spectrometry (HPLC-MS/MS) method for quantification ofmajor molnupiravir metabolite (β-D-N4-hydroxycytidine) in human plasma. Biomedicines. 2023;11(9):2356. doi: 10.3390/biomedicines11092356
- Fischer WA 2nd, Eron JJ, Holman W, et al. A phase 2a clinical trial of molnupiravir in patients with COVID-19 shows accelerated SARS-CoV-2 RNA clearance and elimination of infectious virus. Sci Transl Med. 2022;14(628):eabl7430. doi: 10.1126/scitranslmed.abl7430
- Painter WP, Holman W, Bush JA, et al. Human safety, tolerability, and pharmacokinetics of molnupiravir, a novel broad-spectrum oral antiviral agent with activity against SARS-CoV-2. Antimicrob Agents Chemother. 2021;65(5):e02428–20. doi: 10.1128/AAC.02428-20
- Nakamura K, Fujimoto K, Hasegawa C, et al. A phase I, randomized, placebo-controlled study of molnupiravir in healthy Japanese to support special approval in Japan to treat COVID-19. Clin Transl Sci. 2022;15(11):2697–2708. doi: 10.1111/cts.13395