
Abstract
Objective: This study aimed to evaluate improvement of dyspareunia and associated vaginal dryness with a 17β-estradiol softgel vaginal insert (TX-004HR; TherapeuticsMD, Boca Raton, FL, USA) in women with postmenopausal vulvar and vaginal atrophy (VVA).
Methods: Postmenopausal women with VVA and moderate to severe dyspareunia received TX-004HR (4, 10, or 25 μg) or placebo in the 12-week, randomized, double-blind, placebo-controlled, phase 3 REJOICE trial. Post hoc analyses examined improvement levels in dyspareunia and concurrent vaginal dryness with TX-004HR and assessed the effects of patient characteristics on vaginal dryness treatment.
Results: Significantly more women treated with TX-004HR (all doses) than placebo had complete resolution or substantial improvement in dyspareunia or vaginal dryness (concurrent with dyspareunia) by 12 weeks, observed as early as week 2 with most doses. TX-004HR significantly improved both dyspareunia and vaginal dryness at least one level versus placebo by week 12 in women with both symptoms. Subgroup analyses showed TX-004HR improved vaginal dryness associated with dyspareunia regardless of age, body mass index, uterine status, prior pregnancy, and vaginal birth number.
Conclusion: TX-004HR provided clinically meaningful improvements in dyspareunia and vaginal dryness associated with dyspareunia in postmenopausal women with VVA. Clinicians may be able to use this information when discussing patients’ expectations regarding symptom improvement with the estradiol vaginal insert.
摘要
目的:本研究旨在评估患有绝经后外阴和阴道萎缩 (VVA) 的妇女应用17β-雌二醇软胶囊阴道插入物 (TX-004HR, TherapeuticsMD, Boca Raton, FL, USA) 对性交困难以及相关的阴道干燥的改善状况。
方法:在为期12周的随机, 双盲, 安慰剂对照的3期REJOICE试验中, 患有VVA和中度至重度性交困难的绝经后妇女接受TX-004HR (4, 10或25μg) 或安慰剂。事后分析检查了TX-004HR对性交困难和与之并存的阴道干燥的改善程度, 并评估了患者特征对阴道干燥治疗的影响。
结果:接受TX-004HR (所有剂量) 治疗的妇女中在12周时出现性交困难或阴道干燥 (与性交困难并发) 的症状完全缓解或明显改善的数量明显多于安慰剂组, 而服用最多剂量的妇女最早在第2周就出现了这种情况。与安慰剂组相比, 对于同时患有性交困难和阴道干燥的女性, TX-004HR在第12周时至少在一个水平上可以明显改善这两种症状。亚组分析显示TX-004HR改善了与性交困难相关的阴道干燥, 而与年龄, 体重指数, 子宫状态, 既往妊娠和阴道分娩数量无关。
结论:TX-004HR为患有VVA的绝经后妇女的性交困难和与性交困难相关的阴道干燥提供了临床上有意义的改善。在讨论患者对雌二醇阴道插入物改善症状的期望时, 临床医生可能能够使用这些信息。
Introduction
Postmenopausal women can experience various vulvar and vaginal symptoms, including dyspareunia (painful sex) and dryness. These symptoms result from the thinning, drying, and loss of elasticity of the vaginal epithelium also known as vulvar and vaginal atrophy (VVA)Citation1. VVA is associated with a reduction in serum estrogen levels and is a component of the genitourinary syndrome of menopause (GSM)Citation2. Surveys of postmenopausal women have shown that vulvar and vaginal symptoms can occur in almost half of womenCitation3–6, with almost two-thirds of women experiencing moderate to severe symptomsCitation3,Citation4,Citation6,Citation7. Although vulvar and vaginal symptoms can be assessed separately, they may be related and treatment for one may lead to improvement in others.
Two doses (4 and 10 µg) of a 17β-estradiol (E2) softgel vaginal insert (formerly referred to as TX-004HR) were approved by the US Food and Drug Administration (FDA) for the treatment of moderate to severe dyspareunia, a symptom of VVA due to menopause, in May 2018 under the tradename Imvexxy® (TherapeuticsMD, Boca Raton, FL, USA). In the 12-week, phase 3 REJOICE trial, three E2 doses were evaluated (4, 10, and 25 µg) and all showed statistically significantly improvements in the four coprimary endpoints (percentages of superficial and parabasal cells, vaginal pH, and severity of dyspareunia) compared with placebo in postmenopausal women with VVA, with improvements beginning as early as 2 weeks for some dosesCitation8.
Most women in the REJOICE study had mild, moderate, or severe vaginal dryness in addition to moderate to severe dyspareunia at baseline. A prespecified secondary endpoint of the trial was assessment of the improvement in vaginal dryness at weeks 2, 6, 8, and 12. All doses of the E2 softgel vaginal insert significantly improved vaginal dryness after 12 weeks, and as early as 2–6 weeks, depending on the doseCitation8. Furthermore, TX-004HR resulted in negligible to very low systemic estrogen absorptionCitation9. In addition to the efficacy endpoints, a significantly greater percentage of women reported being ‘satisfied’ or ‘very satisfied’ in the active treatment groups compared with placeboCitation10.
The objective of this post hoc analysis of the REJOICE trial was to further examine clinical improvement of vaginal symptoms with the E2 softgel vaginal insert. The percentages of women who showed improvement or complete resolution of their dyspareunia or vaginal dryness associated with dyspareunia were compared between TX-004HR doses and placebo. Changes in vaginal dryness with each dose were also analyzed by patient characteristics (age, body mass index [BMI], uterine status, prior pregnancy, and number of vaginal births).
Methods
Study design and population
The REJOICE trial (clinicaltrials.gov: NCT02253173) was a 12-week, multicenter, double-blind, randomized, placebo-controlled, phase 3 trial conducted in the United States and Canada. The study design and the inclusion and exclusion criteria have been described elsewhereCitation8. Briefly, eligible women were randomized (1:1:1:1) to an E2 softgel vaginal insert (TX-004HR; E2 4, 10, or 25 μg) or matching placebo softgel vaginal insert. Women self-administered one insert into the vagina without an applicator at approximately the same time of day, daily for 2 weeks and then twice weekly for 10 weeks.
Postmenopausal women (age 40–75 years) were included in the study if they had VVA defined as ≤5% superficial cells on vaginal cytological smear and vaginal pH > 5.0, and reported moderate to severe dyspareunia as their most bothersome symptom (MBS). Postmenopausal women with only vaginal dryness were not eligible. Subjects had to anticipate having sexual activity (with vaginal penetration) during conduct of the trial. Exclusion criteria included a history or active presence of clinically important medical disease that might confound the study or be detrimental to their health.
Women could not have used vaginal lubricants or moisturizers within 7 days prior to vaginal pH assessment performed at screening; transdermal hormone products or vaginal hormone products (rings, creams, gels) within 4 weeks; oral products containing estrogens, progestins, androgens, or selective estrogen receptor modulators or intrauterine progestins within 8 weeks; an intrauterine device within 12 weeks; progestin implants/injectables or estrogen pellets/injectables within 6 months; or investigational drugs within 60 days before screening. Concomitant medications were allowed during the study and had to be recorded in diaries, but other investigational drugs, medications containing estrogen, progestin, androgens, or selective estrogen receptor modulators, and prescription and non-prescription medications/remedies used to treat VVA, including vaginal lubricants and moisturizers, were not allowed.
The study was conducted in accordance with applicable laws and regulations including the International Conference on Harmonization Guideline for Good Clinical Practice and the ethical principles that have their origins in the Declaration of Helsinki. An independent institutional review board approved the study protocol and the informed consent form. All subjects gave written informed consent prior to any study activities.
Vaginal symptom assessments
Change in severity of dyspareunia from baseline to week 12 versus placebo was one of the four coprimary endpoints of the study, and change in severity of vaginal dryness at weeks 2, 6, 8, and 12 versus placebo was a secondary endpoint. Women self-assessed the severity of dyspareunia and vaginal dryness with the VVA Symptom Self-Assessment Questionnaire using a 4-point scale (0 = none, 1 = mild, 2 = moderate, and 3 = severe) at baseline and at weeks 2, 6, 8, and 12. Changes from baseline in levels of severity of dyspareunia and vaginal dryness were evaluated at each time point. For example, moderate to severe symptoms could change by –3 points/levels (severe to none) to +1 point/level (moderate to severe).
Three different post hoc analyses were conducted. Firstly, the percentages of women who had a change in severity of dyspareunia or vaginal dryness of 2 points/levels or more (substantial improvement)Citation11, and those who had complete resolution of each symptom (score of 0 = none), with each TX-004HR dose were compared with placebo in the entire study population (women with VVA and MBS of moderate to severe dyspareunia) from weeks 2 to 12. Secondly, improvements in symptom scores of at least 1 point/level were determined for dyspareunia and dyspareunia concurrent with vaginal dryness at week 12 in women who also had mild, moderate, or severe vaginal dryness at baseline. Thirdly, changes from baseline in vaginal dryness for each TX-004HR dose versus placebo were analyzed by patients’ age (≤56 years, 57–61 years, ≥62 years), BMI (≤24 kg/m2, 25–28 kg/m2, ≥29 kg/m2), uterine status (intact uterus or no intact uterus), prior pregnancy (n = 0 or n ≥ 1), and number of vaginal births (n = 0 or n ≥ 1) at each time point.
Statistical analysis
The REJOICE trial was powered to compare each TX-004HR dose with placebo. Analyses were performed on the modified intent-to-treat (MITT) population defined as women who received their assigned treatment, had baseline values for all coprimary endpoints (vaginal superficial and parabasal cells, vaginal pH, dyspareunia severity), and had at least one postbaseline value for any of the four coprimary endpoints.
Statistical analyses were performed using SAS Release 9.2 (SAS Institute Inc., Cary, NC, USA). Baseline and demographic characteristics were descriptively summarized by treatment group. Each TX-004HR dose was compared with placebo in all post hoc analyses using mixed-effects model repeated-measures methods. Comparisons between TX-004HR doses and placebo for percentages of women with symptom improvement and complete symptom resolution were made using the two-sided Fisher’s exact test. Subgroup analyses, within each subgroup versus placebo, were also performed by mixed-effects model repeated-measures methods. Data comparisons were considered statistically significant at an alpha level of 0.05. No last observation carried forward methods for efficacy were used for missing data.
Results
Participant disposition and demographics
Participant demographics and characteristics have been described elsewhereCitation8. A total of 764 women were randomized to TX-004HR 4 µg (n = 191), 10 µg (n = 191), and 25 µg (n = 190) or placebo (n = 192). Overall, 704 (92.1%) participants completed the study. The MITT population consisted of 747 participants (97.8%).
Most women were Caucasian (87%), and had a mean age of 59 years and a mean BMI of 27 kg/m2 (). At baseline, all participants had an MBS of moderate to severe dyspareunia with a mean severity score of 2.7 (range 2–3). Of the 747 women in the MITT population, 733 (98.1%) also had mild, moderate, or severe vaginal dryness, with most (n = 698; 93.4%) having moderate to severe vaginal dryness. The baseline severity score for vaginal dryness was 2.4 (range 0–3). No overall differences between treatment groups were observed for baseline characteristics.
Table 1. Participant demographics and baseline characteristics (MITT population).
Vaginal symptom improvement
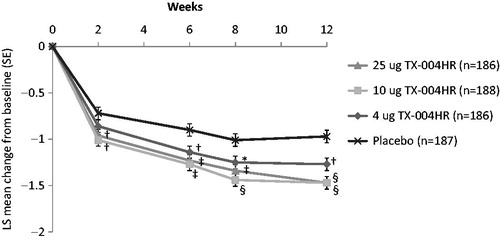
All doses of E2 vaginal insert compared with placebo significantly improved dyspareunia and vaginal dryness severity from baseline to week 12 (p < 0.05 for all), as previously reportedCitation8. shows improvements in vaginal dryness with TX-004HR over time in the entire study population, with significant improvements seen as early as 2 weeks with the 10-µg and 25-µg doses and at 6 weeks with the 4-µg dose.
Figure 1. Least-squares mean change in the severity of vaginal dryness from baseline to week 12 in the MITT population. *p < 0.05, †p < 0.01, ‡p < 0.001, §p < 0.0001 versus placebo. LS, least squares; MITT, modified intent-to-treat; SE, standard error.
When analyzed by level of improvements, significantly more participants had substantial improvement (improved by two levels or more) in dyspareunia when treated with the E2 vaginal insert (4 µg, 41.4%; 10 µg, 47.3%; 25 µg, 55.4%) than when treated with placebo (35.8%) after 12 weeks (p < 0.05 for 4-µg dose and p < 0.001 for the 10-µg and 25-µg doses; . These significant differences were observed as early as 2 weeks for the 10-µg and 25-µg doses and as early as 6 weeks for the 4-µg dose (). Dyspareunia also completely resolved (score of 0 = none) at week 12 in significantly more women using TX-004HR 10 µg (32.4%) and 25 µg (31.2%) than in those using placebo (19.8%; p < 0.05 for each). The proportion of women with complete resolution in those randomized to the 4-µg dose (25.8%) was not significantly different (p = 0.1693) from those using placebo.
Figure 2. Percentage of women who had substantial improvements (two levels or more) in severity of (A) dyspareunia and (B) vaginal dryness over 12 weeks. *p < 0.05, †p < 0.01, ‡p < 0.001 versus placebo in women with moderate to severe vaginal symptoms.
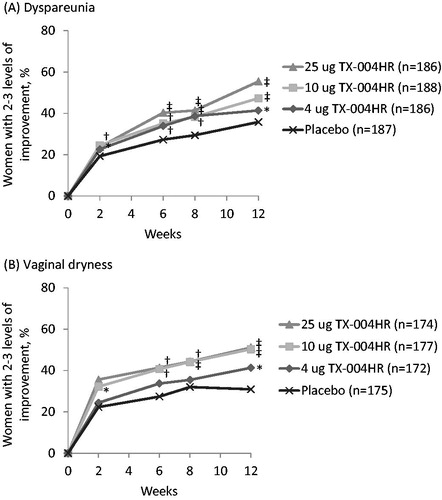
In women with moderate to severe vaginal dryness at baseline (n = 698), vaginal dryness severity also substantially improved (by two or three levels) in significantly more subjects treated with TX-004HR (4 µg, 41.3%; 10 µg, 50.3%; 25 µg, 51.1%) than those with placebo (30.9%) at week 12 (p < 0.05 for 4-µg dose and p < 0.001 for the 10-µg and 25-µg doses; . These significant improvements were observed as early as 2 weeks for the 10-µg dose and at 6 weeks for the 25-µg dose (). At 12 weeks, significantly more women had complete resolution of their vaginal dryness with all TX-004HR doses (30.8–38.3%) than with placebo (16.4%; p < 0.001 for all).
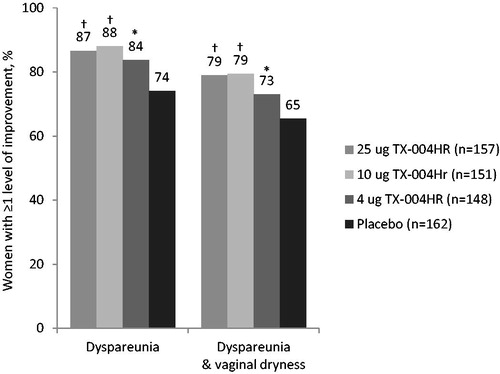
For women with mild, moderate, or severe vaginal dryness in addition to dyspareunia at baseline (n = 733), significantly more had a clinically meaningful improvement (of at least one level) in dyspareunia with all doses of the E2 vaginal insert (83.8–88.1%) versus placebo (74.1%) from baseline to week 12 (). Almost 80% of women in any TX-004HR group had concurrent clinically meaningful improvement of at least one level in both dyspareunia and vaginal dryness compared with 65% in the placebo group after 12 weeks (p < 0.05;).
Figure 3. Percentage of women with at least one level of improvement from baseline in dyspareunia, vaginal dryness, or both at week 12. Women with missing data or who did not have vaginal penetration were not included in the calculation. *p < 0.05, †p < 0.01 versus placebo.
Subgroup analysis for vaginal dryness
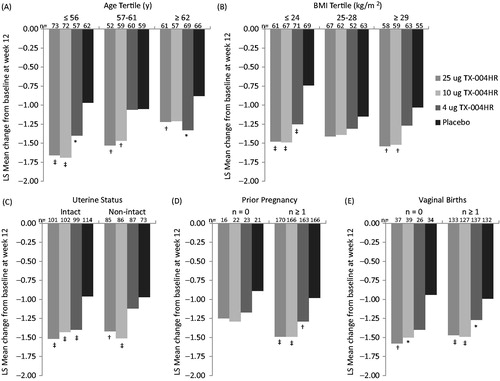
Baseline characteristics showed significant differences in the percentages of parabasal cells and vaginal pH by age as well as BMI, as previously reportedCitation12. Although not powered to evaluate efficacy in subgroups, treatment with 4, 10, and 25 µg of the E2 vaginal insert reduced vaginal dryness from baseline to week 12 in the majority of subgroups for age, BMI, uterine status, prior pregnancy, and number of vaginal births. The least-squares mean changes from baseline to week 12 for vaginal dryness are presented in . Several of the subgroups had significantly greater improvements in vaginal dryness with TX-004HR compared with placebo. Similar patterns of reduction for TX-004HR doses versus placebo were observed for subgroups that did not reach statistical significance.
Figure 4. Least-squares mean change in vaginal dryness score from baseline to week 12 by (A) age, (B) BMI, (C) uterine status, (D) prior pregnancy, and (E) number of vaginal births. *p < 0.05, †p < 0.01, ‡p < 0.001 versus placebo. BMI, body mass index; LS, least squares; y, years.
Discussion
These analyses demonstrate that women treated with an E2 softgel vaginal insert had clinically meaningful improvements in both dyspareunia and vaginal dryness (concurrent with dyspareunia), the two most commonly reported symptoms of VVA. Significantly more women treated with TX-004HR than placebo had complete resolution of their vaginal symptoms (dyspareunia or vaginal dryness) or substantial improvements (two levels or more) by 12 weeks, with effects observed as early as 2 weeks with most doses. Furthermore, in women with both moderate to severe dyspareunia and mild to severe vaginal dryness at baseline, both symptoms clinically meaningfully improved by at least one level with the E2 vaginal insert versus placebo by week 12. TX-004HR was also efficacious in reducing vaginal dryness, regardless of patient’s age, BMI, uterine status, prior pregnancy, and vaginal births.
These symptom data, from a large, rigorously collected and analyzed dataset, are particularly relevant for clinical practice as postmenopausal women with VVA frequently experience dyspareunia and/or vaginal dryness and often want to know when they may begin to feel better. These results provide information for clinicians to enable evidence-based discussions with their patients regarding the time frame and extent of their expected symptom relief (i.e. resolution of dryness or dyspareunia, or both). For example, greater than 80% had at least one level of improvement in their dyspareunia severity and approximately 75% of women had concurrent improvement of at least one level for both dyspareunia and vaginal dryness severity at week 12. Moreover, approximately 40% of women with moderate to severe symptoms who took TX-004HR doses might expect to have almost complete (mild severity score) or complete resolution of their dyspareunia as well as their vaginal dryness by 6 weeks of treatment. Results from the primary analyses also showed that dyspareunia and vaginal dryness significantly improved over time with all TX-004HR doses versus placebo from weeks 2 to 12 (except 4 µg, which significantly improved from weeks 6 to 12 for vaginal dryness)Citation8. However, clinicians should also counsel their patients with realistic expectations as to when they might expect improvement. Some women may respond to treatment as early as 2 weeks, but others may need to be patient and expect that resolution or improvement of their symptoms may not occur until week 12. Patients should also be reminded that VVA is a chronic condition whose symptoms may return after treatment discontinuation, and that adherence and compliance to treatment are very important factors in obtaining optimal results.
Data reported here also support the notion that VVA symptoms are intertwined and improve together rather than individually. Collectively, the primary REJOICE study analyses show that both dyspareunia and vaginal dryness improve with TX-004HR, as early as 2 weeks with most doses, in women with an MBS of moderate to severe dyspareuniaCitation8. These post hoc analyses extend those findings by showing that women with mild to severe vaginal dryness at baseline also had improvement in dyspareunia. These findings are consistent with the fact that local estrogens improve vaginal physiology, which is likely the basis for symptom improvement. Further, the REJOICE study also found that total Female Sexual Function Index scores and the pain and lubrication domain scores of the Female Sexual Function Index improved with 10-µg and 25-µg TX-004HR treatment, which were associated with improvements in vaginal cells and pHCitation13. The coprimary analyses of the REJOICE trial demonstrated that TX-004HR significantly increased the percentage of superficial cells (change from a baseline value of 1.3% to ∼18–24% with TX-004HR vs. 6% for placebo), decreased the percentage of parabasal cells (change from 52% to ∼8–11% vs. 7% for placebo), and lowered vaginal pH (change from 6.3 to ∼4.9 vs. 6 for placebo) at week 12 (p < 0.0001 for all doses at all time points)Citation8.
While the magnitudes of improvement for vaginal dryness severity were similar among most subgroups, showing a consistency of effect with TX-004HR, statistical significance was not observed for all subgroups. These results are similar to what was observed with dyspareunia subgroup analysesCitation12. The REJOICE trial was sufficiently powered to compare differences between treatment groups in the entire study population, but the power was insufficient to detect differences among smaller subgroups.
While data from the 12-week GSM trial from the Menopause Strategies: Finding Lasting Answers for Symptoms and Health (MsFLASH) research group suggest that prescribing vaginal 10-µg E2 tablets and over-the-counter vaginal moisturizer does not provide additional benefit over a placebo vaginal tablet and gel in reducing postmenopausal VVA symptomsCitation14, the trial had several limitations. As noted by Pinkerton et al.Citation15, the trial had a small sample size (n = 302); used a non-conventional placebo gel, which might have contributed to the high placebo response; and did not evaluate a specific MBS (i.e. dyspareunia) as required by the FDA, but rather evaluated many different symptoms as a single entity. Nonetheless, the MsFLASH trial found significantly greater improvement in total Menopause-specific Quality of Life Questionnaire scores with the vaginal E2 tablets compared with the placebo gelCitation16, and objective findings (vaginal pH and vaginal maturation index) as well as patient satisfaction favored the vaginal E2 tablets versus the placebo gelCitation14. Therefore, these results are limited in scope, and clinicians should not disregard the large body of evidence showing that low-dose vaginal estrogen can be useful in the management of GSM.
Pharmacokinetic studies of TX-004HR have shown negligible to very low systemic absorption of E2 compared with placeboCitation9, and significantly less absorption than the currently available vaginal E2 tabletCitation17. Therefore, postmenopausal women with symptomatic VVA whose main concern is the risks of systemic estrogen exposure as the reason why they do not want to take estrogen therapy may now consider this E2 vaginal insertCitation7,Citation18,Citation19. Given its pharmacokinetic profile, TX-004HR may represent an estrogen therapy option for survivors of estrogen-dependent breast cancer in whom non-hormonal treatments are not effective, as supported by the American College of Obstetricians and Gynecologists and the North American Menopause SocietyCitation20,Citation21.
Limitations of this study include the fact that most of the women enrolled in the REJOICE trial were white, in good health, and had an average BMI of 27 kg/m2, and hence may not be representative of all postmenopausal women with VVA. The requirement for having an MBS of moderate to severe dyspareunia may also somewhat limit the population with regard to VVA symptoms; however, this was not the case as most women (93.4%, 698/747) also had moderate to severe vaginal dryness at baseline. Another limitation is that data were only collected at a limited number of time points (between 2 and 12 weeks), and thus onset of effect could not be observed before 2 weeks or between 2 and 6 weeks, which may be useful information for clinicians when counseling their patients on when to expect symptom improvement. Finally, a large effect was observed in the placebo group in this study, most likely due to the use of Miglyol®, a fractionated coconut oil with potential lubricative properties and a component of the softgel capsulesCitation13. However, improvements in vaginal physiology and VVA symptoms occurred with the low doses of E2 in TX-004HR over placebo despite the lubricating effects of the coconut oil.
In conclusion, TX-004HR was shown to provide clinically meaningful improvements in postmenopausal women with VVA and moderate to severe dyspareunia. Improvements in dyspareunia and concurrent vaginal dryness were observed as early as 2 weeks and were maintained over the 12-week study period. Improvements in vaginal dryness were also observed independent of baseline characteristics. Clinicians may find this information useful when discussing patients’ expectations with regard to symptom improvement with this E2 softgel vaginal insert.
Potential conflict of interest
Dr Simon has served (within the past year, or current) as a consultant/advisor to AbbVie, Allergan plc, AMAG, Amgen, Ascend Therapeutics, Bayer Healthcare, CEEK Enterprises, Covance, Dare Bioscience, Duchesnay, Hologic, KaNDy/NeRRe Therapeutics, Mitsubishi Tanage, ObsEva SA, Palatin Technologies, Sanofi SA, Shionogi, Sprout, and TherapeuticsMD; has received (within the past year, or current) grant/research support from AbbVie, Agile Therapeutics, Allergan plc, Bayer Healthcare, Endocuetics, GTx, Ipsen, Myovant Sciences, New England Research Institute, ObsEva SA, Palatin Technologies, Symbio Research, TherapeuticsMD, and Viveve Medical; has also served (within the past year, or current) on the speaker's bureaus of AbbVie, AMAG, Duchesnay, Novo Nordisk, Shionogi, and TherapeuticsMD; and is a stockholder (direct purchase) in Sermonix Pharmaceuticals. Dr Kagan is a consultant to or on the advisory boards of Allergan, Cooper Surgical, Duchesnay, Lupin, Noven, Proctor & Gamble, Radius Health, and TherapeuticsMD; and has served on the speakers’ bureau of AMAG, Cooper Surgical, and TherapeuticsMD. Dr Archer serves as a consultant for AbbVie, Actavis, Agile Therapeutics, Bayer Healthcare, Endoceutics, Exeltis, InnovaGyn, Merck, Pfizer, Radius Health, Sermonix, Shionogi, Teva Women’s Healthcare, and TherapeuticsMD; and has received research support from Actavis, Bayer Healthcare, Endoceutics, Glenmark, Merck, Radius Health, Shionogi, and TherapeuticsMD. Dr Constantine consults to multiple pharmaceutical companies, including but not limited to TherapeuticsMD, and has stock options with TherapeuticsMD. Dr Bernick is a board member and an employee of TherapeuticsMD with stock/stock options. Dr Graham and Dr Mirkin are employees of TherapeuticsMD with stock/stock options.
Acknowledgements
The authors would like to thank the investigators of the REJOICE Study Group. The authors would also like to acknowledge the medical writing assistance of Dominique Verlaan, PhD, of Precise Publications, LLC, which was supported by TherapeuticsMD.
Additional information
Funding
References
- Mac Bride MB, Rhodes DJ, Shuster LT. Vulvovaginal atrophy. Mayo Clin Proc 2010;85:87–94
- Portman DJ, Gass ML. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women's Sexual Health and the North American Menopause Society. Menopause 2014;21:1063–8
- Nappi RE, Kokot-Kierepa M. Women's voices in the menopause: Results from an international survey on vaginal atrophy. Maturitas 2010;67:233–8
- Nappi RE, Kokot-Kierepa M. Vaginal health: Insights, views & attitudes (VIVA)–results from an international survey. Climacteric 2012;15:36–44
- Krychman M, Kingsberg S. REVEAL: revealing vaginal effects at mid-life: surveys of postmenopausal women and health care professionals who treat postmenopausal women. (Personal Communication) 2016
- Simon JA, Kokot-Kierepa M, Goldstein J, et al. Vaginal health in the United States: results from the Vaginal Health: insights, Views & Attitudes survey. Menopause 2013;20:1043–8
- Kingsberg S, Krychman M, Graham S, et al. The Women’s EMPOWER Survey: Identifying women’s perceptions on vulvar and vaginal atrophy (VVA) and its treatment. J Sex Med 2017;14:413–24
- Constantine G, Simon JA, Pickar JH, et al. The REJOICE trial: A phase 3 randomized, controlled trial evaluating the safety and efficacy of a novel vaginal estradiol soft-gel capsule for symptomatic vulvar and vaginal atrophy. Menopause 2017;24:409–16
- Archer DF, Constantine GD, Simon J, et al. TX-004HR vaginal estradiol has negligible to very low systemic absorption of estradiol. Menopause 2017;24:510–16
- Kingsberg S, Kroll R, Goldstein I, et al. Patient acceptability and satisfaction with a low-dose solubilized vaginal estradiol softgel capsule, TX-004HR. Menopause 2017;24:894–9
- Nappi RE, Panay N, Bruyniks N, et al. The clinical relevance of the effect of ospemifene on symptoms of vulvar and vaginal atrophy. Climacteric 2015;18:233–40
- Constantine GD, Bouchard C, Pickar JH, et al. Consistency of effect with a low-dose, estradiol vaginal capsule (TX-004HR): evaluating improvement in vaginal physiology and moderate-to-severe dyspareunia in subgroups of postmenopausal women. J Womens Health 2017;26:616–23
- Kingsberg S, Derogatis L, Simon JA, et al. TX-004HR improves sexual function as measured by the female sexual function index in postmenopausal women with vulvovaginal vulvar and vaginal atrophy: the REJOICE Trial. J Sex Med 2016;13:1930–7
- Mitchell CM, Reed SD, Diem S, et al. Efficacy of vaginal estradiol or vaginal moisturizer vs placebo for treating postmenopausal vulvovaginal symptoms: a randomized clinical trial. JAMA Intern Med 2018;178:681–90
- Pinkerton JV, Kaunitz AM, Manson JE. Not time to abandon use of local vaginal hormone therapies. Menopause 2018;25:855–8
- Diem SJ, Guthrie KA, Mitchell CM, et al. Effects of vaginal estradiol tablets and moisturizer on menopause-specific quality of life and mood in healthy postmenopausal women with vaginal symptoms: a randomized clinical trial. Menopause 2018;25:1086–93
- Pickar JH, Amadio JM, Bernick BA, et al. Pharmacokinetic studies of solubilized estradiol given vaginally in a novel softgel capsule. Climacteric 2016;19:181–7
- Nappi RE, Palacios S, Panay N, et al. Vulvar and vaginal atrophy in four European countries: evidence from the European REVIVE Survey. Climacteric 2016;19:188–97
- Kingsberg SA, Wysocki S, Magnus L, et al. Vulvar and vaginal atrophy in postmenopausal women: findings from the REVIVE (REal Women's VIews of Treatment Options for Menopausal Vaginal ChangEs) survey. J Sex Med 2013;10:1790–9
- The use of vaginal estrogen in women with a history of estrogen-dependent breast cancer. Committee Opinion No. 659. American College of Obstetricians and Gynecologists. Obstet Gynecol 2016;127:e93–e96
- Management of symptomatic vulvovaginal atrophy: 2013 position statement of The North American Menopause Society. Menopause 2013;20:888–902