
Abstract
Objective
This study aimed to measure safety, systemic pharmacokinetics and preliminary efficacy of a vaginal tamoxifen capsule (DARE-VVA1) among postmenopausal women with moderate-to-severe vulvovaginal atrophy.
Methods
This was a randomized, placebo-controlled, double-blind, phase 1/2 study of DARE-VVA1, in four doses (1, 5, 10 and 20 mg).
Results
Seventeen women were enrolled and 14 completed the 8-week treatment. DARE-VVA1 was safe. All adverse events were of mild or moderate severity and distributed similarly among active and placebo groups. Plasma tamoxifen concentrations were highest among women using DARE-VVA1 20 mg, but the maximum mean (standard deviation) plasma tamoxifen concentrations on day 1 (2.66 ± 0.85 ng/ml) and day 56 (5.69 ± 1.87 ng/ml) were <14% of those measured after one oral tamoxifen dose. Active study product users had significant decreases from pre-treatment baseline in vaginal pH and proportion of vaginal parabasal cells (p = 0.04 for both endpoints), with women randomized to the 10 mg or 20 mg dose experiencing the largest treatment impact. The severity of vaginal dryness and dyspareunia decreased significantly from baseline with active study product use (p = 0.02 for both endpoints).
Conclusions
DARE-VVA1 is safe and results in minimal systemic exposure to tamoxifen. Preliminary efficacy data support further development of this product.
摘要
目的: 本研究旨在判定阴道他莫昔芬胶囊(DARE-VVA1)在绝经后中度至重度外阴阴道萎缩妇女中的安全性、系统药代动力学和初步疗效。
方法: 这是一项DARE-VVA1的随机、安慰剂对照、双盲、1/2期研究, 分为四个剂量(1、5、10和20mg)。
结果: 17名女性入选, 14名完成了为期8周的治疗。DARE-VVA1是安全的。所有不良事件均为轻度或中度, 在活跃组和安慰剂组中的分布相似。在使用DARE-VVA1 20 mg的女性中, 血浆他莫昔芬浓度最高, 但第1天(2.66 ± 0.85 ng/ml)和第56天(5.69 ± 1.87 ng/ml)的最大平均(标准差)血浆他莫昔芬浓度小于一次口服他莫昔芬后测得的浓度的14%。与治疗前基线相比, 活跃的研究产品使用者的阴道pH值和阴道副基底细胞比例显著下降(两个终点p = 0.04), 随机接受10 mg或20 mg剂量的女性受到的治疗影响最大。积极使用研究产品后, 阴道干燥和性交困难的严重程度较基线显著降低(两个终点p = 0.02)。
结论: DARE-VVA1是安全的, 并导致对他莫昔芬的最小全身暴露。初步疗效数据支持该产品的进一步开发。
Introduction
Vulvovaginal atrophy (VVA) is a common and underreported condition that results from decreased local mucosal estrogenization of the vaginal epithelium [Citation1–3]. The vaginal epithelium has decreased proliferation and glycogen content and becomes thinned and fragile with chronic estrogen deprivation. Typical symptoms include vaginal dryness, itching, burning and painful intercourse, adversely impacting an individual’s quality of life [Citation3]. Clinical findings include the diagnostic presence of a pale, friable and/or dry vulvovaginal mucosa, as well as vaginal pH > 5 [Citation3]. VVA most commonly occurs in the postmenopausal phase, during which the prevalence is more than 50% [Citation2,Citation4]. Localized micro-dose estrogen therapy (e.g. estradiol [E2]-containing vaginal creams, rings and tablets) is effective in treating the underlying physiologic issue, and is the most commonly prescribed treatment for VVA [Citation5]. However, the majority of health-care providers remain reluctant to prescribe vaginal E2 therapy in women with hormone receptor-positive (HR+) breast cancer and in women who have contraindications to systemic estrogen therapy [Citation6] because the approved package labeling notes that this therapeutic approach should not be used for these women. Although systemic E2 exposure is negligible with micro-dose vaginal E2 therapy, the exposure concern persists among health-care providers and women diagnosed with breast cancer [Citation6]. In addition, some women would like a vaginal, non-hormonal, non-estrogen option for VVA treatment. Although an oral, non-estrogen VVA treatment (Ospemifene®) was recently approved for use, it has not been studied in women with HR + breast cancer [Citation7].
Tamoxifen is an estrogen agonist/antagonist that elicits tissue-dependent responses in hormone-responsive tissues and organs, such as the vagina, breast and endometrium [Citation8]. Studies of oral tamoxifen conducted over the last 40 years have documented estrogen agonist effects of this oral selective estrogen receptor modulator (SERM) on the vaginal epithelium in postmenopausal women, but an estrogen antagonist action on the vaginal epithelium in premenopausal users [Citation9–16]. There remains a large, unmet need for a novel, non-hormonal, SERM, non-E2, local VVA treatment.
The primary objectives of this first-in-woman study were to characterize the local and systemic safety and systemic plasma pharmacokinetics (PK) of DARE-VVA1 vaginal inserts in four doses (1, 5, 10 and 20 mg) versus placebo, when dosed over a 56-day treatment window. The secondary objectives of this study were to evaluate the preliminary efficacy and pharmacodynamics (PD) of DARE-VVA1 by describing changes from pre-treatment baseline in the severity of the most bothersome genitourinary symptom (MBS) and changes in the vaginal maturation index (VMI) and vaginal pH.
Methods
Clinical study
This was a phase 1/2, randomized, double-blind, placebo-controlled study among postmenopausal women with moderate-to-severe VVA at two centers in Australia. The study was approved by the ethics board for each study site (Central Adelaide Local Health Network Human Research Ethics Committee Reference Number 2021/HRE00239) and registered with ClinicalTrials.gov (NCT05378269). Briefly, the sites enrolled healthy women, aged 40–75 years, who were not taking exogenous hormones, and were menopausal as defined by 12 months of spontaneous amenorrhea or 6 months of spontaneous amenorrhea with a serum follicle stimulating hormone (FSH) concentration of 40 mIU/ml or higher. On a 4-point Likert scale of no, mild, moderate or severe symptoms, women had to self-report that at least one of the following symptoms was moderate (level 2) or severe (level 3) in intensity: vaginal dryness; vaginal and/or vulvar irritation/itching; dysuria; or vaginal pain with sexual activity (dyspareunia). Alternatively, women with vaginal bleeding associated with sexual activity were also eligible for the study. Volunteers had to have vaginal cellular cytology with ≤5% superficial cells and vaginal pH > 5 at screening. For women with an intact uterus, the endometrial thickness had to be ≤4 mm on transvaginal ultrasound. Subjects had to have a normal mammogram within 2 years of the screening visit, normal cervical cytology cancer screening, a normal baseline 12-lead electrocardiogram and baseline laboratory values either within normal limits or accepted by the investigator and medical monitor as not clinically significant. Participants were excluded if they had significant cardiovascular, renal, pulmonary, neurological or hepatic diseases preventing compliance with the study, if they were currently taking anticoagulant drugs or if they had uncontrolled hypertension. Finally, for this first-in-woman study, women could not have a history of cancer in the past 5 years, undiagnosed vaginal bleeding or a known or suspected estrogen-dependent neoplasia.
Women self-administered a vaginal soft gel capsule once a day for the first 2 weeks and then twice weekly for 6 weeks (approximately 26 doses), which is the normal dosing frequency for many micro-dose vaginal E2 VVA creams and vaginal tablet regimens [Citation5]. They were seen over a total of approximately 10 visits where safety, PK, PD and acceptability endpoints were collected (see Supplemental Table 1).
Randomization
In this parallel-group randomized study, women who met the study entry criteria were randomly assigned, in a 1:1:1:1:1 ratio, to one of the five treatment groups (DARE-VVA1 1 mg or 5 mg or 10 mg or 20 mg dose or matching placebo). The randomization schedule was computer-generated using a permuted block algorithm. The study center was not a blocking factor in the randomization schedule. The randomization numbers were assigned sequentially by an un-blinded member of the ICON Clinical Research Team who was not otherwise involved in the study. No one involved in the study performance had access to the randomization schedule before the official un-blinding of treatment assignment.
Blinding and un-blinding treatment assignment
The different tamoxifen-dose DARE-VVA1 vaginal capsules and placebo capsules are indistinguishable by appearance, smell and feel. All participants, investigators and study personnel involved in the conduct of the study, including data management, were blinded to treatment assignment with the exception of a specified un-blinded statistician from ICON Clinical Research who had access to the randomization code. The un-blinded study personnel did not participate in study procedures or data analysis prior to un-blinding of the study data to all study-related personnel.
Study product
DARE-VVA1 was supplied as 1, 5, 10 and 20 mg vaginal capsules or matching placebo capsules by Catalent Pharma Solutions (St. Petersburg, FL, USA). All study products were stored at room temperature.
Safety assessments
Safety was assessed primarily through treatment emergent adverse events (TEAEs), which were graded as to their severity and relationship to product use. Any vaginal or vulvar irritation, vaginal discharge or pelvic pain was reported as an adverse event (AE) using standardized Medical Dictionary for Regulatory Activities (MedDRA) codes and graded for severity. Adverse drug reactions (ADRs) were defined as all noxious and unintended responses to a medicinal product, for which a causal relationship between a medicinal product and an AE is at least a reasonable possibility (i.e. the relationship cannot be ruled out). All AEs judged by either the reporting investigator or the sponsor as having a reasonable causal relationship to an investigational product (IP) qualify as ADRs. An unexpected ADR was defined as an ADR for which the nature or severity is not consistent with the applicable product information (e.g. Investigator Brochure for an unapproved IP).
Safety was also measured by changes from screening in clinical laboratory assessments (hematology, serum chemistry, coagulation panel, urinalysis), vital signs, 12-lead electrocardiograms, physical examinations and endometrial stripe width measurements by transvaginal ultrasound. During each vaginal speculum examination, local erythema and edema were graded by the investigator on a 4-point Likert scale of none/absent (score 0), mild (score 1), moderate (score 2) and severe (score 3).
Pharmacokinetic evaluations
After baseline evaluations including the time 0 venous plasma sample, the day 1 DARE-VVA1 dose or matching placebo was self-administered in the clinic and serial peripheral venous samples were drawn from an indwelling line at 0.5, 1, 2, 4, 5, 8, 12 and 24 h. Trough samples were collected 24 h after administration of inserts on days 4, 7, 10, 18, 28, 42 and 56 (end of treatment) and a final sample was collected at follow-up on day 63, approximately 7 days since the last dose. On the last day of dosing (day 56), serial peripheral venous samples were repeated at 0.5, 1, 2, 4, 5, 8, 12 and 24 h.
Plasma concentrations of tamoxifen and three tamoxifen metabolites (N-desmethyltamoxifen [NDT], 4-hydroxytamoxifen [4-OHT] and N-desmethyl-4-hydroxytamoxifen [endoxifen]) were analyzed by Agilex Laboratories using dual tandem liquid chromatography and mass spectrometry (LC/MS-MS). The lower limit of quantitation was 0.1 ng/ml (tamoxifen), 0.1 ng/ml (NDT), 0.05 ng/ml (4-OHT) and 0.05 ng/ml (endoxifen). For PK characterization, concentrations for each dose were calculated using non-compartmental analysis. Concentrations that were below the lower limit of quantitation (BLQ) were estimated as 0.5 × lower limit of quantitation. The PK parameter estimates were completed using WinNonlin (Pharsight Corporation). The actual sampling time was used for all parameter estimations. Standard PK parameters assessed included measures of the extent of absorption using estimates of the area under the plasma concentration–time curve, the maximum observed drug concentration and the time to reach the maximum drug concentration.
Preliminary pharmacodynamics and preliminary treatment efficacy
Local estrogenic effect measurements included obtaining vaginal epithelial cells from the lateral vaginal sidewall and determining the VMI. The VMI is determined by categorizing the ratio (proportion in 100 cells) of the three types of vaginal epithelial cell types (parabasal, intermediate and superficial). A total VMI score was calculated as: % superficial cells + (0.5 × % intermediate cells) + (0 × % parabasal cells). The VMI and vaginal pH were measured at screening and baseline, and subsequently every 2 weeks during treatment and 1 week after treatment.
At the screening visit, participants rated on a 4-item Likert scale (not present [none], mild, moderate or severe) the severity of the following vaginal symptoms: vaginal dryness; vaginal and/or vulvar irritation/itching; dysuria; and vaginal pain associated with sexual activity (dyspareunia). Additionally, participants indicated whether vaginal bleeding associated with vaginal activity was present or absent. They also selected one of the five symptoms as their MBS. All five symptoms were evaluated again during later study visits and the change in severity from baseline was determined.
Sample size and statistical analysis
As is the case for first-in-woman safety and PK studies, the primary endpoints of this study were primarily descriptive in nature. The sample size for this first-in-woman safety and PK study was originally estimated to include approximately eight participants in each of the five dosing groups. However, study recruitment was adversely impacted by the COVID-19 pandemic and was halted once approximately half of the enrollment goal was met after a blinded interim analysis of some select secondary endpoints revealed potential treatment efficacy potential.
The following three analysis populations were described: safety population, which included all participants who were enrolled and received any amount of planned IP; intent-to-treat population, which included all randomized participants, who were used for the analysis of secondary efficacy endpoints; and PK population, which included all participants who received at least one dose of IP and provided at least one quantifiable PK plasma sample.
Summary statistics were described for each dosing cohort and for the active combined doses (1 mg and 5 mg and 10 mg and 20 mg) versus placebo. For primary safety and PK endpoints, data for each dosing cohort were described separately. Because the sample size for each active dosing cohort was small (n = 3–4 participants), comparisons between all active product users versus placebo were used for the secondary endpoints of preliminary product efficacy (vaginal pH, VMI and MBS). Continuous variables included the number of participants, mean, standard deviation, median, minimum and maximum. For categorical variables, we present the number and percentage of participants in each category. The denominator for the percentage is based on the number of participants appropriate for the purpose of analysis. For all calculations of change from baseline, the last observation recorded before the first administration of the insert (DARE-VVA1 or placebo) was considered as the baseline observation. Paired changes from baseline were compared using a Wilcoxon signed rank sum test, as all continuous variables were not normally distributed. Comparison of categorical variables was done by Fisher’s exact test.
Systemic plasma PK/local vaginal PD endpoint correlations were done using the Spearman correlation coefficient. Plasma concentrations of tamoxifen obtained at days 0, 1, 18, 28, 42, 57 and 63 were correlated with vaginal pH measurements and VMI parameters obtained at the same time point for all DARE-VVA1 users combined. p-Values <0.05 were considered statistically significant.
Results
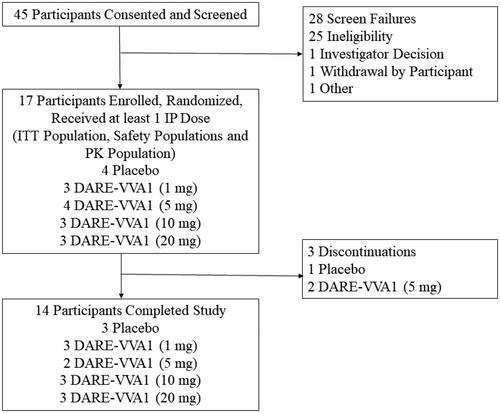
As shown in , 45 participants were screened for the study and 28 participants were screen failures. We therefore enrolled 17 women, all of whom had an intact uterus and were randomized and dosed with IP. A total of 14 women completed the study. The three participants who discontinued the study did so due to protocol deviations (n = 1) and non-compliance with the study medication (n = 2).
Figure 1. Disposition of participants. IP, investigational product; ITT, intent to treat; PK, pharmacokinetic.
presents the demographics of the intent-to-treat population. All enrolled women had a normal screening thyroid stimulating hormone (TSH) concentration, normal Pap smear results and a normal screening mammogram. All participants were menopausal based on the presence of spontaneous amenorrhea for 12 months or longer.
Table 1. Demographics of the intent-to-treat population.
Safety data
presents the TEAE data and Supplemental Table 2 presents the total exposure to tamoxifen (mg), number of days of dosing, protocol adherence per participant self-reported diary entries and the system organ preferred term for product-related TEAEs. As already noted, 14 of the 17 randomized participants completed the entire 56-day treatment period. The majority of participants were greater than 95% compliant with the dosing regimen, per their diary report.
Table 2. Safety data.
The were no severe adverse events and no TEAEs which led to study product or study discontinuation. The majority of participants who received at least one dose of the study IP (15/17, 88.2%) reported at least one TEAE. Of the 72 reported TEAEs for all participants, the highest number of reported TEAEs occurred in the 20 mg DARE-VVA1 dosing group (n = 20 TEAEs), with the second highest reported by the placebo group (n = 15 total reported TEAEs). All TEAEs were mild or moderate in severity.
Almost half (8/17, 47%) of the participants reported at least one TEAE which was deemed as possibly related or related to study IP use ( and see Supplemental Table 2). Of the 22 TEAEs reported by the 17 participants that were deemed possibly related or related to study IP use, the highest number (n = 8) was reported by the placebo users (). Finally, 5/17 (29%) participants reported at least one ADR, with a total of nine ADRs being reported in the study. There were no unexpected ADRs reported in this study.
Of the 15 participants who reported at least one TEAE, nine reported a TEAE related to the reproductive system, with vulvovaginal discomfort (n = 5 participant reports) and vulvovaginal pruritus (n = 4 participant reports) being the most common organ system preferred term (see Supplemental Table 2).
The mean local erythema scores at all visits, for all dosing groups, were in the none/absent (score 0) to mild (score 1) range, with a few outliers in the moderate (score 2) grading, with no discernible pattern or correlation to group. All local edema scores at all visits, for all dosing groups, were graded as none (score 0) or mild (score 1). All endometrial stripe measurements were normal at baseline and at 57 days of treatment, with the maximum measurement not exceeding 4.0 mm. All vaginal speculum examinations were reported as normal or had an abnormality or finding that was deemed not clinically significant by the investigator.
Finally, there were isolated, not clinically significant changes in serum chemistry, hematology and coagulation parameters and concentrations, and urinalyses, with no discernible pattern or dose relationship. There were no clinically significant electrocardiogram findings or changes from baseline during the study.
Systemic plasma pharmacokinetics
PK parameters for tamoxifen and NDT are presented in Supplemental Table 3, and –l shows the mean (standard deviation) plasma concentrations by time for tamoxifen and the three metabolites (ng/ml). As expected, the highest median plasma concentrations of tamoxifen occurred in the 20 mg dosing group. The highest median plasma tamoxifen concentration measured in the study was 8.29 ng/ml, measured on day 10 of dosing in the 20 mg dosing group. The single maximum plasma tamoxifen concentration observed in the study was 12.0 ng/ml, from a participant randomized to the 20 mg dose at the same time point (see Supplemental Table 3).
Figure 2. (a) Plasma tamoxifen 1 mg dosing group. (b) Plasma tamoxifen 5 mg dosing group. (c) Plasma tamoxifen 10 mg dosing group. (d) Plasma tamoxifen 20 mg dosing group. (e) Plasma NDT 1 mg dosing group. (f) Plasma NDT 5 mg dosing group. (g) Plasma NDT 10 mg dosing group. (h) Plasma NDT 20 mg dosing group. (i) Plasma endoxifen 10 mg dosing group. (j) Plasma endoxifen 20 mg dosing group. (k) Plasma 4-OHT 10 mg dosing group. (l) Plasma 4-OHT 20 mg dosing group. EOT, end of treatment; LLOQ, lower limit of quantitation; NDT, N-desmethyltamoxifen; 4-OHT, 4-hydroxytamoxifen.
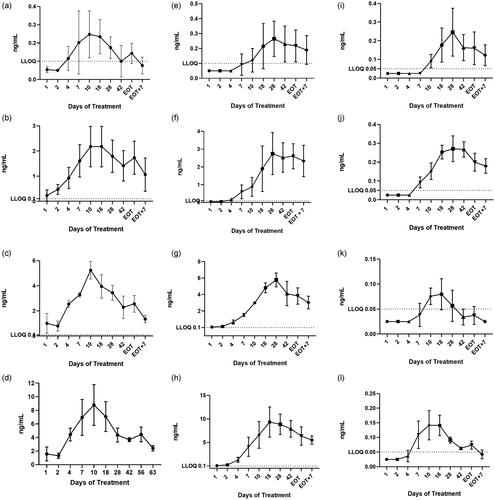
The major metabolite of orally dosed tamoxifen is NDT [Citation17]. As with the tamoxifen plasma PK profile, VVA1 20 mg users had the highest concentrations of NDT. The highest median plasma concentration of NDT was 8.43 ng/ml, measured on day 18 of dosing, with a single maximum plasma concentration of NDT recorded at 12.9 ng/ml on the same day. The mean and median PK parameters of plasma tamoxifen and NDT are presented in .
Table 3. Pharmacokinetic parameters for plasma tamoxifen and N-desmethyl tamoxifen (NDT).
For endoxifen concentrations, DARE-VVA1 1 mg users had BLQ concentrations of this metabolite at all sampling points. DARE-VVA1 5 mg users had BLQ concentrations of this metabolite at all time points except day 42 where median endoxifen concentrations were 0.056 ng/ml (range 0.03–0.14 ng/ml). Supplemental Table 3 and demonstrate that DARE-VVA1 10 mg users had BLQ concentrations of plasma endoxifen until day 10 of dosing, while DARE-VVA1 20 mg users had BLQ concentrations of plasma endoxifen until day 7 of dosing.
DARE-VVA1 1 mg and 5 mg users had concentrations of the metabolite 4-OHT that were BLQ at all time points measured. As shown in Supplemental Table 3 and , DARE-VVA1 10 mg users had BLQ concentrations of this metabolite until day 10 of dosing, and plasma 4-OHT concentrations were then low but measurable between days 10 and 42, when concentrations returned to BLQ levels. As shown in Supplemental Table 3 and , DARE-VVA1 20 mg users had BLQ concentrations of 4-OHT until day 7 of dosing and had very low concentrations of this metabolite throughout the observation period.
Vaginal pH and vaginal maturation index
Both of these preliminary assessments of product efficacy were secondary endpoints, as the sample size was not powered to demonstrate significant differences for individual dosing groups. When all active DARE-VVA1 doses were combined into one group, there was a significant decrease in vaginal pH between baseline and end of treatment (day 56), with the DARE-VVA1 20 mg cohort experiencing the largest decrease in median vaginal pH (–0.7) (). The 1 mg and 5 mg dosing cohorts experienced a median decrease in vaginal pH of −0.4 and 0, respectively.
Table 4. Vaginal pH and vaginal cytology indices at baseline and end of treatment by dosing group.
Similar to the data for vaginal pH, the overall VMI increased with treatment, although not significantly so for any individual dosing group (all p > 0.09) (). The largest increase in total VMI, reflecting the largest shift to healthy, superficial cells, occurred in the 20 mg VVA1 dosing group. This dosing group also had the largest increase in superficial cells, from a median of 0% at baseline to a median of 54% at the end of treatment (p = 0.06). There was a statistically significant decrease in vaginal parabasal cells, from a median of 75% at baseline to a median of 1% at end of treatment (p = 0.04), for all active study product users ().
Systemic plasma tamoxifen pharmacokinetic/local vaginal Pharmacodynamic correlations
Plasma tamoxifen concentrations were significantly and negatively correlated with vaginal pH (Spearman R = −0.51, p < 0.01) and % vaginal parabasal cells (Spearman R = −0.53, p < 0.01). Plasma tamoxifen concentrations were significantly and positively correlated with % vaginal superficial cells (Spearman R = 0.45, p < 0.01), % vaginal intermediate cells (Spearman R = 0.45, p < 0.01) and total VMI (Spearman R = 0.62, p < 0.01).
Most bothersome genitourinary symptom and severity of symptom
Of the 17 enrolled participants, seven reported that vaginal dryness was their MBS and 10 reported that dyspareunia was their MBS at screening. presents the severity of the MBS reported by participants in the placebo group versus all active VVA1 users combined for this secondary endpoint. Among active VVA1 users, there was a significant decrease in the severity of vaginal dryness and dyspareunia from baseline at the end of treatment (p = 0.02 for both indices). Placebo users did not experience significant changes in the severity of their MBS from baseline (p = 0.17 for vaginal dryness and p = 0.33 for dyspareunia). Due to sample size, the data were not analyzed by individual active dosing group.
Table 5. Frequency of most bothersome genitourinary symptom and severity by dosing group at pretreatmentbaseline and end of treatment.
Discussion
Tamoxifen is a SERM which acts as an estrogen antagonist in the breast; it binds to alpha estrogen receptors in breast tissue, effectively blocking natural estrogen from binding to these sites, and is therefore an effective HR + breast cancer treatment [Citation17,Citation18]. Tamoxifen was approved by the US Federal Drug Administration (FDA) in oral dosage forms (tablet and solution) for the prevention and treatment of estrogen receptor-positive breast cancer in 1977 [Citation19]. The tissue-specific effects of tamoxifen, importantly, are dependent on the patient’s age and endogenous hormonal profile, and currently, oral tamoxifen is primarily given to premenopausal women diagnosed with breast cancer. Oral, systemic tamoxifen is known to cause VVA in premenopausal women by working at the vaginal tissue level to block estrogen activity [Citation20,Citation21]. Postmenopausal breast cancer patients are often treated with oral aromatase inhibitors, which may amplify VVA by blocking any peripheral conversion of steroid hormones to E2 centrally [Citation22]. Thus, the prevalence of VVA in both premenopausal and postmenopausal HR + breast cancer survivors is estimated to be 70% [Citation23,Citation24].
A previously conducted, exploratory, proof-of-concept clinical study, where four postmenopausal women with moderate-to-severe VVA symptoms were treated with a preliminary formulation of DARE-VVA1 (approximately 13 mg tamoxifen citrate vaginal suppository, produced by a compounding pharmacy) intra-vaginally once daily for a week and then twice weekly for 3 months found decreases in vaginal pH and improvements in patient-reported assessments of vaginal dryness [Citation25].
This current first-in-woman study of DARE-VVA1 at four dosing strengths (1, 5, 10 and 20 mg) demonstrated that during 8 weeks of use (daily for 2 weeks and twice weekly for 6 weeks), dosed in the same dosing frequency as several other approved micro-dose VVA E2 therapies [Citation5], DARE-VVA1, in all strengths, was safe and had an AE profile similar to placebo. All TEAEs were mild to moderate, with no participant experiencing a TEAE that was severe or required study drug discontinuation. Vaginal speculum examinations were normal, with most assessments of vaginal edema or erythema graded as not present or mild.
Due to ovulatory concentrations of serum progesterone in premenopausal women, the risk of endometrial hyperplasia with oral tamoxifen use is no different than in the general premenopausal population [Citation26]. However, among postmenopausal women taking daily oral tamoxifen for at least 4 years, the risk of simple hyperplasia is approximately 12%, of complex hyperplasia 3% and of endometrial carcinoma is 2% [Citation27,Citation28], which is about two to three times that of the general postmenopausal population. We assessed the risk of endometrial hyperplasia in this first-in-woman study by transvaginal ultrasound measurement of the endometrial stripe width and found that all participants had an endometrial stripe width of ≤4 mm. Although this is reassuring, we plan to obtain endometrial tissue biopsies at pre-treatment baseline and the end of treatment in future studies of DARE-VVA1.
The low-risk systemic safety profile seen in this study was likely due to the minimal systemic drug exposure experienced by active product users, even among women randomized to the highest DARE-VVA1 dose (20 mg). After a single oral dose of tamoxifen (20 mg), the mean maximum plasma concentration of tamoxifen is 40 ng/ml (range 35–45 ng/ml) [Citation19]. Mean steady-state concentrations of orally administered tamoxifen (20 mg dose) are 122 ng/ml (range 71–183) [Citation19]. Participants in the highest DARE-VVA1 dosing group experienced median plasma maximum observed drug concentration and tamoxifen concentrations that were at least 10-fold less than those seen with systemic, oral tamoxifen use [Citation19].
Orally dosed tamoxifen is metabolized in the liver by the cytochrome P450 system [Citation20]. The first metabolite of tamoxifen after CYP3A 4/5 degradation is NDT, which is approximately 1.7× more potent systemically than tamoxifen [Citation20]. When dosed orally, the mean peak plasma concentration of NDT is 15 ng/ml (range 10–20 ng/ml) [Citation19]. The average steady-state plasma concentration of NDT after 3 months of oral administration of daily 20 mg tamoxifen is 353 ng/ml (range 152–706 ng/ml) [Citation19]. During 8 weeks of VVA1 use, we found that the major metabolite of tamoxifen, NDT, was present in plasma at concentrations which were less than 2% of that seen with oral tamoxifen use [Citation19]. NDT is further metabolized into endoxifen by the CYP2D6 enzyme and this enzyme also metabolizes tamoxifen to 4-OHT [Citation20]. Both of these secondary tamoxifen metabolites are approximately 100× more potent than tamoxifen [Citation20], and were found mostly in the BLQ range for all DARE-VVA1 dosing groups, suggesting minimal impact on systemic exposure. Our current PK findings are consistent with the initial proof-of-concept study, which showed median plasma tamoxifen concentrations of 5.8 ng/ml (range 1.0–10.0 ng/ml) measured after 2 months of use with the 13 mg intravaginal dose [Citation25].
Although the assessment of preliminary efficacy and PD were secondary endpoints, this study demonstrated that topical delivery of tamoxifen resulted in statistically significant decreases in vaginal pH with active product use (all active doses combined), with the 10 mg and 20 mg dosing groups showing the most improvement in vaginal pH over the 8-week treatment. We recognize that the median end of treatment vaginal pH levels (range 5.0–5.3) remained in what would be considered the menopausal range (≥5.0), which is higher than the vaginal pH levels normally achieved after 4 weeks of micro-dose E2 treatment (<5.0, premenopausal range) [Citation5]. This is likely due to the small overall sample size, particularly the fact that only six of the participants who completed the study used either the 10 mg (n = 3) or the 20 mg (n = 3) dose. Vaginal pH is likely the best marker of vaginal health [Citation24] and because plasma tamoxifen concentrations had a statistically significant inverse relationship with vaginal pH, we plan to move forward with the 10 mg and 20 mg doses.
Similarly, even with micro-dose vaginal therapy, the VMI showed overall improvement with active product use, although the increase in the overall VMI was not statistically significant. The largest increase in total VMI, reflecting a shift to healthy, superficial cells, occurred in the 20 mg DARE-VVA1 dosing group. There was a non-significant increase in superficial cells among active product users, again with the 20 mg dosing cohort experiencing the highest increase in healthy, differentiated superficial cells. However, all active product users had a significant decrease in parabasal cells (p = 0.04), with the 20 mg dosing cohort showing the largest decrease in this cell type, which is characteristic of a fragile, atrophic vaginal epithelium. We found that increasing plasma tamoxifen concentrations were significantly positively correlated with local increases in healthy superficial cells, intermediate cells and overall VMI score. Increasing plasma tamoxifen concentrations were also statistically and inversely correlated with decreasing proportions of parabasal cells. Thus, systemic PK/local PD correlations with VMI data also support forwarding the 10 mg and 20 mg dosing strengths for further testing.
At baseline, participants reported two genitourinary symptoms as being the most bothersome, vaginal dryness and dyspareunia. It is notable that the self-reported severity of both symptoms changed with active product use (p = 0.02 for both symptoms), while there was no statistically significant change in the severity of these two symptoms with placebo use. We recognize that this was a small sample size for this endpoint, but the Fisher exact test was used for these comparisons, which is not dependent on sample size.
This study was limited by the adverse impact of COVID-19 on participant recruitment and thus did not achieve the sample size which was estimated prior to study start. However, both primary endpoints of safety and PK were primarily descriptive in nature, and we found no discernible safety differences between active and placebo product, and the PK data were consistent, with the highest dosing group having the highest systemic tamoxifen and metabolite exposure. We will confirm the safety and efficacy data in future studies of larger sample sizes. We recognize that combining all active study doses for comparison of preliminary efficacy endpoints is not typical, but we did this due to the low sample size and the fact that these endpoints were secondary endpoints and were specified as preliminary efficacy assessments. The preliminary efficacy data complement the PK and safety data and serve to support our selection of the 10 mg and 20 mg doses for further evaluation. We did not document the presence or absence of systemic hypo-estrogenism symptoms (e.g. vasomotor symptoms, night sweats) among the participants at baseline. Although vasomotor symptoms are associated with oral tamoxifen use, we did not note this as a common TEAE among DARE-VVA1 users, but we acknowledge that any changes from baseline status could not be measured. Given the low systemic exposure, we anticipate that this will not be a common TEAE with the topical formulation.
In conclusion, this first-in-woman study of DARE-VVA1 in healthy postmenopausal women demonstrated that DARE-VVA1 was safe, resulted in minimal systemic tamoxifen or tamoxifen metabolite exposure and had TEAEs that were mostly localized to the vagina and were mild or moderate. Although the study was not powered to measure efficacy of the four individual dosing groups, the vaginal pH, vaginal cytology and subjective reports of the severity of MBS support that the 10 mg and 20 mg doses show promise for treating VVA and support continued study of these doses in women with moderate to severe VVA.
Potential conflict of interest
The authors report the following conflicts of interest: Dr Thurman, Dr Mauck, Dr Friend and Mrs Hatheway are current employees of Dare Bioscience. Mrs Zack was a former employee of Dare Bioscience. Dr Hull and Dr Stuckey received funding from the sponsor of the study, Dare Bioscience, for study-related activities.
The authors alone are responsible for the content and writing of the article.
Source of funding
Dare Bioscience sponsored the study.
Supplemental Material
Download MS Word (41.6 KB)References
- Portman DJ, Gass ML. Vulvovaginal atrophy terminology consensus conference P. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the international society for the study of women’s sexual health and the North American menopause society. Maturitas. 2014;79(3):349–354.
- Gandhi J, Chen A, Dagur G, et al. Genitourinary syndrome of menopause: an overview of clinical manifestations, pathophysiology, etiology, evaluation, and management. Am J Obstet Gynecol. 2016;215(6):704–711.
- Benini V, Ruffolo AF, Casiraghi A, et al. New innovations for the treatment of vulvovaginal atrophy: an up-to-Date review. Medicina. 2022;58(6):770.
- Santoro N, Komi J. Prevalence and impact of vaginal symptoms among postmenopausal women. J Sex Med. 2009;6(8):2133–2142.
- Suckling J, Lethaby A, Kennedy R. Local oestrogen for vaginal atrophy in postmenopausal women. Cochrane Database Syst Rev. 2006;8(4):CD001500.
- Biglia N, Bounous VE, D’Alonzo M, et al. Vaginal atrophy in breast cancer survivors: attitude and approaches among oncologists. Clin Breast Cancer. 2017;17(8):611–617.
- Duchesnay. Approved Labeling Osphena Tablets. https://wwwaccessdatafdagov/drugsatfda_docs/label/2019/203505s015lblpdf. 2019.
- Pinkerton JV, Stanczyk FZ. Clinical effects of selective estrogen receptor modulators on vulvar and vaginal atrophy. Menopause. 2014;21(3):309–319.
- Ferrazzi E, Cartei G, Mattarazzo R, et al. Oestrogen-like effect of tamoxifen on vaginal epithelium. Br Med J. 1977;1(6072):1351–1352.
- Boccardo F, Bruzzi P, Rubagotti A, et al. Estrogen-like action of tamoxifen on vaginal epithelium in breast cancer patients. Oncology. 1981;38(5):281–285.
- Miodrag A, Ekelund P, Burton R, et al. Tamoxifen and partial oestrogen agonism in postmenopausal women. Age Ageing. 1991;20(1):52–54.
- Lahti E, Vuopala S, Kauppila A, et al. Maturation of vaginal and endometrial epithelium in postmenopausal breast cancer patients receiving long-term tamoxifen. Gynecol Oncol. 1994;55(3 Pt 1):410–414.
- Love RR, Kurtycz DF, Dumesic DA, et al. The effects of tamoxifen on the vaginal epithelium in postmenopausal women. J Womens Health Gend Based Med. 2000;9(5):559–563.
- Shiota A, Igarashi T, Kurose T, et al. Reciprocal effects of tamoxifen on hormonal cytology in postmenopausal women. Acta Cytol. 2002;46(3):499–506.
- Vardy MD, Lindsay R, Scotti RJ, et al. Short-term urogenital effects of raloxifene, tamoxifen, and estrogen. Am J Obstet Gynecol. 2003;189(1):81–88.
- Wardell SE, Nelson ER, McDonnell DP. From empirical to mechanism-based discovery of clinically useful selective estrogen receptor modulators (SERMs). Steroids. 2014;90:30–38.
- Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2(3):205–213.
- Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for the prevention of breast cancer: current status of the national surgical adjuvant breast and bowel project P-1 study. J Natl Cancer Inst. 2005;97(22):1652–1662.
- Astra Zenaca. Approved Labeling for Nolvadex (Tamoxifen Citrate Tablets) NDA 017970. https://wwwaccessdatafdagov/drugsatfda_docs/label/2005/17970s053lblpdf. 2009.
- Jordan VC. New insights into the metabolism of tamoxifen and its role in the treatment and prevention of breast cancer. Steroids. 2007;72(13):829–842.
- Fan EM, Zimmern PE. Challenges of managing lower urinary tract symptoms in women with tamoxifen use. Womens Health Rep. 2022;3(1):430–436.
- Kallak TK, Baumgart J, Goransson E, et al. Aromatase inhibitors affect vaginal proliferation and steroid hormone receptors. Menopause. 2014;21(4):383–390.
- Faubion SS, Larkin LC, Stuenkel CA, et al. Management of genitourinary syndrome of menopause in women with or at high risk for breast cancer: consensus recommendations from the North American menopause society and the international society for the study of women’s sexual health. Menopause. 2018;25(6):596–608.
- Nappi RE, Kokot-Kierepa M. Vaginal health: insights, views & attitudes (VIVA) – results from an international survey. Climacteric. 2012;15(1):36–44.
- Chollet J, Mermelstein F, Rocamboli SC, et al. Vaginal tamoxifen for treatment of vulvar and vaginal atrophy: pharmacokinetics and local tolerance in a rabbit model over 28 days. Int J Pharm. 2019;570:118691.
- ACOG committee opinion no. 601: tamoxifen and uterine cancer. Obstet Gynecol. 2014;123(6):1394–1397.
- Wickerham DL, Costantino JP, Vogel VG, et al. The use of tamoxifen and raloxifene for the prevention of breast cancer. Recent Results Cancer Res. 2009;181:113–119.
- Wickerham DL, O’Connell MJ, Costantino JP, et al. The half century of clinical trials of the national surgical adjuvant breast and bowel project. Semin Oncol. 2008;35(5):522–529.